Нубека
УкраинаСодержание
ИНСТРУКЦИЯ ПО ПРИМЕНЕНИЮ ЛЕКАРСТВЕННОГО СРЕДСТВА НУБЕКА (NUBEQA)
Состав:
действующее вещество: даролутамид;
1 таблетка, покрытая пленочной оболочкой, содержит 300 мг даролутамида;
вспомогательные вещества: кальция гидрофосфат, натрия кроскармеллоза, лактозы моногидрат, магния стеарат, повидон К 30; пленочная оболочка: лак белый (гипромеллоза 15 cP, лактозы моногидрат, макрогол 3350, диоксид титана (Е 171)).
Лекарственная форма. Таблетки, покрытые пленочной оболочкой.
Основные физико-химические свойства: овальные таблетки, покрытые пленочной оболочкой, белого или почти белого цвета, с маркировкой «BAYER» с одной стороны и «300» — с другой.
Фармакотерапевтическая группа. Средства, применяемые для гормональной терапии. Антагонисты гормонов и родственные средства. Антиандрогенные средства. Даролутамид.
Код АТХ L02B B06.
Фармакологические свойства
Фармакодинамика
Механизм действия. Даролутамид является ингибитором андрогенного рецептора (АР) с гибкой полярно замещённой структурой пиразола, который связывается с высокой аффинностью непосредственно с лиганд-связывающим доменом рецептора.
Даролутамид конкурентно подавляет связывание андрогенов, ядерную транслокацию АР и опосредованную АР транскрипцию. Основной его метаболит — кето-даролутамид in vitro продемонстрировал активность, схожую с активностью даролутамида. Лечение даролутамидом уменьшает пролиферацию клеток опухоли предстательной железы, что приводит к выраженной противоопухолевой активности.
Фармакодинамические эффекты. Не наблюдалось удлинение среднего интервала QTcF (т.е. более 10 мс) после приёма внутрь 600 мг даролутамида дважды в день по сравнению с соответствующим показателем при применении плацебо.
Клиническая эффективность и безопасность. Эффективность и безопасность были установлены в двух рандомизированных плацебо-контролируемых многоцентровых исследованиях фазы III с участием пациентов с неметастатическим кастрационно-резистентным раком предстательной железы (нмКРРПЖ) (исследование ARAMIS) и метастатическим гормоночувствительным раком предстательной железы (мГЧРПЖ) (исследование ARASENS). Все пациенты одновременно получали аналог лютеинизирующего гормона рилизинг-гормона (ЛГРГ) или имели двустороннюю орхиэктомию.
Неметастатический кастрационно-резистентный рак предстательной железы (нмКРРПЖ)
Эффективность и безопасность применения даролутамида оценивали в рандомизированном двойном слепом плацебо-контролируемом многоцентровом исследовании фазы III (ARAMIS) у пациентов с неметастатическим (по данным стандартных методов КТ, сцинтиграфии костей, МРТ) кастрационно-резистентным раком предстательной железы со временем удвоения простат-специфического антигена (ЧУПСА) ≤ 10 месяцев.
Пациенты включались в исследование, если у них было зафиксировано 3 повышения уровня простат-специфического антигена (ПСА) после достижения минимального уровня с интервалом не менее одной недели во время андрогенной депривационной терапии, уровень ПСА ≥ 2 нг/мл на момент скрининга и уровень посткастрационного тестостерона в сыворотке крови < 1,7 нмоль/л.
Пациенты с анамнезом судорог были допущены к участию в исследовании. В группе даролутамида было зарегистрировано 12 пациентов (0,21 %), имевших в анамнезе судороги.
Пациенты с неконтролируемой гипертензией или недавним (в течение последних 6 месяцев) инсультом, инфарктом миокарда, тяжёлой/нестабильной стенокардией, шунтированием коронарной/периферической артерии, застойной сердечной недостаточностью III или IV класса по классификации Нью-Йоркской ассоциации сердца были исключены из исследования.
Пациенты, ранее получавшие ингибиторы андрогенного рецептора (АР) второго поколения, такие как энзалутамид, апалутамид и даролутамид, или ингибиторы фермента CYP17, такие как абиратерона ацетат, а также пациенты, получавшие системные кортикостероиды в дозе, превышающей эквивалент 10 мг преднизона в день в течение 28 дней до рандомизации, были исключены из исследования.
Всего 1509 пациентов были рандомизированы в соотношении 2:1 для получения 600 мг даролутамида перорально дважды в день (n = 955) или плацебо (n = 554).
Пациенты с наличием тазовых лимфатических узлов < 2 см по короткой оси ниже бифуркации аорты были включены в исследование. Отсутствие или наличие метастазов устанавливалось с помощью независимой централизованной лучевой оценки. Такая оценка была проведена у 89 пациентов, у которых ретроспективно были выявлены метастазы на момент начала исследования. Рандомизация была стратифицирована по ЧУПСА (≤ 6 месяцев или > 6 месяцев) и по применению таргетной терапии, направленной на остеокласты, на момент начала исследования (да или нет).
Демографические характеристики пациентов и характеристики заболевания были сбалансированы между группами лечения. Средний возраст пациентов составлял 74 года (диапазон 48–95), 9 % пациентов были в возрасте 85 лет и старше; 79 % пациентов составляли представители европеоидной расы, 13 % — азиатского происхождения и 3 % — с тёмным цветом кожи. Большинство пациентов имели 7 баллов и выше по шкале Глисона на момент постановки диагноза (73 %). Медиана ЧУПСА составляла 4,5 месяца. Девять процентов (9 %) пациентов имели предшествующую орхиэктомию, 25 % — предшествующую простатэктомию, а 50 % — по крайней мере одну предшествующую лучевую терапию. Семьдесят шесть процентов (76 %) пациентов получали более одного предшествующего антигормонального лечения. Пациенты имели 0 баллов (69 %) или 1 балл (31 %) по шкале эффективности Восточной онкологической кооперативной группы (ECOG PS).
Лечение даролутамидом продолжалось до прогрессирования заболевания по данным визуализации (КТ, сцинтиграфия костей, МРТ), оцениваемого с помощью слепой централизованной оценки, неприемлемой токсичности или отказа пациента от продолжения участия в исследовании.
Первичной конечной точкой эффективности было выживание без метастазов (ВБМ). Вторичными конечными точками были общая выживаемость (ОВ), время до прогрессирования боли, время до начала первой цитотоксической химиотерапии рака предстательной железы и время до первых симптоматических поражений скелета (определяется как появление любого из следующего: наружная лучевая терапия для облегчения скелетных симптомов, новый симптоматический патологический перелом костей, компрессия спинного мозга или связанное с опухолью ортопедическое хирургическое вмешательство).
Лечение даролутамидом привело к улучшению ВБМ по сравнению с показателем при применении плацебо (см. таблицу 1 и рис. 1).
Показатели ВБМ были сопоставимы во всех подгруппах пациентов независимо от ЧУПСА, предыдущего применения агентов, влияющих на костную ткань, или локально-регионального заболевания. Дополнительные подгруппы с согласованными результатами ВБМ включали уровень ПСА на начало исследования, балл по шкале Глисона на момент постановки диагноза, возраст пациента, географический регион, балл по ECOG PS на начало исследования, расовую принадлежность и количество предшествующих курсов гормональной терапии.
После первичного анализа ВБМ, как только исследование было раскрыто, пациентам, получавшим плацебо, было предложено лечение даролутамидом (перекрёстный вариант). Из 554 пациентов, рандомизированных в группу плацебо, 170 (31 %) перешли на лечение даролутамидом. Анализ общей выживаемости не был скорректирован с учётом влияния перекрёстного перехода.
На момент окончательного анализа лечение даролутамидом привело к статистически значимому улучшению показателя общей выживаемости по сравнению с показателем при применении плацебо (медиана не была достигнута ни в одной из групп, см. таблицу 1 и рис. 2). Также наблюдалось статистически значимое увеличение продолжительности времени до прогрессирования боли, до начала первой цитотоксической химиотерапии и до первой симптоматической скелетной события по сравнению с соответствующими показателями при применении плацебо (см. таблицу 1).
На момент окончательного анализа средняя продолжительность лечения пациентов, получавших даролутамид, составляла 33,3 месяца (диапазон от 0,0 до 74,0 месяца) в течение комбинированного двойного слепого и открытого периода.
Все анализы проводились в полном наборе исследований.
Таблица 1. Результаты эффективности по данным исследования ARAMIS
| Параметр эффективности |
Количество (%) пациентов с событиями |
Медиана (месяцы) (95 % ДИ) |
Отношение рисков (ОР)b (95 % доверительный интервал [ДИ]) p‑значение (двустороннее) |
||
| Даролутамид (N = 955) |
Плацебоa (N = 554) |
Даролутамид (N = 955) |
Плацебоa (N = 554) |
||
| Выживание без метастазовc |
221 (23,1 %) |
216 (39,0 %) |
40,4 (34,3; НД) |
18,4 (15,5; 22,3) |
0,413 (0,341; 0,500) < 0,000001 |
| Общее выживание |
148 (15,5 %) |
106 (19,1 %) |
НД (56,1; НД) |
НД (46,9; НД) |
0,685 (0,533; 0,881) 0,003048 |
| Время до прогрессирования болиc, d |
251 (26,3 %) |
178 (32,1 %) |
40,3 (33,2; 41,2) |
25,4 (19,1; 29,6) |
0,647 (0,533; 0,785) 0,000008 |
| Время до начала первой цитотоксической химиотерапии |
127 (13,3 %) |
98 (17,7 %) |
НД (НД, НД) |
НД |
0,579 (0,444; 0,755) 0,000044 |
| Время до первой симптоматической скелетной события |
29 (3,0 %) |
28 (5,1 %) |
НД (НД, НД) |
(НД, НД) |
0,484 (0,287; 0,815) 0,005294 |
а В том числе 170 пациентов, которые перешли на даролутамид без периода ослепления.
b Отношение риска < 1 в отношении даролутамида.
c Для ВБМ и времени до прогрессирования боли первичный анализ считается окончательным анализом.
d Результаты, сообщаемые пациентом, по оценке опросника «Краткое описание боли — Краткая форма опросника».
НД — не достигнуто.
Результатом лечения даролутамидом было более длительное выживание без прогрессирования (ВБП, медиана 36,8 против 14,8 месяца, ОР = 0,380, номинальное p < 0,000001) и время до прогрессирования ПСА (медиана 29,5 против 7,2 месяца, ОР = 0,164, номинальное p < 0,000001). Стойкость эффекта наблюдалась по всем показателям выживания (ВБМ, ОВ и ПСА).
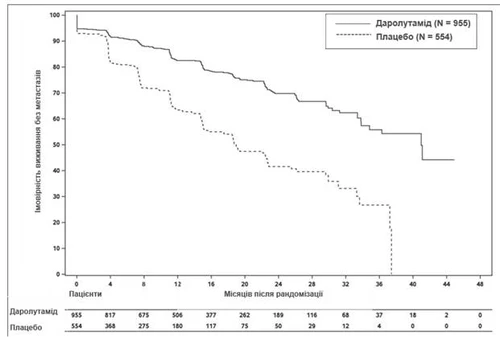
Рис. 1. Кривые Каплана – Мейера выживания без метастазов (ARAMIS)
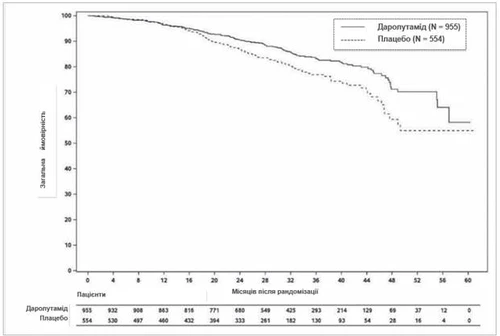
Рис. 2. Кривые Каплана – Мейера общего выживания (ARAMIS)
Пациенты, получавшие даролутамид в исследовании ARAMIS (период двойного слепого), продемонстрировали значительно более высокую подтвержденную частоту ответа по уровню ПСА (определяемая как снижение на ≥ 50 % от исходного уровня) по сравнению с пациентами, получавшими плацебо: 84,0 % против 7,9 % (разница = 76,1 %, p < 0,000001 (номинальное значение p, только для информации)).
Метастатический гормончувствительный рак предстательной железы (мГЧРПЖ)
Эффективность и безопасность применения даролутамида в комбинации с доцетакселом оценивали в многоцентровом двойном слепом плацебо-контролируемом исследовании III фазы (ARASENS) у пациентов с мГЧРПЖ. Всего 1306 пациентов были рандомизированы 1:1 для получения 600 мг даролутамида перорально два раза в день (n = 651) или плацебо (n = 655) одновременно с 75 мг/м² доцетаксела в течение 6 циклов. Лечение даролутамидом или плацебо продолжалось до прогрессирования симптомов заболевания, изменения противоопухолевой терапии, неприемлемой токсичности, смерти или выбытия пациента из исследования.
Наличие метастазов оценивали с помощью независимой централизованной радиологической оценки. Пациенты с поражением только регионарных лимфатических узлов (M0) были исключены из исследования. Рандомизация была стратифицирована по стадии заболевания (только метастазы в нерегионарные лимфатические узлы (M1a), метастазы в кости с метастазами в лимфатические узлы или без них (M1b) или висцеральные метастазы с метастазами в лимфатические узлы или без них, или с метастазами в кости или без них (M1c)) и по уровню щелочной фосфатазы (< или ≥ верхней границы нормы) на начало исследования. Пациентов с метастазами в головной мозг разрешалось включать в исследование, однако таких пациентов не было.
Демографические характеристики пациентов и характеристики заболевания были сбалансированы между группами лечения. Средний возраст составлял 67 лет (диапазон 41–89), 0,5 % пациентов были в возрасте от 85 лет, 52 % — европеоидной расы, 36 % — азиатского происхождения и 4 % участников исследования имели темный цвет кожи. Большинство пациентов имели оценку по шкале Глисона 8 или более баллов на момент постановки диагноза (78 %). 71 % пациентов имели оценку ECOG PS 0 баллов, а 29 % — 1 балл. Было 86,1 % пациентов с заболеванием de novo и 12,9 % пациентов с рецидивом. На начало исследования 3 % пациентов имели M1a, 79,5 % имели M1b и 17,5 % имели M1c; щелочная фосфатаза была < ВГН у 44,5 % пациентов и ≥ ВГН у 55,5 % пациентов; средний исходный уровень ПСА составлял 30,3 мкг/л и 24,2 мкг/л в группах даролутамида и плацебо соответственно. Пациенты с анамнезом судорог были допущены к участию в исследовании, и 4 пациента (0,6 %) были включены в группу даролутамид + доцетаксел.
77,0 % пациентов имели заболевание большого объема, а 23,0 % — заболевания малого объема. Заболевание большого объема определялось как наличие висцеральных метастазов или 4 или более поражений костей, по крайней мере, 1 метастаз за пределами позвоночника и тазовых костей. Примерно 25 % пациентов получали сопутствующую терапию бисфосфонатами или деносумабом.
Основной конечной точкой эффективности было общее выживание (ОВ). Вторичными конечными точками были время до развития кастрационно-резистентного рака предстательной железы, время до прогрессирования боли, время выживания без симптомов скелетного поражения, время до первой симптоматической скелетной события, время до начала следующей противоопухолевой терапии, время до ухудшения физических симптомов, связанных с заболеванием, и время до начала применения опиоидов в течение ≥ 7 дней подряд. Прогрессирование боли оценивали по сообщаемым пациентом результатам по опроснику «Краткое описание боли — Краткая форма», что определялось как ухудшение как минимум на 2 балла от исходного уровня и начало приема опиоидов короткого или длительного действия для купирования боли в течение ≥ 7 дней подряд.
Средняя продолжительность лечения составляла 41,0 месяца (диапазон от 0,1 до 56,5 месяца) у пациентов, получавших даролутамид + доцетаксел, и 16,7 месяца (диапазон от 0,3 до 55,8 месяца) у пациентов, получавших плацебо + доцетаксел. 87,6 % и 85,5 % пациентов получили полные 6 циклов доцетаксела, а 1,5 % и 2,0 % пациентов не получали доцетаксел в группе даролутамид + доцетаксел и плацебо + доцетаксел соответственно.
Таблица 2. Результаты эффективности исследования ARASENS
| Параметр эффективности |
Количество (%) пациентов с событиями |
Медиана (месяцы) (95 % ДИ) |
Отношение рисков (ОР)b (95 % доверительный интервал [ДИ]) р-значение (одностороннее)c |
||
| Даролутамид + доцетаксель (N = 651) |
Плацебо + доцетаксель (N = 654)а |
Даролутамид + доцетаксель (N = 651) |
Плацебо + доцетаксель (N = 654)а |
||
| Общая выживаемостьd |
229 (35,2%) |
304 (46,5%) |
НД (НД, НД) |
48,9 (44,4; НД) |
0,675 (0,568; 0,801) <0,0001 |
а Один пациент в группе плацебо был исключён из всех анализов.
b Отношение рисков < 1 в отношении даролутамида.
c На основании стратифицированного логарифмического рангового теста.
d Результаты ОВ были одинаковыми во всех подгруппах пациентов, включая степень заболевания и уровни щелочной фосфатазы.
НД – не достигнуто.
Вторичные конечные точки эффективности, по которым показано статистически значимое преимущество пациентов группы даролутамид + доцетаксел по сравнению с пациентами группы плацебо + доцетаксел: время до развития кастрационно-резистентного рака предстательной железы (медиана НД против 19,1 месяца; ОР = 0,357, p < 0,0001); время до первой симптоматической скелетной события (медиана НД против НД; ОР = 0,712, p = 0,0081); время до начала следующей противоопухолевой химиотерапии (медиана НД против 25,3 месяца; ОР = 0,388, p < 0,0001); время до прогрессирования боли (медиана НД против 27,5 месяца; ОР = 0,792, p = 0,0058); время выживания без симптоматических скелетных событий (медиана 51,2 против 39,7 месяца; ОР = 0,609, p < 0,0001).
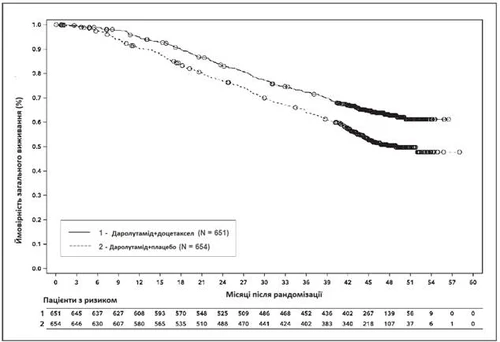
Рис. 3. Кривые общего выживания Каплана – Мейера (ARASENS)a
а ОВ через 36 месяцев составляла 72,3 % (95 % ДИ, 68,8–75,8) в группе даролутамид + доцетаксел против 63,8 % (95 % ДИ, 60,1–67,6) в группе плацебо + доцетаксел.
Частота ОВ через 48 месяцев составляла 62,7 % (95 % ДИ, 58,7–66,7) в группе даролутамид + доцетаксел против 50,4 % (95 % ДИ, 46,3–54,6) в группе плацебо + доцетаксел.
Фармакокинетика
Даролутамид состоит из двух диастереомеров [(S, R)-даролутамид и (S, S)-даролутамид], которые взаимопревращаются через основной циркулирующий метаболит — кето-даролутамид. In vitro все три вещества демонстрируют одинаковую фармакологическую активность. Даролутамид плохо растворяется в водных растворителях в широком диапазоне рН и, как правило, лучше растворяется в органических растворителях.
Всасывание
После перорального приёма 600 мг (2 таблетки по 300 мг) дважды в сутки пиковые концентрации даролутамида в плазме в равновесном состоянии составляли 4,79 мг/л (коэффициент вариации 30,9 %) у пациентов с нмКРРПЖ в исследовании ARAMIS и 3,84 мг/л (коэффициент вариации 35,6 %) у пациентов с мГЧРПЖ в исследовании ARASENS. Среднее время достижения максимальной концентрации в плазме составляло 3–4 часа. Соотношение двух диастереомеров, (S, R)-даролутамида к (S, S)-даролутамиду, изменилось с 1:1 в таблетке до приблизительно 1:9 в плазме на основании показателя AUC0–12 в равновесном состоянии. После перорального применения вместе с пищей равновесное состояние достигается через 2–5 дней повторного приёма дважды в день.
Абсолютная биодоступность после перорального приёма натощак таблетки Нубека, содержащей 300 мг даролутамида, составляет приблизительно 30 % от таковой при внутривенной инъекции.
Биодоступность даролутамида повышалась в 2,0–2,5 раза при приёме с пищей. Подобное увеличение экспозиции наблюдалось для основного метаболита — кето-даролутамида.
Распределение
Видимый объём распределения даролутамида после внутривенного введения составляет 119 л, что указывает на широкое распределение даролутамида в организме как во внутриклеточном, так и во внеклеточном жидкостном пространстве.
Даролутамид умеренно (92 %) связывается с белками плазмы человека, при этом различий между двумя диастереомерами не наблюдается. Основной метаболит даролутамида — кето-даролутамид — в значительной степени (99,8 %) связывается с белками плазмы.
Прохождение даролутамида через гематоэнцефалический барьер клинически не изучалось. Однако влияние даролутамида на головной мозг с точки зрения AUC0–24 является очень низким: 4,5 % экспозиции в плазме после однократной дозы у крыс и 1,9–3,9 % после повторной дозы у мышей. Это указывает на низкое проникновение даролутамида через интактный гематоэнцефалический барьер у крыс и мышей и на низкую вероятность того, что даролутамид пересекает интактный гематоэнцефалический барьер у людей в клинически значимой степени.
Биотрансформация
Диастереомеры (S, R)-даролутамид и (S, S)-даролутамид способны взаимопревращаться с помощью метаболита кето-даролутамида с преимуществом (S, S)-даролутамида.
После однократного перорального приёма 300 мг 14C-даролутамида в виде перорального раствора кето-даролутамид является единственным основным метаболитом с приблизительно в 2 раза более высокой общей экспозицией в плазме по сравнению с даролутамидом. На даролутамид и кето-даролутамид вместе приходится 87,4 % 14C-радиоактивности в плазме, что указывает на то, что все остальные метаболиты являются незначительными.
Даролутамид метаболизируется главным образом путём окислительного метаболизма, опосредованного преимущественно CYP3A4, а также путём прямой глюкуронизации, опосредованной преимущественно UGT1A9 и UGT1A1. Кроме того, было показано, что преимущественно изоформы AKR1C катализируют восстановление кето-даролутамида до диастереомеров вещества.
Выведение
Эффективный период полувыведения даролутамида и кето-даролутамида в плазме крови пациентов составляет приблизительно 18–20 часов. Из двух диастереомеров, входящих в состав даролутамида, (S, R)-даролутамид имеет более короткий эффективный период полураспада — 9 часов по сравнению с (S, S)-даролутамидом, эффективный период полураспада которого составляет 22 часа. Клиренс даролутамида после внутривенного введения составлял 116 мл/мин (CV 39,7 %). В целом 63,4 % даролутамида и его метаболитов выводится с мочой (приблизительно 7 % в неизменённом виде), 32,4 % выводится с калом. Более 95 % дозы было выведено в течение 7 дней после приёма.
Линейность/нелинейность
В диапазоне доз от 100 до 700 мг (после однократной дозы и в равновесном состоянии) экспозиция двух диастереомеров и основного метаболита кето-даролутамида возрастает линейно почти пропорционально дозе. В связи с насыщением всасывания дальнейшего увеличения экспозиции даролутамида при дозе 900 мг дважды в день не наблюдалось.
Особые группы пациентов
Пациенты пожилого возраста. Клинически значимых различий в фармакокинетике даролутамида не наблюдалось (65–95 лет).
Нарушение функции почек. В клиническом фармакокинетическом исследовании AUC и Cmax даролутамида были в 2,5 и 1,6 раза выше у пациентов с тяжёлой почечной недостаточностью (рассчитанная скорость клубочковой фильтрации [рСКФ] от 15 до 29 мл/мин/1,73 м²) по сравнению со здоровыми добровольцами.
Популяционный фармакокинетический анализ указывает на 1,1-, 1,3- и приблизительно 1,5-кратно более высокую экспозицию (AUC) даролутамида у пациентов с лёгкой, умеренной и тяжёлой степенью нарушения функции почек (рСКФ от 15 до 89 мл/мин/1,73 м²) по сравнению с пациентами с нормальной функцией почек.
Фармакокинетика даролутамида не изучалась у пациентов с терминальной стадией почечной недостаточности, находящихся на диализе (рСКФ < 15 мл/мин/1,73 м²).
Нарушение функции печени. В клиническом фармакокинетическом исследовании Cmax и AUC даролутамида были в 1,5 и 1,9 раза выше у пациентов с умеренной печеночной недостаточностью (класс B по классификации Child–Pugh) по сравнению со здоровыми добровольцами. Отсутствуют данные по пациентам с тяжёлой печеночной недостаточностью (класс С по классификации Child–Pugh).
Этнические различия. Не наблюдалось никаких клинически значимых различий в фармакокинетике даролутамида в зависимости от этнической принадлежности пациента (пациенты европеоидной расы, пациенты японского происхождения, пациенты азиатского, но не японского происхождения, пациенты с тёмным цветом кожи или афроамериканского происхождения). Популяционный фармакокинетический анализ показал увеличение среднего геометрического влияния (AUC) до 1,56 раза (90 % ДИ 1,43–1,70) у пациентов японского происхождения по сравнению с пациентами из других регионов в исследованиях ARAMIS и ARASENS.
Доклинические данные по безопасности
Системная токсичность. В исследованиях токсичности повторных доз на животных основными находками были изменения в репродуктивных органах самцов (уменьшение массы органа с атрофией предстательной железы и придатков семенников). Эти эффекты возникали при системном воздействии в диапазоне ожидаемого воздействия на человека или ниже (на основании сравнения AUC). Дополнительные изменения в репродуктивных тканях включали минимальное увеличение вакуолизации гипофиза, атрофию и снижение секреторной активности семенных пузырьков и молочных желёз у крыс, а также гипоспермию яичек, расширение семявыносящих канальцев и дегенерацию у собак. Изменения в мужских репродуктивных органах у обоих видов соответствовали фармакологической активности даролутамида и исчезали или частично исчезали после 4–8-недельного периода восстановления.
Эмбриотоксичность/тератогенность. Исследования токсического влияния на плод не проводились.
Репродуктивная токсичность. Исследования репродуктивной токсичности не проводились. Однако фертильность самцов, вероятно, будет нарушена с учётом результатов исследований токсичности повторных доз у животных, которые согласуются с фармакологической активностью даролутамида.
Генотоксичность и канцерогенность. Даролутамид не индуцировал мутации в анализе микробного мутагенеза (Ames). При высоких концентрациях даролутамид индуцировал структурные хромосомные аберрации in vitro в культивируемых лимфоцитах человека. Однако в комбинированном микроядерном тесте костного мозга in vivo и анализе Comet в печени и двенадцатиперстной кишке крысы генотоксичность не наблюдалась при экспозициях, превышающих максимальную экспозицию для человека.
Пероральное введение даролутамида самцам трансгенных мышей rasH2 в течение 6 месяцев не выявило канцерогенного потенциала при применении в дозах до 1000 мг/кг/сут, что превышает клиническую экспозицию (AUC) даролутамида в 0,9–1,3 раза и кето-даролутамида в 2,1–2,3 раза при применении в рекомендованной клинической суточной дозе 1200 мг/сут. На основании этого исследования нельзя полностью исключить канцерогенный риск при применении даролутамида.
Клинические характеристики
Показания
Лекарственное средство Нубека показано для лечения у взрослых мужчин:
- неметастатического кастрационно-резистентного рака предстательной железы (нмКРРПЖ) с высоким риском метастазирования (см. раздел «Фармакологические свойства»);
- метастатического гормончувствительного рака предстательной железы (мГЧРПЖ) — в комбинации с доцетакселом и андрогендепривационной терапией (см. раздел «Фармакологические свойства»).
Противопоказания
Повышенная чувствительность к действующему веществу или вспомогательным компонентам.
Противопоказан беременным и женщинам детородного возраста.
Взаимодействие с другими лекарственными средствами и другие виды взаимодействий
Влияние других лекарственных средств на даролутамид
Индукторы CYP3A4 и P-gp
Даролутамид является субстратом CYP3A4 и Р-гликопротеина (P-gp).
Использование сильных и умеренных индукторов CYP3A4 и индукторов P-gp (например, карбамазепина, фенобарбитала, зверобоя, фенитоина и рифампицина) во время лечения даролутамидом не рекомендуется, за исключением случаев, когда нет терапевтической альтернативы. Следует рассмотреть возможность выбора альтернативного сопутствующего лекарственного средства, которое не индуцирует или имеет слабый потенциал индукции CYP3A4 или P-gp.
Одновременный приём рифампицина (600 мг), мощного индуктора CYP3A4 и P-gp, с однократной дозой даролутамида (600 мг) во время еды приводил к снижению средней экспозиции (AUC0-72) на 72 % и снижению Cmax даролутамида на 52 %.
Ингибиторы CYP3A4, P-gp и BCRP
Даролутамид является субстратом CYP3A4, P-gp и белка резистентности рака молочной железы (БРРМЖ).
При применении ингибиторов CYP3A4, P-gp или БРРМЖ клинически значимого взаимодействия между препаратами не ожидается. Даролутамид можно назначать одновременно с ингибиторами CYP3A4, P-gp или БРРМЖ. Одновременное применение даролутамида с комбинированным ингибитором P-gp и мощным ингибитором CYP3A4 увеличивает экспозицию даролутамида, что может повысить риск побочных реакций на даролутамид. Рекомендуется чаще контролировать состояние пациента на предмет появления побочных реакций на даролутамид и при необходимости корректировать дозу даролутамида.
Применение итраконазола (200 мг дважды в день в 1-й день и один раз в день в течение последующих 7 дней), мощного ингибитора CYP3A4, P-gp и БРРМЖ, с однократной дозой даролутамида (600 мг на 5-й день вместе с едой) привело к 1,7-кратному увеличению средней экспозиции (AUC0-72) и 1,4-кратному увеличению Cmax даролутамида.
Ингибиторы UGT1A9
Даролутамид является субстратом UGT1A9. При применении ингибитора UGT1A9 клинически значимого взаимодействия между лекарственными средствами не ожидается. Даролутамид можно назначать одновременно с ингибиторами UGT1A9.
Популяционный фармакокинетический анализ показал, что одновременное применение ингибиторов UGT1A9 с даролутамидом приводило к 1,2-кратному увеличению экспозиции (AUC0-72) даролутамида.
Доцетаксел
Применение даролутамида в комбинации с доцетакселом не приводило к клинически значимым изменениям фармакокинетики даролутамида у пациентов с мГЧРПЖ (см. раздел «Фармакологические свойства»).
Влияние даролутамида на другие лекарственные средства
Субстраты БРРМЖ, OATP1B1 и OATP1B3
Даролутамид является ингибитором белка резистентности рака молочной железы и полипептидов, транспортирующих органические анионы (OATP) 1B1 и 1B3.
Следует избегать одновременного применения розувастатина, за исключением случаев, когда нет терапевтической альтернативы. Следует рассмотреть возможность выбора альтернативного сопутствующего лекарственного средства с меньшим потенциалом ингибирования БРРМЖ, OATP1B1 и OATP1B3.
Применение даролутамида (600 мг дважды в день в течение 5 дней) перед однократным приёмом розувастатина (5 мг) вместе с едой привело к приблизительно 5-кратному увеличению средней экспозиции (AUC) и Cmax розувастатина.
Если возможно, следует избегать одновременного применения даролутамида с другими субстратами БРРМЖ. Одновременное применение даролутамида может повысить концентрацию других сопутствующих препаратов — субстратов БРРМЖ, OATP1B1 и OATP1B3 (таких как метотрексат, сульфасалазин, флувастатин, аторвастатин, питавастатин) в плазме крови. Поэтому рекомендуется наблюдать за состоянием пациентов на предмет возникновения побочных реакций при применении субстратов БРРМЖ, OATP1B1 и OATP1B3. Кроме того, при одновременном применении с даролутамидом следует соблюдать соответствующие рекомендации, изложенные в информации об этих субстратах.
Субстраты P-gp
При введении субстрата P-gp клинически значимого взаимодействия между лекарственными средствами не ожидается. Даролутамид можно назначать одновременно с субстратами P-gp (например, с дигоксином, верапамилом или нифедипином). Одновременное применение даролутамида с чувствительным субстратом P-gp дабигатраном этексилатом не привело к повышению экспозиции (AUC и Cmax) дабигатрана.
Субстраты CYP3A4
Даролутамид является слабым индуктором CYP3A4.
При введении субстрата CYP клинически значимого взаимодействия между лекарственными средствами не ожидается. Даролутамид можно назначать одновременно с субстратами CYP (например, с варфарином, L-тироксином, омепразолом).
Применение даролутамида (600 мг дважды в день в течение 9 дней) перед однократным приёмом однократной дозы чувствительного субстрата CYP3A4 мидазолама (1 мг) вместе с едой привело к снижению средней экспозиции (AUC) и Сmax мидазолама на 29 % и 32 % соответственно. Даролутамид in vitro в клинически значимых концентрациях не подавлял метаболизм отдельных субстратов CYP.
Доцетаксел
Применение даролутамида в комбинации с доцетакселом не приводило к клинически значимым изменениям фармакокинетики доцетаксела у пациентов с мГЧРПЖ (см. раздел «Фармакологические свойства»).
Лекарственные средства, удлиняющие интервал QT
Поскольку андрогендепривационная терапия может удлинять интервал QT, следует тщательно оценивать одновременное применение с лекарственными средствами, которые, как известно, удлиняют интервал QT, или с лекарственными средствами, которые могут индуцировать torsade de pointes. К ним относятся такие препараты, как антиаритмические средства класса IA (например, хинидин, дизопирамид) или класса III (например, амиодарон, соталол, дофетилид, ибутилид), метадон, моксифлоксацин и антипсихотические средства (например, галоперидол).
Особенности применения
Нарушение функции почек
Данные по применению у пациентов с тяжелой почечной недостаточностью ограничены.
Поскольку экспозиция может быть повышена, за состоянием таких пациентов следует тщательно наблюдать в отношении возникновения побочных реакций (см. раздел «Особенности применения» и подраздел «Фармакокинетика»).
Нарушение функции печени
Данные по применению у пациентов с умеренным нарушением функции печени ограничены. Применение даролутамида у пациентов с тяжелым нарушением функции печени не изучалось.
Поскольку экспозиция может быть повышена, за состоянием пациентов следует тщательно наблюдать в отношении побочных реакций (см. раздел «Особенности применения» и подраздел «Фармакокинетика»).
Недавнее сердечно-сосудистое заболевание
Пациенты с клинически значимыми сердечно-сосудистыми заболеваниями в течение последних 6 месяцев, включая инсульт, инфаркт миокарда, тяжелую/нестабильную стенокардию, шунтирование коронарной/периферической артерии и симптоматическую сердечную недостаточность, были исключены из клинических исследований. Таким образом, безопасность применения даролутамида у таких пациентов не установлена.
При назначении лекарственного средства Нубека пациенту с клинически значимым сердечно-сосудистым заболеванием лечение этого заболевания следует проводить в соответствии с установленными рекомендациями.
Гепатотоксичность
При выявлении отклонений показателей функции печени, указывающих на идиосинкратическое поражение печени, вызванное применением лекарственных средств, следует окончательно прекратить лечение даролутамидом (см. раздел «Побочные реакции»).
Одновременное применение с другими лекарственными средствами
Применение сильных индукторов CYP3A4 и P-gp во время лечения даролутамидом может снизить концентрацию даролутамида в плазме крови, поэтому не рекомендуется, за исключением случаев, когда нет терапевтической альтернативы. Следует рассмотреть возможность выбора альтернативного сопутствующего лекарственного средства с меньшим потенциалом индукции CYP3A4 или P-gp (см. раздел «Взаимодействие с другими лекарственными средствами и другие виды взаимодействий»).
За состоянием пациентов следует наблюдать в отношении возникновения побочных реакций при применении субстратов BCRP, OATP1B1 и OATP1B3, поскольку одновременное применение с даролутамидом может повысить концентрацию этих субстратов в плазме.
Следует избегать одновременного применения с розувастатином, за исключением случаев, когда нет терапевтической альтернативы (см. раздел «Взаимодействие с другими лекарственными средствами и другие виды взаимодействий»).
Терапия депривации андрогенов может удлинять интервал QT
У пациентов с факторами риска удлинения интервала QT в анамнезе и у пациентов, одновременно получающих лекарственные средства, способные удлинять интервал QT (см. раздел «Взаимодействие с другими лекарственными средствами и другие виды взаимодействий»), врачи должны оценить соотношение польза/риск, включая вероятность возникновения torsade de pointes, перед началом терапии лекарственным средством Нубека.
Информация о вспомогательных веществах
Лекарственное средство Нубека содержит лактозу. Пациентам с редкими наследственными нарушениями, такими как непереносимость галактозы, дефицит лактазы или нарушение всасывания глюкозы-галактозы, не следует принимать это лекарственное средство.
Применение в период беременности или кормления грудью
Этот лекарственный препарат противопоказан женщинам репродуктивного возраста. Он не должен применяться беременным женщинам или женщинам, которые кормят грудью (см. разделы «Показания» и «Противопоказания»).
Женщины репродуктивного возраста / контрацепция для мужчин и женщин
Неизвестно, присутствуют ли даролутамид или его метаболиты в сперме. Если пациент ведет половую жизнь с женщиной репродуктивного возраста, во время и в течение 1 недели после завершения терапии лекарственным средством Нубека для предотвращения беременности следует использовать высокоэффективный метод контрацепции (< 1 % частоты неудач в год).
Беременность
С учетом механизма действия даролутамид может нанести вред плоду. Доклинические исследования репродуктивной токсичности не проводились (см. раздел «Доклинические данные по безопасности»).
Неизвестно, присутствуют ли даролутамид или его метаболиты в сперме. Если пациент ведет половую жизнь с беременной женщиной, во время и в течение 1 недели после завершения терапии лекарственным средством Нубека следует использовать презерватив. Следует избегать воздействия ингибиторов андрогеновых рецепторов на плод через передачу семенной жидкости беременной женщине, поскольку это может повлиять на развитие плода.
Кормление грудью
Неизвестно, выделяется ли даролутамид или его метаболиты в грудное молоко. Исследования на животных для оценки выделения даролутамида или его метаболитов с молоком не проводились (см. раздел «Доклинические данные по безопасности»). Нельзя исключить риск для ребенка, находящегося на грудном вскармливании.
Фертильность
Нет данных о влиянии даролутамида на фертильность человека.
Согласно исследованиям на животных, Нубека может ухудшать фертильность у мужчин с репродуктивным потенциалом (см. раздел «Доклинические данные по безопасности»).
Способность влиять на скорость реакции при управлении автотранспортом или другими механизмами
Лекарственное средство Нубека не влияет или оказывает незначительное влияние на скорость реакции при управлении автотранспортом или другими механизмами.
Способ применения и дозы
Лечение должен начинать и контролировать врач, имеющий опыт лечения рака предстательной железы.
Дозировка
Рекомендуемая доза составляет 600 мг даролутамида (две таблетки по 300 мг) дважды в сутки, что эквивалентно общей суточной дозе 1200 мг (см. подраздел «Фармакокинетика»).
Применение даролутамида следует продолжать до прогрессирования заболевания или неприемлемой токсичности.
У пациентов, не подвергшихся хирургической кастрации, следует продолжать медикаментозную кастрацию с помощью аналога ЛГРГ.
Метастатический гормонально-чувствительный рак предстательной железы (мГЧРПЖ)
Пациенты с мГЧРПЖ должны начинать лечение даролутамидом в комбинации с доцетакселом (см. раздел «Фармакологические свойства»). Первый из 6 циклов доцетаксела следует назначить в течение 6 недель после начала терапии даролутамидом. Следует соблюдать рекомендации, указанные в инструкции по медицинскому применению доцетаксела. Лечение даролутамидом следует продолжать до прогрессирования заболевания или неприемлемой токсичности, даже если цикл применения доцетаксела отложен, прерван или прекращён.
Пропущенная доза
Пропущенную дозу следует принять, как только пациент вспомнит, но только в тот же день. Пациент не должен принимать две дозы, чтобы компенсировать пропущенную дозу.
Изменение дозировки
Если у пациента наблюдается токсичность ≥ 3-й степени или непереносимая побочная реакция, связанная с применением даролутамида (см. разделы «Особенности применения» и «Побочные реакции»), дозу не следует применять или следует уменьшить дозировку до 300 мг дважды в сутки до облегчения симптомов. В дальнейшем лечение можно возобновить в дозе 600 мг дважды в сутки. Снижение дозы ниже 300 мг дважды в сутки не рекомендуется, поскольку эффективность такого применения не установлена.
Особые группы пациентов
Пациенты пожилого возраста. Коррекция дозы для пациентов пожилого возраста не требуется (см. подраздел «Фармакокинетика»).
Нарушение функции почек. Пациентам с лёгкой или умеренной степенью нарушения функции почек коррекция дозы не требуется. Для пациентов с тяжёлой почечной недостаточностью (СКФ 15–29 мл/мин/1,73 м²), не находящихся на гемодиализе, рекомендуемая начальная доза составляет 300 мг дважды в сутки (см. раздел «Особенности применения» и подраздел «Фармакокинетика»).
Нарушение функции печени. Пациентам с лёгкой степенью печеночной недостаточности коррекция дозы не требуется. Данные о фармакокинетике даролутамида при умеренной печеночной недостаточности ограничены. Применение даролутамида у пациентов с тяжёлой печеночной недостаточностью не изучалось.
Для пациентов с умеренной и тяжёлой печеночной недостаточностью (классы B и C по Чайлду – Пью) рекомендуемая начальная доза составляет 300 мг дважды в сутки (см. раздел «Особенности применения» и подраздел «Фармакокинетика»).
Способ применения
Лекарственное средство Нубека предназначено для перорального применения.
Таблетки следует принимать целиком во время еды (см. подраздел «Фармакокинетика»).
Дети
Отсутствует опыт применения даролутамида у педиатрических пациентов.
Передозировка
Наиболее высокая доза даролутамида, изученная клинически, составляла 900 мг дважды в день, что эквивалентно общей суточной дозе 1800 мг. При этой дозе не наблюдалось токсических эффектов, ограничивающих дозу. Учитывая насыщаемую абсорбцию (см. подраздел «Фармакокинетика») и отсутствие признаков острой токсичности, не ожидается, что приём дозы даролутамида, превышающей рекомендованную, приведёт к токсическим эффектам.
В случае приёма дозы, превышающей рекомендованную, лечение даролутамидом можно продолжить следующей дозой по графику.
Специфического антидота для даролутамида не существует, и симптомы передозировки не установлены.
Побочные реакции
Наиболее частые побочные реакции, наблюдавшиеся у пациентов с:
- нмКРПРЗ, получавших даролутамид, — утомление/астенические состояния (15,8 %);
- мКНРПРЗ, получавших даролутамид в комбинации с доцетакселом, — сыпь (16,6 %) и гипертензия (13,8 %).
Для получения дополнительной информации о безопасности при комбинированном применении даролутамида необходимо обратиться к инструкции по медицинскому применению отдельных лекарственных средств.
Побочные реакции, наблюдавшиеся у пациентов с нмКРПРЗ, получавших даролутамид, приведены в таблице 3. Побочные реакции, наблюдавшиеся у пациентов с мКНРПРЗ, получавших даролутамид в комбинации с доцетакселом, приведены в таблице 4.
Побочные реакции классифицируются в соответствии с системами органов. Они сгруппированы в зависимости от их частоты. Частота возникновения побочных реакций определяется следующим образом: очень часто (≥ 1/10), часто (≥ 1/100 до < 1/10), нечасто (≥ 1/1000 до < 1/100), редко (≥ 1/10000 до < 1/1000), очень редко (< 1/10000), неизвестно (нельзя оценить по имеющимся данным). В каждой группе по частоте побочные реакции представлены в порядке уменьшения тяжести.
Таблица 3. Побочные реакции, зарегистрированные в исследовании ARAMISa
| Класс системы органов (MedDRA) |
Очень часто |
Часто |
| Заболевания сердца |
Ишемическая болезнь сердцаb, сердечная недостаточностьc |
|
| Заболевания кожи и подкожной клетчатки |
Сыпьd |
|
| Заболевания костно-мышечной системы и соединительной ткани |
Боль в конечностях, костно-мышечная боль, переломы |
|
| Общие расстройства и реакции в месте введения |
Утомление/астенические состоянияe |
|
| Исследованияf |
Снижение количества нейтрофилов, повышение уровня билирубина в крови, повышение АСТ |
а Средняя продолжительность воздействия составляла 14,8 месяца (диапазон от 0,0 до 44,3 месяца) у пациентов, получавших даролутамид, и 11,0 месяца (диапазон от 0,1 до 40,5 месяца) у пациентов, получавших плацебо.
b Включает атеросклероз коронарной артерии, ишемическую болезнь коронарной артерии, окклюзию коронарной артерии, стеноз коронарной артерии, острый коронарный синдром, острый инфаркт миокарда, стенокардию, нестабильную стенокардию, инфаркт миокарда, ишемию миокарда.
c Включает сердечную недостаточность, острую сердечную недостаточность, хроническую сердечную недостаточность, застойную сердечную недостаточность, кардиогенный шок.
d Включает сыпь, макулярную сыпь, макулопапулёзную сыпь, папулёзную сыпь, пустулёзную сыпь, эритему, дерматит.
e Включает утомление и астению, вялость и недомогание.
f Общие терминологические критерии побочных реакций, версия 4.03. Частота основана на значениях, сообщаемых как отклонения лабораторных показателей.
Таблица 4. Побочные реакции, зарегистрированные у пациентов с мГЧРПЗ, получавших даролутамид в комбинации с доцетакселом в исследовании ARASENSа, b
| Класс системы органов (MedDRA) |
Очень часто |
Часто |
| Нарушения со стороны сосудов |
Артериальная гипертензияc |
|
| Нарушения со стороны кожи и подкожной клетчатки |
Сыпьd, e |
|
| Нарушения со стороны костно-мышечной системы и соединительной ткани |
Переломы |
|
| Нарушения со стороны репродуктивной системы и молочных желез |
Гинекомастия |
|
| Исследованияf |
Снижение количества нейтрофилов, повышение уровня билирубина в крови, повышение АЛТ, повышение АСТ |
а Средняя продолжительность воздействия составила 41,0 месяца (диапазон от 0,1 до 56,5 месяца) у пациентов, получавших даролутамид + доцетаксель, и 16,7 месяца (диапазон от 0,3 до 55,8 месяца) у пациентов, получавших плацебо + доцетаксель.
b Частота побочных реакций не может быть связана отдельно с даролутамидом, но может зависеть от других лекарственных средств, применяемых в комбинации.
c Включает артериальную гипертензию, повышение артериального давления, гипертонический криз.
d Включает сыпь, лекарственную сыпь, эритематозную сыпь, фолликулярную сыпь, макулярную сыпь, макулопапулезную сыпь, папулезную сыпь, зуд, пустулезную сыпь, везикулярную сыпь, эритему, дерматит.
e Частота была самой высокой в течение первых 6 месяцев лечения.
f Общие терминологические критерии побочных реакций, версия 4.03. Частота основана на значениях, сообщаемых как отклонения лабораторных показателей.
Описание отдельных побочных реакций
Показатели функции печени
При лечении даролутамидом сообщалось о случаях идиосинкратического поражения печени, вызванного приемом лекарственных средств, с повышением аланинаминотрансферазы (АЛТ) и/или аспартатаминотрансферазы (АСТ) до ≥ 5 и ≥ 20 раз от верхней границы нормы (ВГН) 3 и 4 степени, включая повышение уровня трансаминаз с одновременным повышением общего билирубина до 2 × ВГН. Время до начала варьировало от 1 месяца до 12 месяцев после начала применения даролутамидом. Во многих случаях повышение АЛТ и АСТ было обратимым после прекращения приема даролутамидом. Рекомендации см. в разделе «Особенности применения».
Неметастатический кастрационно-резистентный рак предстательной железы (нКРРПЖ)
Утомляемость
Сообщалось о слабости/астенических состояниях у 15,8 % пациентов, получавших даролутамид, и у 11,4 % пациентов, получавших плацебо. События с наихудшей степенью 3 были зарегистрированы у 0,6 % пациентов, получавших даролутамид, и у 1,1 % пациентов, получавших плацебо. Утомляемость (не включая астению, вялость или недомогание) наблюдалась в большинстве случаев (12,1 % пациентов, получавших даролутамид, и 8,7 % пациентов, получавших плацебо).
Переломы
Переломы возникли у 4,2 % пациентов, получавших даролутамид, и у 3,6 % пациентов, получавших плацебо.
Ишемическая болезнь сердца и сердечная недостаточность
Ишемическая болезнь сердца возникла у 3,2 % пациентов, получавших даролутамид, и у 2,5 % пациентов, получавших плацебо. События 5 степени наблюдались у 0,3 % пациентов, получавших даролутамид, и у 0,2 % пациентов, получавших плацебо. Сердечная недостаточность возникла у 1,9 % пациентов, получавших даролутамид, и у 0,9 % пациентов, получавших плацебо.
Снижение количества нейтрофилов
Снижение количества нейтрофилов наблюдалось как отклонение лабораторных показателей у 19,6 % пациентов, получавших даролутамид, и у 9,4 % пациентов, получавших плацебо. Среднее время до достижения наинизшего уровня составило 256 дней.
Отклонения лабораторных показателей в основном были 1–2 степени интенсивности. Снижение количества нейтрофилов 3 и 4 степени наблюдалось у 3,5 % и 0,5 % пациентов соответственно. Только один пациент окончательно прекратил прием даролутамидом из-за нейтропении. Нейтропения была временной или обратимой (88 % пациентов) и не была связана с какими-либо клинически значимыми признаками или симптомами.
Повышение уровня билирубина в крови
Сообщалось о повышении уровня билирубина как об отклонении лабораторных показателей у 16,4 % пациентов, получавших даролутамид, и у 6,9 % пациентов, получавших плацебо. Эпизоды были в основном 1 или 2 степени интенсивности, не связаны с какими-либо клинически значимыми признаками или симптомами и были обратимыми после прекращения применения даролутамидом. Повышение уровня билирубина 3 степени наблюдалось у 0,1 % пациентов, получавших даролутамид, и у 0 % пациентов, получавших плацебо. В группе даролутамидом среднее время до первого начала повышения уровня билирубина составило 153 дня, а средняя продолжительность первого эпизода — 182 дня. Ни у одного пациента лечение не было прекращено из-за повышения уровня билирубина.
Повышение АСТ
Сообщалось о повышении АСТ как об отклонении лабораторных показателей у 22,5 % пациентов, получавших даролутамид, и у 13,6 % пациентов, получавших плацебо. Эпизоды были в основном 1 или 2 степени интенсивности, не связаны с какими-либо клинически значимыми признаками или симптомами и были обратимыми после прекращения применения даролутамидом. Повышение АСТ 3 степени наблюдалось у 0,5 % пациентов, получавших даролутамид, и у 0,2 % пациентов, получавших плацебо. В группе даролутамидом среднее время до первого начала повышения АСТ составило 258 дней, а средняя продолжительность первого эпизода — 118 дней. Ни у одного пациента лечение не было прекращено из-за повышения АСТ.
Метастатический гормоночувствительный рак предстательной железы (мГЧРПЖ)
Артериальная гипертензия
В исследовании ARASENS о артериальной гипертензии сообщалось у 13,8 % пациентов, получавших даролутамид + доцетаксель, и у 9,4 % пациентов, получавших плацебо + доцетаксель.
Артериальная гипертензия 3 степени была зарегистрирована у 6,4 % пациентов, получавших даролутамид + доцетаксель, по сравнению с 3,5 % пациентов, получавших плацебо + доцетаксель. По одному пациенту с гипертензией 4 степени было в каждой группе лечения.
Сообщался один случай гипертензии 5 степени с артериосклерозом 5 степени в группе даролутамид + доцетаксель. У этого пациента была давняя история гипертензии и курения, и случай произошел более чем через 3 года после начала лечения даролутамидом. О случаях артериальной гипертензии сообщалось чаще у пациентов без анамнеза гипертензии в обеих группах лечения.
Переломы
Переломы возникали у 7,5 % пациентов, получавших даролутамид + доцетаксель, и у 5,1 % пациентов, получавших плацебо + доцетаксель.
Снижение количества нейтрофилов
Сообщалось о снижении количества нейтрофилов как об отклонении лабораторных показателей у 50,6 % пациентов, получавших даролутамид + доцетаксель, и у 45,5 % пациентов, получавших плацебо + доцетаксель. Снижение количества нейтрофилов 3 и 4 степени наблюдалось у 34,4 % пациентов, получавших даролутамид + доцетаксель, и у 31,4 % пациентов, получавших плацебо + доцетаксель. В обеих группах лечения количество нейтрофилов снижалось, а нейтропения была наиболее выраженной в первые месяцы лечения, после чего частота и тяжесть событий уменьшились.
Повышение уровня билирубина в крови
Повышение уровня билирубина в крови наблюдалось как отклонение лабораторных показателей у 19,6 % пациентов, получавших даролутамид + доцетаксель, и у 10,0 % пациентов, получавших плацебо + доцетаксель. События были преимущественно 1 или 2 степени интенсивности. Повышение билирубина 3 и 4 степени наблюдалось у 0,5 % пациентов, получавших даролутамид + доцетаксель, и у 0,3 % пациентов, получавших плацебо + доцетаксель.
Повышение АЛТ и АСТ
Сообщалось о повышении АЛТ как об отклонении лабораторных показателей у 42,3 % пациентов, получавших даролутамид + доцетаксель, и у 38,0 % пациентов, получавших плацебо + доцетаксель. Сообщалось о повышении АСТ как об отклонении лабораторных показателей у 43,9 % пациентов, получавших даролутамид + доцетаксель, и у 39,3 % пациентов, получавших плацебо + доцетаксель. Повышение АЛТ и АСТ было преимущественно 1 степени интенсивности. Повышение АЛТ 3 и 4 степени наблюдалось у 3,7 % пациентов, получавших даролутамид + доцетаксель, и у 3,0 % пациентов, получавших плацебо + доцетаксель. Повышение АСТ 3 и 4 степени наблюдалось у 3,6 % пациентов, получавших даролутамид + доцетаксель, и у 2,3 % пациентов, получавших плацебо + доцетаксель.
Сообщение о подозреваемых побочных реакциях
Сообщение о побочных реакциях после регистрации лекарственного средства имеет важное значение. Это позволяет проводить мониторинг соотношения польза/риск при применении данного лекарственного средства. Медицинским и фармацевтическим работникам, а также пациентам или их законным представителям следует сообщать обо всех случаях подозреваемых побочных реакций и отсутствия эффективности лекарственного средства через Автоматизированную информационную систему фармаконадзора по ссылке: https://aisf.dec.gov.ua.
Срок годности. 3 года.
Условия хранения
Не требует специальных условий хранения. Хранить в недоступном для детей месте.
Упаковка
По 16 таблеток в блистере, по 7 блистеров в картонной пачке.
Категория отпуска. По рецепту.
Производитель. Орион Корпорейшн, Орион Фарма.
Местонахождение производителя и его адрес места осуществления деятельности
Джоенсуункату 7, 24100 Сало, Финляндия.