Nubeka
UkrainaSpis treści
INSTRUKCJA dot. stosowania leku Nubeka (NUBEQA)
Skład:
substancja czynna: darolutamid;
1 tabletka powlekana powłoką filmową zawiera 300 mg darolutamidu;
substancje pomocnicze: wapnia hydrofosforan, sodowa krzemionka karboksymetylowa, laktoza jednowodna, stearynian magnezu, powidon K 30; powłoka filmowa: biała farba (hipromeloza 15 cP, laktoza jednowodna, makrogol 3350, dwutlenek tytanu (E 171)).
Postać farmaceutyczna. Tabletka powlekana powłoką filmową.
Podstawowe właściwości fizykochemiczne: owalne tabletki powlekane powłoką filmową, od białego do niemal białego koloru, z oznaczeniem „BAYER” po jednej stronie i „300” po drugiej.
Grupa farmakoterapeutyczna. Leki stosowane w hormonoterapii. Antagoniści hormonów i związki pokrewne. Leki przeciwandrogenowe. Darolutamid.
Kod ATC L02B B06.
Właściwości farmakodynamiczne
Farmakodynamika
Mechanizm działania. Darolutamid jest inhibitorem receptora androgenowego (AR) o giętkiej strukturze z grupą polarną pochodzenia pirazolu, który wiąże się z wysokim powinowactwem bezpośrednio z domeną wiązania ligandu receptora.
Darolutamid konkurencyjnie hamuje wiązanie androgenów, translokację jądrową receptora AR oraz zależną od AR transkrypcję. Główny metabolit darolutamidu, keto-darolutamid, in vitro wykazał aktywność podobną do aktywności darolutamidu. Leczenie darolutamidem zmniejsza proliferację komórek nowotworowych gruczołu krokowego, co prowadzi do silnego działania przeciwnowotworowego.
Efekty farmakodynamiczne. Nie zaobserwowano wydłużenia średniego przedziału QTcF (tj. ponad 10 ms) po doustnym przyjęciu 600 mg darolutamidu dwa razy dziennie w porównaniu z placebo.
Skuteczność i bezpieczeństwo kliniczne. Skuteczność i bezpieczeństwo zostały potwierdzone w dwóch randomizowanych, placebo-kontrolowanych, wieloośrodkowych badaniach fazy III z udziałem pacjentów z niemetastatycznym rakiem gruczołu krokowego opornym na kastrację (nmCRPC) (badanie ARAMIS) oraz z metastatycznym nowotworem czułym na hormony (mHRPC) (badanie ARASENS). Wszyscy pacjenci jednocześnie otrzymywali analog hormonu uwalniającego gonadotropinę (LHRH) lub poddali się orchiectomii dwustronnej.
Niemetastatyczny rak gruczołu krokowego oporny na kastrację (nmCRPC)
Skuteczność i bezpieczeństwo stosowania darolutamidu oceniano w randomizowanym, podwójnie ślepym, placebo-kontrolowanym, wieloośrodkowym badaniu fazy III (ARAMIS) u pacjentów z niemetastatycznym (na podstawie standardowych metod: tomografia komputerowa, scyntygrafia kości, rezonans magnetyczny) rakiem gruczołu krokowego opornym na kastrację, z okresem podwojenia antygenu specyficznego dla prostaty (PSA) ≤ 10 miesięcy.
Pacjenci byli włączani do badania, jeśli mieli trzy wzrosty poziomu antygenu specyficznego dla prostaty (PSA) po osiągnięciu najniższego poziomu, z co najmniej tygodniową przerwą podczas terapii deprywacji androgenów, poziom PSA ≥ 2 ng/ml podczas skriningu oraz poziom testosteronu po kastracji w surowicy < 1,7 nmol/l.
Pacjenci z wywiadem padaczki byli dopuszczeni do udziału w badaniu. W grupie otrzymującej darolutamid zarejestrowano 12 pacjentów (0,21%), którzy mieli wywiad padaczki.
Pacjenci z niekontrolowaną nadciśnieniem tętniczym lub niedawnym (w ciągu ostatnich 6 miesięcy) udarem mózgu, zawałem mięśnia sercowego, ciężką/niestabilną dławicą piersiową, wszczepieniem pomostu wieńcowego/obwodowego, niewydolnością serca klasy III lub IV według klasyfikacji New York Heart Association, zostali wykluczeni z badania.
Wykluczono również pacjentów, którzy wcześniej otrzymywali inhibitory receptora androgenowego (AR) drugiej generacji, takie jak enzalutamid, apalutamid i darolutamid, lub inhibitory enzymu CYP17, takie jak octan abirateronu, a także pacjentów, którzy otrzymywali systemowe kortykosteroidy w dawce przekraczającej równowartość 10 mg prednizolu dziennie w ciągu 28 dni przed randomizacją.
Łącznie 1509 pacjentów zostało randomizowanych w stosunku 2:1 w celu otrzymania 600 mg darolutamidu doustnie dwa razy dziennie (n = 955) lub placebo (n = 554).
Pacjenci z obecnością węzłów chłonnych miednicy < 2 cm w osi krótkiej poniżej bifurkacji aorty zostali włączeni do badania. Brak lub obecność przerzutów oceniano za pomocą niezależnej, scentralizowanej oceny radiologicznej. Ocena ta została przeprowadzona u 89 pacjentów, u których retrospektywnie wykryto przerzuty na początku badania. Randomizację zastosowano z uwzględnieniem okresu podwojenia PSA (≤ 6 miesięcy lub > 6 miesięcy) oraz stosowania terapii celowanej skierowanej przeciwko osteoklastom na początku badania (tak lub nie).
Charakterystyki demograficzne pacjentów i cechy choroby były zrównoważone między grupami leczenia. Średni wiek pacjentów wynosił 74 lata (zakres 48–95), 9% pacjentów miało 85 lat lub więcej; 79% stanowiło osoby rasy europejskiej, 13% pochodzenia azjatyckiego i 3% osoby o ciemnej skórze. Większość pacjentów miała 7 punktów lub więcej według skali Gleasona w momencie postawienia diagnozy (73%). Mediana okresu podwojenia PSA wynosiła 4,5 miesiąca. Dziewięć procent (9%) pacjentów poddano wcześniejszej orchiectomii, 25% – wcześniejszej prostatektomii, a 50% – co najmniej jednej poprzedniej terapii promieniowej. Siedemdziesiąt sześć procent (76%) pacjentów otrzymało więcej niż jedno poprzednie leczenie antyhomonalne. Pacjenci mieli 0 punktów (69%) lub 1 punkt (31%) według skali sprawności Eastern Cooperative Oncology Group (ECOG PS).
Leczenie darolutamidem kontynuowano do postępu choroby ocenianego radiologicznie, co oceniano za pomocą tradycyjnej wizualizacji (TK, scyntygrafia kości, rezonans magnetyczny) poprzez ślepe, scentralizowane ocenianie, do wystąpienia nieakceptowalnej toksyczności lub odmowy kontynuowania udziału w badaniu przez pacjenta.
Pierwotnym punktem końcowym skuteczności była przeżycie bez przerzutów (MFS). Punktami końcowymi wtórnymi były całkowite przeżycie (OS), czas do nasilenia bólu, czas do rozpoczęcia pierwszej cytotoksycznej chemioterapii raka gruczołu krokowego oraz czas do pierwszego wystąpienia objawowych zmian w kościach (określanych jako pojawienie się jednego z następujących objawów: zewnętrzna terapia promieniowaniem w celu złagodzenia objawów kostnych, nowy objawowy patologiczny złamany kości, ucisk rdzenia kręgowego lub związane z nowotworem zabiegi ortopedyczne).
Leczenie darolutamidem skutkowało poprawą MFS w porównaniu z placebo (patrz tabela 1 i rys. 1).
Wyniki MFS były spójne we wszystkich podgrupach pacjentów niezależnie od okresu podwojenia PSA, wcześniejszego stosowania leków wpływających na tkankę kostną lub lokalnego/regionarnego zaawansowania choroby. Dodatkowe podgrupy zgodne pod względem wyników MFS obejmowały wartość PSA na początku badania, punktację według skali Gleasona w momencie postawienia diagnozy, wiek pacjenta, region geograficzny, punktację według ECOG PS na początku badania, przynależność rasową oraz liczbę poprzednich cykli terapii hormonalnej.
Po pierwotnej analizie MFS, gdy tylko badanie zostało odsłonięte, pacjentom otrzymującym placebo zaproponowano leczenie darolutamidem (opcja krzyżowa). Spośród 554 pacjentów randomizowanych do grupy placebo, 170 (31%) przeszło na leczenie darolutamidem. Analiza OS nie uwzględniała wpływu przejścia krzyżowego.
W momencie końcowej analizy leczenie darolutamidem skutkowało statystycznie istotną poprawą całkowitego przeżycia w porównaniu z placebo (mediana nie została osiągnięta w żadnej grupie, patrz tabela 1 i rys. 2). Zaobserwowano również statystycznie istotne wydłużenie czasu do nasilenia bólu, do rozpoczęcia pierwszej cytotoksycznej chemioterapii oraz do pierwszego objawowego zdarzenia kostnego w porównaniu z placebo (patrz tabela 1).
W momencie końcowej analizy średnia długość leczenia u pacjentów otrzymujących darolutamid wynosiła 33,3 miesiąca (zakres od 0,0 do 74,0 miesiąca) w okresie podwójnie ślepych i otwartych.
Wszystkie analizy przeprowadzono w pełnej populacji badawczej.
Tabela 1. Wyniki skuteczności z badania ARAMIS
| Parametr skuteczności |
Liczba (%) pacjentów z zdarzeniami |
Mediana (miesiące) (95 % CI) |
Stosunek ryzyka (HR)b (95 % przedział ufności [CI]) p-wartość (dwustronna) |
||
| Darolutamid (N = 955) |
Placeboa (N = 554) |
Darolutamid (N = 955) |
Placeboa (N = 554) |
||
| Przeżycie bez przerzutówc |
221 (23,1 %) |
216 (39,0 %) |
40,4 (34,3; ND) |
18,4 (15,5; 22,3) |
0,413 (0,341; 0,500) < 0,000001 |
| Całkowite przeżycie |
148 (15,5 %) |
106 (19,1 %) |
ND (56,1; ND) |
ND (46,9; ND) |
0,685 (0,533; 0,881) 0,003048 |
| Czas do nasilenia bóluc, d |
251 (26,3 %) |
178 (32,1 %) |
40,3 (33,2; 41,2) |
25,4 (19,1; 29,6) |
0,647 (0,533; 0,785) 0,000008 |
| Czas do pierwszej cytotoksycznej chemioterapii |
127 (13,3 %) |
98 (17,7 %) |
ND (ND, ND) |
ND |
0,579 (0,444; 0,755) 0,000044 |
| Czas do pierwszego objawowego zdarzenia kostnego |
29 (3,0 %) |
28 (5,1 %) |
ND (ND, ND) |
(ND, ND) |
0,484 (0,287; 0,815) 0,005294 |
a W tym przypadku 170 pacjentów przeszło na darolutamid bez okresu zasłonięcia.
b Stosunek ryzyka < 1 dla darolutamidu.
c Dla OSW i czasu do postępu bólu analiza pierwotna jest uznawana za ostateczną analizę.
d Wyniki zgłaszane przez pacjenta zgodnie z oceną kwestionariusza Skrócony opis bólu – Skrócona forma kwestionariusza.
ND – nie osiągnięto.
Leczenie darolutamidem skutkowało dłuższym okresem przeżycia bez postępu (PFS, mediana 36,8 vs 14,8 miesiąca, HR = 0,380, nominalne p < 0,000001) oraz czasem do postępu PSA (mediana 29,5 vs 7,2 miesiąca, HR = 0,164, nominalne p < 0,000001). Spójność efektu obserwowano we wszystkich punktach końcowych przeżycia (OSW, OZ, PSA).
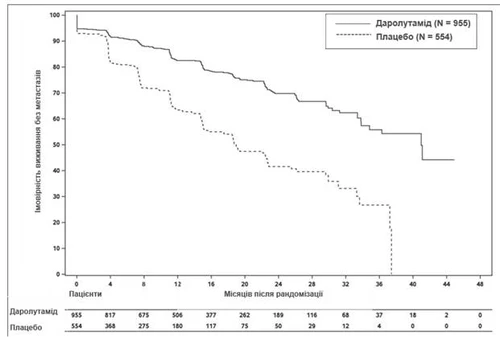
Rys. 1. Krzywe przeżycia Kaplana-Meiera bez przerzutów (ARAMIS)
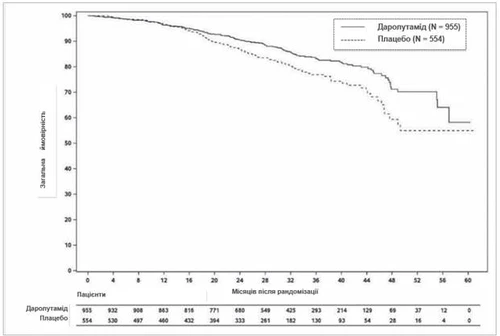
Rys. 2. Krzywe przeżycia Kaplana-Meiera ogólnego przeżycia (ARAMIS)
Pacjenci otrzymujący darolutamid w badaniu ARAMIS (okres podwójnie ślepy) wykazali istotnie wyższą potwierdzoną częstość odpowiedzi co do poziomu PSA (określana jako obniżenie o ≥ 50 % w stosunku do wartości wyjściowej) w porównaniu z pacjentami otrzymującymi placebo – 84,0 % vs 7,9 % (różnica = 76,1 %, p < 0,000001 (nominalna wartość p, wyłącznie w celach informacyjnych)).
Rak prostaty wrażliwy na hormony z przerzutami (mCRPC)
Skuteczność i bezpieczeństwo stosowania darolutamidu w połączeniu z doksorubycyną oceniano w wieloośrodkowym, podwójnie ślepym, kontrolowanym placebo badaniu fazy III (ARASENS) u pacjentów z mCRPC. Ogółem 1306 pacjentów zostało randomizowanych w stosunku 1:1 w celu otrzymania 600 mg darolutamidu doustnie dwa razy dziennie (n = 651) lub placebo (n = 655) jednocześnie z 75 mg/m² doksorubycyny w ciągu 6 cykli. Leczenie darolutamidem lub placebo kontynuowano do postępu objawów choroby, zmiany terapii przeciwnowotworowej, nieakceptowalnej toksyczności, śmierci lub wycofania pacjenta z badania.
Obecność przerzutów oceniano za pomocą niezależnej, scentralizowanej oceny radiologicznej. Pacjentów z zaangażowaniem wyłącznie regionalnych węzłów chłonnych (M0) wykluczono z badania. Randomizację przeprowadzono z uwzględnieniem warstwowania według zaawansowania choroby (wyłącznie przerzuty do nieregionalnych węzłów chłonnych (M1a), przerzuty do kości z przerzutami do węzłów chłonnych lub bez nich (M1b) lub przerzuty do narządów z przerzutami do węzłów chłonnych lub bez nich, lub z przerzutami do kości lub bez nich (M1c)) oraz według poziomu fosfatazy alkalicznej (< lub ≥ górnej granicy normy) na początku badania. Pacjentów z przerzutami do mózgu można było włączać do badania, jednak takich pacjentów nie było.
Charakterystyki demograficzne pacjentów i cechy choroby były zrównoważone między grupami leczenia. Średni wiek wynosił 67 lat (zakres 41–89), 0,5 % pacjentów miało 85 lat lub więcej, 52 % stanowili pacjenci rasy europejskiej, 36 % pochodzenia azjatyckiego i 4 % uczestników badania mieli ciemną karnację skóry. Większość pacjentów miała 8 lub więcej punktów w skali Gleasona w momencie postawienia diagnozy (78 %). 71 % pacjentów miało ocenę ECOG PS 0 punktów, a 29 % – 1 punkt. Było 86,1 % pacjentów z chorobą de novo oraz 12,9 % pacjentów z nawrotem. Na początku badania 3 % pacjentów miało M1a, 79,5 % miało M1b i 17,5 % miało M1c; fosfataza alkaliczna była < GGN u 44,5 % pacjentów i ≥ GGN u 55,5 % pacjentów; średni wyjściowy poziom PSA wynosił odpowiednio 30,3 μg/l i 24,2 μg/l w grupach darolutamidu i placebo. Pacjentów z wywiadem drgawek dopuszczono do udziału w badaniu, a 4 pacjentów (0,6 %) zostało włączonych do grupy darolutamid + doksorubycyna.
77,0 % pacjentów miało chorobę o dużym obciążeniu, a 23,0 % miało chorobę o małym obciążeniu. Chorobę o dużym obciążeniu definiowano jako obecność przerzutów do narządów lub 4 lub więcej zmian w kościach, w tym co najmniej 1 przerzut poza kręgosłup i kości miednicy. Ocenę objawów bólowych oceniano zgodnie z wynikami zgłaszanymi przez pacjenta zgodnie z kwestionariuszem Skrócony opis bólu – Skrócona forma, co zdefiniowano jako pogorszenie o co najmniej 2 punkty od poziomu wyjściowego oraz rozpoczęcie stosowania opioidów o krótkim lub długim działaniu w celu złagodzenia bólu przez ≥ 7 dni z rzędu.
Średni czas trwania leczenia wynosił 41,0 miesiąca (zakres od 0,1 do 56,5 miesiąca) u pacjentów otrzymujących darolutamid + doksorubycynę oraz 16,7 miesiąca (zakres od 0,3 do 55,8 miesiąca) u pacjentów otrzymujących placebo + doksorubycynę. 87,6 % i 85,5 % pacjentów otrzymało pełne 6 cykli doksorubycyny, a 1,5 % i 2,0 % pacjentów nie otrzymało doksorubycyny w grupie darolutamid + doksorubycyna i placebo + doksorubycyna odpowiednio.
Tabela 2. Wyniki skuteczności badania ARASENS
| Parametr skuteczności |
Liczba (%) pacjentów z zdarzeniami |
Mediana (miesiące) (95 % przedział ufności) |
Stosunek ryzyka (SR)b (95 % przedział ufności) Wartość p (jednostronna)c |
||
| Darolutamid + doksotaksel (N = 651) |
Placebo + doksotaksel (N = 654)a |
Darolutamid + doksotaksel (N = 651) |
Placebo + doksotaksel (N = 654)a |
||
| Przeżycie ogólned |
229 (35,2%) |
304 (46,5%) |
N.D. (N.D., N.D.) |
48,9 (44,4; N.D.) |
0,675 (0,568; 0,801) <0,0001 |
a Jeden pacjent w grupie placebo został wykluczony ze wszystkich analiz.
b Stosunek ryzyka < 1 w odniesieniu do darolutamidu.
c Na podstawie zstratifikowanego testu logarytmu rangi.
d Wyniki OS były jednorodne we wszystkich podgrupach pacjentów, w tym w zależności od stopnia zaawansowania choroby i poziomu fosfatazy alkalicznej.
ND – nie osiągnięto.
Wtórne punkty końcowe skuteczności, dla których wykazano istotną statystycznie przewagę grupy darolutamid + doksetaksel w porównaniu z grupą placebo + doksetaksel: czas do rozwoju raka gruczołu krokowego opornego na kastrację (mediana ND vs 19,1 miesiąca; HR = 0,357, p < 0,0001); czas do pierwszego objawowego zdarzenia kostnego (mediana ND vs ND; HR = 0,712, p = 0,0081); czas do rozpoczęcia kolejnej chemioterapii przeciwnowotworowej (mediana ND vs 25,3 miesiąca; HR = 0,388, p < 0,0001); czas do nasilenia bólu (mediana ND vs 27,5 miesiąca; HR = 0,792, p = 0,0058); czas przeżycia wolny od objawowych zdarzeń kostnych (mediana 51,2 vs 39,7 miesiąca; HR = 0,609, p < 0,0001).
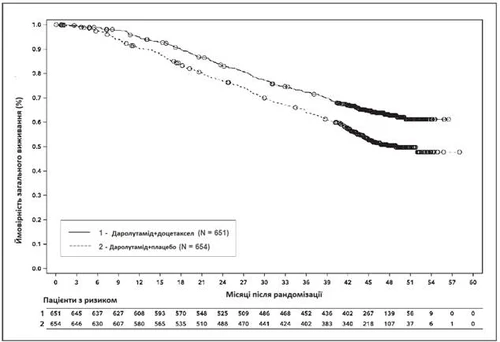
Rys. 3. Krzywe przeżycia całkowitego Kaplana-Meiera (ARASENS)a
a OS po 36 miesiącach wynosiła 72,3% (95% CI, 68,8–75,8) w grupie darolutamid + doksetaksel vs 63,8% (95% CI, 60,1–67,6) w grupie placebo + doksetaksel.
Częstość OS po 48 miesiącach wynosiła 62,7% (95% CI, 58,7–66,7) w grupie darolutamid + doksetaksel vs 50,4% (95% CI, 46,3–54,6) w grupie placebo + doksetaksel.
Farmakokinetyka
Darolutamid składa się z dwóch diastereoizomerów [(S,R)-darolutamid i (S,S)-darolutamid], które mogą się przekształcać wzajemnie poprzez główny cyrkulujący metabolit keto-darolutamid. In vitro wszystkie trzy substancje wykazują taką samą aktywność farmakologiczną. Darolutamid słabo rozpuszcza się w roztworach wodnych w szerokim zakresie pH i zazwyczaj lepiej rozpuszcza się w rozpuszczalnikach organicznych.
Wchłanianie
Po doustnym przyjęciu 600 mg (2 tabletki po 300 mg) dwa razy dziennie, stężenia szczytowe darolutamidu w osoczu w stanie stacjonarnym wynosiły 4,79 mg/l (współczynnik zmienności 30,9%) u pacjentów z nmCRPC w badaniu ARAMIS oraz 3,84 mg/l (współczynnik zmienności 35,6%) u pacjentów z mCRPC w badaniu ARASENS. Średni czas osiągnięcia maksymalnego stężenia w osoczu wynosił 3–4 godziny. Stosunek dwóch diastereoizomerów, (S,R)-darolutamidu do (S,S)-darolutamidu, zmienił się z 1:1 w tabletkach do około 1:9 w osoczu, na podstawie wskaźnika AUC0-12 w stanie stacjonarnym. Po doustnym podaniu razem z posiłkiem stan stacjonarny osiągany jest po 2–5 dniach powtarzalnego przyjmowania dwa razy dziennie.
Absolutna biodostępność po doustnym przyjęciu na czczo tabletki Nubeka zawierającej 300 mg darolutamidu wynosi około 30% w porównaniu z wstrzyknięciem dożylnym.
Biodostępność darolutamidu zwiększała się 2,0–2,5-krotnie przy podawaniu z posiłkiem. Podobny wzrost ekspozycji obserwowano dla głównego metabolitu – keto-darolutamidu.
Rozkład
Objętość urojona rozkładu darolutamidu po wstrzyknięciu dożylnym wynosi 119 l, co wskazuje na szerokie rozprzestrzenienie się darolutamidu w organizmie zarówno w przestrzeni wewnątrzkomórkowej, jak i zewnątrzkomórkowej.
Darolutamid umiarkowanie (92%) wiąże się z białkami osocza ludzkiego, bez różnic między dwoma diastereoizomerami. Główny metabolit darolutamidu, keto-darolutamid, wiąże się w znacznym stopniu (99,8%) z białkami osocza.
Przejście darolutamidu przez barierę krew-mózg nie było klinicznie badane. Jednak wpływ darolutamidu na mózg pod względem AUC0-24 jest bardzo niski: 4,5% ekspozycji w osoczu po pojedynczej dawce u szczurów i 1,9–3,9% po dawkowaniu powtarzalnym u myszy. Wskazuje to na niską zdolność przechodzenia darolutamidu przez nietkniętą barierę krew-mózg u szczurów i myszy oraz na niskie prawdopodobieństwo, że darolutamid przenika tę barierę u ludzi w klinicznie istotnym stopniu.
Biotransformacja
Diastereoizomery (S,R)-darolutamid i (S,S)-darolutamid mogą przekształcać się wzajemnie za pośrednictwem metabolitu keto-darolutamidu, z przewagą (S,S)-darolutamidu.
Po pojedynczym doustnym przyjęciu 300 mg 14C-darolutamidu w postaci roztworu doustnego, keto-darolutamid jest jedynym głównym metabolitem, z około dwukrotnie wyższą całkowitą ekspozycją w osoczu w porównaniu z darolutamidem. Na darolutamid i keto-darolutamid razem przypada 87,4% radioaktywności 14C w osoczu, co wskazuje, że wszystkie inne metabolity są nieistotne.
Darolutamid jest metabolizowany głównie poprzez metabolizm oksydacyjny, głównie za pośrednictwem CYP3A4, a także poprzez bezpośrednią glukuronidację, głównie za pośrednictwem UGT1A9 i UGT1A1. Ponadto wykazano, że izoformy AKR1C głównie katalizują redukcję keto-darolutamidu do diastereoizomerów substancji.
Wydalanie
Efektywny okres półtrwania darolutamidu i keto-darolutamidu w osoczu krwi pacjentów wynosi około 18–20 godzin. Z dwóch diastereoizomerów wchodzących w skład darolutamidu, (S,R)-darolutamid ma krótszy efektywny okres półtrwania – 9 godzin w porównaniu z (S,S)-darolutamidem, którego efektywny okres półtrwania wynosi 22 godziny. Klirens darolutamidu po wstrzyknięciu dożylnym wynosił 116 ml/min (CV 39,7%). Ogółem 63,4% darolutamidu i jego metabolitów wydala się z moczem (około 7% w niezmienionej formie), 32,4% wydala się z kałem. Ponad 95% dawki zostało wydalone w ciągu 7 dni po podaniu.
Liniowość/nieliniowość
W zakresie dawek od 100 do 700 mg (po pojedynczej dawce i w stanie stacjonarnym) ekspozycja dwóch diastereoizomerów i głównego metabolitu keto-darolutamidu wzrasta prawie liniowo w zależności od dawki. Ze względu na nasycenie wchłaniania, dalszego wzrostu ekspozycji darolutamidu przy dawce 900 mg dwa razy dziennie nie zaobserwowano.
Grupy specjalne pacjentów
Pacjenci w podeszłym wieku. Nie zaobserwowano klinicznie istotnych różnic w farmakokinetyce darolutamidu (65–95 lat).
Naruszenie funkcji nerek. W klinicznym badaniu farmakokinetycznym AUC i Cmax darolutamidu były 2,5 i 1,6 razy wyższe u pacjentów z ciężką niewydolnością nerek (oszacowana szybkość filtracji kłębuszkowej [eGFR] od 15 do 29 ml/min/1,73 m²) w porównaniu ze zdrowymi ochotnikami.
Analiza populacyjna farmakokinetyki wskazuje na około 1,1-, 1,3- i 1,5-krotnie wyższą ekspozycję (AUC) darolutamidu u pacjentów z łagodnym, umiarkowanym i ciężkim zaburzeniem funkcji nerek (eGFR od 15 do 89 ml/min/1,73 m²) w porównaniu z pacjentami z normalną funkcją nerek.
Farmakokinetyka darolutamidu nie była badana u pacjentów z niewydolnością nerek w stadium końcowym poddawanych dializie (eGFR < 15 ml/min/1,73 m²).
Naruszenie funkcji wątroby. W klinicznym badaniu farmakokinetycznym Cmax i AUC darolutamidu były 1,5 i 1,9 razy wyższe u pacjentów z umiarkowaną niewydolnością wątroby (klasa B wg klasyfikacji Childa-Pugha) w porównaniu ze zdrowymi ochotnikami. Brak danych dotyczących pacjentów z ciężką niewydolnością wątroby (klasa C wg klasyfikacji Childa-Pugha).
Różnice etniczne. Nie zaobserwowano żadnych klinicznie istotnych różnic w farmakokinetyce darolutamidu w zależności od przynależności etnicznej pacjenta (pacjenci kaukazoidalni, pacjenci pochodzenia japońskiego, pacjenci azjatyccy, ale nie japońscy, pacjenci o ciemnej skórze lub pochodzenia afroamerykańskiego). Analiza populacyjna farmakokinetyki wykazała zwiększenie średniego geometrycznego wpływu (AUC) do 1,56 razy (90% CI 1,43–1,70) u pacjentów pochodzenia japońskiego w porównaniu z pacjentami z innych regionów w badaniach ARAMIS i ARASENS.
Dane przedkliniczne dotyczące bezpieczeństwa
Toksykologia ogólnoustrojowa. W badaniach toksyczności wielokrotnej dawki na zwierzętach głównymi ustaleniami były zmiany w narządach rozrodczych samców (zmniejszenie masy narządu z atrofią gruczołu krokowego i nasieniowodów). Te efekty występowały przy ekspozycji systemowej w zakresie oczekiwanej ekspozycji u ludzi lub niższej (na podstawie porównania AUC). Dodatkowe zmiany w tkankach rozrodczych obejmowały minimalne zwiększenie wakuolizacji przysadki, atrofię i zmniejszenie wydzielania pęcherzyków nasiennych i gruczołów mlecznych u szczurów oraz hipospermię jąder, poszerzenie kanalików nasieniowych i degenerację u psów. Zmiany w męskich narządach rozrodczych u obu gatunków odpowiadały farmakologicznej aktywności darolutamidu i ustępowały lub częściowo ustępowały po 4–8-tygodniowym okresie regeneracji.
Toksykologia płodu/teratogenność. Badania toksycznego wpływu na płód nie przeprowadzono.
Toksykologia rozrodcza. Badania toksyczności rozrodczej nie przeprowadzono. Jednak płodność samców prawdopodobnie będzie zaburzona, biorąc pod uwagę wyniki badań toksyczności wielokrotnej dawki u zwierząt, które są zgodne z farmakologiczną aktywnością darolutamidu.
Genotoksyczność i rakotwórczość. Darolutamid nie indukował mutacji w analizie mikrobiologicznego mutagenezu (Ames). W wysokich stężeniach darolutamid indukował strukturalne aberracje chromosomowe in vitro w hodowanych limfocytach ludzkich. Jednak w połączonym teście mikrojąder szpiku kostnego in vivo oraz analizie Comet w wątrobie i dwunastnicy szczura nie zaobserwowano genotoksyczności przy ekspozycjach przekraczających maksymalną ekspozycję u ludzi.
Doustne podawanie darolutamidu samcom transgenicznych myszy rasH2 przez 6 miesięcy nie wykazało potencjału rakotwórczego przy dawkach do 1000 mg/kg/dzień, co przekracza ekspozycję kliniczną (AUC) darolutamidu u ludzi o 0,9–1,3 razy oraz keto-darolutamidu o 2,1–2,3 razy przy zastosowaniu zalecanej klinicznej dawki dobowej 1200 mg/dzień. Na podstawie tego badania nie można całkowicie wykluczyć ryzyka rakotwórczego przy stosowaniu darolutamidu.
Charakterystyka kliniczna
Wskazania
Preparat leczniczy Nubeka jest wskazany w leczeniu dorosłych mężczyzn:
- niemetastatycznego raka prostaty opornego na kastrację (nCRPC) z wysokim ryzykiem wystąpienia przerzutów (patrz punkt „Właściwości farmakologiczne”);
- metastatycznego raka prostaty wrażliwego na hormony (mHSPC) – w połączeniu z doksetakselą i terapią deprywacji androgenów (patrz punkt „Właściwości farmakologiczne”).
Przeciwwskazania
Podwyższona wrażliwość na substancję czynną lub na substancje pomocnicze.
Przeciwwskazany w ciąży i kobietom w wieku rozrodczym.
Interakcje z innymi lekami i inne rodzaje interakcji
Wpływ innych leków na darolutamid
Induktory CYP3A4 i P-gp
Darolutamid jest substytutem CYP3A4 i glikoproteiny P (P-gp).
Nie zaleca się stosowania silnych i umiarkowanych induktorów CYP3A4 oraz induktorów P-gp (np. karbamazepiny, fenylobutyrazolu, zioła św. Jana, fenytoiny i ryfampicyny) podczas leczenia darolutamidem, chyba że nie ma dostępnej alternatywy terapeutycznej. Należy rozważyć możliwość zastosowania alternatywnego leku towarzyszącego, który nie indukuje lub ma słaby potencjał indukowania CYP3A4 lub P-gp.
Powtarzane podawanie ryfampicyny (600 mg), silnego induktora CYP3A4 i P-gp, w połączeniu z pojedynczą dawką darolutamidu (600 mg) podawaną podczas posiłku, prowadziło do zmniejszenia średniej ekspozycji (AUC0-72) o 72% i zmniejszenia Cmax darolutamidu o 52%.
Inhibitory CYP3A4, P-gp i BCRP
Darolutamid jest substytutem CYP3A4, P-gp i białka oporności raka piersi (BCRP).
W przypadku stosowania inhibitorów CYP3A4, P-gp lub BCRP nie przewiduje się klinicznie istotnej interakcji między lekami. Darolutamid można stosować jednocześnie z inhibitorami CYP3A4, P-gp lub BCRP. Jednoczesne stosowanie darolutamidu z kombinowanym inhibitorem P-gp i silnym inhibitorem CYP3A4 może zwiększyć ekspozycję na darolutamid, co może zwiększyć ryzyko wystąpienia działań niepożądanych. Zaleca się częstsze monitorowanie pacjenta pod kątem wystąpienia działań niepożądanych po zastosowaniu darolutamidu oraz, w razie potrzeby, dostosowanie dawki darolutamidu.
Stosowanie itrakonazolu (200 mg dwa razy dziennie w dniu 1 i raz dziennie w kolejnych 7 dniach), silnego inhibitora CYP3A4, P-gp i BCRP, w połączeniu z pojedynczą dawką darolutamidu (600 mg w dniu 5 podawaną z posiłkiem) prowadziło do 1,7-krotnego zwiększenia średniej ekspozycji (AUC0-72) i 1,4-krotnego zwiększenia Cmax darolutamidu.
Inhibitory UGT1A9
Darolutamid jest substytutem UGT1A9. W przypadku stosowania inhibitora UGT1A9 nie przewiduje się klinicznie istotnej interakcji między lekami. Darolutamid można stosować jednocześnie z inhibitorami UGT1A9.
Populacyjna analiza farmakokinetyczna wykazała, że jednoczesne stosowanie inhibitorów UGT1A9 z darolutamidem prowadziło do 1,2-krotnego zwiększenia ekspozycji (AUC0-72) darolutamidu.
Doksateksel
Stosowanie darolutamidu w połączeniu z doksatekselą nie prowadziło do klinicznie istotnych zmian farmakokinetyki darolutamidu u pacjentów z mHSPC (patrz punkt „Właściwości farmakologiczne”).
Wpływ darolutamidu na inne leki
Substraty BCRP, OATP1B1 i OATP1B3
Darolutamid jest inhibitorem białka oporności raka piersi (BCRP) oraz organicznych transporterów anionów (OATP) 1B1 i 1B3.
Należy unikać jednoczesnego stosowania rosuwastatyny, chyba że nie ma dostępnej alternatywy terapeutycznej. Należy rozważyć możliwość zastosowania alternatywnego leku towarzyszącego o mniejszym potencjale hamowania BCRP, OATP1B1 i OATP1B3.
Stosowanie darolutamidu (600 mg dwa razy dziennie przez 5 dni) przed pojedynczą dawką rosuwastatyny (5 mg) podawaną z posiłkiem prowadziło do około 5-krotnego zwiększenia średniej ekspozycji (AUC) i Cmax rosuwastatyny.
Jeśli to możliwe, należy unikać jednoczesnego stosowania darolutamidu z innymi substratami BCRP. Jednoczesne stosowanie darolutamidu może zwiększyć stężenie innych towarzyszących leków – substratów BCRP, OATP1B1 i OATP1B3 (takich jak metotreksat, sulfasalazyna, flawastatyna, atorwastatyna, pitawastatyna) we krwi. Dlatego zaleca się obserwację pacjentów pod kątem wystąpienia działań niepożądanych po zastosowaniu substratów BCRP, OATP1B1 i OATP1B3. Ponadto, przy jednoczesnym stosowaniu z darolutamidem, należy przestrzegać odpowiednich zaleceń zawartych w informacjach dotyczących tych substratów.
Substraty P-gp
W przypadku podawania substratu P-gp nie przewiduje się klinicznie istotnej interakcji między lekami. Darolutamid można stosować jednocześnie z substratami P-gp (np. z cyklosporyną, werapamilem lub nifedypinem). Jednoczesne stosowanie darolutamidu z wrażliwym substratem P-gp – dabigatranu eteksylatu – nie prowadziło do zwiększenia ekspozycji (AUC i Cmax) dabigatranu.
Substraty CYP3A4
Darolutamid jest słabym induktorem CYP3A4.
W przypadku podawania substratu CYP nie przewiduje się klinicznie istotnej interakcji między lekami. Darolutamid można stosować jednocześnie z substratami CYP (np. z warfaryną, L-tyroksyną, omeprazolem).
Stosowanie darolutamidu (600 mg dwa razy dziennie przez 9 dni) przed pojedynczą dawką wrażliwego substratu CYP3A4 – midazolamu (1 mg) – podawaną z posiłkiem prowadziło do zmniejszenia średniej ekspozycji (AUC) i Cmax midazolamu odpowiednio o 29% i 32%. Darolutamid nie hamował metabolizmu pojedynczych substratów CYP in vitro w klinicznie istotnych stężeniach.
Doksateksel
Stosowanie darolutamidu w połączeniu z doksatekselą nie prowadziło do klinicznie istotnych zmian farmakokinetyki doksatekselu u pacjentów z mHSPC (patrz punkt „Właściwości farmakologiczne”).
Leki wydłużające odcinek QT
Ponieważ terapia deprywacji androgenów może wydłużać odcinek QT, należy dokładnie ocenić jednoczesne stosowanie z lekami, które znane są z wydłużania odcinka QT lub lekami, które mogą indukować torsade de pointes. Do takich leków należą leki przeciwarytmiczne klasy IA (np. chinidyna, dysopyramid) lub klasy III (np. amiodaron, sotalol, dofetylid, ibutylyd), metadon, moxifloksacyna oraz leki przeciwpsychotyczne (np. haloperidol).
Właściwości stosowania
Upośledzenie funkcji nerek
Dane dotyczące stosowania u pacjentów z ciężką niewydolnością nerek są ograniczone.
Ponieważ ekspozycja może być zwiększona, należy dokładnie monitorować stan takich pacjentów pod kątem wystąpienia działań niepożądanych (patrz punkt „Właściwości stosowania” oraz podrozdział „Farmakokinetyka”).
Upośledzenie funkcji wątroby
Dane dotyczące stosowania u pacjentów z umiarkowanym upośledzeniem funkcji wątroby są ograniczone. Nie prowadzono badań dotyczących stosowania darolutamidu u pacjentów z ciężkim upośledzeniem funkcji wątroby.
Ponieważ ekspozycja może być zwiększona, należy dokładnie monitorować stan pacjentów pod kątem wystąpienia działań niepożądanych (patrz punkt „Właściwości stosowania” oraz podrozdział „Farmakokinetyka”).
Niedawne choroby układu sercowo-naczyniowego
Pacjenci z klinicznie istotnymi chorobami układu sercowo-naczyniowego w ciągu ostatnich 6 miesięcy, w tym z udarem, zawałem mięśnia serca, ciężką/niestabilną dławicą piersiową, wszczepieniem pomostu tętniczego wieńcowego/obwodowego lub z objawową niewydolnością serca, byli wykluczani z badań klinicznych. W związku z tym bezpieczeństwo stosowania darolutamidu u tych pacjentów nie zostało ustalone.
W przypadku przepisywania leku Nubeka pacjentowi z klinicznie istotną chorobą układu sercowo-naczyniowego, należy leczyć tę chorobę zgodnie z obowiązującymi zaleceniami.
Hepatotoksyczność
W przypadku stwierdzenia odchyleń wskaźników funkcji wątroby wskazujących na idiosynkrazję wątrobę spowodowaną stosowaniem leków, należy całkowicie przerwać leczenie darolutamidem (patrz punkt „Działania niepożądane”).
Jednoczesne stosowanie z innymi lekami
Stosowanie silnych induktorów CYP3A4 i P-gp podczas leczenia darolutamidem może obniżyć stężenie darolutamidu w osoczu, dlatego nie jest zalecane, chyba że nie ma dostępnej alternatywy terapeutycznej. Należy rozważyć możliwość zastosowania alternatywnego leku towarzyszącego o niższym potencjale indukcji CYP3A4 lub P-gp (patrz punkt „Interakcje z innymi lekami i inne formy interakcji”).
Należy monitorować pacjentów pod kątem wystąpienia działań niepożądanych podczas stosowania substratów BCRP, OATP1B1 i OATP1B3, ponieważ jednoczesne stosowanie z darolutamidem może zwiększyć stężenie tych substratów w osoczu.
Należy unikać jednoczesnego stosowania z rosuwastatyną, chyba że nie ma dostępnej alternatywy terapeutycznej (patrz punkt „Interakcje z innymi lekami i inne formy interakcji”).
Leczenie deprivacji androgenów może wydłużać odstęp QT
U pacjentów z czynnikami ryzyka wydłużenia odstępu QT w wywiadzie oraz u pacjentów otrzymujących jednocześnie leki mogące wydłużać odstęp QT (patrz punkt „Interakcje z innymi lekami i inne formy interakcji”), lekarze powinni ocenić stosunek korzyści do ryzyka, w tym prawdopodobieństwo wystąpienia torsade de pointes, przed rozpoczęciem terapii lekiem Nubeka.
Informacja o substancjach pomocniczych
Lek Nubeka zawiera laktozę. Pacjentom z rzadkimi dziedzicznymi zaburzeniami, takimi jak nietolerancja galaktozy, niedobór laktoazy lub zaburzenia wchłaniania glukozy-galaktozy, nie należy podawać tego leku.
Stosowanie w czasie ciąży lub karmienia piersią
Ten lek jest przeciwwskazany u kobiet w wieku rozrodczym. Nie należy go stosować kobietom w ciąży ani kobietom karmiącym piersią (patrz punkty „Wskazania” i „Przeciwwskazania”).
Kobiety w wieku rozrodczym / antykoncepcja dla mężczyzn i kobiet
Nie wiadomo, czy darolutamid lub jego metabolity występują w nasieniu. Jeśli pacjent prowadzi aktywne życie seksualne z kobietą w wieku rozrodczym, w trakcie i przez 1 tydzień po zakończeniu terapii lekiem Nubeka, w celu zapobiegania ciążę należy stosować skuteczną metodę antykoncepcji (< 1 % częstości niepowodzeń rocznie).
Ciąża
Ze względu na mechanizm działania darolutamid może szkodzić płodowi. Badania przedkliniczne dotyczące toksyczności rozrodczej nie były prowadzone (patrz punkt „Dane przedkliniczne dotyczące bezpieczeństwa”).
Nie wiadomo, czy darolutamid lub jego metabolity występują w nasieniu. Jeśli pacjent prowadzi aktywne życie seksualne z ciężarną kobietą, w trakcie i przez 1 tydzień po zakończeniu terapii lekiem Nubeka należy stosować prezerwatywę. Należy unikać wpływu inhibitorów receptorów androgenowych na rozwój płodu poprzez przekazanie nasienia ciężarnej kobiecie, ponieważ może to wpłynąć na rozwój płodu.
Karmienie piersią
Nie wiadomo, czy darolutamid lub jego metabolity wydzielają się w mleko matki. Nie przeprowadzono badań na zwierzętach w celu oceny wydzielania darolutamidu lub jego metabolitów z mlekiem (patrz punkt „Dane przedkliniczne dotyczące bezpieczeństwa”). Nie można wykluczyć ryzyka dla niemowlęcia karmionego piersią.
Fertylność
Nie ma danych dotyczących wpływu darolutamidu na płodność człowieka.
Zgodnie z badaniami na zwierzętach Nubeka może pogarszać płodność u mężczyzn posiadających potencjał rozrodczy (patrz punkt „Dane przedkliniczne dotyczące bezpieczeństwa”).
Wpływ na zdolność prowadzenia pojazdów lub obsługi mechanizmów
Lek Nubeka nie wpływa lub ma nieznaczny wpływ na szybkość reakcji podczas prowadzenia pojazdów lub obsługi mechanizmów.
Sposób stosowania i dawki
Leczenie powinno rozpoczynać i kontrolować się przez lekarza posiadającego doświadczenie w leczeniu raka gruczołu krokowego.
Dawkowanie
Zalecana dawka wynosi 600 mg darolutamidu (dwie tabletki po 300 mg) dwa razy dziennie, co odpowiada całkowitej dawce dobowej 1200 mg (patrz podrozdział „Farmakokinetyka”).
Stosowanie darolutamidu należy kontynuować aż do postępu choroby lub wystąpienia nieakceptowalnej toksyczności.
U pacjentów, którzy nie byli kastratyzowani chirurgicznie, należy kontynuować kastrację farmakologiczną przy użyciu analogu LH-RH.
Rak gruczołu krokowego wrażliwy na hormony z przerzutami (mHRPCZ)
Pacjenci z mHRPCZ powinni rozpocząć leczenie darolutamidem w połączeniu z doksorubicyną (patrz sekcja „Właściwości farmakologiczne”). Pierwszy z 6 cykli doksorubicyny należy podać w ciągu 6 tygodni od rozpoczęcia terapii darolutamidem. Należy przestrzegać zaleceń zawartych w ulotce do doksorubicyny przeznaczonej do stosowania medycznego. Leczenie darolutamidem należy kontynuować aż do postępu choroby lub wystąpienia nieakceptowalnej toksyczności, nawet jeśli cykl stosowania doksorubicyny zostanie odłożony, przerwany lub zakończony.
Pominięta dawka
Pominiętą dawkę należy przyjąć tak szybko, jak tylko pacjent sobie przypomni, ale tylko w tym samym dniu. Pacjent nie powinien przyjmować dwóch dawek jednocześnie, aby nadrobić pominiętą dawkę.
Zmiana dawkowania
W przypadku wystąpienia toksyczności ≥ stopnia 3 lub nieznośnej reakcji niepożądanej związanej ze stosowaniem darolutamidu (patrz sekcje „Szczególne ostrzeżenia i środki ostrożności” oraz „Reakcje niepożądane”), należy pominąć dawkę lub zmniejszyć dawkowanie do 300 mg dwa razy dziennie do czasu ustąpienia objawów. Następnie leczenie można wznowić w dawce 600 mg dwa razy dziennie. Zmniejszanie dawki poniżej 300 mg dwa razy dziennie nie jest zalecane, ponieważ skuteczność takiego stosowania nie została ustalona.
Osobliwe grupy pacjentów
Pacjenci w podeszłym wieku. U pacjentów w podeszłym wieku nie jest wymagana korekta dawki (patrz podrozdział „Farmakokinetyka”).
Naruszenie funkcji nerek. U pacjentów z łagodnym lub umiarkowanym zaburzeniem funkcji nerek korekta dawki nie jest wymagana. U pacjentów z ciężką niewydolnością nerek (eGFR 15–29 ml/min/1,73 m²), którzy nie poddają się hemodializie, zalecana dawka początkowa wynosi 300 mg dwa razy dziennie (patrz sekcja „Szczególne ostrzeżenia i środki ostrożności” oraz podrozdział „Farmakokinetyka”).
Naruszenie funkcji wątroby. U pacjentów z łagodnym zaburzeniem funkcji wątroby korekta dawki nie jest wymagana. Dane dotyczące farmakokinetyki darolutamidu u pacjentów z umiarkowanym zaburzeniem funkcji wątroby są ograniczone. Stosowanie darolutamidu u pacjentów z ciężkim zaburzeniem funkcji wątroby nie zostało zbadane.
U pacjentów z umiarkowanym i ciężkim zaburzeniem funkcji wątroby (klasy B i C wg skali Childa-Pugh) zalecana dawka początkowa wynosi 300 mg dwa razy dziennie (patrz sekcja „Szczególne ostrzeżenia i środki ostrożności” oraz podrozdział „Farmakokinetyka”).
Sposób stosowania
Lek Nubeka przeznaczony jest do doustnego stosowania.
Tabletki należy przyjmować w całości podczas posiłku (patrz podrozdział „Farmakokinetyka”).
Dzieci
Brak doświadczenia w stosowaniu darolutamidu u pacjentów pediatrycznych.
Przedawkowanie
Najwyższa dawka darolutamidu zbadana klinicznie wynosiła 900 mg dwa razy dziennie, co odpowiada całkowitej dawce dobowej 1800 mg. Przy tej dawce nie zaobserwowano efektów toksycznych ograniczających dawkę. Ze względu na nasycenie procesu absorpcji (patrz podrozdział „Farmakokinetyka”) oraz brak dowodów ostrej toksyczności, nie przewiduje się, że przyjęcie dawki darolutamidu przekraczającej zalecaną dawkę spowoduje efekty toksyczne.
W przypadku przyjęcia dawki przekraczającej zalecaną, leczenie darolutamidem może być kontynuowane według harmonogramu następną dawką.
Nie istnieje specyficzny antydotum na darolutamid i objawy przedawkowania nie zostały ustalone.
Efekty uboczne
Najczęstsze efekty uboczne obserwowane u pacjentów z:
- nmCRPC leczonych darolutamidem – zmęczenie/stany asteniczne (15,8%);
- mCRPC leczonych darolutamidem w połączeniu z doucetakselem – wysypka (16,6%) oraz nadciśnienie (13,8%).
Aby uzyskać dodatkowe informacje dotyczące bezpieczeństwa stosowania darolutamidu w połączeniu z innymi lekami, należy zapoznać się z instrukcją do stosowania poszczególnych leków.
Efekty uboczne obserwowane u pacjentów z nmCRPC leczonych darolutamidem przedstawiono w tabeli 3. Efekty uboczne obserwowane u pacjentów z mCRPC leczonych darolutamidem w połączeniu z doucetakselem przedstawiono w tabeli 4.
Efekty uboczne sklasyfikowano zgodnie z układami narządów. Zestawiono je według częstości występowania. Częstość występowania efektów ubocznych określono następująco: bardzo często (≥ 1/10), często (≥ 1/100 do < 1/10), rzadko (≥ 1/1000 do < 1/100), rzadko (≥ 1/10000 do < 1/1000), bardzo rzadko (< 1/10000), nieznane (nie można oszacować na podstawie dostępnych danych). W każdej grupie według częstości efekty uboczne wymieniono w kolejności zmniejszania się nasilenia.
Tabela 3. Efekty uboczne zarejestrowane w badaniu ARAMISa
| Klasa układu narządów (MedDRA) |
Bardzo często |
Często |
| Zaburzenia serca |
Choroba wieńcowa b, niewydolność serca c |
|
| Zaburzenia skóry i tkanki podskórnej |
Wysypka d |
|
| Zaburzenia układu mięśniowo-szkieletowego i tkanki łącznej |
Ból kończyn, ból mięśniowo-szkieletowy, złamania |
|
| Zaburzenia ogólne i reakcje w miejscu podania |
Znużenie/stany asteniczne e |
|
| Badania f |
Obniżenie liczby neutrofili, podwyższenie stężenia bilirubiny we krwi, podwyższenie AST |
a Średnia długość wpływu wynosiła 14,8 miesiąca (zakres od 0,0 do 44,3 miesiąca) u pacjentów otrzymujących darolutamid oraz 11,0 miesiąca (zakres od 0,1 do 40,5 miesiąca) u pacjentów otrzymujących placebo.
b W tym zespół wieńcowy miażdżycowy, chorobę niedokrwienną tętnic wieńcowych, zamknięcie tętnicy wieńcowej, zwężenie tętnicy wieńcowej, zespół wieńcowy ostry, zawał mięśnia sercowego ostry, dławicę piersiową, dławicę piersiową niestabilną, zawał mięśnia sercowego, niedokrwienie mięśnia sercowego.
c W tym niewydolność serca, ostrą niewydolność serca, przewlekłą niewydolność serca, niewydolność serca zastoinową, szok kardiogenny.
d W tym wysypkę, wysypkę makularną, wysypkę makulopapularną, wysypkę papularną, wysypkę pustularną, zaczerwienienie, zapalenie skóry.
e W tym zmęczenie i osłabienie, letarg i niedomaganie.
f Ogólne kryteria nomenklatury zaburzeń niepożądanych, wersja 4.03. Częstość oparta na wartościach zgłaszanych jako odchylenia od normy badań laboratoryjnych.
Tabela 4. Reakcje niepożądane zaobserwowane u pacjentów z mCRPC otrzymujących darolutamid w połączeniu z doksotakselem w badaniu ARASENSa, b
| Klasa układu narządów (MedDRA) |
Bardzo często |
Często |
| Zaburzenia naczyniowe |
Przewlekłe nadciśnienie tętniczec |
|
| Zaburzenia skóry i tkanki podskórnej |
Wysypkad, e |
|
| Zaburzenia układu mięśniowo-szkieletowego i tkanki łącznej |
Przerwy |
|
| Zaburzenia układu rozrodczego i gruczołów mlekowych |
Ginekomastia |
|
| Badaniaf |
Obniżenie liczby neutrofili, podwyższenie poziomu bilirubiny we krwi, podwyższenie ALAT, podwyższenie ASPAT |
a Średni czas działania wynosił 41,0 miesiąca (zakres od 0,1 do 56,5 miesiąca) u pacjentów otrzymujących darolutamid + doksetaksel oraz 16,7 miesiąca (zakres od 0,3 do 55,8 miesiąca) u pacjentów otrzymujących placebo + doksetaksel.
b Częstotliwość działań niepożądanych nie może być oddzielnie powiązana z darolutamidem, ale może zależeć od innych leków stosowanych w połączeniu.
c Obejmuje nadciśnienie tętnicze, podwyższenie ciśnienia tętniczego, nadciśnieniowy kryzys.
d Obejmuje wysypkę, wysypkę lekową, wysypkę rumieniową, wysypkę grzybiastą, wysypkę makularną, wysypkę makulopapularną, wysypkę grudkową, świąd, wysypkę pęcherzykową, wysypkę pęcherzykową, zaczerwienienie, zapalenie skóry.
e Częstotliwość była najwyższa w pierwszych 6 miesiącach leczenia.
f Ogólne Kryteria Terminologiczne działań niepożądanych, wersja 4.03. Częstotliwość oparta jest na wartościach zgłaszanych jako odchylenia wyników laboratoryjnych.
Opis wybranych działań niepożądanych
Wskazniki funkcji wątroby
Podczas leczenia darolutamidem zgłaszano przypadki idiosyncratycznego uszkodzenia wątroby spowodowanego lekami, z podwyższeniem alaninotransaminazy (ALT) i/lub asparaginianotransaminazy (AST) do ≥ 5 i ≥ 20-krotności górnej granicy normy (GGN) w stopniu 3 i 4, w tym podwyższenie poziomu transaminaz z jednoczesnym wzrostem bilirubiny ogólnej do 2 × GGN. Czas do wystąpienia wynosił od 1 do 12 miesięcy od rozpoczęcia stosowania darolutamidu. W wielu przypadkach podwyższenie ALT i AST było odwracalne po odstawieniu darolutamidu. Zobacz zalecenia w sekcji „Szczególne środki ostrożności”.
Niemetastatyczny rakiem prostaty opornym na kastrację (nmCRPC)
Zmęczenie
Zgłaszano uczucie zmęczenia/astenii u 15,8 % pacjentów otrzymujących darolutamid i u 11,4 % pacjentów otrzymujących placebo. Zdarzenia najwyższego stopnia 3 odnotowano u 0,6 % pacjentów otrzymujących darolutamid i u 1,1 % pacjentów otrzymujących placebo. Zmęczenie (bez astenii, osłabienia lub niedowolności) występowało w większości przypadków (12,1 % pacjentów otrzymujących darolutamid i 8,7 % pacjentów otrzymujących placebo).
Złamania
Złamania wystąpiły u 4,2 % pacjentów otrzymujących darolutamid i u 3,6 % pacjentów otrzymujących placebo.
Choroba niedokrwienna serca i niewydolność serca
Choroba niedokrwienna serca wystąpiła u 3,2 % pacjentów otrzymujących darolutamid i u 2,5 % pacjentów otrzymujących placebo. Zdarzenia stopnia 5 zaobserwowano u 0,3 % pacjentów otrzymujących darolutamid i u 0,2 % pacjentów otrzymujących placebo. Niewydolność serca wystąpiła u 1,9 % pacjentów otrzymujących darolutamid i u 0,9 % pacjentów otrzymujących placebo.
Zmniejszenie liczby neutrofili
Zmniejszenie liczby neutrofili obserwowano jako odchylenie wyników laboratoryjnych u 19,6 % pacjentów otrzymujących darolutamid i u 9,4 % pacjentów otrzymujących placebo. Średni czas do osiągnięcia najniższego poziomu wynosił 256 dni.
Odchylenia laboratoryjne występowały głównie w stopniu 1–2. Zmniejszenie liczby neutrofili w stopniu 3 i 4 zaobserwowano odpowiednio u 3,5 % i 0,5 % pacjentów. Tylko jeden pacjent całkowicie odstawił darolutamid z powodu neutropenii. Neutropenia była tymczasowa lub odwracalna (88 % pacjentów) i nie była związana z żadnymi klinicznie istotnymi objawami lub objawami towarzyszącymi.
Podwyższenie poziomu bilirubiny we krwi
Zgłaszano podwyższenie poziomu bilirubiny jako odchylenie wyników laboratoryjnych u 16,4 % pacjentów otrzymujących darolutamid i u 6,9 % pacjentów otrzymujących placebo. Epizody były głównie stopnia 1 lub 2, nie związane z żadnymi klinicznie istotnymi objawami lub objawami towarzyszącymi i były odwracalne po odstawieniu darolutamidu. Podwyższenie bilirubiny w stopniu 3 zaobserwowano u 0,1 % pacjentów otrzymujących darolutamid i u 0 % pacjentów otrzymujących placebo. W grupie darolutamidu średni czas do pierwszego wzrostu poziomu bilirubiny wynosił 153 dni, a średnia długość pierwszego epizodu wynosiła 182 dni. Żaden pacjent nie odstawił leczenia z powodu podwyższenia poziomu bilirubiny.
Podwyższenie AST
Zgłaszano podwyższenie AST jako odchylenie wyników laboratoryjnych u 22,5 % pacjentów otrzymujących darolutamid i u 13,6 % pacjentów otrzymujących placebo. Epizody były głównie stopnia 1 lub 2, nie związane z żadnymi klinicznie istotnymi objawami lub objawami towarzyszącymi i były odwracalne po odstawieniu darolutamidu. Podwyższenie AST w stopniu 3 zaobserwowano u 0,5 % pacjentów otrzymujących darolutamid i u 0,2 % pacjentów otrzymujących placebo. W grupie darolutamidu średni czas do pierwszego wzrostu AST wynosił 258 dni, a średnia długość pierwszego epizodu wynosiła 118 dni. Żaden pacjent nie odstawił leczenia z powodu podwyższenia AST.
Metastatyczny rakiem prostaty wrażliwym na hormony (mCRPC)
Nadciśnienie tętnicze
W badaniu ARASENS o nadciśnieniu tętniczym zgłaszano u 13,8 % pacjentów otrzymujących darolutamid + doksetaksel i u 9,4 % pacjentów otrzymujących placebo + doksetaksel.
Nadciśnienie tętnicze stopnia 3 zarejestrowano u 6,4 % pacjentów otrzymujących darolutamid + doksetaksel w porównaniu z 3,5 % pacjentów otrzymujących placebo + doksetaksel. Po jednym przypadku nadciśnienia stopnia 4 odnotowano w każdej grupie leczenia.
Zgłoszono jeden przypadek nadciśnienia stopnia 5 z miażdżycą stopnia 5 w grupie darolutamid + doksetaksel. Pacjent ten miał długą historię nadciśnienia i palenia tytoniu, a incydent wystąpił ponad 3 lata po rozpoczęciu leczenia darolutamidem. O przypadkach nadciśnienia tętniczego zgłaszano częściej u pacjentów bez historii nadciśnienia w obu grupach leczenia.
Złamania
Złamania występowały u 7,5 % pacjentów otrzymujących darolutamid + doksetaksel i u 5,1 % pacjentów otrzymujących placebo + doksetaksel.
Zmniejszenie liczby neutrofili
Zgłaszano zmniejszenie liczby neutrofili jako odchylenie wyników laboratoryjnych u 50,6 % pacjentów otrzymujących darolutamid + doksetaksel i u 45,5 % pacjentów otrzymujących placebo + doksetaksel. Zmniejszenie liczby neutrofili w stopniu 3 i 4 zaobserwowano u 34,4 % pacjentów otrzymujących darolutamid + doksetaksel i u 31,4 % pacjentów otrzymujących placebo + doksetaksel. W obu grupach leczenia liczba neutrofili spadła, a neutropenia była najwyższa w pierwszych miesiącach leczenia, po czym częstość i ciężkość zdarzeń zmniejszyły się.
Podwyższenie poziomu bilirubiny we krwi
Podwyższenie poziomu bilirubiny we krwi obserwowano jako odchylenie wyników laboratoryjnych u 19,6 % pacjentów otrzymujących darolutamid + doksetaksel i u 10,0 % pacjentów otrzymujących placebo + doksetaksel. Zdarzenia były głównie stopnia 1 lub 2. Podwyższenie bilirubiny w stopniu 3 i 4 zaobserwowano u 0,5 % pacjentów otrzymujących darolutamid + doksetaksel i u 0,3 % pacjentów otrzymujących placebo + doksetaksel.
Podwyższenie ALT i AST
Zgłaszano podwyższenie ALT jako odchylenie wyników laboratoryjnych u 42,3 % pacjentów otrzymujących darolutamid + doksetaksel i u 38,0 % pacjentów otrzymujących placebo + doksetaksel. Zgłaszano podwyższenie AST jako odchylenie wyników laboratoryjnych u 43,9 % pacjentów otrzymujących darolutamid + doksetaksel i u 39,3 % pacjentów otrzymujących placebo + doksetaksel. Podwyższenie ALT i AST było głównie stopnia 1. Podwyższenie ALT w stopniu 3 i 4 zaobserwowano u 3,7 % pacjentów otrzymujących darolutamid + doksetaksel i u 3,0 % pacjentów otrzymujących placebo + doksetaksel. Podwyższenie AST w stopniu 3 i 4 zaobserwowano u 3,6 % pacjentów otrzymujących darolutamid + doksetaksel i u 2,3 % pacjentów otrzymujących placebo + doksetaksel.
Zgłaszanie podejrzewanych działań niepożądanych
Zgłaszanie działań niepożądanych po rejestracji leku ma istotne znaczenie. Umożliwia to monitorowanie stosunku korzyści do ryzyka w przypadku stosowania tego leku. Osoby medyczne i farmaceutyczne, a także pacjenci lub ich prawni przedstawiciele, powinni zgłaszać wszystkie przypadki podejrzewanych działań niepożądanych oraz braku skuteczności leku poprzez Automatyczny System Informacyjny nadzoru farmakologicznego pod adresem: https://aisf.dec.gov.ua.
Okres ważności. 3 lata.
Warunki przechowywania
Nie wymaga specjalnych warunków przechowywania. Przechowywać w miejscu niedostępnym dla dzieci.
Opakowanie
Po 16 tabletek w blistrze, 7 blisterów w tekturowym pudełku.
Kategoria wydawania. Na receptę.
Producent. Orion Corporation, Orion Pharma.
Adres producenta i miejsce prowadzenia działalności
Joensuunkatu 7, 24100 Salo, Finlandia.