Nubeka
UkraineTable of Contents
INSTRUCTIONS FOR MEDICAL USE OF THE MEDICINAL PRODUCT NUBECA (NUBEQA)
Composition:
active substance: darolutamide;
1 film-coated tablet contains 300 mg of darolutamide;
excipients: calcium hydrogen phosphate, sodium croscarmellose, lactose monohydrate, magnesium stearate, povidone K 30; film coating: white lacquer (hypromellose 15 cP, lactose monohydrate, macrogol 3350, titanium dioxide (E 171)).
Pharmaceutical form. Film-coated tablets.
Basic physicochemical properties: oval, film-coated tablets, white to almost white, with the imprint «BAYER» on one side and «300» on the other.
Pharmacotherapeutic group. Agents used in hormonal therapy. Hormone antagonists and related agents. Antiandrogenic agents. Darolutamide.
ATC code L02B B06.
Pharmacological Properties
Pharmacodynamics
Mechanism of action. Darolutamide is an androgen receptor (AR) inhibitor with a flexible, polar-substituted pyrazole structure, which binds with high affinity directly to the ligand-binding domain of the receptor.
Darolutamide competitively inhibits androgen binding, AR nuclear translocation, and AR-mediated transcription. Its major metabolite, keto-darolutamide, demonstrated in vitro activity similar to that of darolutamide. Treatment with darolutamide reduces prostate tumor cell proliferation, resulting in potent antitumor activity.
Pharmacodynamic effects. No mean QTcF interval prolongation (i.e., exceeding 10 ms) was observed after oral administration of 600 mg darolutamide twice daily compared to placebo.
Clinical efficacy and safety. Efficacy and safety were established in two randomized, placebo-controlled, multicenter Phase III trials involving patients with non-metastatic castration-resistant prostate cancer (nmCRPC) (study ARAMIS) and metastatic hormone-sensitive prostate cancer (mHSPC) (study ARASENS). All patients concurrently received a gonadotropin-releasing hormone (GnRH) analogue or had undergone bilateral orchidectomy.
Non-metastatic castration-resistant prostate cancer (nmCRPC)
The efficacy and safety of darolutamide were evaluated in a randomized, double-blind, placebo-controlled, multicenter Phase III trial (ARAMIS) in patients with non-metastatic (as assessed by conventional CT, bone scan, and MRI) castration-resistant prostate cancer and a prostate-specific antigen doubling time (PSADT) ≤ 10 months.
Patients were eligible for inclusion if they had three consecutive rises in prostate-specific antigen (PSA) levels from the nadir, with at least a one-week interval between measurements during androgen deprivation therapy, a screening PSA ≥ 2 ng/mL, and a post-castration serum testosterone level < 1.7 nmol/L.
Patients with a history of seizures were permitted to participate in the study. In the darolutamide group, 12 patients (0.21%) had a history of seizures.
Patients with uncontrolled hypertension or recent (within the past 6 months) stroke, myocardial infarction, severe/unstable angina, coronary/peripheral artery bypass grafting, or New York Heart Association (NYHA) Class III or IV congestive heart failure were excluded from the study.
Patients who had previously received second-generation androgen receptor (AR) inhibitors such as enzalutamide, apalutamide, or darolutamide, or CYP17 enzyme inhibitors such as abiraterone acetate, as well as patients who had received systemic corticosteroids at a dose exceeding 10 mg prednisone equivalent per day for more than 28 days prior to randomization, were excluded from the study.
Overall, 1509 patients were randomized 2:1 to receive 600 mg darolutamide orally twice daily (n = 955) or placebo (n = 554).
Patients with pelvic lymph nodes < 2 cm in short axis located below the aortic bifurcation were included in the study. The absence or presence of metastases was determined by independent centralized radiological assessment. This assessment was performed in 89 patients, in whom metastases were retrospectively identified at baseline. Randomization was stratified by PSADT (≤ 6 months or > 6 months) and by use of bone-targeted therapy at baseline (yes or no).
Demographic characteristics and disease features were balanced between treatment groups. The median age of patients was 74 years (range 48–95); 9% of patients were aged 85 years or older. 79% of patients were of Caucasian race, 13% were of Asian origin, and 3% were Black. The majority of patients had a Gleason score of 7 or higher at diagnosis (73%). The median PSADT was 4.5 months. Nine percent (9%) of patients had prior orchidectomy, 25% had prior prostatectomy, and 50% had received at least one prior radiotherapy. Seventy-six percent (76%) of patients had received more than one prior anti-hormonal therapy. Patients had an Eastern Cooperative Oncology Group (ECOG PS) performance status of 0 (69%) or 1 (31%).
Treatment with darolutamide continued until radiologically confirmed disease progression, as assessed by traditional imaging (CT, bone scan, MRI) via blinded centralized evaluation, unacceptable toxicity, or patient withdrawal from the study.
The primary efficacy endpoint was metastasis-free survival (MFS). Secondary endpoints included overall survival (OS), time to pain progression, time to initiation of first cytotoxic chemotherapy for prostate cancer, and time to first symptomatic skeletal event (defined as occurrence of any of the following: external beam radiation therapy for palliation of skeletal symptoms, new symptomatic pathological bone fracture, spinal cord compression, or tumor-related orthopedic surgical intervention).
Treatment with darolutamide resulted in improved MFS compared to placebo (see Table 1 and Fig. 1).
MFS outcomes were consistent across all patient subgroups regardless of PSADT, prior use of bone-targeting agents, or locoregional disease. Additional subgroups with consistent MFS results included baseline PSA level, Gleason score at diagnosis, patient age, geographic region, ECOG PS at baseline, race, and number of prior hormonal therapy regimens.
After the primary MFS analysis, once the study was unblinded, patients receiving placebo were offered darolutamide treatment (crossover option). Of the 554 patients randomized to the placebo group, 170 (31%) crossed over to darolutamide treatment. The OS analysis was not adjusted for the impact of crossover.
At the time of final analysis, treatment with darolutamide resulted in a statistically significant improvement in overall survival compared to placebo (median not reached in either group; see Table 1 and Fig. 2). Statistically significant increases in the time to pain progression, time to initiation of first cytotoxic chemotherapy, and time to first symptomatic skeletal event were also observed compared to placebo (see Table 1).
At the time of final analysis, the median duration of treatment for patients receiving darolutamide was 33.3 months (range: 0.0 to 74.0 months) during the combined double-blind and open-label periods.
All analyses were performed in the full analysis set.
Table 1. Efficacy results from the ARAMIS study
| Measure of efficacy |
Number (%) of patients with events |
Median (months) (95 % CI) |
Hazard ratio (HR)b (95 % confidence interval [CI]) p-value (two-sided) |
||
| Enzalutamide (N = 955) |
Placeboa (N = 554) |
Enzalutamide (N = 955) |
Placeboa (N = 554) |
||
| Metastasis-free survivalc |
221 (23.1 %) |
216 (39.0 %) |
40.4 (34.3; NR) |
18.4 (15.5; 22.3) |
0.413 (0.341; 0.500) < 0.000001 |
| Overall survival |
148 (15.5 %) |
106 (19.1 %) |
NR (56.1; NR) |
NR (46.9; NR) |
0.685 (0.533; 0.881) 0.003048 |
| Time to pain progressionc, d |
251 (26.3 %) |
178 (32.1 %) |
40.3 (33.2; 41.2) |
25.4 (19.1; 29.6) |
0.647 (0.533; 0.785) 0.000008 |
| Time to initiation of first cytotoxic chemotherapy |
127 (13.3 %) |
98 (17.7 %) |
NR (NR, NR) |
NR |
0.579 (0.444; 0.755) 0.000044 |
| Time to first symptomatic skeletal event |
29 (3.0 %) |
28 (5.1 %) |
NR (NR, NR) |
(NR, NR) |
0.484 (0.287; 0.815) 0.005294 |
a Including 170 patients who transitioned to darolutamide without a blinded period.
b Hazard ratio < 1 for darolutamide.
c For MFS and time to pain progression, the primary analysis is considered the final analysis.
d Patient-reported outcomes according to the Brief Pain Inventory – Short Form questionnaire.
NR – not reached.
Treatment with darolutamide resulted in a longer median metastasis-free survival (MFS) (36.8 vs 14.8 months, HR = 0.380, nominal p < 0.000001) and time to PSA progression (29.5 vs 7.2 months, HR = 0.164, nominal p < 0.000001). The effect was consistent across all survival endpoints (MFS, OS, and PSA).
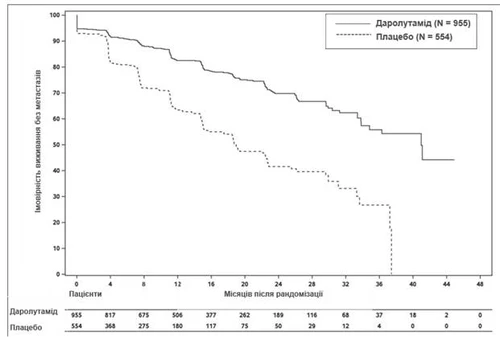
Fig. 1. Kaplan–Meier curves for metastasis-free survival (ARAMIS)
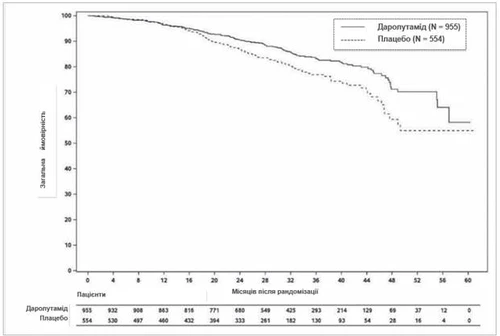
Fig. 2. Kaplan–Meier curves for overall survival (ARAMIS)
Patients receiving darolutamide in the ARAMIS study (during the double-blind period) demonstrated a significantly higher confirmed response rate in PSA levels (defined as a decrease of ≥ 50% from baseline) compared to those receiving placebo: 84.0% vs 7.9% (difference = 76.1%, p < 0.000001 (nominal p-value, for informational purposes only)).
Metastatic hormone-sensitive prostate cancer (mHSPC)
The efficacy and safety of darolutamide in combination with docetaxel were evaluated in a multicenter, double-blind, placebo-controlled Phase III study (ARASENS) in patients with mHSPC. A total of 1306 patients were randomized 1:1 to receive either 600 mg of darolutamide orally twice daily (n = 651) or placebo (n = 655), both in combination with 75 mg/m² docetaxel for 6 cycles. Treatment with darolutamide or placebo was continued until symptomatic disease progression, change in anticancer therapy, unacceptable toxicity, death, or patient withdrawal from the study.
Presence of metastases was assessed by independent centralized radiological evaluation. Patients with disease limited to regional lymph nodes (M0) were excluded from the study. Randomization was stratified by disease stage (metastases only in non-regional lymph nodes (M1a), bone metastases with or without lymph node involvement (M1b), or visceral metastases with or without lymph node or bone involvement (M1c)) and by baseline alkaline phosphatase level (< or ≥ upper limit of normal) at study entry. Patients with brain metastases were allowed to enroll, but none were included.
Demographic and disease characteristics were balanced between treatment groups. The median age was 67 years (range 41–89); 0.5% of patients were aged 85 years or older, 52% were Caucasian, 36% were of Asian origin, and 4% of study participants were Black. Most patients had a Gleason score of 8 or higher at diagnosis (78%). 71% of patients had an ECOG PS of 0, and 29% had an ECOG PS of 1. There were 86.1% of patients with de novo disease and 12.9% with recurrent disease. At baseline, 3% of patients had M1a, 79.5% had M1b, and 17.5% had M1c; alkaline phosphatase was < ULN in 44.5% of patients and ≥ ULN in 55.5% of patients; the median baseline PSA level was 30.3 µg/L and 24.2 µg/L in the darolutamide and placebo groups, respectively. Patients with a history of seizures were allowed to participate, and 4 patients (0.6%) were included in the darolutamide + docetaxel group.
77.0% of patients had high-volume disease, and 23.0% had low-volume disease. High-volume disease was defined as the presence of visceral metastases or ≥4 bone lesions, with at least one metastasis outside the spine and pelvis. Approximately 25% of patients received concomitant treatment with bisphosphonates or denosumab.
The primary efficacy endpoint was overall survival (OS). Secondary endpoints included time to development of castration-resistant prostate cancer, time to pain progression, time to symptomatic skeletal event-free survival, time to first symptomatic skeletal event, time to initiation of next anticancer therapy, time to worsening of disease-related physical symptoms, and time to initiation of opioid use for ≥7 consecutive days. Pain progression was assessed by patient-reported outcomes using the Brief Pain Inventory – Short Form questionnaire, defined as at least a 2-point worsening from baseline and initiation of short- or long-acting opioids for pain relief for ≥7 consecutive days.
The median duration of treatment was 41.0 months (range: 0.1 to 56.5 months) in patients receiving darolutamide + docetaxel and 16.7 months (range: 0.3 to 55.8 months) in patients receiving placebo + docetaxel. 87.6% and 85.5% of patients completed all 6 cycles of docetaxel, and 1.5% and 2.0% of patients did not receive docetaxel in the darolutamide + docetaxel and placebo + docetaxel groups, respectively.
Table 2. Efficacy results from the ARASENS study
| Efficiency parameter |
Number (%) of patients with events |
Median (months) (95% CI) |
Hazard ratio (HR)b (95% confidence interval [CI]) p-value (one-sided)c |
||
| Darolutamide + docetaxel (N = 651) |
Placebo + docetaxel (N = 654)a |
Darolutamide + docetaxel (N = 651) |
Placebo + docetaxel (N = 654)a |
||
| Overall survivald |
229 (35.2%) |
304 (46.5%) |
NR (NR, NR) |
48.9 (44.4; NR) |
0.675 (0.568; 0.801) <0.0001 |
a One patient in the placebo group was excluded from all analyses.
b Risk ratio < 1 in favor of darolutamide.
c Based on stratified log-rank test.
d OS results were consistent across all patient subgroups, including disease extent and alkaline phosphatase levels.
NR – not reached.
Secondary efficacy endpoints showing statistically significant benefit for patients in the darolutamide + docetaxel group compared to the placebo + docetaxel group: time to development of castration-resistant prostate cancer (median NR vs. 19.1 months; HR = 0.357, p < 0.0001); time to first symptomatic skeletal event (median NR vs. NR; HR = 0.712, p = 0.0081); time to initiation of subsequent antineoplastic chemotherapy (median NR vs. 25.3 months; HR = 0.388, p < 0.0001); time to pain progression (median NR vs. 27.5 months; HR = 0.792, p = 0.0058); survival without symptomatic skeletal events (median 51.2 vs. 39.7 months; HR = 0.609, p < 0.0001).
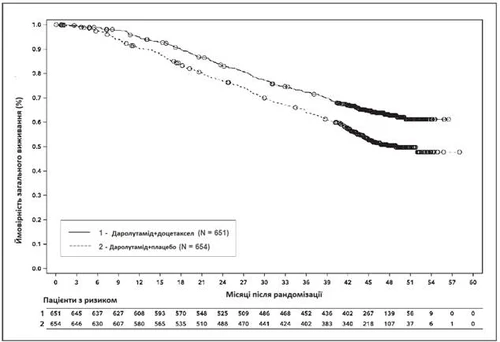
Fig. 3. Kaplan–Meier overall survival curves (ARASENS)a
a OS at 36 months was 72.3% (95% CI, 68.8–75.8) in the darolutamide + docetaxel group versus 63.8% (95% CI, 60.1–67.6) in the placebo + docetaxel group.
OS at 48 months was 62.7% (95% CI, 58.7–66.7) in the darolutamide + docetaxel group versus 50.4% (95% CI, 46.3–54.6) in the placebo + docetaxel group.
Pharmacokinetics
Darolutamide consists of two diastereomers [(S,R)-darolutamide and (S,S)-darolutamide], which interconvert via the major circulating metabolite keto-darolutamide. In vitro, all three compounds show similar pharmacological activity. Darolutamide is poorly soluble in aqueous solvents over a wide pH range and generally dissolves better in organic solvents.
Absorption
Following oral administration of 600 mg (2 tablets of 300 mg) twice daily, peak plasma concentrations of darolutamide at steady state were 4.79 mg/L (coefficient of variation 30.9%) in patients with nmCRPC in the ARAMIS trial and 3.84 mg/L (coefficient of variation 35.6%) in patients with mHSPC in the ARASENS trial. The mean time to reach maximum plasma concentration was 3–4 hours. The ratio of the two diastereomers, (S,R)-darolutamide to (S,S)-darolutamide, changed from 1:1 in the tablet to approximately 1:9 in plasma based on steady-state AUС0–12. At steady state, plasma concentrations are achieved within 2–5 days of repeated twice-daily dosing when administered with food.
The absolute bioavailability after oral administration of a Nubeqa tablet containing 300 mg darolutamide under fasting conditions is approximately 30% of that after intravenous administration.
The bioavailability of darolutamide increased by 2.0–2.5 times when administered with food. A similar increase in exposure was observed for the major metabolite keto-darolutamide.
Distribution
The apparent volume of distribution of darolutamide after intravenous administration is 119 L, indicating extensive distribution throughout the body, both intracellularly and extracellularly.
Darolutamide is moderately (92%) bound to human plasma proteins, with no difference between the two diastereomers. The major metabolite of darolutamide, keto-darolutamide, is highly (99.8%) bound to plasma proteins.
Clinical evaluation of darolutamide penetration across the blood-brain barrier has not been conducted. However, the impact of darolutamide on the brain, in terms of AUC0–24, is very low: 4.5% of plasma exposure after a single dose in rats and 1.9–3.9% after repeated dosing in mice. This indicates low penetration of darolutamide across the intact blood-brain barrier in rats and mice and suggests a low likelihood that darolutamide crosses the intact blood-brain barrier in humans to a clinically significant extent.
Metabolism
The diastereomers (S,R)-darolutamide and (S,S)-darolutamide are capable of interconversion via the metabolite keto-darolutamide, with a preference for (S,S)-darolutamide.
After a single oral dose of 300 mg 14C-darolutamide as an oral solution, keto-darolutamide is the sole major metabolite, with approximately twice the total plasma exposure compared to darolutamide. Darolutamide and keto-darolutamide together account for 87.4% of 14C-radioactivity in plasma, indicating that all other metabolites are insignificant.
Darolutamide is primarily metabolized via oxidative metabolism mediated predominantly by CYP3A4, as well as by direct glucuronidation mediated primarily by UGT1A9 and UGT1A1. Additionally, AKR1C isoforms have been shown to predominantly catalyze the reduction of keto-darolutamide back to the substance diastereomers.
Elimination
The effective half-life of darolutamide and keto-darolutamide in patient plasma is approximately 18–20 hours. Of the two diastereomers comprising darolutamide, (S,R)-darolutamide has a shorter effective half-life of 9 hours compared to (S,S)-darolutamide, which has an effective half-life of 22 hours. The clearance of darolutamide after intravenous administration was 116 mL/min (CV 39.7%). Overall, 63.4% of darolutamide and its metabolites are excreted in urine (approximately 7% unchanged), and 32.4% are excreted in feces. Over 95% of the dose was eliminated within 7 days after administration.
Linearity/Non-linearity
Within the dose range of 100 to 700 mg (after single dose and at steady state), exposure of the two diastereomers and the major metabolite keto-darolutamide increases almost linearly with dose. Due to saturated absorption, no further increase in darolutamide exposure was observed at a dose of 900 mg twice daily.
Special patient populations
Elderly patients. No clinically relevant differences in darolutamide pharmacokinetics were observed (65–95 years of age).
Renal impairment. In a clinical pharmacokinetic study, AUC and Cmax of darolutamide were 2.5 and 1.6 times higher, respectively, in patients with severe renal impairment (estimated glomerular filtration rate [eGFR] of 15–29 mL/min/1.73 m²) compared to healthy volunteers.
Population pharmacokinetic analysis indicates approximately 1.1-, 1.3-, and 1.5-fold higher exposure (AUC) of darolutamide in patients with mild, moderate, and severe renal impairment (eGFR 15–89 mL/min/1.73 m²), respectively, compared to patients with normal renal function.
Pharmacokinetics of darolutamide have not been studied in patients with end-stage renal disease on dialysis (eGFR < 15 mL/min/1.73 m²).
Hepatic impairment. In a clinical pharmacokinetic study, Cmax and AUC of darolutamide were 1.5 and 1.9 times higher, respectively, in patients with moderate hepatic impairment (Child–Pugh class B) compared to healthy volunteers. No data are available for patients with severe hepatic impairment (Child–Pugh class C).
Ethnic differences. No clinically relevant differences in darolutamide pharmacokinetics were observed based on patient ethnicity (Caucasian patients, Japanese patients, non-Japanese Asian patients, Black or African American patients). Population pharmacokinetic analysis showed a 1.56-fold (90% CI 1.43–1.70) increase in geometric mean exposure (AUC) in Japanese patients compared to patients from other regions in the ARAMIS and ARASENS trials.
Preclinical safety data
Systemic toxicity. In repeat-dose toxicity studies in animals, the main findings were effects on male reproductive organs (organ weight reduction with atrophy of the prostate and epididymides). These effects occurred at systemic exposures within or below the expected human exposure (based on AUC comparison). Additional reproductive tissue changes included minimal increase in pituitary vacuolization, atrophy and reduced secretory activity of seminal vesicles and mammary glands in rats, and testicular hypospermia, dilatation of seminiferous tubules, and degeneration in dogs. Changes in male reproductive organs in both species were consistent with the pharmacological activity of darolutamide and resolved or partially resolved after a 4–8 week recovery period.
Embryotoxicity/Teratogenicity. Embryo-fetal toxicity studies have not been conducted.
Reproductive toxicity. Reproductive toxicity studies have not been conducted. However, male fertility is likely to be impaired based on results from repeat-dose toxicity studies in animals, which are consistent with the pharmacological activity of darolutamide.
Genotoxicity and carcinogenicity. Darolutamide did not induce mutations in a microbial mutagenicity assay (Ames test). At high concentrations, darolutamide induced structural chromosomal aberrations in vitro in cultured human lymphocytes. However, no genotoxicity was observed in vivo in a combined bone marrow micronucleus test and Comet assay in the liver and duodenum of rats at exposures exceeding the maximum human exposure.
Oral administration of darolutamide to male transgenic rasH2 mice for 6 months revealed no carcinogenic potential at doses up to 1000 mg/kg/day, corresponding to 0.9–1.3 times the clinical exposure (AUC) of darolutamide and 2.1–2.3 times that of keto-darolutamide at the recommended clinical daily dose of 1200 mg/day. Based on this study, a carcinogenic risk with darolutamide cannot be fully excluded.
Clinical characteristics
Indications
The medicinal product Nubeqa is indicated for the treatment of adult men with:
- non-metastatic castration-resistant prostate cancer (nmCRPC) at high risk of metastasis (see section "Pharmacological properties");
- metastatic hormone-sensitive prostate cancer (mHSPC) – in combination with docetaxel and androgen deprivation therapy (see section "Pharmacological properties").
Contraindications
Hypersensitivity to the active substance or to any of the excipients.
Contraindicated in pregnant women and women of reproductive potential.
Interaction with other medicinal products and other forms of interaction
Effect of other medicinal products on darolutamide
Inducers of CYP3A4 and P-gp
Darolutamide is a substrate of CYP3A4 and P-glycoprotein (P-gp).
The use of strong and moderate inducers of CYP3A4 and inducers of P-gp (e.g. carbamazepine, phenobarbital, St. John's wort, phenytoin, and rifampicin) during darolutamide treatment is not recommended, except when there are no therapeutic alternatives. Consideration should be given to selecting an alternative concomitant medicinal product that does not induce or has a weak potential to induce CYP3A4 or P-gp.
Repeated administration of rifampicin (600 mg), a potent inducer of CYP3A4 and P-gp, together with a single dose of darolutamide (600 mg) with food resulted in a 72% reduction in mean exposure (AUC0-72) and a 52% reduction in Cmax of darolutamide.
Inhibitors of CYP3A4, P-gp and BCRP
Darolutamide is a substrate of CYP3A4, P-gp and breast cancer resistance protein (BCRP).
When inhibitors of CYP3A4, P-gp or BCRP are used, a clinically relevant drug interaction is not expected. Darolutamide may be administered concomitantly with inhibitors of CYP3A4, P-gp or BCRP. Concomitant use of darolutamide with a combined P-gp and potent CYP3A4 inhibitor increases darolutamide exposure, which may increase the risk of adverse reactions to darolutamide. It is recommended to monitor patients more frequently for adverse reactions to darolutamide and adjust the darolutamide dose if necessary.
Administration of itraconazole (200 mg twice daily on day 1 and once daily for the following 7 days), a strong inhibitor of CYP3A4, P-gp and BCRP, with a single dose of darolutamide (600 mg on day 5 with food) resulted in a 1.7-fold increase in mean exposure (AUC0-72) and a 1.4-fold increase in Cmax of darolutamide.
Inhibitors of UGT1A9
Darolutamide is a substrate of UGT1A9. When an inhibitor of UGT1A9 is used, a clinically relevant drug interaction is not expected. Darolutamide may be administered concomitantly with inhibitors of UGT1A9.
Population pharmacokinetic analysis showed that concomitant use of inhibitors of UGT1A9 with darolutamide led to a 1.2-fold increase in exposure (AUC0-72) of darolutamide.
Docetaxel
Administration of darolutamide in combination with docetaxel did not result in clinically relevant changes in the pharmacokinetics of darolutamide in patients with mHSPC (see section "Pharmacological properties").
Effect of darolutamide on other medicinal products
Substrates of BCRP, OATP1B1 and OATP1B3
Darolutamide is an inhibitor of breast cancer resistance protein (BCRP) and organic anion transporting polypeptides (OATP) 1B1 and 1B3.
Concomitant use of rosuvastatin should be avoided, except when there are no therapeutic alternatives. Consideration should be given to selecting an alternative concomitant medicinal product with a lower potential to inhibit BCRP, OATP1B1 and OATP1B3.
Administration of darolutamide (600 mg twice daily for 5 days) prior to a single dose of rosuvastatin (5 mg) with food resulted in approximately a 5-fold increase in mean exposure (AUC) and Cmax of rosuvastatin.
If possible, concomitant use of darolutamide with other BCRP substrates should be avoided. Concomitant use of darolutamide may increase plasma concentrations of other concomitant drugs that are substrates of BCRP, OATP1B1 and OATP1B3 (such as methotrexate, sulfasalazine, fluvastatin, atorvastatin, pitavastatin). Therefore, patients should be monitored for adverse reactions associated with the use of BCRP, OATP1B1 and OATP1B3 substrates. In addition, when used concomitantly with darolutamide, appropriate recommendations provided in the information for these substrates should be followed.
Substrates of P-gp
When administering a P-gp substrate, a clinically relevant drug interaction is not expected. Darolutamide may be administered concomitantly with P-gp substrates (e.g. digoxin, verapamil, or nifedipine). Concomitant use of darolutamide with the sensitive P-gp substrate dabigatran etexilate did not lead to increased exposure (AUC and Cmax) of dabigatran.
Substrates of CYP3A4
Darolutamide is a weak inducer of CYP3A4.
When administering a CYP substrate, a clinically relevant drug interaction is not expected. Darolutamide may be administered concomitantly with CYP substrates (e.g. warfarin, L-thyroxine, omeprazole).
Administration of darolutamide (600 mg twice daily for 9 days) prior to a single dose of the sensitive CYP3A4 substrate midazolam (1 mg) with food resulted in a 29% and 32% reduction in mean exposure (AUC) and Cmax of midazolam, respectively. Darolutamide did not inhibit the metabolism of individual CYP substrates in vitro at clinically relevant concentrations.
Docetaxel
Administration of darolutamide in combination with docetaxel did not result in clinically relevant changes in the pharmacokinetics of docetaxel in patients with mHSPC (see section "Pharmacological properties").
Medicinal products that prolong the QT interval
Since androgen deprivation therapy may prolong the QT interval, concomitant use with medicinal products known to prolong the QT interval or those that may induce torsade de pointes should be carefully evaluated. These include medicinal products such as class IA antiarrhythmics (e.g. quinidine, disopyramide) or class III (e.g. amiodarone, sotalol, dofetilide, ibutilide), methadone, moxifloxacin, and antipsychotics (e.g. haloperidol).
Special precautions for use
Renal impairment
Data on use in patients with severe renal impairment are limited.
Since exposure may be increased, patients should be closely monitored for the occurrence of adverse reactions (see section "Special precautions for use" and subsection "Pharmacokinetics").
Hepatic impairment
Data on use in patients with moderate hepatic impairment are limited. The use of darolutamide in patients with severe hepatic impairment has not been studied.
Since exposure may be increased, patients should be closely monitored for adverse reactions (see section "Special precautions for use" and subsection "Pharmacokinetics").
Recent cardiovascular disease
Patients with clinically significant cardiovascular diseases within the last 6 months, including stroke, myocardial infarction, severe/unstable angina, coronary/peripheral artery bypass grafting, and symptomatic congestive heart failure, were excluded from clinical trials. Therefore, the safety of darolutamide use in these patients has not been established.
If prescribing Nubeqa to a patient with clinically significant cardiovascular disease, the condition should be managed according to established guidelines.
Hepatotoxicity
If liver function test abnormalities indicating drug-induced idiosyncratic liver injury are detected, darolutamide treatment should be permanently discontinued (see section "Adverse reactions").
Concomitant use with other medicinal products
Concomitant use of strong CYP3A4 and P-gp inducers during darolutamide treatment may reduce darolutamide plasma concentrations and is therefore not recommended, except when no therapeutic alternative exists. Consideration should be given to selecting an alternative concomitant medicinal product with lower potential for CYP3A4 or P-gp induction (see section "Interaction with other medicinal products and other forms of interaction").
Patients should be monitored for adverse reactions when substrates of BCRP, OATP1B1, and OATP1B3 are used concomitantly, as co-administration with darolutamide may increase plasma concentrations of these substrates.
Concomitant use with rosuvastatin should be avoided, except when no therapeutic alternative exists (see section "Interaction with other medicinal products and other forms of interaction").
Androgen deprivation therapy may prolong the QT interval
In patients with risk factors for QT interval prolongation and in patients receiving concomitant medicinal products capable of prolonging the QT interval (see section "Interaction with other medicinal products and other forms of interaction"), physicians should assess the benefit-risk ratio, including the risk of torsade de pointes, before initiating treatment with Nubeqa.
Information on excipients
Nubeqa contains lactose. Patients with rare hereditary disorders such as galactose intolerance, lactase deficiency, or glucose-galactose malabsorption should not take this medicinal product.
Use during pregnancy or breastfeeding
This medicinal product is contraindicated in women of reproductive potential. It should not be used in pregnant women or women who are breastfeeding (see sections "Indications" and "Contraindications").
Women of reproductive potential / contraception in men and women
It is unknown whether darolutamide or its metabolites are present in semen. If a patient has sexual intercourse with a woman of reproductive potential, a highly effective method of contraception (< 1% failure rate per year) should be used during treatment and for 1 week after discontinuation of Nubeqa to prevent pregnancy.
Pregnancy
Due to its mechanism of action, darolutamide may cause harm to the fetus. Reproductive toxicity studies have not been conducted in non-clinical settings (see section "Non-clinical safety data").
It is unknown whether darolutamide or its metabolites are present in semen. If a patient has sexual intercourse with a pregnant woman, a condom should be used during treatment and for 1 week after discontinuation of Nubeqa. Exposure of the fetus to androgen receptor inhibitors via seminal fluid should be avoided, as this may affect fetal development.
Breastfeeding
It is unknown whether darolutamide or its metabolites are excreted in human milk. Animal studies to assess excretion of darolutamide or its metabolites in milk have not been conducted (see section "Non-clinical safety data"). A risk to the breastfed child cannot be excluded.
Fertility
There are no data on the effect of darolutamide on human fertility.
Animal studies indicate that Nubeqa may impair fertility in men with reproductive potential (see section "Non-clinical safety data").
Ability to affect reaction speed when driving or operating machinery
Nubeqa has no effect or has a negligible effect on the ability to drive or operate machinery.
Method of Administration and Dosage
Treatment should be initiated and monitored by a physician experienced in the treatment of prostate cancer.
Dosage
The recommended dose is 600 mg of darolutamide (two 300 mg tablets) twice daily, equivalent to a total daily dose of 1200 mg (see section "Pharmacokinetics").
Treatment with darolutamide should be continued until disease progression or unacceptable toxicity.
Patients who have not undergone surgical castration should continue medical castration with a GnRH analogue during treatment.
Metastatic hormone-sensitive prostate cancer (mHSPC)
Patients with mHSPC should initiate treatment with darolutamide in combination with docetaxel (see section "Pharmacological Properties"). The first of 6 cycles of docetaxel should be administered within 6 weeks after starting darolutamide therapy. The recommendations provided in the prescribing information for docetaxel should be followed. Darolutamide treatment should be continued until disease progression or unacceptable toxicity, even if administration of docetaxel is delayed, interrupted, or discontinued.
Missed Dose
If a dose is missed, it should be taken as soon as the patient remembers, but only on the same day. The patient should not take two doses to compensate for a missed dose.
Dose Modifications
If a patient experiences toxicity ≥ grade 3 or an intolerable adverse reaction related to darolutamide (see sections "Special Warnings and Precautions for Use" and "Adverse Reactions"), the dose should be withheld or reduced to 300 mg twice daily until symptoms improve. Treatment may subsequently be resumed at a dose of 600 mg twice daily. Dose reduction below 300 mg twice daily is not recommended, as efficacy at lower doses has not been established.
Special Patient Populations
Elderly patients. Dose adjustment is not required for elderly patients (see section "Pharmacokinetics").
Renal impairment. Dose adjustment is not required for patients with mild or moderate renal impairment. For patients with severe renal impairment (eGFR 15–29 mL/min/1.73 m²) not receiving hemodialysis, the recommended initial dose is 300 mg twice daily (see section "Special Warnings and Precautions for Use" and subsection "Pharmacokinetics").
Hepatic impairment. Dose adjustment is not required for patients with mild hepatic impairment. Pharmacokinetic data in patients with moderate hepatic impairment are limited. The use of darolutamide has not been studied in patients with severe hepatic impairment.
For patients with moderate and severe hepatic impairment (Child-Pugh classes B and C), the recommended initial dose is 300 mg twice daily (see section "Special Warnings and Precautions for Use" and subsection "Pharmacokinetics").
Method of Administration
The medicinal product Nubeqa is intended for oral administration.
Tablets should be taken whole with food (see subsection "Pharmacokinetics").
Children
Experience with darolutamide in pediatric patients is lacking.
Overdose
The highest dose of darolutamide clinically investigated was 900 mg twice daily, equivalent to a total daily dose of 1800 mg. At this dose, no dose-limiting toxic effects were observed. Due to saturable absorption (see subsection "Pharmacokinetics") and lack of evidence of acute toxicity, administration of a dose exceeding the recommended dose is not expected to result in toxic effects.
In the event of an overdose, darolutamide treatment may be continued with the next scheduled dose.
There is no specific antidote for darolutamide, and symptoms of overdose have not been established.
Adverse Reactions
The most common adverse reactions observed in patients with:
- nmCRPC receiving darolutamide were fatigue/asthenic conditions (15.8%);
- mHSPC receiving darolutamide in combination with docetaxel were rash (16.6%) and hypertension (13.8%).
For additional safety information regarding the combined use of darolutamide, refer to the package leaflet of the individual medicinal products.
Adverse reactions observed in patients with nmCRPC receiving darolutamide are listed in Table 3. Adverse reactions observed in patients with mHSPC receiving darolutamide in combination with docetaxel are listed in Table 4.
Adverse reactions are classified by organ system. They are grouped according to their frequency. The frequency of adverse reactions is defined as follows: very common (≥ 1/10), common (≥ 1/100 to < 1/10), uncommon (≥ 1/1000 to < 1/100), rare (≥ 1/10,000 to < 1/1000), very rare (< 1/10,000), not known (cannot be estimated from available data). Within each frequency group, adverse reactions are listed in order of decreasing severity.
Table 3. Adverse reactions recorded in the ARAMIS studya
| System organ class (MedDRA) |
Very common |
Common |
| Cardiac disorders |
Ischaemic heart diseaseb, heart failurec |
|
| Skin and subcutaneous tissue disorders |
Rashd |
|
| Musculoskeletal and connective tissue disorders |
Limb pain, musculoskeletal pain, fractures |
|
| General disorders and administration site conditions |
Fatigue/asthenice |
|
| Investigationsf |
Decreased neutrophil count, increased blood bilirubin, increased AST |
a The median duration of exposure was 14.8 months (range from 0.0 to 44.3 months) in patients receiving darolutamide and 11.0 months (range from 0.1 to 40.5 months) in patients receiving placebo.
b Includes coronary artery atherosclerosis, coronary artery ischemic disease, coronary artery occlusion, coronary artery stenosis, acute coronary syndrome, acute myocardial infarction, angina pectoris, unstable angina, myocardial infarction, myocardial ischemia.
c Includes heart failure, acute heart failure, chronic heart failure, congestive heart failure, cardiogenic shock.
d Includes rash, macular rash, maculopapular rash, papular rash, pustular rash, erythema, dermatitis.
e Includes fatigue and asthenia, lethargy and malaise.
f Common Terminology Criteria for Adverse Events, version 4.03. Frequency is based on values reported as laboratory parameter abnormalities.
Table 4. Adverse reactions observed in patients with mCRPC receiving darolutamide in combination with docetaxel in the ARASENS studya,b
| System organ class (MedDRA) |
Very common |
Common |
| Vascular disorders |
Arterial hypertensionc |
|
| Skin and subcutaneous tissue disorders |
Rashd, e |
|
| Musculoskeletal and connective tissue disorders |
Fractures |
|
| Reproductive system and breast disorders |
Gynaecomastia |
|
| Investigationsf |
Decreased neutrophil count, increased blood bilirubin, increased ALT, increased AST |
a The median duration of effect was 41.0 months (range: 0.1 to 56.5 months) in patients receiving darolutamide + docetaxel, and 16.7 months (range: 0.3 to 55.8 months) in patients receiving placebo + docetaxel.
b The frequency of adverse reactions cannot be attributed specifically to darolutamide alone but may depend on other concomitant medications.
c Includes arterial hypertension, increased blood pressure, hypertensive crisis.
d Includes rash, drug eruption, erythematous rash, follicular rash, macular rash, maculopapular rash, papular rash, pruritus, pustular rash, vesicular rash, erythema, dermatitis.
e The frequency was highest during the first 6 months of treatment.
f Common Terminology Criteria for Adverse Events, version 4.03. Frequency is based on values reported as laboratory parameter abnormalities.
Description of selected adverse reactions
Liver function parameters
Cases of drug-induced idiosyncratic liver injury associated with elevated alanine aminotransferase (ALT) and/or aspartate aminotransferase (AST) levels to ≥ 5 and ≥ 20 times the upper limit of normal (ULN), grade 3 and 4, including transaminase elevations with concurrent increases in total bilirubin to 2 × ULN, have been reported during darolutamide treatment. Time to onset ranged from 1 month to 12 months after initiation of darolutamide. In many cases, ALT and AST elevations were reversible upon discontinuation of darolutamide. For recommendations, see section "Special precautions for use".
Non-metastatic castration-resistant prostate cancer (nmCRPC)
Fatigue
Fatigue/asthenic conditions were reported in 15.8% of patients receiving darolutamide and in 11.4% of patients receiving placebo. Events of grade 3 severity were recorded in 0.6% of patients receiving darolutamide and in 1.1% of patients receiving placebo. Fatigue (excluding asthenia, lethargy, or malaise) was observed in the majority of cases (12.1% of patients receiving darolutamide and 8.7% of patients receiving placebo).
Fractures
Fractures occurred in 4.2% of patients receiving darolutamide and in 3.6% of patients receiving placebo.
Ischemic heart disease and heart failure
Ischemic heart disease occurred in 3.2% of patients receiving darolutamide and in 2.5% of patients receiving placebo. Grade 5 events were observed in 0.3% of patients receiving darolutamide and in 0.2% of patients receiving placebo. Heart failure occurred in 1.9% of patients receiving darolutamide and in 0.9% of patients receiving placebo.
Decreased neutrophil count
Decreased neutrophil count was observed as a laboratory abnormality in 19.6% of patients receiving darolutamide and in 9.4% of patients receiving placebo. The median time to nadir was 256 days.
Laboratory abnormalities were predominantly of grade 1–2 intensity. Decreased neutrophil count of grade 3 and 4 was observed in 3.5% and 0.5% of patients, respectively. Only one patient permanently discontinued darolutamide due to neutropenia. Neutropenia was transient or reversible (88% of patients) and was not associated with any clinically significant signs or symptoms.
Increased blood bilirubin levels
Increased bilirubin levels were reported as laboratory abnormalities in 16.4% of patients receiving darolutamide and in 6.9% of patients receiving placebo. Episodes were predominantly of grade 1 or 2 intensity, not associated with any clinically significant signs or symptoms, and were reversible after discontinuation of darolutamide. Grade 3 bilirubin elevation was observed in 0.1% of patients receiving darolutamide and in 0% of patients receiving placebo. In the darolutamide group, the median time to first increase in bilirubin was 153 days, and the median duration of the first episode was 182 days. No patient discontinued treatment due to elevated bilirubin levels.
Increased AST levels
Increased AST levels were reported as laboratory abnormalities in 22.5% of patients receiving darolutamide and in 13.6% of patients receiving placebo. Episodes were predominantly of grade 1 or 2 intensity, not associated with any clinically significant signs or symptoms, and were reversible after discontinuation of darolutamide. Grade 3 AST elevation was observed in 0.5% of patients receiving darolutamide and in 0.2% of patients receiving placebo. In the darolutamide group, the median time to first increase in AST was 258 days, and the median duration of the first episode was 118 days. No patient discontinued treatment due to increased AST levels.
Metastatic hormone-sensitive prostate cancer (mHSPC)
Arterial hypertension
In the ARASENS trial, arterial hypertension was reported in 13.8% of patients receiving darolutamide + docetaxel and in 9.4% of patients receiving placebo + docetaxel.
Grade 3 arterial hypertension was recorded in 6.4% of patients receiving darolutamide + docetaxel compared to 3.5% of patients receiving placebo + docetaxel. One patient with grade 4 hypertension was reported in each treatment group.
One case of grade 5 hypertension with grade 5 arteriosclerosis was reported in the darolutamide + docetaxel group. This patient had a long history of hypertension and smoking, and the event occurred more than 3 years after initiation of darolutamide treatment. Cases of arterial hypertension were more frequently reported in patients without a history of hypertension in both treatment groups.
Fractures
Fractures occurred in 7.5% of patients receiving darolutamide + docetaxel and in 5.1% of patients receiving placebo + docetaxel.
Decreased neutrophil count
Decreased neutrophil count was reported as a laboratory abnormality in 50.6% of patients receiving darolutamide + docetaxel and in 45.5% of patients receiving placebo + docetaxel. Grade 3 and 4 neutrophil count decreases were observed in 34.4% of patients receiving darolutamide + docetaxel and in 31.4% of patients receiving placebo + docetaxel. In both treatment groups, neutrophil counts decreased, and neutropenia was highest during the first months of treatment, after which the frequency and severity of events decreased.
Increased blood bilirubin levels
Increased blood bilirubin levels were observed as laboratory abnormalities in 19.6% of patients receiving darolutamide + docetaxel and in 10.0% of patients receiving placebo + docetaxel. Events were predominantly of grade 1 or 2 intensity. Grade 3 and 4 bilirubin elevations were observed in 0.5% of patients receiving darolutamide + docetaxel and in 0.3% of patients receiving placebo + docetaxel.
Increased ALT and AST levels
Increased ALT levels were reported as laboratory abnormalities in 42.3% of patients receiving darolutamide + docetaxel and in 38.0% of patients receiving placebo + docetaxel. Increased AST levels were reported in 43.9% of patients receiving darolutamide + docetaxel and in 39.3% of patients receiving placebo + docetaxel. ALT and AST elevations were predominantly of grade 1 intensity. Grade 3 and 4 ALT elevations were observed in 3.7% of patients receiving darolutamide + docetaxel and in 3.0% of patients receiving placebo + docetaxel. Grade 3 and 4 AST elevations were observed in 3.6% of patients receiving darolutamide + docetaxel and in 2.3% of patients receiving placebo + docetaxel.
Reporting of suspected adverse reactions
Reporting suspected adverse reactions after marketing authorization is important. It allows continued monitoring of the benefit-risk balance of the medicinal product. Healthcare professionals and patients, as well as their legal representatives, should report all suspected adverse reactions and lack of efficacy through the Automated Pharmacovigilance Information System at: https://aisf.dec.gov.ua.
Shelf life. 3 years.
Storage conditions
No special storage conditions required. Keep out of the reach of children.
Packaging
16 tablets in a blister, 7 blisters in a cardboard pack.
Prescription status. Prescription only.
Manufacturer. Orion Corporation, Orion Pharma.
Manufacturer's address
Joensuunkatu 7, 24100 Salo, Finland