Nubeka
UcraniaContenido
INSTRUCCIONES PARA USO MÉDICO DEL MEDICAMENTO NUBEKA (NUBEQA)
Composición:
principio activo: darolutamida;
1 tableta recubierta con película contiene 300 mg de darolutamida;
excipientes: fosfato cálcico, carboximetilcelulosa sódica, lactosa monohidrato, estearato de magnesio, povidona K 30; recubrimiento de película: lacado blanco (hipromelosa 15 cP, lactosa monohidrato, macrogol 3350, dióxido de titanio (E 171)).
Forma farmacéutica. Tabletas recubiertas con película.
Características físico-químicas principales: tabletas ovaladas, recubiertas con película, de color blanco a casi blanco, con la inscripción «BAYER» en un lado y «300» en el otro.
Grupo farmacoterapéutico. Agentes utilizados en terapia hormonal. Antagonistas hormonales y agentes relacionados. Agentes antiandrógenos. Darolutamida.
Código ATC L02B B06.
Propiedades farmacológicas
Farmacodinámica
Mecanismo de acción. Darolutamida es un inhibidor del receptor de andrógenos (AR) con una estructura de pirazol sustituida polarmente y flexible, que se une con alta afinidad directamente al dominio de unión del ligando del receptor.
Darolutamida inhibe competitivamente la unión de andrógenos, la translocación nuclear del AR y la transcripción mediada por AR. El metabolito principal, keto-darolutamida, demostró in vitro una actividad similar a la de darolutamida. El tratamiento con darolutamida reduce la proliferación de células tumorales de la próstata, lo que resulta en una potente actividad antitumoral.
Efectos farmacodinámicos. No se observó prolongación del intervalo medio QTcF (es decir, superior a 10 ms) tras la administración oral de 600 mg de darolutamida dos veces al día en comparación con el placebo.
Eficacia y seguridad clínicas. La eficacia y la seguridad se evaluaron en dos estudios multicéntricos aleatorizados, controlados con placebo, de fase III, que incluyeron pacientes con cáncer de próstata no metastásico resistente a la castración (nmCRPC) (estudio ARAMIS) y cáncer de próstata metastásico sensible a hormonas (mCSPH) (estudio ARASENS). Todos los pacientes recibieron simultáneamente un análogo del hormona liberadora de gonadotropina (LHRH) o se sometieron a orquiectomía bilateral.
Cáncer de próstata no metastásico resistente a la castración (nmCRPC)
La eficacia y seguridad del uso de darolutamida se evaluaron en un estudio aleatorizado, doble ciego, controlado con placebo, multicéntrico de fase III (ARAMIS) en pacientes con cáncer de próstata resistente a la castración no metastásico (según evaluación mediante métodos convencionales de TC, gammagrafía ósea e IRM) y con un tiempo de duplicación del antígeno prostático específico (tPSA) ≤ 10 meses.
Los pacientes fueron incluidos en el estudio si presentaban tres elevaciones del nivel del antígeno prostático específico (PSA) tras el valor más bajo, con intervalos de al menos una semana durante la terapia de privación androgénica, un nivel de PSA ≥ 2 ng/ml en el momento del cribado y un nivel de testosterona poscastración en suero < 1,7 nmol/l.
Los pacientes con antecedentes de convulsiones fueron admitidos en el estudio. En el grupo de darolutamida se registraron 12 pacientes (0,21 %) con antecedentes de convulsiones.
Se excluyeron del estudio pacientes con hipertensión no controlada o con ictus reciente (en los últimos 6 meses), infarto de miocardio, angina inestable grave, derivación arterial coronaria o periférica, insuficiencia cardíaca congestiva clase III o IV según la clasificación de la New York Heart Association.
Se excluyeron también los pacientes que previamente habían recibido inhibidores del receptor de andrógenos (AR) de segunda generación, como enzalutamida, apalutamida y darolutamida, o inhibidores de la enzima CYP17, como el acetato de abiraterona, así como aquellos que habían recibido corticosteroides sistémicos en dosis superiores al equivalente de 10 mg de prednisona al día durante los 28 días previos a la aleatorización.
En total, 1509 pacientes fueron aleatorizados en una proporción 2:1 para recibir 600 mg de darolutamida vía oral dos veces al día (n = 955) o placebo (n = 554).
Se incluyeron en el estudio pacientes con ganglios linfáticos pélvicos presentes de menos de 2 cm en el eje corto por debajo de la bifurcación de la aorta. La ausencia o presencia de metástasis se determinó mediante evaluación radiológica centralizada e independiente. Esta evaluación se realizó en 89 pacientes en quienes retrospectivamente se detectaron metástasis al inicio del estudio. La aleatorización se estratificó según el tPSA (≤ 6 meses o > 6 meses) y según el uso de terapia dirigida contra los osteoclastos al inicio del estudio (sí o no).
Las características demográficas de los pacientes y las características de la enfermedad estuvieron equilibradas entre los grupos de tratamiento. La edad media de los pacientes fue de 74 años (rango de 48 a 95), el 9 % de los pacientes tenían 85 años o más; el 79 % eran de raza caucásica, el 13 % de origen asiático y el 3 % eran afrodescendientes. La mayoría de los pacientes tenían un puntaje de Gleason de 7 o superior al momento del diagnóstico (73 %). La mediana del tPSA fue de 4,5 meses. El 9 % de los pacientes habían sido sometidos previamente a orquiectomía, el 25 % a prostatectomía previa y el 50 % habían recibido al menos un tratamiento previo con radioterapia. El 76 % de los pacientes habían recibido más de un tratamiento antiandrogénico previo. Los pacientes tenían un puntaje de 0 (69 %) o 1 (31 %) en la escala de estado funcional del grupo cooperativo de oncología del este (ECOG PS).
El tratamiento con darolutamida continuó hasta la progresión de la enfermedad según evaluación radiológica, determinada mediante imágenes convencionales (TC, gammagrafía ósea, IRM) mediante evaluación centralizada enmascarada, toxicidad inaceptable o retirada del paciente del estudio.
El punto final primario de eficacia fue la supervivencia libre de metástasis (SLM). Los puntos finales secundarios incluyeron la supervivencia global (SG), el tiempo hasta la progresión del dolor, el tiempo hasta el inicio de la primera quimioterapia citotóxica para el cáncer de próstata y el tiempo hasta el primer evento esquelético sintomático (definido como la aparición de cualquiera de los siguientes: radioterapia externa para aliviar síntomas esqueléticos, nueva fractura patológica sintomática, compresión medular o intervención quirúrgica ortopédica relacionada con el tumor).
El tratamiento con darolutamida condujo a una mejora de la SLM en comparación con el placebo (véase la tabla 1 y la fig. 1).
Los resultados de SLM fueron consistentes en todos los subgrupos de pacientes, independientemente del tPSA, del uso previo de agentes que afectan al hueso o de la enfermedad local-regional. Subgrupos adicionales con resultados coherentes de SLM incluyeron el valor de PSA al inicio del estudio, el puntaje de Gleason al diagnóstico, la edad del paciente, la región geográfica, el puntaje ECOG PS al inicio del estudio, la raza y el número de tratamientos hormonales previos.
Tras el análisis primario de SLM, tan pronto como se reveló el estudio, se ofreció a los pacientes que recibían placebo el tratamiento con darolutamida (variante cruzada). De los 554 pacientes aleatorizados al grupo placebo, 170 (31 %) pasaron al tratamiento con darolutamida. El análisis de la SG no se ajustó para tener en cuenta el impacto del cruce.
En el momento del análisis final, el tratamiento con darolutamida mostró una mejora estadísticamente significativa en la supervivencia global en comparación con el placebo (la mediana no se alcanzó en ninguno de los grupos, véase la tabla 1 y la fig. 2). También se observó un aumento estadísticamente significativo en el tiempo hasta la progresión del dolor, hasta el inicio de la primera quimioterapia citotóxica y hasta el primer evento esquelético sintomático en comparación con el placebo (véase la tabla 1).
En el momento del análisis final, la duración media del tratamiento en los pacientes que recibieron darolutamida fue de 33,3 meses (rango de 0,0 a 74,0 meses) durante el período combinado ciego y abierto.
Todos los análisis se realizaron en el conjunto completo de análisis.
Tabla 1. Resultados de eficacia del estudio ARAMIS
| Parámetro de eficacia |
Número (%) de pacientes con eventos |
Mediana (meses) (95 % IC) |
Relación de riesgos (RR)b (95 % intervalo de confianza [IC]) valor p (bilateral) |
||
| Darolutamida (N = 955) |
Placeboa (N = 554) |
Darolutamida (N = 955) |
Placeboa (N = 554) |
||
| Supervivencia libre de metástasisc |
221 (23,1 %) |
216 (39,0 %) |
40,4 (34,3; ND) |
18,4 (15,5; 22,3) |
0,413 (0,341; 0,500) < 0,000001 |
| Supervivencia global |
148 (15,5 %) |
106 (19,1 %) |
ND (56,1; ND) |
ND (46,9; ND) |
0,685 (0,533; 0,881) 0,003048 |
| Tiempo hasta la progresión del dolorc, d |
251 (26,3 %) |
178 (32,1 %) |
40,3 (33,2; 41,2) |
25,4 (19,1; 29,6) |
0,647 (0,533; 0,785) 0,000008 |
| Tiempo hasta la primera quimioterapia citotóxica |
127 (13,3 %) |
98 (17,7 %) |
ND (ND, ND) |
ND |
0,579 (0,444; 0,755) 0,000044 |
| Tiempo hasta el primer evento esquelético sintomático |
29 (3,0 %) |
28 (5,1 %) |
ND (ND, ND) |
(ND, ND) |
0,484 (0,287; 0,815) 0,005294 |
a Incluyendo 170 pacientes que pasaron al darolutamida sin período de enmascaramiento.
b Riesgo relativo < 1 para darolutamida.
c Para la SVLM y el tiempo hasta la progresión del dolor, el análisis primario se considera el análisis final.
d Resultados informados por el paciente según la evaluación del cuestionario Brief Pain Inventory – Short Form.
ND – no alcanzado.
El resultado del tratamiento con darolutamida fue un período más prolongado de supervivencia libre de progresión (SLP, mediana 36,8 frente a 14,8 meses, HR = 0,380, p nominal < 0,000001) y tiempo hasta la progresión del PSA (mediana 29,5 frente a 7,2 meses, HR = 0,164, p nominal < 0,000001). La consistencia del efecto se observó en todos los parámetros de supervivencia (SLM, SV y PSA).
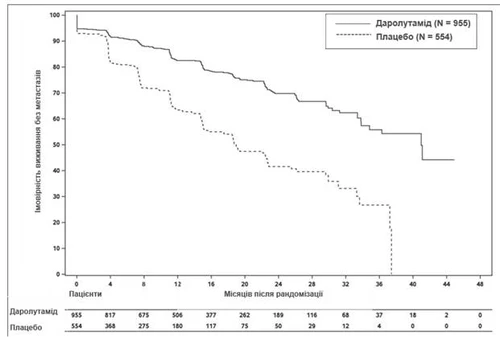
Fig. 1. Curvas de Kaplan-Meier de supervivencia libre de metástasis (ARAMIS)
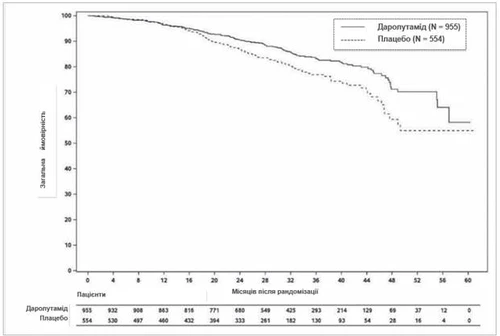
Fig. 2. Curvas de Kaplan-Meier de supervivencia global (ARAMIS)
Los pacientes que recibieron darolutamida en el estudio ARAMIS (período doble ciego) mostraron una tasa significativamente mayor de respuesta confirmada respecto al nivel de PSA (definida como una reducción ≥ 50 % respecto al valor basal) en comparación con los pacientes que recibieron placebo, 84,0 % frente a 7,9 % (diferencia = 76,1 %, p < 0,000001 (valor p nominal, solo para información)).
Cáncer de próstata sensible a hormonas metastásico (CSHM)
La eficacia y seguridad del uso de darolutamida en combinación con docetaxel se evaluaron en un estudio multicéntrico, doble ciego, controlado con placebo, de fase III (ARASENS) en pacientes con CSHM. En total, 1306 pacientes fueron aleatorizados 1:1 para recibir 600 mg de darolutamida vía oral dos veces al día (n = 651) o placebo (n = 655), simultáneamente con 75 mg/m² de docetaxel durante 6 ciclos. El tratamiento con darolutamida o placebo continuó hasta la progresión sintomática de la enfermedad, el cambio de terapia antineoplásica, toxicidad inaceptable, muerte o abandono del estudio por parte del paciente.
La presencia de metástasis se evaluó mediante evaluación radiológica centralizada e independiente. Se excluyeron del estudio los pacientes con afectación únicamente de ganglios linfáticos regionales (M0). La aleatorización se estratificó según la extensión de la enfermedad (solo metástasis en ganglios linfáticos no regionales (M1a), metástasis óseas con o sin metástasis en ganglios linfáticos (M1b), o metástasis viscerales con o sin metástasis en ganglios linfáticos o en huesos (M1c)) y según el nivel de fosfatasa alcalina (< o ≥ límite superior normal) al inicio del estudio. Se permitió la inclusión de pacientes con metástasis cerebrales, aunque no se incluyó ninguno.
Las características demográficas de los pacientes y las características de la enfermedad fueron equilibradas entre los grupos de tratamiento. La edad media fue de 67 años (rango 41–89), el 0,5 % de los pacientes tenían 85 años o más, el 52 % eran de raza caucásica, el 36 % de origen asiático y el 4 % de los participantes en el estudio tenían piel oscura. La mayoría de los pacientes tenían un puntaje de Gleason de 8 o más al momento del diagnóstico (78 %). El 71 % de los pacientes tenían una puntuación ECOG PS de 0 y el 29 % de 1. Hubo un 86,1 % de pacientes con enfermedad de novo y un 12,9 % con recidiva. Al inicio del estudio, el 3 % de los pacientes tenían M1a, el 79,5 % tenían M1b y el 17,5 % tenían M1c; la fosfatasa alcalina era < LSN en el 44,5 % de los pacientes y ≥ LSN en el 55,5 %; el nivel medio basal de PSA fue de 30,3 µg/l y 24,2 µg/l en los grupos de darolutamida y placebo, respectivamente. Se permitió la inclusión de pacientes con antecedentes de convulsiones, y 4 pacientes (0,6 %) fueron incluidos en el grupo de darolutamida + docetaxel.
El 77,0 % de los pacientes tenían enfermedad de gran volumen y el 23,0 % tenían enfermedad de pequeño volumen. La enfermedad de gran volumen se definió como la presencia de metástasis viscerales o de 4 o más lesiones óseas, con al menos una metástasis fuera de la columna vertebral y los huesos pélvicos. Aproximadamente el 25 % de los pacientes recibieron tratamiento concomitante con bifosfonatos o denosumab.
El punto final primario de eficacia fue la supervivencia global (SV). Los puntos finales secundarios incluyeron el tiempo hasta el desarrollo de cáncer de próstata resistente a la castración, el tiempo hasta la progresión del dolor, el tiempo de supervivencia libre de eventos esqueléticos sintomáticos, el tiempo hasta el primer evento esquelético sintomático, el tiempo hasta el inicio del siguiente tratamiento antineoplásico, el tiempo hasta el empeoramiento de los síntomas físicos relacionados con la enfermedad y el tiempo hasta el inicio del uso de opioides durante ≥ 7 días consecutivos. La progresión del dolor se evaluó mediante resultados informados por el paciente según el cuestionario Brief Pain Inventory – Short Form, definido como un empeoramiento de al menos 2 puntos desde el valor basal y el inicio del uso de opioides de acción corta o prolongada para el alivio del dolor durante ≥ 7 días consecutivos.
La duración media del tratamiento fue de 41,0 meses (rango de 0,1 a 56,5 meses) en los pacientes que recibieron darolutamida + docetaxel y de 16,7 meses (rango de 0,3 a 55,8 meses) en los pacientes que recibieron placebo + docetaxel. El 87,6 % y el 85,5 % de los pacientes recibieron los 6 ciclos completos de docetaxel, y el 1,5 % y el 2,0 % de los pacientes no recibieron docetaxel en los grupos de darolutamida + docetaxel y placebo + docetaxel, respectivamente.
Tabla 2. Resultados de eficacia del estudio ARASENS
| Parámetro de eficacia |
Número (%) de pacientes con eventos |
Mediana (meses) (IC del 95 %) |
Relación de riesgos (RR)b (IC del 95 %) Valor de p (unilateral)c |
||
| Darolutamida + docetaxel (N = 651) |
Placebo + docetaxel (N = 654)a |
Darolutamida + docetaxel (N = 651) |
Placebo + docetaxel (N = 654)a |
||
| Sobrevida globald |
229 (35,2 %) |
304 (46,5 %) |
NA (NA, NA) |
48,9 (44,4; NA) |
0,675 (0,568; 0,801) <0,0001 |
a Un paciente en el grupo de placebo fue excluido de todos los análisis.
b Relación de riesgo < 1 a favor de darolutamida.
c Basado en la prueba logarítmica estratificada.
d Los resultados de SG fueron consistentes en todos los subgrupos de pacientes, incluyendo el estadio de la enfermedad y los niveles de fosfatasa alcalina.
ND – no alcanzado.
Puntos finales secundarios de eficacia en los que se demostró una ventaja estadísticamente significativa para los pacientes del grupo darolutamida + docetaxel frente a los pacientes del grupo placebo + docetaxel: tiempo hasta el desarrollo de cáncer de próstata resistente a la castración (mediana ND frente a 19,1 meses; HR = 0,357, p < 0,0001); tiempo hasta el primer evento esquelético sintomático (mediana ND frente a ND; HR = 0,712, p = 0,0081); tiempo hasta el inicio del siguiente tratamiento quimioterápico antineoplásico (mediana ND frente a 25,3 meses; HR = 0,388, p < 0,0001); tiempo hasta la progresión del dolor (mediana ND frente a 27,5 meses; HR = 0,792, p = 0,0058); tiempo hasta la supervivencia libre de eventos esqueléticos sintomáticos (mediana 51,2 frente a 39,7 meses; HR = 0,609, p < 0,0001).
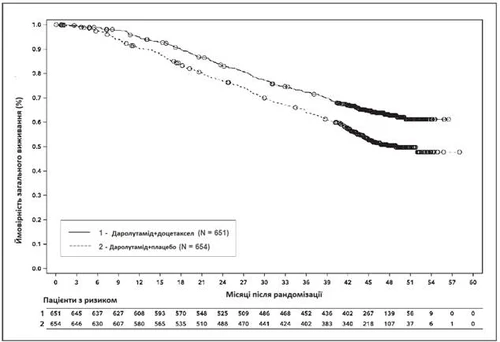
Fig. 3. Curvas de supervivencia global de Kaplan-Meier (ARASENS)a
a La SG a los 36 meses fue del 72,3 % (IC del 95 %, 68,8–75,8) en el grupo darolutamida + docetaxel frente al 63,8 % (IC del 95 %, 60,1–67,6) en el grupo placebo + docetaxel.
La tasa de SG a los 48 meses fue del 62,7 % (IC del 95 %, 58,7–66,7) en el grupo darolutamida + docetaxel frente al 50,4 % (IC del 95 %, 46,3–54,6) en el grupo placebo + docetaxel.
Farmacocinética
Darolutamida está compuesta por dos diastereómeros [(S,R)-darolutamida y (S,S)-darolutamida], que se interconvierten a través del principal metabolito circulante, la keto-darolutamida. In vitro, las tres sustancias muestran una actividad farmacológica similar. La darolutamida es poco soluble en disolventes acuosos en un amplio rango de pH y generalmente se disuelve mejor en disolventes orgánicos.
Absorción
Tras la administración oral de 600 mg (2 comprimidos de 300 mg) dos veces al día, las concentraciones máximas de darolutamida en plasma en estado estacionario fueron de 4,79 mg/l (coeficiente de variación del 30,9 %) en pacientes con CPNPnm en el estudio ARAMIS y de 3,84 mg/l (coeficiente de variación del 35,6 %) en pacientes con CPNPm en el estudio ARASENS. El tiempo medio para alcanzar la concentración máxima en plasma fue de 3–4 horas. La proporción entre los dos diastereómeros, (S,R)-darolutamida y (S,S)-darolutamida, cambió de 1:1 en el comprimido a aproximadamente 1:9 en plasma, basado en el parámetro AUC0-12 en estado estacionario. Tras la administración oral con alimentos, se alcanza el estado estacionario tras 2–5 días de administración repetida dos veces al día.
La biodisponibilidad absoluta tras la administración oral en ayunas de un comprimido de Nubeqa que contiene 300 mg de darolutamida es aproximadamente del 30 % respecto a la administración intravenosa.
La biodisponibilidad de darolutamida aumentó entre 2,0 y 2,5 veces cuando se administró con alimentos. Se observó un aumento similar en la exposición para el metabolito principal, la keto-darolutamida.
Distribución
El volumen aparente de distribución de darolutamida tras administración intravenosa es de 119 l, lo que indica que la darolutamida se distribuye ampliamente en el organismo, tanto en el espacio líquido intracelular como extracelular.
Darolutamida se une moderadamente (92 %) a las proteínas del plasma humano, sin diferencias entre los dos diastereómeros. El metabolito principal de darolutamida, la keto-darolutamida, se une en gran medida (99,8 %) a las proteínas del plasma.
La atravesamiento de la barrera hematoencefálica por darolutamida no se ha estudiado clínicamente. Sin embargo, el impacto de darolutamida en el cerebro, en términos de AUC0-24, es muy bajo: 4,5 % de la exposición en plasma tras una dosis única en ratas y 1,9–3,9 % tras dosis repetidas en ratones. Esto indica un bajo paso de darolutamida a través de la barrera hematoencefálica intacta en ratas y ratones y una baja probabilidad de que darolutamida atraviese la barrera hematoencefálica intacta en humanos de forma clínicamente significativa.
Biotransformación
Los diastereómeros (S,R)-darolutamida y (S,S)-darolutamida pueden interconvertirse mediante el metabolito keto-darolutamida, con predominio del (S,S)-darolutamida.
Tras una dosis oral única de 300 mg de darolutamida marcada con 14C en forma de solución oral, la keto-darolutamida es el único metabolito principal, con una exposición total en plasma aproximadamente dos veces mayor que la de darolutamida. Juntas, darolutamida y keto-darolutamida representan el 87,4 % de la radiactividad de 14C en plasma, lo que indica que todos los demás metabolitos son insignificantes.
Darolutamida se metaboliza principalmente mediante vía de metabolismo oxidativo mediado principalmente por CYP3A4, así como por glucuronidación directa, mediada principalmente por UGT1A9 y UGT1A1. Además, se ha demostrado que las isoformas AKR1C catalizan predominantemente la reducción de la keto-darolutamida a los diastereómeros de la sustancia.
Eliminación
La semivida efectiva de darolutamida y de keto-darolutamida en plasma en pacientes es de aproximadamente 18–20 horas. De los dos diastereómeros que componen darolutamida, el (S,R)-darolutamida tiene una semivida efectiva más corta (9 horas) en comparación con el (S,S)-darolutamida, cuya semivida efectiva es de 22 horas. El aclaramiento de darolutamida tras administración intravenosa fue de 116 ml/min (CV 39,7 %). En general, el 63,4 % de darolutamida y sus metabolitos se excreta por orina (aproximadamente el 7 % sin cambios), y el 32,4 % se excreta por heces. Más del 95 % de la dosis se eliminó dentro de los 7 días posteriores a la administración.
Linealidad/no linealidad
En el rango de dosis de 100 a 700 mg (tras dosis única y en estado estacionario), la exposición de los dos diastereómeros y del metabolito principal, keto-darolutamida, aumenta linealmente casi en proporción a la dosis. Debido a la saturación de la absorción, no se observó un aumento adicional de la exposición de darolutamida con una dosis de 900 mg dos veces al día.
Grupos de pacientes especiales
Pacientes de edad avanzada. No se observaron diferencias clínicamente relevantes en la farmacocinética de darolutamida (65–95 años).
Alteración de la función renal. En un estudio clínico farmacocinético, el AUC y la Cmax de darolutamida fueron 2,5 y 1,6 veces más altos en pacientes con insuficiencia renal grave (filtración glomerular estimada [eFG] de 15 a 29 ml/min/1,73 m²) en comparación con voluntarios sanos.
El análisis farmacocinético poblacional indica una exposición (AUC) de darolutamida aproximadamente 1,1, 1,3 y 1,5 veces mayor en pacientes con disfunción renal leve, moderada y grave (eFG de 15 a 89 ml/min/1,73 m²) en comparación con pacientes con función renal normal.
La farmacocinética de darolutamida no se ha estudiado en pacientes con insuficiencia renal en estadio terminal en diálisis (eFG < 15 ml/min/1,73 m²).
Alteración de la función hepática. En un estudio clínico farmacocinético, la Cmax y el AUC de darolutamida fueron 1,5 y 1,9 veces más altos en pacientes con insuficiencia hepática moderada (Clase B según la clasificación de Child-Pugh) en comparación con voluntarios sanos. No hay datos disponibles para pacientes con insuficiencia hepática grave (Clase C según la clasificación de Child-Pugh).
Diferencias étnicas. No se observaron diferencias clínicamente relevantes en la farmacocinética de darolutamida según la etnia del paciente (pacientes de raza caucásica, pacientes de origen japonés, pacientes asiáticos no japoneses, pacientes de piel oscura o de origen afroamericano). El análisis farmacocinético poblacional mostró un aumento en el efecto medio geométrico (AUC) de hasta 1,56 veces (IC del 90 %, 1,43–1,70) en pacientes de origen japonés en comparación con pacientes de otras regiones en los estudios ARAMIS y ARASENS.
Datos preclínicos de seguridad
Toxicidad sistémica. En estudios de toxicidad con dosis repetidas en animales, los principales hallazgos fueron cambios en los órganos reproductores masculinos (disminución del peso orgánico con atrofia de la próstata y el epidídimo). Estos efectos ocurrieron con exposiciones sistémicas dentro o por debajo del rango de exposición esperado en humanos (basado en comparaciones de AUC). Cambios adicionales en tejidos reproductivos incluyeron una mínima vacuolización de la hipófisis, atrofia y disminución de la secreción de las vesículas seminales y glándulas mamarias en ratas, así como hipospermia de los testículos, dilatación de los conductos seminíferos y degeneración en perros. Los cambios en los órganos reproductores masculinos en ambas especies correspondieron a la actividad farmacológica de darolutamida y desaparecieron o disminuyeron parcialmente tras un período de recuperación de 4–8 semanas.
Embriotoxicidad/teratogenicidad. No se han realizado estudios sobre toxicidad fetal.
Toxicidad reproductiva. No se han realizado estudios de toxicidad reproductiva. Sin embargo, es probable que la fertilidad de los hombres se vea afectada, según los resultados de estudios de toxicidad con dosis repetidas en animales, que son coherentes con la actividad farmacológica de darolutamida.
Genotoxicidad y carcinogenicidad. Darolutamida no indujo mutaciones en el ensayo de mutagénesis microbiana (Ames). A altas concentraciones, darolutamida indujo aberraciones cromosómicas estructurales in vitro en linfocitos humanos cultivados. Sin embargo, no se observó genotoxicidad en el ensayo combinado in vivo de micronúcleos en médula ósea y en el ensayo Comet en hígado e intestino delgado de ratas, incluso con exposiciones superiores a la exposición máxima en humanos.
La administración oral de darolutamida a machos de ratones transgénicos rasH2 durante 6 meses no reveló potencial carcinogénico a dosis de hasta 1000 mg/kg/día, lo que excede la exposición clínica (AUC) de darolutamida en 0,9–1,3 veces y la de keto-darolutamida en 2,1–2,3 veces respecto a la dosis clínica recomendada de 1200 mg/día. A partir de este estudio, no se puede excluir completamente el riesgo carcinogénico con el uso de darolutamida.
Características clínicas
Indicaciones
El medicamento Nubeqa está indicado para el tratamiento en hombres adultos:
- de cáncer de próstata resistente a la castración no metastásico (nmCRPC) con alto riesgo de metástasis (ver sección «Propiedades farmacológicas»);
- de cáncer de próstata sensible a hormonas metastásico (mCHSPC) — en combinación con docetaxel y terapia de privación androgénica (ver sección «Propiedades farmacológicas»).
Contraindicaciones
Hipersensibilidad al principio activo o a los excipientes.
Está contraindicado en mujeres embarazadas y mujeres en edad fértil.
Interacción con otros medicamentos y otras formas de interacción
Efecto de otros medicamentos sobre darolutamida
Inductores de CYP3A4 y P-gp
Darolutamida es sustrato de CYP3A4 y de la glucoproteína P (P-gp).
No se recomienda el uso de inductores potentes y moderados de CYP3A4 y de inductores de P-gp (por ejemplo, carbamazepina, fenobarbital, hipérico, fenitoína y rifampicina) durante el tratamiento con darolutamida, salvo cuando no exista alternativa terapéutica. Se debe considerar la posibilidad de elegir un medicamento concomitante alternativo que no induzca o que tenga un potencial débil de inducción de CYP3A4 o P-gp.
La administración repetida de rifampicina (600 mg), un inductor potente de CYP3A4 y P-gp, junto con una dosis única de darolutamida (600 mg) con alimentos, provocó una reducción del 72 % en la exposición media (AUC0-72) y una reducción del 52 % en la Cmax de darolutamida.
Inhibidores de CYP3A4, P-gp y BCRP
Darolutamida es sustrato de CYP3A4, P-gp y de la proteína de resistencia del cáncer de mama (BCRP).
No se espera una interacción clínicamente significativa entre medicamentos cuando se administren inhibidores de CYP3A4, P-gp o BCRP. Darolutamida puede administrarse simultáneamente con inhibidores de CYP3A4, P-gp o BCRP. La administración concomitante de darolutamida con un inhibidor potente combinado de CYP3A4 y P-gp aumenta la exposición a darolutamida, lo que podría incrementar el riesgo de reacciones adversas a darolutamida. Se recomienda un control más frecuente del paciente para detectar la aparición de reacciones adversas a darolutamida y, si es necesario, ajustar la dosis de darolutamida.
La administración de itraconazol (200 mg dos veces al día el día 1 y una vez al día durante los siguientes 7 días), un inhibidor potente de CYP3A4, P-gp y BCRP, junto con una dosis única de darolutamida (600 mg el día 5 con alimentos), provocó un aumento promedio de 1,7 veces en la exposición (AUC0-72) y un aumento de 1,4 veces en la Cmax de darolutamida.
Inhibidores de UGT1A9
Darolutamida es sustrato de UGT1A9. No se espera una interacción clínicamente significativa entre medicamentos cuando se administren inhibidores de UGT1A9. Darolutamida puede administrarse simultáneamente con inhibidores de UGT1A9.
Un análisis farmacocinético poblacional mostró que la administración concomitante de inhibidores de UGT1A9 con darolutamida provocó un aumento de 1,2 veces en la exposición (AUC0-72) de darolutamida.
Docetaxel
La administración de darolutamida en combinación con docetaxel no provocó cambios clínicamente significativos en la farmacocinética de darolutamida en pacientes con mCHSPC (ver sección «Propiedades farmacológicas»).
Efecto de darolutamida sobre otros medicamentos
Sustratos de BCRP, OATP1B1 y OATP1B3
Darolutamida es un inhibidor de la proteína de resistencia del cáncer de mama (BCRP) y de los péptidos transportadores de aniones orgánicos (OATP) 1B1 y 1B3.
Se debe evitar la administración concomitante de rosuvastatina, salvo cuando no exista alternativa terapéutica. Se debe considerar la posibilidad de elegir un medicamento concomitante alternativo con menor potencial de inhibición de BCRP, OATP1B1 y OATP1B3.
La administración de darolutamida (600 mg dos veces al día durante 5 días) antes de una dosis única de rosuvastatina (5 mg) con alimentos provocó un aumento aproximado de 5 veces en la exposición media (AUC) y en la Cmax de rosuvastatina.
Siempre que sea posible, se debe evitar la administración concomitante de darolutamida con otros sustratos de BCRP. La administración concomitante de darolutamida puede aumentar la concentración plasmática de otros medicamentos concomitantes que sean sustratos de BCRP, OATP1B1 y OATP1B3 (como metotrexato, sulfasalazina, fluvastatina, atorvastatina, pitavastatina). Por lo tanto, se recomienda observar al paciente para detectar la aparición de reacciones adversas relacionadas con el uso de estos sustratos. Además, al administrar conjuntamente con darolutamida, se deben seguir las recomendaciones pertinentes incluidas en la información de estos medicamentos.
Sustratos de P-gp
No se espera una interacción clínicamente significativa entre medicamentos al administrar sustratos de P-gp. Darolutamida puede administrarse simultáneamente con sustratos de P-gp (por ejemplo, digoxina, verapamilo o nifedipino). La administración concomitante de darolutamida junto con dabigatrán etexilato, un sustrato sensible de P-gp, no provocó un aumento en la exposición (AUC y Cmax) de dabigatrán.
Sustratos de CYP3A4
Darolutamida es un inductor débil de CYP3A4.
No se espera una interacción clínicamente significativa entre medicamentos al administrar sustratos de CYP. Darolutamida puede administrarse simultáneamente con sustratos de CYP (por ejemplo, warfarina, L-tiroxina, omeprazol).
La administración de darolutamida (600 mg dos veces al día durante 9 días) antes de una dosis única de midazolam (1 mg), un sustrato sensible de CYP3A4, con alimentos, provocó una reducción del 29 % en la exposición media (AUC) y del 32 % en la Cmax de midazolam, respectivamente. Darolutamida no inhibió el metabolismo de sustratos individuales de CYP in vitro en concentraciones clínicamente relevantes.
Docetaxel
La administración de darolutamida en combinación con docetaxel no provocó cambios clínicamente significativos en la farmacocinética de docetaxel en pacientes con mCHSPC (ver sección «Propiedades farmacológicas»).
Medicamentos que prolongan el intervalo QT
Dado que la terapia de privación androgénica puede prolongar el intervalo QT, se debe evaluar cuidadosamente la administración concomitante con medicamentos que se sabe prolongan el intervalo QT o con medicamentos que pueden inducir torsade de pointes. Entre ellos se incluyen medicamentos como antiarrítmicos de clase IA (por ejemplo, quinidina, disopiramida) o clase III (por ejemplo, amiodarona, sotalol, dofetilida, ibutilida), metadona, moxifloxacino y agentes antipsicóticos (por ejemplo, haloperidol).
Características de uso
Alteración de la función renal
Los datos disponibles sobre el uso en pacientes con insuficiencia renal grave son limitados.
Dado que la exposición puede aumentar, se debe observar cuidadosamente a estos pacientes en busca de reacciones adversas (ver sección «Características de uso» y subsección «Farmacocinética»).
Alteración de la función hepática
Los datos disponibles sobre el uso en pacientes con alteración hepática moderada son limitados. No se ha estudiado el uso de darolutamida en pacientes con alteración hepática grave.
Dado que la expos游戏副本
Vía de administración y dosis
El tratamiento debe iniciarse y controlarse por un médico con experiencia en el tratamiento del cáncer de próstata.
Dosificación
La dosis recomendada es de 600 mg de darolutamida (dos tabletas de 300 mg) dos veces al día, lo que equivale a una dosis diaria total de 1200 mg (ver sección «Farmacocinética»).
El tratamiento con darolutamida debe continuar hasta la progresión de la enfermedad o toxicidad inaceptable.
Durante el tratamiento de pacientes no castrados quirúrgicamente, debe mantenerse la castración farmacológica mediante un análogo de la LH-RH.
Cáncer de próstata sensible a hormonas metastásico (CPSHM)
Los pacientes con CPSHM deben iniciar el tratamiento con darolutamida en combinación con docetaxel (ver sección «Propiedades farmacológicas»). El primer ciclo de los 6 ciclos de docetaxel debe administrarse dentro de las 6 semanas posteriores al inicio del tratamiento con darolutamida. Deben seguirse las recomendaciones indicadas en el prospecto del docetaxel. El tratamiento con darolutamida debe continuar hasta la progresión de la enfermedad o toxicidad inaceptable, incluso si la administración de docetaxel se retrasa, interrumpe o suspende.
Dosificación olvidada
Si se olvida una dosis, debe tomarse tan pronto como el paciente lo recuerde, pero solo durante el mismo día. El paciente no debe tomar dos dosis para compensar la dosis olvidada.
Ajuste de la dosis
Si el paciente presenta toxicidad de grado ≥ 3 o una reacción adversa insoportable relacionada con darolutamida (ver secciones «Precauciones de uso» y «Reacciones adversas»), se debe omitir la dosis o reducir la dosificación a 300 mg dos veces al día hasta la mejora de los síntomas. Posteriormente, el tratamiento puede reiniciarse a la dosis de 600 mg dos veces al día. No se recomienda reducir la dosis por debajo de 300 mg dos veces al día, ya que la eficacia de esta dosificación no ha sido establecida.
Grupos de pacientes especiales
Pacientes de edad avanzada. No se requiere ajuste de dosis en pacientes de edad avanzada (ver subsección «Farmacocinética»).
Alteración de la función renal. No se requiere ajuste de dosis en pacientes con alteración renal leve o moderada. En pacientes con insuficiencia renal grave (TFG 15–29 ml/min/1,73 m²) que no estén en hemodiálisis, la dosis inicial recomendada es de 300 mg dos veces al día (ver sección «Precauciones de uso» y subsección «Farmacocinética»).
Alteración de la función hepática. No se requiere ajuste de dosis en pacientes con insuficiencia hepática leve. Los datos disponibles sobre la farmacocinética de darolutamida en insuficiencia hepática moderada son limitados. No se ha estudiado el uso de darolutamida en pacientes con insuficiencia hepática grave.
En pacientes con insuficiencia hepática moderada o grave (clases B y C según la clasificación de Child-Pugh), la dosis inicial recomendada es de 300 mg dos veces al día (ver sección «Precauciones de uso» y subsección «Farmacocinética»).
Vía de administración
El medicamento Nubeqa está indicado para administración oral.
Las tabletas deben tomarse enteras durante las comidas (ver subsección «Farmacocinética»).
Niños
No existe experiencia en el uso de darolutamida en pacientes pediátricos.
Sobredosis
La dosis más alta de darolutamida estudiada clínicamente fue de 900 mg dos veces al día, equivalente a una dosis diaria total de 1800 mg. A esta dosis, no se observaron efectos tóxicos limitantes de la dosis. Debido a la absorción de saturación (ver subsección «Farmacocinética») y a la ausencia de evidencia de toxicidad aguda, no se espera que la ingesta de una dosis superior a la recomendada provoque efectos tóxicos.
En caso de ingesta de una dosis superior a la recomendada, el tratamiento con darolutamida puede continuar con la siguiente dosis según el horario previsto.
No existe un antídoto específico para darolutamida y no se han identificado síntomas específicos de sobredosis.
Reacciones adversas
Las reacciones adversas más frecuentes observadas en pacientes con:
- CPRCnm que recibieron darolutamida fueron fatiga/estados asténicos (15,8 %);
- CPRCm que recibieron darolutamida en combinación con docetaxel fueron erupción cutánea (16,6 %) e hipertensión (13,8 %).
Para obtener información adicional sobre la seguridad del uso combinado de darolutamida, debe consultarse el prospecto de los medicamentos individuales.
Las reacciones adversas observadas en pacientes con CPRCnm que recibieron darolutamida se presentan en la Tabla 3. Las reacciones adversas observadas en pacientes con CPRCm que recibieron darolutamida en combinación con docetaxel se presentan en la Tabla 4.
Las reacciones adversas se clasifican según los sistemas orgánicos. Se agrupan según su frecuencia. La frecuencia de aparición de las reacciones adversas se define de la siguiente manera: muy frecuentes (≥ 1/10), frecuentes (≥ 1/100 hasta < 1/10), poco frecuentes (≥ 1/1000 hasta < 1/100), raras (≥ 1/10000 hasta < 1/1000), muy raras (< 1/10000), frecuencia desconocida (no puede determinarse con los datos disponibles). Dentro de cada grupo por frecuencia, las reacciones adversas se presentan en orden decreciente de gravedad.
Tabla 3. Reacciones adversas registradas en el estudio ARAMISa
| Clase de sistema de órganos (MedDRA) |
Muy frecuentes |
Frecuentes |
| Trastornos cardiacos |
Enfermedad cardíaca isquémicab, insuficiencia cardíacac |
|
| Trastornos de la piel y del tejido subcutáneo |
Erupción cutánead |
|
| Trastornos del sistema musculoesquelético y del tejido conjuntivo |
Dolor en las extremidades, dolor musculoesquelético, fracturas |
|
| Trastornos generales y condiciones en el sitio de administración |
Debilidad/fatigae |
|
| Exploracionesf |
Disminución del recuento de neutrófilos, aumento del nivel sanguíneo de bilirrubina, aumento de AST |
a La duración media del efecto fue de 14,8 meses (rango de 0,0 a 44,3 meses) en pacientes que recibieron darolutamida y de 11,0 meses (range de 0,1 a 40,5 meses) en pacientes que recibieron placebo.
b Incluye aterosclerosis de la arteria coronaria, enfermedad isquémica de la arteria coronaria, oclusión de la arteria coronaria, estenosis de la arteria coronaria, síndrome coronario agudo, infarto agudo de miocardio, angina de pecho, angina inestable, infarto de miocardio e isquemia de miocardio.
c Incluye insuficiencia cardíaca, insuficiencia cardíaca aguda, insuficiencia cardíaca crónica, insuficiencia cardíaca congestiva y shock cardiógeno.
d Incluye erupción cutánea, erupción maculosa, erupción maculopapular, erupción papular, erupción pustulosa, eritema y dermatitis.
e Incluye fatiga y astenia, letargo y malestar.
f Criterios Terminológicos Comunes para Eventos Adversos (CTCAE), versión 4.03. La frecuencia se basa en valores notificados como desviaciones de los parámetros de laboratorio.
Tabla 4. Reacciones adversas registradas en pacientes con CPNMm que recibieron darolutamida en combinación con docetaxel en el estudio ARASENSa, b
| Clase de sistema de órganos (MedDRA) |
Muy frecuentes |
Frecuentes |
| Trastornos vasculares |
Hipertensión arterialc |
|
| Trastornos de la piel y del tejido subcutáneo |
Erupción cutánead, e |
|
| Trastornos del sistema musculoesquelético y del tejido conjuntivo |
Fracturas |
|
| Trastornos del sistema reproductivo y de las glándulas mamarias |
Ginecomastia |
|
| Estudiosf |
Disminución del recuento de neutrófilos, aumento del nivel de bilirrubina en sangre, aumento de ALT, aumento de AST |
a La duración media de la exposición fue de 41,0 meses (rango de 0,1 a 56,5 meses) en pacientes que recibieron darolutamida + docetaxel y de 16,7 meses (rango de 0,3 a 55,8 meses) en pacientes que recibieron placebo + docetaxel.
b La frecuencia de reacciones adversas no puede atribuirse exclusivamente a la darolutamida, sino que puede depender de otros medicamentos utilizados en combinación.
c Incluye hipertensión arterial, aumento de la presión arterial y crisis hipertensiva.
d Incluye erupción cutánea, erupción medicamentosa, erupción eritematosa, erupción folicular, erupción maculosa, erupción maculopapular, erupción papular, prurito, erupción pustulosa, erupción vesicular, eritema y dermatitis.
e La frecuencia fue más alta durante los primeros 6 meses de tratamiento.
f Criterios Comunes de Terminología para Eventos Adversos (CTCAE), versión 4.03. La frecuencia se basa en valores reportados como desviaciones de los parámetros de laboratorio.
Descripción de reacciones adversas individuales
Función hepática
Se han notificado casos de hepatotoxicidad idiosincrásica inducida por medicamentos con aumento de alanina aminotransferasa (ALT) y/o aspartato aminotransferasa (AST) hasta ≥ 5 y ≥ 20 veces el límite superior normal (LSN), de grado 3 y 4, incluyendo aumento de transaminasas con aumento simultáneo de bilirrubina total hasta 2 × LSN, durante el tratamiento con darolutamida. El tiempo hasta el inicio varió entre 1 mes y 12 meses tras el inicio del tratamiento con darolutamida. En muchos casos, el aumento de ALT y AST fue reversible tras la interrupción de la darolutamida. Para recomendaciones, véase la sección «Precauciones de uso».
Cáncer de próstata no metastásico resistente a la castración (nmCRPC)
Fatiga
Se notificó fatiga/estado asténico en el 15,8 % de los pacientes que recibieron darolutamida y en el 11,4 % de los que recibieron placebo. Los eventos de grado más alto (grado 3) se registraron en el 0,6 % de los pacientes que recibieron darolutamida y en el 1,1 % de los que recibieron placebo. La fatiga (excluyendo astenia, letargo o malestar) se observó en la mayoría de los casos (12,1 % de los pacientes que recibieron darolutamida y 8,7 % de los que recibieron placebo).
Fracturas
Las fracturas ocurrieron en el 4,2 % de los pacientes que recibieron darolutamida y en el 3,6 % de los que recibieron placebo.
Enfermedad isquémica cardiaca e insuficiencia cardíaca
La enfermedad isquémica cardiaca ocurrió en el 3,2 % de los pacientes que recibieron darolutamida y en el 2,5 % de los que recibieron placebo. Los eventos de grado 5 se observaron en el 0,3 % de los pacientes que recibieron darolutamida y en el 0,2 % de los que recibieron placebo. La insuficiencia cardíaca ocurrió en el 1,9 % de los pacientes que recibieron darolutamida y en el 0,9 % de los que recibieron placebo.
Disminución del recuento de neutrófilos
La disminución del recuento de neutrófilos se observó como desviación de parámetros de laboratorio en el 19,6 % de los pacientes que recibieron darolutamida y en el 9,4 % de los que recibieron placebo. El tiempo medio hasta el valor más bajo fue de 256 días.
Las alteraciones de laboratorio fueron principalmente de grado 1-2 de intensidad. La disminución del recuento de neutrófilos de grado 3 y 4 se observó en el 3,5 % y 0,5 % de los pacientes, respectivamente. Solo un paciente interrumpió definitivamente el tratamiento con darolutamida debido a neutropenia. La neutropenia fue temporal o reversible (88 % de los pacientes) y no se asoció con signos ni síntomas clínicamente significativos.
Aumento de la bilirrubina en sangre
Se notificó aumento de bilirrubina como desviación de parámetros de laboratorio en el 16,4 % de los pacientes que recibieron darolutamida y en el 6,9 % de los que recibieron placebo. Los episodios fueron principalmente de grado 1 o 2, no asociados con signos ni síntomas clínicamente significativos y fueron reversibles tras la interrupción del tratamiento con darolutamida. El aumento de bilirrubina de grado 3 se observó en el 0,1 % de los pacientes que recibieron darolutamida y en el 0 % de los que recibieron placebo. En el grupo de darolutamida, el tiempo medio hasta el primer aumento de bilirrubina fue de 153 días, y la duración media del primer episodio fue de 182 días. En ningún paciente se interrumpió el tratamiento por aumento de bilirrubina.
Aumento de AST
Se notificó aumento de AST como desviación de parámetros de laboratorio en el 22,5 % de los pacientes que recibieron darolutamida y en el 13,6 % de los que recibieron placebo. Los episodios fueron principalmente de grado 1 o 2, no asociados con signos ni síntomas clínicamente significativos y fueron reversibles tras la interrupción del tratamiento con darolutamida. El aumento de AST de grado 3 se observó en el 0,5 % de los pacientes que recibieron darolutamida y en el 0,2 % de los que recibieron placebo. En el grupo de darolutamida, el tiempo medio hasta el primer aumento de AST fue de 258 días, y la duración media del primer episodio fue de 118 días. En ningún paciente se interrumpió el tratamiento por aumento de AST.
Cáncer de próstata metastásico sensible a hormonas (mCPRH)
Hipertensión arterial
En el estudio ARASENS, se notificó hipertensión arterial en el 13,8 % de los pacientes que recibieron darolutamida + docetaxel y en el 9,4 % de los que recibieron placebo + docetaxel.
La hipertensión arterial de grado 3 se registró en el 6,4 % de los pacientes que recibieron darolutamida + docetaxel, en comparación con el 3,5 % de los pacientes que recibieron placebo + docetaxel. Un paciente con hipertensión de grado 4 se notificó en cada grupo de tratamiento.
Se notificó un caso de hipertensión de grado 5 con arteriosclerosis de grado 5 en el grupo de darolutamida + docetaxel. Este paciente tenía antecedentes de hipertensión y tabaquismo, y el evento ocurrió más de 3 años después del inicio del tratamiento con darolutamida. Se notificaron más casos de hipertensión arterial en pacientes sin antecedentes de hipertensión en ambos grupos de tratamiento.
Fracturas
Las fracturas ocurrieron en el 7,5 % de los pacientes que recibieron darolutamida + docetaxel y en el 5,1 % de los que recibieron placebo + docetaxel.
Disminución del recuento de neutrófilos
Se notificó disminución del recuento de neutrófilos como desviación de parámetros de laboratorio en el 50,6 % de los pacientes que recibieron darolutamida + docetaxel y en el 45,5 % de los que recibieron placebo + docetaxel. La disminución del recuento de neutrófilos de grado 3 y 4 se observó en el 34,4 % de los pacientes que recibieron darolutamida + docetaxel y en el 31,4 % de los que recibieron placebo + docetaxel. En ambos grupos de tratamiento, el recuento de neutrófilos disminuyó, y la neutropenia fue más frecuente durante los primeros meses de tratamiento, tras lo cual la frecuencia y gravedad de los eventos disminuyeron.
Aumento de la bilirrubina en sangre
El aumento de la bilirrubina en sangre se observó como desviación de parámetros de laboratorio en el 19,6 % de los pacientes que recibieron darolutamida + docetaxel y en el 10,0 % de los que recibieron placebo + docetaxel. Los eventos fueron principalmente de grado 1 o 2. El aumento de bilirrubina de grado 3 y 4 se observó en el 0,5 % de los pacientes que recibieron darolutamida + docetaxel y en el 0,3 % de los que recibieron placebo + docetaxel.
Aumento de ALT y AST
Se notificó aumento de ALT como desviación de parámetros de laboratorio en el 42,3 % de los pacientes que recibieron darolutamida + docetaxel y en el 38,0 % de los que recibieron placebo + docetaxel. Se notificó aumento de AST como desviación de parámetros de laboratorio en el 43,9 % de los pacientes que recibieron darolutamida + docetaxel y en el 39,3 % de los que recibieron placebo + docetaxel. El aumento de ALT y AST fue principalmente de grado 1. El aumento de ALT de grado 3 y 4 se observó en el 3,7 % de los pacientes que recibieron darolutamida + docetaxel y en el 3,0 % de los que recibieron placebo + docetaxel. El aumento de AST de grado 3 y 4 se observó en el 3,6 % de los pacientes que recibieron darolutamida + docetaxel y en el 2,3 % de los que recibieron placebo + docetaxel.
Notificación de reacciones adversas sospechosas
La notificación de reacciones adversas tras la autorización del medicamento es importante. Permite una vigilancia continua de la relación beneficio-riesgo del medicamento. Los profesionales médicos y farmacéuticos, así como los pacientes o sus representantes legales, deben notificar cualquier caso sospechoso de reacción adversa o falta de eficacia del medicamento a través del Sistema Automatizado de Información de Farmacovigilancia en el siguiente enlace: https://aisf.dec.gov.ua.
Período de validez. 3 años.
Condiciones de conservación
No requiere condiciones especiales de conservación. Mantener en un lugar fuera del alcance y de la vista de los niños.
Envase
16 comprimidos en blíster, 7 blísteres en estuche de cartón.
Categoría de dispensación. Bajo receta médica.
Fabricante. Orion Corporation, Orion Pharma.
Domicilio del fabricante y dirección del lugar de actividad
Joensuunkatu 7, 24100 Salo, Finlandia.