Nubeka
UcrainaIndice
ISTRUZIONI PER L'USO DI NUBEKA (NUBEQA)
Composizione:
principio attivo: darolutamide;
1 compressa rivestita con film contiene 300 mg di darolutamide;
eccipienti: fosfato bicalcico, croscarmellosa sodica, lattosio monoidrato, magnesio stearato, povidone K 30; rivestimento filmogeno: lacca bianca (idrossipropilmetilcellulosa 15 cP, lattosio monoidrato, macrogol 3350, biossido di titanio (E 171)).
Forma farmaceutica. Compresse rivestite con film.
Caratteristiche fisico-chimiche principali: compresse ovali rivestite con film, di colore bianco o quasi bianco, con incisione «BAYER» su un lato e «300» sull'altro.
Gruppo farmacoterapeutico. Farmaci utilizzati nella terapia ormonale. Antagonisti ormonali e sostanze correlate. Agenti antiandrogeni. Darolutamide.
Codice ATC L02B B06.
Proprietà farmacodinamiche
Farmacodinamica
Meccanismo d'azione. Darolutamide è un inibitore del recettore degli androgeni (AR) con una struttura del pirazolo flessibile e polarmente sostituita, che si lega con elevata affinità direttamente al dominio di legame del ligando del recettore.
Darolutamide inibisce competitivamente il legame degli androgeni, la traslocazione nucleare dell'AR e la trascrizione mediata dall'AR. Il suo principale metabolita, cheto-darolutamide, ha dimostrato in vitro un'attività simile a quella di darolutamide. Il trattamento con darolutamide riduce la proliferazione delle cellule tumorali della prostata, determinando una potente attività antitumorale.
Effetti farmacodinamici. Non è stato osservato un allungamento medio dell'intervallo QTcF (ovvero superiore a 10 ms) dopo somministrazione orale di 600 mg di darolutamide due volte al giorno, rispetto al valore corrispondente con placebo.
Efficacia e sicurezza clinica. L'efficacia e la sicurezza sono state stabilite in due studi randomizzati, controllati con placebo, multicentrici di Fase III che hanno coinvolto pazienti con carcinoma della prostata non metastatico resistente alla castrazione (nmCRPC) (studio ARAMIS) e carcinoma della prostata metastatico sensibile agli ormoni (mCSPC) (studio ARASENS). Tutti i pazienti ricevevano contemporaneamente un analogo dell'ormone rilasciante della gonadotropina luteinizzante (LHRH) o avevano subito orchiettomia bilaterale.
Carcinoma della prostata non metastatico resistente alla castrazione (nmCRPC)
L'efficacia e la sicurezza dell'uso di darolutamide sono state valutate in uno studio randomizzato, in doppio cieco, controllato con placebo, multicentrico di Fase III (ARAMIS) su pazienti con carcinoma della prostata resistente alla castrazione non metastatico (valutato mediante metodiche di imaging convenzionali: TC, scintigrafia ossea, RM) con tempo di raddoppio dell'antigene specifico della prostata (PSA) ≤ 10 mesi.
I pazienti sono stati inclusi nello studio se avevano presentato 3 aumenti dei livelli di antigene specifico della prostata (PSA) dopo il valore più basso, misurati con un intervallo di almeno una settimana durante la terapia di deprivazione androgenica, con PSA ≥ 2 ng/ml allo screening e livelli sierici post-castrazione di testosterone < 1,7 nmol/l.
I pazienti con anamnesi di convulsioni sono stati ammessi allo studio. Nel gruppo darolutamide sono stati registrati 12 pazienti (0,21%) con anamnesi di convulsioni.
Sono stati esclusi dallo studio i pazienti con ipertensione non controllata o con ictus recente (negli ultimi 6 mesi), infarto miocardico, angina instabile/grave, bypass coronarico o periferico, scompenso cardiaco di classe III o IV secondo la classificazione della New York Heart Association.
Sono stati esclusi dallo studio i pazienti che avevano precedentemente ricevuto inibitori del recettore degli androgeni (AR) di seconda generazione, come enzalutamide, apalutamide e darolutamide, o inibitori dell'enzima CYP17, come l'acetato di abiraterone, nonché i pazienti che avevano ricevuto corticosteroidi sistemici in dosi superiori all'equivalente di 10 mg di prednisone al giorno nei 28 giorni precedenti la randomizzazione.
Complessivamente, 1509 pazienti sono stati randomizzati nel rapporto 2:1 per ricevere 600 mg di darolutamide per via orale due volte al giorno (n = 955) o placebo (n = 554).
Sono stati inclusi nello studio pazienti con linfonodi pelvici presenti ma con diametro minore di 2 cm sull'asse corto al di sotto della biforcazione dell'aorta. L'assenza o la presenza di metastasi è stata determinata mediante valutazione radiologica centralizzata indipendente. Tale valutazione è stata effettuata su 89 pazienti nei quali, retrospettivamente, sono state riscontrate metastasi all'inizio dello studio. La randomizzazione è stata stratificata in base al tempo di raddoppio del PSA (≤ 6 mesi o > 6 mesi) e all'uso di terapia mirata contro gli osteoclasti all'inizio dello studio (sì/no).
Le caratteristiche demografiche dei pazienti e quelle della malattia erano bilanciate tra i gruppi di trattamento. L'età media dei pazienti era di 74 anni (intervallo 48–95), il 9% aveva 85 anni o più; il 79% dei pazienti era di razza caucasica, il 13% di origine asiatica e il 3% di colore scuro della pelle. La maggior parte dei pazienti aveva un punteggio di Gleason di 7 o superiore al momento della diagnosi (73%). La mediana del tempo di raddoppio del PSA era di 4,5 mesi. Il 9% dei pazienti aveva subito orchiettomia precedente, il 25% prostatectomia precedente e il 50% almeno un precedente trattamento radioterapico. Il 76% dei pazienti aveva ricevuto più di un precedente trattamento antiormonale. I pazienti avevano un punteggio ECOG PS di 0 (69%) o 1 (31%).
Il trattamento con darolutamide è proseguito fino alla progressione della malattia documentata per via radiologica, valutata mediante imaging convenzionale (TC, scintigrafia ossea, RM) tramite valutazione centralizzata in cieco, fino a tossicità inaccettabile, ritiro del consenso o interruzione dello studio da parte del paziente.
Il punto finale primario di efficacia era la sopravvivenza libera da metastasi (SLM). I punti finali secondari erano la sopravvivenza globale (SG), il tempo alla progressione del dolore, il tempo all'inizio della prima chemioterapia citotossica per carcinoma della prostata e il tempo alla prima manifestazione scheletrica sintomatica (definita come l'insorgenza di uno qualsiasi dei seguenti eventi: radioterapia esterna per il sollievo dei sintomi scheletrici, nuova frattura patologica sintomatica, compressione del midollo spinale o intervento ortopedico chirurgico correlato al tumore).
Il trattamento con darolutamide ha determinato un miglioramento della SLM rispetto al placebo (vedi Tabella 1 e Fig. 1).
I risultati della SLM sono stati coerenti in tutti i sottogruppi di pazienti, indipendentemente dal tempo di raddoppio del PSA, dall'uso precedente di agenti che agiscono sul tessuto osseo o dalla malattia locale-regionale. Altri sottogruppi con risultati coerenti della SLM includevano il valore di PSA all'inizio dello studio, il punteggio di Gleason al momento della diagnosi, l'età del paziente, la regione geografica, il punteggio ECOG PS all'inizio dello studio, l'appartenenza razziale e il numero di cicli precedenti di terapia ormonale.
Dopo l'analisi primaria della SLM, non appena lo studio è stato svelato, ai pazienti che ricevevano placebo è stato offerto il trattamento con darolutamide (opzione di crossover). Dei 554 pazienti randomizzati al gruppo placebo, 170 (31%) sono passati al trattamento con darolutamide. L'analisi della sopravvivenza globale non è stata aggiustata per tenere conto dell'impatto del crossover.
All'analisi finale, il trattamento con darolutamide ha determinato un miglioramento statisticamente significativo della sopravvivenza globale rispetto al placebo (la mediana non è stata raggiunta in nessun gruppo, vedi Tabella 1 e Fig. 2). Sono stati inoltre osservati aumenti statisticamente significativi del tempo alla progressione del dolore, del tempo all'inizio della prima chemioterapia citotossica e del tempo alla prima manifestazione scheletrica sintomatica rispetto al placebo (vedi Tabella 1).
All'analisi finale, la durata media del trattamento nei pazienti che ricevevano darolutamide era di 33,3 mesi (intervallo da 0,0 a 74,0 mesi) durante il periodo combinato in doppio cieco e aperto.
Tutte le analisi sono state effettuate sul Full Analysis Set.
Tabella 1. Risultati di efficacia dallo studio ARAMIS
| Parametro di efficacia |
Numero (%) di pazienti con eventi |
Mediana (mesi) (95 % IC) |
Rapporto dei rischi (HR)b (95 % intervallo di confidenza [IC]) Valore p (bilateralе) |
||
| Darolutamide (N = 955) |
Placeboa (N = 554) |
Darolutamide (N = 955) |
Placeboa (N = 554) |
||
| Sopravvivenza libera da metastasi c |
221 (23,1 %) |
216 (39,0 %) |
40,4 (34,3; NM) |
18,4 (15,5; 22,3) |
0,413 (0,341; 0,500) < 0,000001 |
| Sopravvivenza globale |
148 (15,5 %) |
106 (19,1 %) |
NM (56,1; NM) |
NM (46,9; NM) |
0,685 (0,533; 0,881) 0,003048 |
| Tempo fino alla progressione del dolorec, d |
251 (26,3 %) |
178 (32,1 %) |
40,3 (33,2; 41,2) |
25,4 (19,1; 29,6) |
0,647 (0,533; 0,785) 0,000008 |
| Tempo fino all'inizio della prima chemioterapia citotossica |
127 (13,3 %) |
98 (17,7 %) |
NM (NM, NM) |
NM |
0,579 (0,444; 0,755) 0,000044 |
| Tempo fino al primo evento scheletrico sintomatico |
29 (3,0 %) |
28 (5,1 %) |
NM (NM, NM) |
(NM, NM) |
0,484 (0,287; 0,815) 0,005294 |
a Compresi 170 pazienti che sono passati al darolutamide senza periodo di cieco.
b Rapporto di rischio < 1 per il darolutamide.
c Per la SMN e il tempo alla progressione del dolore, l'analisi primaria è considerata l'analisi finale.
d Risultati riferiti dal paziente secondo la valutazione del questionario Brief Pain Inventory – Short Form.
ND – non raggiunto.
Il risultato del trattamento con darolutamide è stato un più lungo tempo di sopravvivenza libera da progressione (SLP, mediana 36,8 vs 14,8 mesi, HR = 0,380, p nominale < 0,000001) e tempo alla progressione del PSA (mediana 29,5 vs 7,2 mesi, HR = 0,164, p nominale < 0,000001). La coerenza dell'effetto è stata osservata per tutti gli endpoint di sopravvivenza (SLM, SG e PSA).
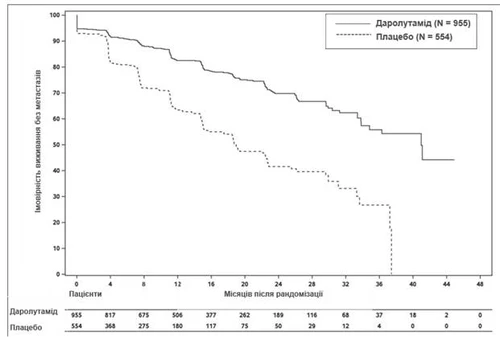
Fig. 1. Curve di Kaplan-Meier della sopravvivenza libera da metastasi (ARAMIS)
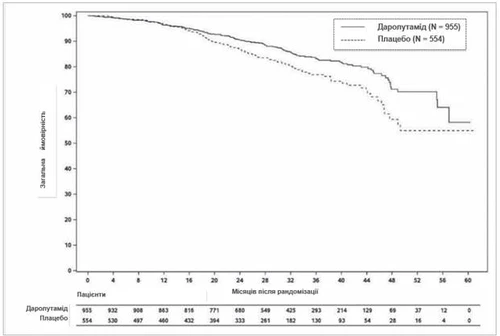
Fig. 2. Curve di Kaplan-Meier della sopravvivenza globale (ARAMIS)
I pazienti che hanno ricevuto darolutamide nello studio ARAMIS (periodo in doppio cieco) hanno mostrato una frequenza confermata significativamente più alta di risposta in termini di livello di PSA (definita come riduzione ≥ 50 % rispetto al valore basale) rispetto ai pazienti che hanno ricevuto placebo: 84,0 % vs 7,9 % (differenza = 76,1 %, p < 0,000001 (valore p nominale, solo a scopo informativo)).
Carcinoma prostatico metastatico sensibile agli ormoni (mCRPC)
L'efficacia e la sicurezza dell'uso del darolutamide in combinazione con docetaxel sono state valutate in uno studio multicentrico di fase III in doppio cieco controllato con placebo (ARASENS) su pazienti con mCRPC. Complessivamente, 1306 pazienti sono stati randomizzati in rapporto 1:1 per ricevere 600 mg di darolutamide per via orale due volte al giorno (n = 651) o placebo (n = 655) contemporaneamente al docetaxel 75 mg/m² per 6 cicli. Il trattamento con darolutamide o placebo è proseguito fino alla progressione sintomatica della malattia, al cambio di terapia antitumorale, a tossicità inaccettabile, alla morte o all'uscita del paziente dallo studio.
La presenza di metastasi è stata valutata mediante valutazione radiologica centralizzata indipendente. I pazienti con coinvolgimento esclusivo dei linfonodi regionali (M0) sono stati esclusi dallo studio. La randomizzazione è stata stratificata per grado di malattia (solo metastasi ai linfonodi extraregionali (M1a), metastasi ossee con o senza metastasi linfonodali (M1b), o metastasi viscerali con o senza metastasi linfonodali o ossee (M1c)) e per livello di fosfatasi alcalina (< o ≥ limite superiore della norma) all'inizio dello studio. Era consentita l'inclusione di pazienti con metastasi cerebrali, ma nessun paziente di questo tipo è stato arruolato.
Le caratteristiche demografiche e quelle della malattia erano bilanciate tra i gruppi di trattamento. L'età media era di 67 anni (intervallo 41–89), lo 0,5 % dei pazienti aveva un'età ≥ 85 anni, il 52 % era di razza caucasica, il 36 % di origine asiatica e il 4 % dei partecipanti allo studio aveva la pelle scura. La maggior parte dei pazienti aveva un punteggio di Gleason pari o superiore a 8 al momento della diagnosi (78 %). Il 71 % dei pazienti aveva un punteggio ECOG PS di 0, il 29 % di 1. L'86,1 % dei pazienti aveva una malattia de novo e il 12,9 % una recidiva. All'inizio dello studio, il 3 % dei pazienti aveva M1a, il 79,5 % M1b e il 17,5 % M1c; la fosfatasi alcalina era < LSN nel 44,5 % dei pazienti e ≥ LSN nel 55,5 %; il livello medio basale di PSA era di 30,3 µg/l e 24,2 µg/l rispettivamente nei gruppi darolutamide e placebo. I pazienti con anamnesi di convulsioni potevano partecipare allo studio, e 4 pazienti (0,6 %) sono stati inclusi nel gruppo darolutamide + docetaxel.
Il 77,0 % dei pazienti aveva una malattia ad alto volume e il 23,0 % una malattia a basso volume. La malattia ad alto volume è stata definita come presenza di metastasi viscerali o di 4 o più lesioni ossee, con almeno una metastasi al di fuori della colonna vertebrale e dell'osso pelvico. Circa il 25 % dei pazienti ha ricevuto trattamento concomitante con bifosfonati o denosumab.
L'endpoint primario di efficacia era la sopravvivenza globale (SG). Gli endpoint secondari comprendevano il tempo allo sviluppo di carcinoma prostatico resistente alla castrazione, il tempo alla progressione del dolore, il tempo di sopravvivenza libera da sintomi da coinvolgimento scheletrico, il tempo alla prima evento scheletrico sintomatico, il tempo all'inizio della successiva terapia antitumorale, il tempo al peggioramento dei sintomi fisici correlati alla malattia e il tempo all'inizio dell'uso di oppioidi per ≥ 7 giorni consecutivi. La progressione del dolore è stata valutata tramite i risultati riferiti dal paziente secondo il questionario Brief Pain Inventory – Short Form, definita come un peggioramento di almeno 2 punti rispetto al valore basale e l'inizio dell'assunzione di oppioidi a breve o lunga durata d'azione per il controllo del dolore per ≥ 7 giorni consecutivi.
La durata media del trattamento è stata di 41,0 mesi (intervallo da 0,1 a 56,5 mesi) nei pazienti trattati con darolutamide + docetaxel e di 16,7 mesi (intervallo da 0,3 a 55,8 mesi) nei pazienti trattati con placebo + docetaxel. L'87,6 % e l'85,5 % dei pazienti hanno completato tutti e 6 i cicli di docetaxel, mentre l'1,5 % e il 2,0 % dei pazienti non hanno ricevuto docetaxel rispettivamente nei gruppi darolutamide + docetaxel e placebo + docetaxel.
Tabella 2. Risultati di efficacia dello studio ARASENS
| Parametro di efficacia |
Numero (%) di pazienti con eventi |
Mediana (mesi) (IC 95%) |
Rapporto di rischio (HR)b (intervallo di confidenza [IC] al 95%) Valore p (unilaterale)c |
||
| Darolutamide + docetaxel (N = 651) |
Placebo + docetaxel (N = 654)а |
Darolutamide + docetaxel (N = 651) |
Placebo + docetaxel (N = 654)а |
||
| Sopravvivenza globaled |
229 (35,2%) |
304 (46,5%) |
ND (ND, ND) |
48,9 (44,4; ND) |
0,675 (0,568; 0,801) <0,0001 |
a Un paziente nel gruppo placebo è stato escluso da tutte le analisi.
b Rapporto di rischio < 1 a favore di darolutamide.
c Basato su un test log-rank stratificato.
d I risultati di SLD sono stati coerenti in tutti i sottogruppi di pazienti, compreso lo stadio della malattia e i livelli di fosfatasi alcalina.
ND – non raggiunto.
Endpoint secondari di efficacia in cui è stata dimostrata una superiorità statisticamente significativa per i pazienti del gruppo darolutamide + docetaxel rispetto ai pazienti del gruppo placebo + docetaxel: tempo alla progressione verso un carcinoma della prostata resistente alla castrazione (mediana ND vs 19,1 mesi; HR = 0,357, p < 0,0001); tempo alla prima evento scheletrico sintomatico (mediana ND vs ND; HR = 0,712, p = 0,0081); tempo all’inizio della successiva chemioterapia antitumorale (mediana ND vs 25,3 mesi; HR = 0,388, p < 0,0001); tempo alla progressione del dolore (mediana ND vs 27,5 mesi; HR = 0,792, p = 0,0058); tempo alla sopravvivenza libera da eventi scheletrici sintomatici (mediana 51,2 vs 39,7 mesi; HR = 0,609, p < 0,0001).
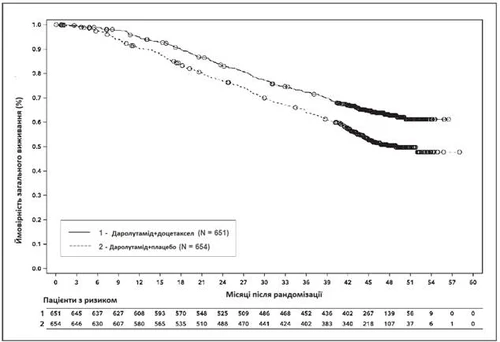
Fig. 3. Curve di sopravvivenza globale di Kaplan-Meier (ARASENS)a
a La SLD a 36 mesi è stata del 72,3 % (IC 95 %, 68,8–75,8) nel gruppo darolutamide + docetaxel rispetto al 63,8 % (IC 95 %, 60,1–67,6) nel gruppo placebo + docetaxel.
La SLD a 48 mesi è stata del 62,7 % (IC 95 %, 58,7–66,7) nel gruppo darolutamide + docetaxel rispetto al 50,4 % (IC 95 %, 46,3–54,6) nel gruppo placebo + docetaxel.
Farmacocinetica
Darolutamide è composta da due diastereomeri [(S,R)-darolutamide e (S,S)-darolutamide], che si interconvertono tramite il principale metabolita circolante cheto-darolutamide. In vitro, le tre sostanze mostrano attività farmacologica simile. Darolutamide ha scarsa solubilità nei solventi acquosi in un ampio intervallo di pH ed è generalmente più solubile nei solventi organici.
Assorbimento
Dopo somministrazione orale di 600 mg (2 compresse da 300 mg) due volte al giorno, le concentrazioni plasmatiche di picco di darolutamide allo steady state sono state di 4,79 mg/l (coefficiente di variazione 30,9 %) nei pazienti con nmCRPC nello studio ARAMIS e di 3,84 mg/l (coefficiente di variazione 35,6 %) nei pazienti con mHSPC nello studio ARASENS. Il tempo medio per raggiungere la concentrazione massima nel plasma è stato di 3–4 ore. Il rapporto tra i due diastereomeri, (S,R)-darolutamide e (S,S)-darolutamide, è cambiato da 1:1 nella compressa a circa 1:9 nel plasma, basato sull’indice AUC0-12 allo steady state. Dopo somministrazione orale con cibo, lo steady state viene raggiunto dopo 2–5 giorni di somministrazione ripetuta due volte al giorno.
La biodisponibilità assoluta dopo somministrazione orale a digiuno della compressa di Nubeka contenente 300 mg di darolutamide è di circa il 30 % rispetto a quella dopo somministrazione endovenosa.
La biodisponibilità di darolutamide aumentava da 2,0 a 2,5 volte quando assunta con il cibo. Un aumento simile dell’esposizione è stato osservato per il metabolita principale, cheto-darolutamide.
Distribuzione
Il volume di distribuzione apparente di darolutamide dopo somministrazione endovenosa è di 119 l, indicando una diffusa distribuzione del farmaco nell’organismo sia nello spazio intracellulare che extracellulare.
Darolutamide si lega in modo moderato (92 %) alle proteine plasmatiche umane, senza differenze tra i due diastereomeri. Il principale metabolita di darolutamide, cheto-darolutamide, si lega in larga misura (99,8 %) alle proteine plasmatiche.
Il passaggio di darolutamide attraverso la barriera ematoencefalica non è stato clinicamente studiato. Tuttavia, l’impatto di darolutamide sul cervello in termini di AUC0-24 è molto basso: 4,5 % dell’esposizione plasmatica dopo una dose singola nei ratti e 1,9–3,9 % dopo dosi ripetute nei topi. Ciò indica un basso passaggio di darolutamide attraverso la barriera ematoencefalica intatta nei ratti e nei topi e una bassa probabilità che darolutamide attraversi la barriera ematoencefalica intatta negli esseri umani in misura clinicamente rilevante.
Biotrasformazione
I diastereomeri (S,R)-darolutamide e (S,S)-darolutamide possono interconvertirsi tramite il metabolita cheto-darolutamide, con un predominio di (S,S)-darolutamide.
Dopo una dose orale singola di 300 mg di 14C-darolutamide in forma di soluzione orale, cheto-darolutamide è l’unico metabolita principale, con un’esposizione plasmatica totale circa doppia rispetto a darolutamide. Insieme, darolutamide e cheto-darolutamide rappresentano l’87,4 % della radioattività 14C nel plasma, indicando che tutti gli altri metaboliti sono trascurabili.
Darolutamide è metabolizzata principalmente attraverso il metabolismo ossidativo mediato principalmente dal CYP3A4, nonché attraverso la glucuronizzazione diretta mediata principalmente da UGT1A9 e UGT1A1. Inoltre, è stato dimostrato che le isoforme AKR1C catalizzano principalmente la riduzione di cheto-darolutamide ai diastereomeri della sostanza.
Eliminazione
L’emivita effettiva di darolutamide e cheto-darolutamide nel plasma dei pazienti è di circa 18–20 ore. Tra i due diastereomeri che compongono darolutamide, (S,R)-darolutamide ha un’emivita effettiva più breve – 9 ore – rispetto a (S,S)-darolutamide, il cui emivita effettiva è di 22 ore. La clearance di darolutamide dopo somministrazione endovenosa è stata di 116 ml/min (CV 39,7 %). Nel complesso, il 63,4 % di darolutamide e dei suoi metaboliti viene escreto nelle urine (circa il 7 % in forma invariata), il 32,4 % viene escreto con le feci. Oltre il 95 % della dose è stato eliminato entro 7 giorni dall’assunzione.
Linearità/non linearità
Nel range di dosi da 100 a 700 mg (dopo dose singola e allo steady state), l’esposizione ai due diastereomeri e al metabolita principale cheto-darolutamide aumenta linearmente quasi in funzione della dose. A causa di un assorbimento saturabile, non è stato osservato un ulteriore aumento dell’esposizione a darolutamide alla dose di 900 mg due volte al giorno.
Gruppi di pazienti particolari
Pazienti anziani. Non sono state osservate differenze clinicamente significative nella farmacocinetica di darolutamide (65–95 anni).
Alterazioni della funzionalità renale. In uno studio clinico di farmacocinetica, AUC e Cmax di darolutamide sono risultati rispettivamente 2,5 e 1,6 volte più elevati nei pazienti con insufficienza renale grave (velocità stimata di filtrazione glomerulare [eGFR] da 15 a 29 ml/min/1,73 m2) rispetto ai volontari sani.
Un’analisi farmacocinetica di popolazione indica un’esposizione (AUC) a darolutamide rispettivamente 1,1, 1,3 e circa 1,5 volte maggiore nei pazienti con compromissione renale lieve, moderata e grave (eGFR da 15 a 89 ml/min/1,73 m2) rispetto ai pazienti con funzionalità renale normale.
La farmacocinetica di darolutamide non è stata studiata nei pazienti con insufficienza renale terminale in trattamento dialitico (eGFR < 15 ml/min/1,73 m2).
Alterazioni della funzionalità epatica. In uno studio clinico di farmacocinetica, Cmax e AUC di darolutamide sono risultati rispettivamente 1,5 e 1,9 volte più elevati nei pazienti con insufficienza epatica moderata (classe B secondo la classificazione di Child-Pugh) rispetto ai volontari sani. Non sono disponibili dati per pazienti con insufficienza epatica grave (classe C secondo la classificazione di Child-Pugh).
Differenze etniche. Non sono state osservate differenze clinicamente significative nella farmacocinetica di darolutamide in base all’etnia del paziente (pazienti di razza caucasica, pazienti di origine giapponese, pazienti asiatici non giapponesi, pazienti con pelle scura o di origine afroamericana). Un’analisi farmacocinetica di popolazione ha mostrato un aumento dell’esposizione media geometrica (AUC) fino a 1,56 volte (IC 90 % 1,43–1,70) nei pazienti di origine giapponese rispetto ai pazienti di altre regioni negli studi ARAMIS e ARASENS.
Dati preclinici di sicurezza
Toxicità sistemica. Negli studi di tossicità con dosi ripetute negli animali, i principali effetti osservati riguardavano gli organi riproduttivi maschili (riduzione della massa degli organi con atrofia della prostata e dell’epididimo). Questi effetti si sono verificati con esposizioni sistemiche nell’intervallo o inferiori a quelle previste nell’uomo (in base al confronto dell’AUC). Ulteriori alterazioni nei tessuti riproduttivi includevano un minimo aumento della vacuolizzazione dell’ipofisi, atrofia e ridotta secrezione delle vescicole seminali e delle ghiandole mammarie nei ratti, nonché ipospermia, dilatazione dei tubuli seminiferi e degenerazione nei cani. Le alterazioni negli organi riproduttivi maschili in entrambe le specie erano correlate all’attività farmacologica di darolutamide e si sono risolte o parzialmente risolte dopo un periodo di recupero di 4–8 settimane.
Embrotossicità/teratogenicità. Non sono stati condotti studi sulla tossicità per il feto.
Tossicità riproduttiva. Non sono stati condotti studi sulla tossicità riproduttiva. Tuttavia, la fertilità maschile potrebbe essere compromessa, in base ai risultati degli studi di tossicità con dosi ripetute negli animali, coerenti con l’attività farmacologica di darolutamide.
Genotossicità e cancerogenicità. Darolutamide non ha indotto mutazioni nel test di mutagenesi microbica (Ames). A concentrazioni elevate, darolutamide ha indotto aberrazioni cromosomiche strutturali in vitro in linfociti umani coltivati. Tuttavia, non è stata osservata genotossicità nel test combinato dei micronuclei nel midollo osseo in vivo e nell’analisi Comet nel fegato e nel duodeno del ratto, a esposizioni superiori all’esposizione massima nell’uomo.
La somministrazione orale di darolutamide a maschi di topi transgenici rasH2 per 6 mesi non ha evidenziato potenziale cancerogeno alle dosi fino a 1000 mg/kg/giorno, che superano l’esposizione clinica (AUC) di darolutamide da 0,9 a 1,3 volte e di cheto-darolutamide da 2,1 a 2,3 volte rispetto alla dose clinica raccomandata di 1200 mg/giorno. Sulla base di questo studio, non può essere escluso completamente il rischio cancerogeno con l’uso di darolutamide.
Caratteristiche cliniche
Indicazioni
Il medicinale Nubeka è indicato per il trattamento negli uomini adulti:
- di carcinoma della prostata resistente alla castrazione non metastatico (nmCRPC) con alto rischio di metastasi (vedere il paragrafo «Proprietà farmacologiche»);
- di carcinoma della prostata ormono-sensibile metastatico (mHSPC) – in combinazione con docetaxel e terapia di deprivazione androgenica (vedere il paragrafo «Proprietà farmacologiche»).
Controindicazioni
Ipersensibilità alla sostanza attiva o agli eccipienti.
È controindicato in donne in gravidanza e in donne in età fertile.
Interazioni con altri medicinali e altre forme di interazione
Effetto di altri medicinali su darolutamide
Induttori di CYP3A4 e P-gp
Darolutamide è un substrato del CYP3A4 e della glicoproteina-P (P-gp).
L’uso concomitante di induttori forti e moderati di CYP3A4 e induttori di P-gp (ad esempio carbamazepina, fenobarbital, erba di San Giovanni, fenitoina e rifampicina) durante il trattamento con darolutamide non è raccomandato, salvo nei casi in cui non esista un’alternativa terapeutica. Si deve considerare la possibilità di scegliere un medicinale concomitante alternativo che non induca o abbia un potenziale debole di induzione di CYP3A4 o P-gp.
L’assunzione ripetuta di rifampicina (600 mg), un potente induttore di CYP3A4 e P-gp, in combinazione con una dose singola di darolutamide (600 mg) assunta con cibo ha determinato una riduzione media dell’esposizione (AUC0-72) del 72% e una riduzione della Cmax di darolutamide del 52%.
Inibitori di CYP3A4, P-gp e BCRP
Darolutamide è un substrato del CYP3A4, P-gp e della proteina di resistenza al cancro al seno (BCRP).
Quando si utilizzano inibitori di CYP3A4, P-gp o BCRP, non ci si aspetta un’interazione clinicamente significativa tra i farmaci. Darolutamide può essere somministrato contemporaneamente a inibitori di CYP3A4, P-gp o BCRP. L’uso concomitante di darolutamide con un inibitore potente combinato di CYP3A4 e P-gp aumenta l’esposizione a darolutamide, il che potrebbe aumentare il rischio di reazioni avverse. Si raccomanda un monitoraggio più frequente del paziente per la comparsa di reazioni avverse a darolutamide e, se necessario, un aggiustamento della dose di darolutamide.
L’uso di itraconazolo (200 mg due volte al giorno al giorno 1 e una volta al giorno nei successivi 7 giorni), un inibitore potente di CYP3A4, P-gp e BCRP, in combinazione con una dose singola di darolutamide (600 mg al giorno 5 assunta con cibo) ha determinato un aumento medio di 1,7 volte dell’esposizione (AUC0-72) e un aumento di 1,4 volte della Cmax di darolutamide.
Inibitori di UGT1A9
Darolutamide è un substrato di UGT1A9. Quando si utilizza un inibitore di UGT1A9, non ci si aspetta un’interazione clinicamente significativa tra i farmaci. Darolutamide può essere somministrato contemporaneamente a inibitori di UGT1A9.
Un’analisi farmacocinetica popolazionale ha mostrato che l’uso concomitante di inibitori di UGT1A9 con darolutamide ha determinato un aumento di 1,2 volte dell’esposizione (AUC0-72) a darolutamide.
Docetaxel
L’uso di darolutamide in combinazione con docetaxel non ha determinato variazioni clinicamente significative della farmacocinetica di darolutamide nei pazienti con mHSPC (vedere il paragrafo «Proprietà farmacologiche»).
Effetto di darolutamide su altri medicinali
Substrati di BCRP, OATP1B1 e OATP1B3
Darolutamide è un inibitore della proteina di resistenza al cancro al seno e dei peptidi trasportatori di anioni organici (OATP) 1B1 e 1B3.
Si deve evitare l’uso concomitante di rosuvastatina, salvo nei casi in cui non esista un’alternativa terapeutica. Si deve considerare la possibilità di scegliere un medicinale concomitante alternativo con minor potenziale di inibizione di BCRP, OATP1B1 e OATP1B3.
L’uso di darolutamide (600 mg due volte al giorno per 5 giorni) prima di una singola dose di rosuvastatina (5 mg) assunta con cibo ha determinato un aumento di circa 5 volte dell’esposizione media (AUC) e della Cmax di rosuvastatina.
Se possibile, si deve evitare l’uso concomitante di darolutamide con altri substrati di BCRP. L’uso concomitante di darolutamide può aumentare la concentrazione plasmatica di altri farmaci concomitanti substrati di BCRP, OATP1B1 e OATP1B3 (come metotrexato, sulfasalazina, fluvastatina, atorvastatina, pitavastatina). Pertanto, si raccomanda di monitorare i pazienti per la comparsa di reazioni avverse legate all’uso di questi substrati. Inoltre, quando si usano questi substrati in concomitanza con darolutamide, si devono seguire le raccomandazioni appropriate riportate nelle informazioni su tali substrati.
Substrati di P-gp
Quando si somministra un substrato di P-gp, non ci si aspetta un’interazione clinicamente significativa tra i farmaci. Darolutamide può essere somministrato contemporaneamente a substrati di P-gp (ad esempio digossina, verapamil o nifedipina). L’uso concomitante di darolutamide con il sensibile substrato di P-gp dabigatran etexilato non ha determinato un aumento dell’esposizione (AUC e Cmax) di dabigatran.
Substrati di CYP3A4
Darolutamide è un debole induttore di CYP3A4.
Quando si somministra un substrato di CYP, non ci si aspetta un’interazione clinicamente significativa tra i farmaci. Darolutamide può essere somministrato contemporaneamente a substrati di CYP (ad esempio warfarin, L-tiroxina, omeprazolo).
L’uso di darolutamide (600 mg due volte al giorno per 9 giorni) prima di una singola dose di midazolam (1 mg), un sensibile substrato di CYP3A4, assunta con cibo, ha determinato una riduzione media dell’esposizione (AUC) e della Cmax di midazolam rispettivamente del 29% e del 32%. Darolutamide non inibisce il metabolismo di singoli substrati di CYP in vitro a concentrazioni clinicamente rilevanti.
Docetaxel
L’uso di darolutamide in combinazione con docetaxel non ha determinato variazioni clinicamente significative della farmacocinetica di docetaxel nei pazienti con mHSPC (vedere il paragrafo «Proprietà farmacologiche»).
Medicinali che prolungano l’intervallo QT
Poiché la terapia di deprivazione androgenica può prolungare l’intervallo QT, si deve valutare attentamente l’uso concomitante con medicinali noti per prolungare l’intervallo QT o medicinali che possono indurre torsade de pointes. Tra questi vi sono medicinali come antiaritmici di classe IA (ad esempio chinidina, disopiramide) o di classe III (ad esempio amiodarone, sotalolo, dofetilide, ibutilide), metadone, moxifloxacina e farmaci antipsicotici (ad esempio aloperidolo).
Caratteristiche particolari di impiego
Compromissione renale
I dati disponibili sull'uso in pazienti con insufficienza renale grave sono limitati.
Poiché l'esposizione può essere aumentata, è necessario monitorare attentamente questi pazienti per la comparsa di reazioni avverse (vedere il paragrafo «Caratteristiche particolari di impiego» e il sottoparagrafo «Farmacocinetica»).
Compromissione epatica
I dati disponibili sull'uso in pazienti con compromissione epatica moderata sono limitati. L'uso di darolutamide non è stato studiato in pazienti con compromissione epatica grave.
Poiché l'esposizione può essere aumentata, è necessario monitorare attentamente questi pazienti per la comparsa di reazioni avverse (vedere il paragrafo «Caratteristiche particolari di impiego» e il sottoparagrafo «Farmacocinetica»).
Recente patologia cardiovascolare
I pazienti con patologie cardiovascolari clinicamente rilevanti negli ultimi 6 mesi, inclusi ictus, infarto miocardico, angina instabile/grave, bypass coronarico o periferico e insufficienza cardiaca congestizia sintomatica, sono stati esclusi dagli studi clinici. Pertanto, la sicurezza dell'uso di darolutamide in questi pazienti non è stata stabilita.
Nel caso in cui il medicinale Nubeka venga prescritto a un paziente con patologia cardiovascolare clinicamente rilevante, la patologia deve essere trattata in conformità con le raccomandazioni stabilite.
Epatotossicità
In caso di alterazioni dei parametri di funzionalità epatica che indicano un danno epatico indotto da farmaci (epatotossicità idiopatica), il trattamento con darolutamide deve essere definitivamente interrotto (vedere il paragrafo «Reazioni avverse»).
Uso concomitante con altri medicinali
L'uso concomitante di forti induttori di CYP3A4 e P-gp durante il trattamento con darolutamide può ridurre la concentrazione plasmatica di darolutamide e pertanto non è raccomandato, salvo nei casi in cui non esista un'alternativa terapeutica. Si deve considerare la possibilità di scegliere un medicinale concomitante alternativo con un potenziale minore di induzione di CYP3A4 o P-gp (vedere il paragrafo «Interazioni con altri medicinali ed altre forme di interazione»).
È necessario monitorare i pazienti per la comparsa di reazioni avverse quando vengono utilizzati substrati di BCRP, OATP1B1 e OATP1B3, poiché l'uso concomitante con darolutamide può aumentare la concentrazione plasmatica di questi substrati.
È necessario evitare l'uso concomitante con rosuvastatina, salvo nei casi in cui non esista un'alternativa terapeutica (vedere il paragrafo «Interazioni con altri medicinali ed altre forme di interazione»).
La terapia di privazione androgenica può prolungare l'intervallo QT
Nei pazienti con fattori di rischio anamnestici di prolungamento dell'intervallo QT e nei pazienti che assumono contemporaneamente medicinali in grado di prolungare l'intervallo QT (vedere il paragrafo «Interazioni con altri medicinali ed altre forme di interazione»), i medici devono valutare il rapporto rischio/beneficio, compresa la probabilità di torsade de pointes, prima di iniziare la terapia con il medicinale Nubeka.
Informazioni sugli eccipienti
Il medicinale Nubeka contiene lattosio. I pazienti con rari problemi ereditari di intolleranza al galattosio, deficit di lattasi o malassorbimento di glucosio-galattosio non devono assumere questo medicinale.
Uso durante la gravidanza o l'allattamento
Questo medicinale è controindicato nelle donne in età fertile. Non deve essere somministrato a donne in gravidanza o che allattano al seno (vedere i paragrafi «Indicazioni» e «Controindicazioni»).
Donne in età fertile / contraccezione per uomini e donne
Non è noto se darolutamide o i suoi metaboliti siano presenti nello sperma. Se il paziente ha rapporti sessuali con una donna in età fertile, durante il trattamento e per 1 settimana dopo la sua conclusione, deve essere utilizzato un metodo contraccettivo ad alta efficacia (< 1% di tasso di fallimento annuo) per prevenire la gravidanza.
Gravidanza
A causa del meccanismo d'azione, darolutamide può arrecare danno al feto. Studi preclinici sulla tossicità riproduttiva non sono stati condotti (vedere il paragrafo «Dati preclinici di sicurezza»).
Non è noto se darolutamide o i suoi metaboliti siano presenti nello sperma. Se il paziente ha rapporti sessuali con una donna incinta, deve utilizzare un preservativo durante il trattamento e per 1 settimana dopo la sua conclusione. È necessario evitare l'esposizione del feto agli inibitori dei recettori androgeni attraverso il trasferimento del liquido seminale a una donna incinta, poiché ciò potrebbe influire sullo sviluppo fetale.
Allattamento
Non è noto se darolutamide o i suoi metaboliti siano escreti nel latte materno. Studi sugli animali per valutare l'escrezione di darolutamide o dei suoi metaboliti nel latte non sono stati condotti (vedere il paragrafo «Dati preclinici di sicurezza»). Non può essere escluso un rischio per il bambino allattato al seno.
Fertilità
Non sono disponibili dati sull'effetto di darolutamide sulla fertilità umana.
Studi sugli animali indicano che Nubeka può compromettere la fertilità negli uomini con potenziale riproduttivo (vedere il paragrafo «Dati preclinici di sicurezza»).
Capacità di guidare veicoli o di usare macchinari
Il medicinale Nubeka non ha alcun effetto oppure ha un effetto trascurabile sulla capacità di guidare veicoli o di usare macchinari.
Modalità e posologia di somministrazione
Il trattamento deve essere iniziato e monitorato da un medico esperto nel trattamento del cancro alla prostata.
Dosaggio
La dose raccomandata è di 600 mg di darolutamide (due compresse da 300 mg) due volte al giorno, pari a una dose giornaliera totale di 1200 mg (vedere il paragrafo «Farmacocinetica»).
Il trattamento con darolutamide deve proseguire fino alla progressione della malattia o a tossicità inaccettabile.
Nei pazienti non castrati chirurgicamente, deve essere continuata la castrazione farmacologica con un analogo del LHRH durante il trattamento.
Carcinoma della prostata ormono-sensibile metastatico (mCSPC)
I pazienti con mCSPC devono iniziare il trattamento con darolutamide in combinazione con docetaxel (vedere il paragrafo «Proprietà farmacologiche»). Il primo dei 6 cicli di docetaxel deve essere somministrato entro 6 settimane dall'inizio della terapia con darolutamide. È necessario seguire le raccomandazioni riportate nel foglio illustrativo del docetaxel. Il trattamento con darolutamide deve proseguire fino alla progressione della malattia o a tossicità inaccettabile, anche qualora la somministrazione di docetaxel venga posticipata, interrotta o sospesa.
Dose mancata
Se una dose viene dimenticata, deve essere assunta non appena il paziente se ne ricorda, ma solo nello stesso giorno. Il paziente non deve assumere due dosi contemporaneamente per compensare la dose dimenticata.
Modifica del dosaggio
In caso di tossicità di grado ≥ 3 o di reazione avversa inaccettabile correlata all’uso di darolutamide (vedere i paragrafi «Precauzioni particolari» e «Effetti indesiderati»), la dose deve essere sospesa o ridotta a 300 mg due volte al giorno fino alla risoluzione dei sintomi. Successivamente, il trattamento può essere ripreso alla dose di 600 mg due volte al giorno. Non è raccomandata una riduzione della dose al di sotto di 300 mg due volte al giorno, poiché l’efficacia di tale regime non è stata stabilita.
Popolazioni particolari
Pazienti anziani. Non è necessaria alcuna modifica del dosaggio nei pazienti anziani (vedere il paragrafo «Farmacocinetica»).
Compromissione renale. Nei pazienti con compromissione renale lieve o moderata non è necessaria alcuna modifica del dosaggio. Nei pazienti con insufficienza renale grave (clearance della creatinina 15–29 ml/min/1,73 m²), che non siano sottoposti a emodialisi, la dose iniziale raccomandata è di 300 mg due volte al giorno (vedere il paragrafo «Precauzioni particolari» e il paragrafo «Farmacocinetia»).
Compromissione epatica. Nei pazienti con insufficienza epatica lieve non è necessaria alcuna modifica del dosaggio. I dati disponibili sulla farmacocinetica della darolutamide in caso di insufficienza epatica moderata sono limitati. L’uso di darolutamide nei pazienti con insufficienza epatica grave non è stato studiato.
Nei pazienti con insufficienza epatica moderata o grave (classi B e C secondo Child-Pugh), la dose iniziale raccomandata è di 300 mg due volte al giorno (vedere il paragrafo «Precauzioni particolari» e il paragrafo «Farmacocinetica»).
Modalità di somministrazione
Il medicinale Nubeka è destinato alla somministrazione orale.
Le compresse devono essere assunte intere durante i pasti (vedere il paragrafo «Farmacocinetica»).
Uso pediatrico
Non vi è esperienza nell’uso di darolutamide nei pazienti pediatrici.
Sovradosaggio
La dose più elevata di darolutamide studiata clinicamente è stata di 900 mg due volte al giorno, pari a una dose giornaliera totale di 1800 mg. A questa dose non sono stati osservati effetti tossici dose-limitanti. Considerata l’assorbimento di saturazione (vedere il paragrafo «Farmacocinetica») e l’assenza di evidenze di tossicità acuta, non ci si aspetta che l’assunzione di una dose superiore a quella raccomandata determini effetti tossici.
In caso di assunzione di una dose superiore a quella raccomandata, il trattamento con darolutamide può proseguire con la successiva dose prevista dal programma terapeutico.
Non esiste un antidoto specifico per darolutamide e non sono stati identificati sintomi specifici di sovradosaggio.
Effetti indesiderati
Gli effetti indesiderati più comuni osservati nei pazienti con:
- CaPNCNM trattati con darolutamide sono stati affaticamento/stati astenici (15,8%);
- CaPAM trattati con darolutamide in combinazione con docetaxel sono stati eruzioni cutanee (16,6%) e ipertensione (13,8%).
Per ulteriori informazioni sulla sicurezza dell'uso combinato di darolutamide, si rimanda al foglio illustrativo dei singoli medicinali.
Gli effetti indesiderati osservati nei pazienti con CaPNCNM trattati con darolutamide sono riportati nella Tabella 3. Gli effetti indesiderati osservati nei pazienti con CaPAM trattati con darolutamide in combinazione con docetaxel sono riportati nella Tabella 4.
Gli effetti indesiderati sono classificati in base ai sistemi e agli organi. Sono raggruppati in base alla frequenza. La frequenza degli effetti indesiderati è definita come segue: molto comune (≥ 1/10), comune (≥ 1/100 fino a < 1/10), non comune (≥ 1/1000 fino a < 1/100), raro (≥ 1/10000 fino a < 1/1000), molto raro (< 1/10000), non noto (non può essere stimato sulla base dei dati disponibili). All'interno di ciascun gruppo per frequenza, gli effetti indesiderati sono elencati in ordine decrescente di gravità.
Tabella 3. Effetti indesiderati registrati nello studio ARAMISa
| Classe di sistema di organi (MedDRA) |
Molto frequente |
Frequente |
| Disturbi cardiaci |
Malattia coronaricab, insufficienza cardiacac |
|
| Disturbi della cute e del tessuto sottocutaneo |
Eruzione cutanead |
|
| Disturbi del sistema muscoloscheletrico e del tessuto connettivo |
Dolore agli arti, dolore muscoloscheletrico, fratture |
|
| Disturbi generali e condizioni inerenti al sito di somministrazione |
Stanchezza/stati astenicie |
|
| Indaginif |
Diminuzione del numero di neutrofili, aumento del livello ematico di bilirubina, aumento dell'AST |
a La durata media dell'effetto è stata di 14,8 mesi (intervallo da 0,0 a 44,3 mesi) nei pazienti trattati con darolutamide e di 11,0 mesi (intervallo da 0,1 a 40,5 mesi) nei pazienti trattati con placebo.
b Include aterosclerosi coronarica, cardiopatia ischemica, occlusione dell'arteria coronaria, stenosi dell'arteria coronaria, sindrome coronarica acuta, infarto miocardico acuto, angina pectoris, angina instabile, infarto del miocardio, ischemia miocardica.
c Include insufficienza cardiaca, insufficienza cardiaca acuta, insufficienza cardiaca cronica, insufficienza cardiaca congestizia, shock cardiogeno.
d Include eruzioni cutanee, eruzione maculare, eruzione maculopapulare, eruzione papulare, eruzione pustolosa, eritema, dermatite.
e Include affaticamento e astenia, letargia e malessere.
f Common Terminology Criteria for Adverse Events, versione 4.03. La frequenza si basa su valori riportati come anomalie nei parametri di laboratorio.
Tabella 4. Reazioni avverse osservate nei pazienti con CPCnm trattati con darolutamide in associazione a docetaxel nello studio ARASENSa, b
| Classe di sistema organico (MedDRA) |
Molto frequente |
Frequente |
| Disturbi vascolari |
Ipertensione arteriosac |
|
| Disturbi della cute e del tessuto sottocutaneo |
Eruzione cutanead, e |
|
| Disturbi del sistema muscoloscheletrico e del tessuto connettivo |
Fratture |
|
| Disturbi del sistema riproduttivo e delle ghiandole mammarie |
Ginecomastia |
|
| Indaginif |
Diminuzione del numero di neutrofili, aumento dei livelli ematici di bilirubina, aumento dell'ALT, aumento dell'AST |
a La durata media dell'effetto è stata di 41,0 mesi (intervallo da 0,1 a 56,5 mesi) nei pazienti trattati con darolutamide + docetaxel e di 16,7 mesi (intervallo da 0,3 a 55,8 mesi) nei pazienti trattati con placebo + docetaxel.
b La frequenza delle reazioni avverse non può essere attribuita esclusivamente alla darolutamide, ma potrebbe dipendere da altri farmaci utilizzati in combinazione.
c Include ipertensione arteriosa, aumento della pressione arteriosa, crisi ipertensiva.
d Include eruzioni cutanee, eruzione da farmaci, eruzione eritematosa, eruzione follicolare, eruzione maculare, eruzione maculopapulare, eruzione papulosa, prurito, eruzione pustolosa, eruzione vescicolare, eritema, dermatite.
e La frequenza è stata più elevata nei primi 6 mesi di trattamento.
f Common Terminology Criteria for Adverse Events, versione 4.03. La frequenza si basa sui valori riportati come anomalie nei parametri di laboratorio.
Descrizione delle singole reazioni avverse
Funzionalità epatica
Durante il trattamento con darolutamide sono stati riportati casi di epatotossicità idiosincrasica indotta da farmaci, con aumento delle alanina aminotransferasi (ALT) e/o aspartato aminotransferasi (AST) a ≥ 5 e ≥ 20 volte il limite superiore della norma (LSN), di grado 3 e 4, inclusi aumenti delle transaminasi con contemporaneo aumento della bilirubina totale fino a 2 × LSN. Il tempo di insorgenza variava da 1 a 12 mesi dall'inizio della darolutamide. In molti casi, l'aumento di ALT e AST si è rivelato reversibile dopo l'interruzione della darolutamide. Per le raccomandazioni, vedere la sezione «Avvertenze speciali e precauzioni di impiego».
Carcinoma della prostata resistente alla castrazione non metastatico (nmCRPC)
Affaticamento
Sono stati riportati affaticamento/stati astenici nel 15,8% dei pazienti trattati con darolutamide e nell'11,4% dei pazienti trattati con placebo. Eventi di grado 3 più severi sono stati registrati nello 0,6% dei pazienti trattati con darolutamide e nell'1,1% di quelli trattati con placebo. L'affaticamento (esclusa astenia, letargia o malessere) è stato osservato nella maggior parte dei casi (12,1% dei pazienti trattati con darolutamide e 8,7% di quelli trattati con placebo).
Fratture
Le fratture si sono verificate nel 4,2% dei pazienti trattati con darolutamide e nel 3,6% di quelli trattati con placebo.
Malattia coronarica e insufficienza cardiaca
La malattia coronarica si è verificata nel 3,2% dei pazienti trattati con darolutamide e nel 2,5% di quelli trattati con placebo. Eventi di grado 5 sono stati osservati nello 0,3% dei pazienti trattati con darolutamide e nello 0,2% di quelli trattati con placebo. L'insufficienza cardiaca si è verificata nell'1,9% dei pazienti trattati con darolutamide e nello 0,9% di quelli trattati con placebo.
Diminuzione del numero di neutrofili
La diminuzione del numero di neutrofili è stata osservata come anomalia di laboratorio nel 19,6% dei pazienti trattati con darolutamide e nel 9,4% di quelli trattati con placebo. Il tempo medio per raggiungere il valore minimo è stato di 256 giorni.
Le anomalie di laboratorio erano prevalentemente di intensità grado 1–2. La diminuzione del numero di neutrofili di grado 3 e 4 è stata osservata rispettivamente nel 3,5% e nello 0,5% dei pazienti. Un solo paziente ha interrotto definitivamente la darolutamide a causa di neutropenia. La neutropenia è stata temporanea o reversibile (88% dei pazienti) e non è stata associata a segni o sintomi clinicamente significativi.
Aumento della bilirubina nel sangue
È stato riportato un aumento della bilirubina come anomalia di laboratorio nel 16,4% dei pazienti trattati con darolutamide e nel 6,9% di quelli trattati con placebo. Gli episodi erano prevalentemente di grado 1 o 2, non associati a segni o sintomi clinicamente significativi e si sono rivelati reversibili dopo l'interruzione della darolutamide. Aumenti di bilirubina di grado 3 sono stati osservati nello 0,1% dei pazienti trattati con darolutamide e nello 0% di quelli trattati con placebo. Nel gruppo darolutamide, il tempo medio per il primo aumento della bilirubina è stato di 153 giorni e la durata media del primo episodio è stata di 182 giorni. Nessun paziente ha interrotto il trattamento a causa dell'aumento della bilirubina.
Aumento dell'AST
È stato riportato un aumento dell'AST come anomalia di laboratorio nel 22,5% dei pazienti trattati con darolutamide e nel 13,6% di quelli trattati con placebo. Gli episodi erano prevalentemente di grado 1 o 2, non associati a segni o sintomi clinicamente significativi e si sono rivelati reversibili dopo l'interruzione della darolutamide. Aumenti dell'AST di grado 3 sono stati osservati nello 0,5% dei pazienti trattati con darolutamide e nello 0,2% di quelli trattati con placebo. Nel gruppo darolutamide, il tempo medio per il primo aumento dell'AST è stato di 258 giorni e la durata media del primo episodio è stata di 118 giorni. Nessun paziente ha interrotto il trattamento a causa dell'aumento dell'AST.
Carcinoma della prostata ormone-sensibile metastatico (mHSPC)
Ipertensione arteriosa
Nello studio ARASENS, l'ipertensione arteriosa è stata riportata nel 13,8% dei pazienti trattati con darolutamide + docetaxel e nel 9,4% di quelli trattati con placebo + docetaxel.
L'ipertensione arteriosa di grado 3 è stata registrata nel 6,4% dei pazienti trattati con darolutamide + docetaxel, rispetto al 3,5% dei pazienti trattati con placebo + docetaxel. Un paziente per gruppo di trattamento ha presentato ipertensione di grado 4.
È stato riportato un caso di ipertensione di grado 5 con arteriosclerosi di grado 5 nel gruppo darolutamide + docetaxel. Questo paziente aveva una lunga storia di ipertensione e di fumo, e l'evento si è verificato più di 3 anni dopo l'inizio del trattamento con darolutamide. I casi di ipertensione arteriosa sono stati riportati più frequentemente nei pazienti senza anamnesi di ipertensione in entrambi i gruppi di trattamento.
Fratture
Le fratture si sono verificate nel 7,5% dei pazienti trattati con darolutamide + docetaxel e nel 5,1% di quelli trattati con placebo + docetaxel.
Diminuzione del numero di neutrofili
È stata riportata una diminuzione del numero di neutrofili come anomalia di laboratorio nel 50,6% dei pazienti trattati con darolutamide + docetaxel e nel 45,5% di quelli trattati con placebo + docetaxel. La diminuzione del numero di neutrofili di grado 3 e 4 è stata osservata nel 34,4% dei pazienti trattati con darolutamide + docetaxel e nel 31,4% di quelli trattati con placebo + docetaxel. In entrambi i gruppi di trattamento si è verificata una riduzione del numero di neutrofili, con neutropenia più elevata nei primi mesi di trattamento, seguita da una riduzione della frequenza e della gravità degli eventi.
Aumento della bilirubina nel sangue
L'aumento della bilirubina nel sangue è stato osservato come anomalia di laboratorio nel 19,6% dei pazienti trattati con darolutamide + docetaxel e nel 10,0% di quelli trattati con placebo + docetaxel. Gli eventi erano prevalentemente di grado 1 o 2. Aumenti di bilirubina di grado 3 e 4 sono stati osservati nello 0,5% dei pazienti trattati con darolutamide + docetaxel e nello 0,3% di quelli trattati con placebo + docetaxel.
Aumento di ALT e AST
È stato riportato un aumento di ALT come anomalia di laboratorio nel 42,3% dei pazienti trattati con darolutamide + docetaxel e nel 38,0% di quelli trattati con placebo + docetaxel. È stato riportato un aumento di AST come anomalia di laboratorio nel 43,9% dei pazienti trattati con darolutamide + docetaxel e nel 39,3% di quelli trattati con placebo + docetaxel. L'aumento di ALT e AST è stato prevalentemente di grado 1. Aumenti di ALT di grado 3 e 4 sono stati osservati nel 3,7% dei pazienti trattati con darolutamide + docetaxel e nel 3,0% di quelli trattati con placebo + docetaxel. Aumenti di AST di grado 3 e 4 sono stati osservati nel 3,6% dei pazienti trattati con darolutamide + docetaxel e nel 2,3% di quelli trattati con placebo + docetaxel.
Segnalazione delle reazioni avverse sospette
La segnalazione delle reazioni avverse sospette dopo l'autorizzazione del medicinale è importante. Permette un monitoraggio continuo del rapporto beneficio/rischio del medicinale. I professionisti sanitari e farmaceutici, nonché i pazienti o i loro rappresentanti legali, devono segnalare tutti i casi sospetti di reazioni avverse e di mancata efficacia del medicinale attraverso il Sistema informativo automatizzato di farmacovigilanza al seguente link: https://aisf.dec.gov.ua.
Periodo di validità. 3 anni.
Condizioni di conservazione
Non richiede condizioni particolari di conservazione. Conservare in un luogo inaccessibile ai bambini.
Confezionamento
16 compresse in un blister, 7 blister in una confezione di cartone.
Categoria di prescrizione. Sotto prescrizione medica.
Produttore. Orion Corporation, Orion Pharma.
Indirizzo del produttore e sede operativa
Joensuunkatu 7, 24100 Salo, Finlandia.