Еврисди
УкраинаСодержание
ИНСТРУКЦИЯ по медицинскому применению лекарственного средства Эврисди® (Evrysdi®)
Состав:
действующее вещество: рисдиплам (risdiplam);
1 флакон содержит 60 мг рисдиплама;
1 мл восстановленного раствора содержит 0,75 мг рисдиплама;
вспомогательные вещества: маннит (Е 421); изомальт (Е 953); ароматизатор клубничный; кислота винная; бензоат натрия (Е 211); полигиэтленгликоль 6000; сахаралоза; кислота аскорбиновая; динатрия эдетат, дигидрат.
Лекарственная форма. Порошок для перорального раствора.
Основные физико-химические свойства: Порошок или порошок с комочками или порошкообразная масса светло-желтого или желтого, или серовато-желтого, или зеленовато-желтого, или светло-зеленого цвета. Восстановленный раствор от зеленовато-желтого до желтого цвета.
Фармакотерапевтическая группа. Средства, влияющие на опорно-двигательный аппарат. Другие средства, применяемые при патологии опорно-двигательного аппарата.
Код АТС M09A X10.
Фармакологические свойства.
Фармакодинамика.
Механизм действия
Рисдиплам является модификатором сплайсинга предшественника матричной РНК (прe-мРНК) гена выживания мотонейронов 2 (SMN2), разработанным для лечения СМА, вызванной мутациями хромосомы 5q, приводящими к дефициту белка SMN. Функциональный дефицит белка SMN является патофизиологическим механизмом СМА всех типов. Рисдиплам корректирует сплайсинг SMN2, изменяя баланс с исключением экзона 7 на включение экзона 7 в транскрипт мРНК, в результате чего увеличивается выработка функционального и стабильного белка SMN. Таким образом, рисдиплам лечит СМА за счёт повышения и поддержания функционального уровня белка SMN.
Электрофизиология сердца
Влияние рисдиплама на интервал QTc оценивали в исследовании с участием 47 здоровых взрослых добровольцев. В пределах терапевтического воздействия рисдиплам не удлинял интервал QTc.
Рисдиплам равномерно распределяется по всем частям тела, включая центральную нервную систему, проникая через гематоэнцефалический барьер, и таким образом увеличивает уровень белка SMN в ЦНС и во всём организме. Концентрация рисдиплама в плазме крови и уровень белка SMN в крови отражают распределение и фармакодинамические эффекты рисдиплама в таких тканях, как головной мозг и мышцы.
В клинических исследованиях FIREFISH, SUNFISH и JEWELFISH, проведённых с участием пациентов с инфантильным началом спинальной мышечной атрофии (СМА) и пациентов с поздним началом СМА, рисдиплам приводил к последовательному и длительному увеличению определяемого в крови уровня белка SMN со сменой медианы более чем вдвое от исходного уровня в течение 4 недель после начала лечения. Такое увеличение уровня белка SMN сохранялось на протяжении всего периода лечения, по крайней мере, 24 месяца (см. подраздел «Клиническая эффективность»).
Клиническая эффективность
Эффективность препарата Еврисди® в лечении пациентов с инфантильной формой СМА (СМА 1 типа) и формой СМА с поздним началом (СМА 2 и 3 типа) была оценена в двух базовых клинических исследованиях — FIREFISH и SUNFISH — и подтверждена дополнительными данными из исследования JEWELFISH. Эффективность препарата Еврисди® в лечении пациентов с пресимптоматической СМА была оценена на основании промежуточного анализа вторичных конечных точек продолжающегося исследования RAINBOWFISH.
Пациенты с клиническим диагнозом СМА 4 типа не участвовали в клинических исследованиях.
В клинических исследованиях была продемонстрирована долгосрочная эффективность на протяжении по меньшей мере 24 месяцев лечения. Существуют ограниченные данные применения препарата Еврисди® более чем в течение 2 лет.
Инфантильная форма СМА
Исследование BP39056 (FIREFISH) — открытое исследование, состоящее из двух частей, по изучению эффективности, безопасности, фармакокинетики и фармакодинамики препарата Еврисди® с участием пациентов с СМА 1 типа и наличием симптомов (все пациенты имели генетически подтверждённое заболевание с наличием 2 копий гена SMN2). Часть 1 исследования FIREFISH была разработана как исследование по поиску дозы. В подтверждающей части 2 исследования FIREFISH оценивалась эффективность препарата Еврисди® в терапевтических дозах, выбранных на основании результатов, полученных в части 1 (см. раздел «Способ применения и дозы»). Пациенты из части 1 не участвовали в части 2.
Всего 62 пациента с СМА 1 типа и наличием симптомов были включены в часть 1 (n = 21) и часть 2 (n = 41) исследования FIREFISH, из которых 58 пациентов получали терапевтическую дозу препарата Еврисди®. Средний возраст на момент появления клинических симптомов составлял 1,5 месяца (0,9–3 месяца). Средний возраст на момент включения в исследование составлял 5,6 месяца (2,2–6,9 месяца), а среднее время между появлением симптомов и получением первой дозы составляло 3,7 месяца (1–6 месяцев). 60 % пациентов были женского пола, 57 % — европеоидной расы и 29 % — азиатами. В начале исследования медианный балл CHOP-INTEND составлял 23 (8–37), а медианный балл HINE-2 — 1 (0–5). Исходные демографические характеристики и характеристики заболевания пациентов, включённых в часть 1 исследования, были сопоставимы с таковыми для части 2 исследования.
Первичной конечной точкой исследования была доля пациентов, способных сидеть без поддержки в течение не менее 5 секунд, как определено в пункте 22 шкалы развития младенцев и детей раннего возраста Бейли — III издание (BSID-III) для оценки общей моторики, через 12 месяцев лечения препаратом Еврисди® в части 2 исследования; и такой результат был достигнут у 29 % пациентов (n = 12/41, 90 % ДИ: 17,8 %, 43,1 %, p <0,0001).
Ключевые результаты эффективности у пациентов, получавших лечение препаратом Еврисди® в исследовании FIREFISH (обобщённые данные, полученные в части 1 и части 2), приведены в таблице 1.
Таблица 1
Резюме ключевых результатов эффективности через 12 и 24 месяца (FIREFISH, часть 1 и часть 2)
| Конечные точки эффективности |
Через 12 месяцев |
Через 24 месяца |
| Доля пациентов (90 % ДИ) |
||
| Этапы моторного развития и двигательная функция |
N = 58a |
|
| BSID-III: сидение без поддержки в течение не менее 5 секунд |
32,8 % |
60,3 % |
| CHOP-INTEND: количество баллов 40 или выше |
56,9 % |
74,1 % |
| Увеличение баллов CHOP-INTEND на ≥ 4 балла от исходного уровня |
89,7 % |
87,9 % |
| HINE-2: пациенты, ответившие согласно критериям развития моторной функцииb |
77,6 % |
82,8 % |
| Выживаемость и выживаемость без событий |
N=62a |
|
| Выживаемость без событийc |
87,1 % |
|
| Выживаемость |
91,9 % |
90,3 % |
| Питание |
N = 58a |
|
| Способность получать пероральное питаниеd |
84,5 % |
82,8 % |
BSID-III — шкала развития младенцев и детей раннего возраста Бейли — III издание;
CHOP-INTEND — тест Детской больницы Филадельфии для оценки двигательных функций при нейромышечных заболеваниях у младенцев;
HINE-2 — модуль 2 неврологического обследования младенцев по Хаммерсмиту.
a Данные о выживаемости и выживаемости без вентиляции были объединены для всех пациентов, получавших любую дозу рисдиплама в части 1 и части 2 исследования (n = 62). Для этапов моторного развития, двигательной функции и питания объединённые данные эффективности относятся ко всем пациентам, получавшим терапевтическую дозу рисдиплама (все пациенты в части 2 исследования и пациенты когорты высокой дозы в части 1; n = 58).
b Определение ответа по критерию HINE-2: ответ в данном анализе определён как увеличение ≥ 2 баллов (или максимально возможный показатель) способности стучать ножками ИЛИ увеличение ≥ 1 балла по таким этапам развития двигательной функции, как контроль удержания головы, перевороты, сидение, ползание, стояние или ходьба, И улучшение по большему числу категорий развития двигательной функции, чем ухудшение.
c Событие, соответствующее конечной точке постоянной вентиляции, определено как трахеостомия или ≥ 16 часов неинвазивной вентиляции лёгких в день, или интубация более 21 дня подряд при отсутствии или после снятия острого обратимого состояния. Четыре пациента соответствовали критериям конечной точки постоянной вентиляции до 24 месяцев. Эти четыре пациента достигли увеличения показателя CHOP-INTEND по крайней мере на 4 балла по сравнению с исходным уровнем.
d Включая пациентов, получавших исключительно пероральное питание (41 пациент через 12 и 24 месяца), и тех пациентов, которые получали пероральное питание в комбинации с зондовым питанием (8 пациентов через 12 месяцев и 7 пациентов через 24 месяца).
Через 24 месяца 40 % (23/58) пациентов, получавших терапевтическую дозу лекарственного средства Эвризди®, могли сидеть без поддержки в течение 30 секунд (BSID-III, пункт 26). Кроме того, пациенты продолжали достигать дополнительные этапы моторного развития по показателю HINE-2 через 24 месяца; 78 % пациентов могли переворачиваться (31 % пациентов могли переворачиваться на бок, 7 % пациентов могли переворачиваться из положения лёжа на животе в положение лёжа на спине и 40 % пациентов могли переворачиваться из положения лёжа на спине в положение лёжа на животе) и 28 % пациентов достигли сидения (16 % удерживали вес и 12 % стояли с поддержкой).
Доля живых пациентов без необходимости постоянной вентиляции (выживаемость без событий) составила 84 % среди всех пациентов через 24 месяца. Шесть младенцев умерло (4 в течение первых 3 месяцев после включения в исследование), и у одного пациента было преждевременно прекращено лечение, а через 3,5 месяца после этого пациент умер. Четырём пациентам была необходима постоянная вентиляция до 24 месяцев.
СМА с поздним течением
Исследование BP39055 (SUNFISH) — многоцентровое исследование, состоящее из двух частей, по изучению эффективности, безопасности, фармакокинетики и фармакодинамики препарата Эвризди® у пациентов с диагнозом СМА 2 или 3 типа в возрасте от 2 до 25 лет. Часть 1 была исследовательской частью по определению дозы, а часть 2 — рандомизированной двойной слепой плацебо-контролируемой подтверждающей частью. Пациенты из части 1 исследования не участвовали в части 2.
Первичной конечной точкой была оценка изменения показателя двигательной функции (MFM32) к 12 месяцу по сравнению с исходным уровнем. MFM32 позволяет оценить широкий диапазон двигательных функций для широкой группы пациентов с СМА. Общий показатель MFM32 выражается в процентах (диапазон: от 0 до 100) максимального возможного показателя, при этом более высокий показатель свидетельствует о большей двигательной функции. MFM32 определяет способность к двигательной функции, касающейся важных ежедневных функций. Незначительные изменения двигательной функции могут привести к значимому улучшению или потере ежедневной функции(-й).
SUNFISH, часть 2
Часть 2 SUNFISH — это рандомизированная двойная слепая плацебо-контролируемая часть исследования с участием 180 пациентов, неспособных ходить, с СМА 2 типа (71 %) или 3 типа (29 %). Пациенты были рандомизированы в соотношении 2:1 для получения препарата Эвризди® в терапевтической дозе (см. раздел «Способ применения и дозы») или плацебо. Рандомизация была стратифицирована по возрасту (от 2 до 5 лет, от 6 до 11 лет, от 12 до 17 лет, от 18 до 25 лет).
Средний возраст пациентов на момент начала лечения составлял 9 лет (диапазон 2–25 лет), медиана времени от появления симптомов СМА до первого лечения составила 102,6 (1–275) месяца. Среди участников исследования 51 % пациентов были женского пола, 67 % — европеоидной расы, 19 % — азиатского происхождения. На момент начала исследования 67 % пациентов имели сколиоз (32 % из них имели тяжёлый сколиоз). Пациенты имели средний исходный показатель МFM32 46,1 и показатель RULM 20,1. В целом исходные демографические характеристики были хорошо сбалансированы между группами приёма препарата Эвризди® и плацебо, за исключением несоответствия количества пациентов со сколиозом (63,3 % пациентов в группе препарата Эвризди® и 73,3 % пациентов в группе плацебо-контроля).
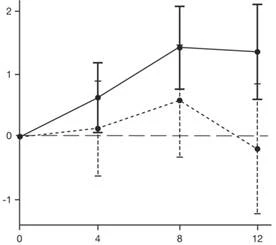
Результаты первичного анализа части 2 исследования SUNFISH по изменению общего показателя MFM32 от исходного уровня через 12 месяцев продемонстрировали клинически значимую и статистически достоверную разницу между группами пациентов, получавших лечение препаратом Эвризди® и плацебо. Результаты первичного анализа и основные вторичные конечные точки приведены в таблице 2 и на рисунке 1.
Таблица 2
Резюме результатов эффективности у пациентов с СМА с поздним началом через 12 месяцев лечения (часть 2 исследования SUNFISH)
| Конечная точка |
Еврисди ® (N = 120) |
Плацебо (N = 60) |
| Первичная конечная точка: |
||
| Изменение общего показателя MFM321 через 12 месяцев по сравнению с исходным уровнем, среднеквадратичное среднее значение (95 %, ДИ) |
1,36 (0,61; 2,11) |
-0,19 (-1,22; 0,84) |
| Разница по сравнению с плацебо (95 % ДИ), значение p2 |
1,55 (0,30; 2,81) 0,0156 |
|
| Вторичные конечные точки |
||
| Доля пациентов со изменением общего показателя MFM321 на 3 или более через 12 месяцев по сравнению с исходным уровнем (95 % ДИ) |
38,3 % (28,9; 47,6) |
23,7 % (12,0; 35,4) |
| Отношение шансов общей ответной реакции (95 % ДИ), скорректированное (нескорректированное) значение p3,4 |
2,35 (1,01; 5,44) 0,0469 (0,0469) |
|
| Изменение общего показателя RULM5 через 12 месяцев по сравнению с исходным уровнем, среднеквадратичное среднее значение (95 % ДИ) |
1,61 (1,00; 2,22) |
0,02 (-0,8; 0,87) |
| Разница по сравнению с плацебо (95 % ДИ), скорректированное (нескорректированное) значение p2,4 |
1,59 (0,55; 2,62) 0,0469 (0,0028) |
|
1 На основании правила отсутствующих данных по MFM32 6 пациентов были исключены из анализа (Eврисди®, n = 115; группа плацебо-контроля, n = 59).
2 Данные анализировались с помощью смешанной модели с повторными измерениями с учётом исходного общего показателя, лечения, визита, возрастной группы, эффекта взаимодействия лечения и визита, а также взаимодействия исходного уровня и визита.
3 Данные анализировались с помощью логистической регрессии с учётом исходного общего показателя, группы лечения и возрастной группы.
4 Скорректированное значение p получено для конечных точек, включённых в иерархическое тестирование, и рассчитано на основе всех значений p для конечных точек в порядке иерархии до текущей конечной точки. Нескорректированное значение p тестировалось при уровне значимости 5 %.
5 На основании правила отсутствующих данных по RULM 3 пациента были исключены из анализа (Eврисди®, n = 119; группа плацебо-контроля, n = 58).
После завершения 12 месяцев лечения 117 пациентов продолжили получать препарат Еврисди®. На момент анализа через 24 месяца пациенты, получавшие лечение в течение 24 месяцев, продемонстрировали дальнейшее улучшение двигательной функции в период между 12 и 24 месяцами терапии. Среднее изменение показателя MFM32 по сравнению с исходным уровнем составило 1,83 (ДИ: 0,74–2,92), а RULM — 2,79 (ДИ: 1,94–3,64).
| Изменение среднеквадратичного среднего значения общего показателя MFM32 |
|
| Месяцы |
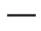
Еврисди® Плацебо
* Планка погрешностей означает 95 % доверительный интервал.
† Общий показатель МФМ был рассчитан в соответствии с руководством для пользователя, выраженный как процент от максимального возможного показателя по шкале (то есть сумму баллов для 32 пунктов делили на 96 и умножали на 100).
Рис. 1. Среднее изменение (LS) общего показателя MFM32 от исходного уровня через 12 месяцев в части 2 исследования SUNFISH.
SUNFISH, часть 1
Эффективность препарата Еврисди® у пациентов с СМА с поздним началом также подтверждается результатами части 1 исследования SUNFISH по определению дозы. В часть 1 было включено 51 пациент с СМА типа 2 и 3 (в том числе 7 пациентов, способных ходить) в возрасте от 2 до 25 лет. Через один год лечения терапевтической дозой (дозой, выбранной для части 2) наблюдалось клинически значимое улучшение двигательной функции по результатам MFM32 со средним изменением от исходного уровня на 2,7 балла (95 % ДИ: 1,5; 3,8). Улучшение MFM32 сохранялось также в течение периода до 2 лет лечения препаратом Еврисди® (среднее изменение 2,7 балла (95 % ДИ: 1,2; 4,2)).
В постулированном анализе двигательная функция, оценённая с помощью MFM, сравнивалась в части 1 исследования SUNFISH и историческом контроле с естественным течением заболевания (с учётом весовых коэффициентов основных прогностических факторов). Изменение общего показателя MFM по сравнению с исходным уровнем через 1 и 2 года было больше у пациентов, получавших Еврисди®, по сравнению с когортой естественного течения (через 1 год: разница в 2,7 балла; p < 0,0001; через два года: разница в 4 балла; p < 0,0001). В когорте естественного течения наблюдалось снижение двигательной функции, что соответствует ожидаемому естественному прогрессированию СМА (среднее изменение через 1 год: –0,6 балла; через 2 года: –2 балла).
Пресимптоматическая СМА
Исследование BN40703 (RAINBOWFISH) — одногрупповое открытое многоцентровое исследование, которое продолжается, по изучению эффективности, безопасности, фармакокинетики и фармакодинамики лекарственного средства Еврисди® у младенцев в возрасте от рождения до 6 недель (на момент получения первой дозы), которым установлен генетический диагноз СМА, однако симптомы ещё отсутствуют.
Эффективность применения лекарственного средства Еврисди® при пресимптоматической СМА оценивали на 12-м месяце у 26 пациентов [популяция, которым было назначено лечение (ITT)]. Медиана возраста этих пациентов при получении первой дозы составляла 25 дней (диапазон от 16 до 41 дня), 62 % составляли пациенты женского пола и 85 % — представители европеоидной расы. У 8 пациентов, 13 пациентов и 5 пациентов было 2, 3 и ≥ 4 копии гена SMN2 соответственно. В начале исследования медианный показатель CHOP-INTEND составлял 51,5 (диапазон от 35,0 до 62,0), медианный показатель HINE-2 составлял 2,5 (диапазон от 0 до 6,0), а медиана амплитуды потенциала действия мышц локтевого нерва (CMAP) составляла 3,6 мВ (диапазон от 0,5 до 6,7 мВ).
Первичная популяция анализа эффективности (N = 5) включала пациентов с 2 копиями гена SMN2 и исходной амплитудой CMAP ≥ 1,5 мВ. У этих пациентов медиана показателя CHOP-INTEND составляла 48,0 (диапазон 36,0–52,0), медиана показателя HINE-2 равнялась 2,0 (диапазон от 1,0 до 3,0), а медиана амплитуды CMAP составляла 2,6 мВ (диапазон от 1,6 до 3,8 мВ) на исходном уровне.
Первичной конечной точкой была доля пациентов в первичной популяции анализа эффективности, которые были способны сидеть без поддержки не менее 5 секунд (шкала грубой моторики BSID-III, пункт 22) на 12-м месяце; статистически значимая и клинически значимая доля пациентов достигла этой вехи по сравнению с заранее определённым критерием эффективности 5 %.
Ключевые конечные точки эффективности у пациентов, получавших препарат Еврисди®, приведены в таблицах 3 и 4 и на рисунке 2.
Таблица 3
Способность сидеть согласно определению пункта 22 шкалы BSID-III для пресимптомных пациентов на 12-м месяце
| Конечная точка оценки эффективности |
Популяция пациентов |
||
| Первичный анализ эффективности (N = 5) |
Пациенты с 2 копиями гена SMN2a (N = 8) |
ITT (N = 26) |
|
| Доля пациентов, которые могли сидеть без поддержки не менее 5 секунд (шкала BSID-III, пункт 22); (90 % ДИ) |
80 % (34,3 %; 99,0 %) |
87,5 % (52,9 %; 99,4 %) |
96,2 % (83,0 %; 99,8 %) |
Сокращение: BSID-III – Шкалы Бейли развития младенцев и малышей, третье издание; ДИ – доверительный интервал; ITT – популяция, которой было назначено лечение.
a Пациенты с 2 копиями гена SMN2 имели среднюю амплитуду CMAP 2,0 (диапазон 0,5–3,8) в начале исследования.
b Значение p основано на одностороннем точном биномиальном критерии. Результат сравнивается с порогом 5 %.
Кроме того, 80 % (4/5) первичной популяции анализа эффективности, 87,5 % (7/8) пациентов с 2 копиями гена SMN2 и 80,8 % (21/26) пациентов популяции ITT достигли способности сидеть без поддержки в течение 30 секунд (BSID-III, пункт 26). Пациенты популяции ITT также достигли этапов двигательной активности, измеряемых по модулю HINE-2 на 12-м месяце (N = 25). В этой популяции 96,0 % пациентов могли сидеть [1 пациент (1/8 пациентов с 2 копиями гена SMN2) достиг стабильного сидения, а 23 пациента (6/8, 13/13, 4/4 пациентов с 2, 3 и ≥ 4 копиями гена SMN2 соответственно) могли поворачиваться/переворачиваться]. Кроме того, 84 % пациентов могли стоять; 32 % (N = 8) пациентов могли стоять с опорой (3/8, 3/13 и 2/4 пациентов с 2, 3 и ≥ 4 копиями гена SMN2 соответственно) и 52 % (N = 13) пациентов могли стоять без посторонней помощи (1/8, 10/13 и 2/4 пациентов с 2, 3 и ≥ 4 копиями гена SMN2 соответственно). Кроме того, 72 % пациентов могли подпрыгивать, ходить с опорой или без; 8 % (N = 2) пациентов могли подпрыгивать (2/8 пациентов с 2 копиями гена SMN2), 16 % (N = 4) могли ходить с опорой (3/13 и 1/4 пациентов с 3 и ≥ 4 копиями гена SMN2 соответственно) и 48 % (N = 12) могли ходить самостоятельно (1/8, 9/13 и 2/4 пациентов с 2, 3 и ≥ 4 копиями гена SMN2 соответственно). Семь пациентов не проходили анализ на 12-м месяце.
Таблица 4
Резюме ключевых конечных точек эффективности для пресимптоматических пациентов на 12-м месяце
| Конечные точки эффективности |
Популяция ITT (N = 26) |
| Двигательная функция |
|
| Доля пациентов, достигших общего балла 50 или выше по шкале CHOP-INTEND (90 ДИ %) |
92 %a (76,9 %; 98,6 %) |
| Доля пациентов, достигших общего балла 60 или выше по шкале CHOP-INTEND (90 ДИ %) |
80 %a (62,5 %; 91,8 %) |
| Питание |
|
| Доля пациентов, способных принимать пищу перорально (90 ДИ %) |
96,2 %b (83,0 %; 99,8 %) |
| Использование ресурсов системы здравоохранения |
|
| Доля пациентов, не требовавших госпитализации (90 ДИ %) |
92,3 % (77,7 %; 98,6 %) |
| Выживаемость без событийd Доля пациентов, характеризовавшихся выживаемостью без событий (90 ДИ %) |
100 % (100 %; 100 %) |
Сокращения: CHOP INTEND – тест детской больницы Филадельфии для оценки двигательных функций при нейромышечных заболеваниях у младенцев; ДИ – доверительный интервал; ITT – популяция, которой было назначено лечение.
a На основании количества N = 25.
b Для одного пациента оценка не проводилась.
c Госпитализации включали все случаи пребывания в больнице, продолжавшиеся не менее двух дней и не связанные с требованиями исследования.
d Событие в данном случае означает смерть или необходимость в постоянной вентиляции; постоянная вентиляция определяется как трахеостомия или ≥ 16 часов неинвазивной вентиляции лёгких в день, или интубация в течение > 21 дня подряд при отсутствии или после исчезновения острого обратимого состояния.
Сокращения: IQR – интерквартильный диапазон; SMN2 – ген выживания мотонейронов 2.
Рис. 2. Медиана общих баллов CHOP-INTEND по визитам и количеству копий гена SMN2 (популяция ITT)
Применение у пациентов с СМА, ранее получавших другие модифицирующие методы лечения
Исследование BP39054 (JEWELFISH) – одногрупповое открытое исследование безопасности, переносимости, фармакокинетики и фармакодинамики препарата Эврисди® с участием пациентов с инфантильной формой СМА или СМА с поздним началом в возрасте от 6 месяцев до 60 лет, ранее получавших лечение по поводу СМА (включая нусинерсен и онасемноген абепарвовек). Из 173 пациентов, получавших препарат Эврисди®, 76 пациентов ранее получали лечение нусинерсеном (9 пациентов с СМА 1 типа, 43 пациента с СМА 2 типа и 24 пациента с СМА 3 типа) и 14 пациентов ранее получали лечение онасемногеном абепарвовеком (4 пациента с СМА 1 типа и 10 пациентов с СМА 2 типа). Средний возраст пациентов на начало лечения препаратом Эврисди® составил 14 лет (диапазон 1–60 лет).
На момент включения в исследование из 168 пациентов в возрасте 2–60 лет 83 % имели сколиоз (39 % – тяжелый сколиоз), и у 63 % показатель по расширенной шкале оценки двигательной функции по Хаммерсмиту (HFMSE) составлял < 10 баллов. В исследование также были включены 15 пациентов, способных передвигаться самостоятельно (в возрасте 5–46 лет).
Показатели эффективности оценивались путем измерения двигательной функции в соответствии с возрастом, включая шкалы MFM-32 и RULM для пациентов в возрасте 2–60 лет, BSID-III и HINE-2 для пациентов в возрасте до 2 лет, а также тест шестиминутной ходьбы (6MWT) для пациентов в возрасте ≥ 6 лет, способных передвигаться самостоятельно. По результатам первичного анализа после 24 месяцев лечения у пациентов в возрасте 2–60 лет наблюдалась общая стабилизация двигательной функции по шкалам MFM-32 и RULM (n = 137 и n = 133 соответственно). Пациенты в возрасте до 2 лет (n = 6) сохранили или улучшили развитие двигательной функции, в частности удержание головы, перевороты, сидение без поддержки. Результаты 6MWT демонстрируют среднее улучшение на 30,88 метра (95 % ДИ: -5,54, 67,29, n = 8). Все пациенты, способные передвигаться самостоятельно, сохранили способность ходить.
Фармакокинетика.
Фармакокинетические параметры рисдиплама были охарактеризованы у здоровых взрослых лиц и у пациентов с СМА.
После приема орального раствора препарата Эврисди® в дозах от 0,6 до 18 мг фармакокинетика рисдиплама была приблизительно линейной. Фармакокинетика рисдиплама наилучшим образом описывалась с помощью популяционной ФК модели с всасыванием из трехкамерного транзита, двухкамерным распределением и выведением первого порядка. Было установлено, что масса тела и возраст пациента оказывают существенное влияние на фармакокинетику препарата.
Расчетная экспозиция (средняя AUC0–24ч) у пациентов с инфантильной формой СМА (в возрасте 2–7 месяцев на момент включения в исследование) при рекомендованной дозе 0,2 мг/кг один раз в сутки составила 1930 нг·ч/мл. Средняя расчетная экспозиция у младенцев в возрасте от 16 дней до < 2 месяцев с пресимптоматической СМА в исследовании RAINBOWFISH составила 2020 нг·ч/мл после 2 недель ежедневного приема в дозе 0,15 мг/кг.
Расчетная экспозиция у пациентов с формой СМА с поздним началом (в возрасте 2–25 лет на момент включения в исследование) в исследовании SUNFISH (часть 2) при терапевтической дозе (0,25 мг/кг один раз в сутки пациентам с массой тела < 20 кг; 5 мг один раз в день пациентам с массой тела ≥ 20 кг) составила 2010 нг·ч/мл. Максимальная концентрация, наблюдавшаяся (средняя Cmax), составила 194 нг/мл при дозировке 0,2 мг/кг в исследовании FIREFISH и 120 нг/мл в части 2 исследования SUNFISH. Средняя расчетная максимальная концентрация при дозировке 0,15 мг/кг в исследовании RAINBOWFISH составляет 111 нг/мл.
Всасывание
Рисдиплам быстро всасывался при пероральном приеме натощак, при этом tmax в плазме крови варьировал от 1 до 4 часов. В клинических исследованиях рисдиплам применяли утром с приемом пищи или после кормления грудью.
Распределение
Рассчитанные популяционные фармакокинетические показатели составили: 98 л – видимый центральный объем распределения, 93 л – периферический объем и 0,68 л/час – межкомпартментный клиренс.
Рисдиплам в основном связывается с альбуминами сыворотки крови, не связываясь с альфа-1-кислыми гликопротеинами, при этом свободная фракция составляет 11 %.
Метаболизм
Рисдиплам в основном метаболизируется с помощью флавинсодержащих монооксигеназ 1 и 3 (FMO1 и FMO3), а также изоферментами CYP 1A1, 2J2, 3A4 и 3A7. Исходное лекарственное средство было основным компонентом, обнаруженным в плазме, и составляло в кровотоке 83 % вещества, связанного с лекарственным средством. Фармакологически неактивный метаболит M1 был идентифицирован как основной циркулирующий метаболит.
Выведение
Популяционный фармакокинетический показатель видимого клиренса (CL/F) рисдиплама составил 2,6 л/час. Эффективный период полувыведения рисдиплама составлял около 50 часов у пациентов с СМА.
Приблизительно 53 % дозы (14 % в виде неизмененного рисдиплама) выводилось с калом и 28 % с мочой (8 % в виде неизмененного рисдиплама).
Особые группы пациентов
Нарушения функции печени
Легкие и умеренные нарушения функции печени не оказывали влияния на ФК рисдиплама. После приема 5 мг рисдиплама средние соотношения Cmax и AUC составили 0,95 и 0,80 у лиц с легким (n = 8) и 1,20 и 1,08 у пациентов с умеренным нарушением функции печени (n = 8) по сравнению с соответствующими показателями у пациентов с нормальной функцией печени (контрольная группа, n = 10). Безопасность и ФК у пациентов с тяжелым нарушением функции печени на сегодняшний день не изучались.
Нарушения функции почек
Исследования ФК рисдиплама у пациентов с нарушением функции почек не проводились. Выведение рисдиплама в виде неизмененного вещества почками является незначительным (8 %).
Пациенты пожилого возраста
Специальные исследования фармакокинетики препарата Эврисди® у пациентов с СМА в возрасте старше 60 лет не проводились. Пациенты с СМА в возрасте до 60 лет были включены в исследование JEWELFISH. Пациенты без СМА в возрасте до 69 лет были включены в клинические исследования ФК.
Дети
Масса тела и возраст пациента были идентифицированы как ковариаты в популяционном ФК анализе. Поэтому дозу корректируют в зависимости от возраста (менее и более 2 месяцев и 2 лет) и массы тела (до 20 кг) с целью получения сопоставимой экспозиции у различных возрастных групп и весовых категорий. Фармакокинетические данные у младенцев в возрасте до 16 дней отсутствуют.
Этническая принадлежность
Фармакокинетика рисдиплама не отличается у японцев и пациентов европеоидной расы.
Клинические характеристики.
Показания.
Лечение 5q-ассоциированной спинальной мышечной атрофии (СМА) у детей и взрослых пациентов.
Противопоказания.
Известная повышенная чувствительность к рисдипламу или к любому из вспомогательных веществ, указанных в разделе «Состав».
Взаимодействие с другими лекарственными средствами и другие виды взаимодействий.
Влияние Еврисди® на другие лекарственные средства
In vitro рисдиплам и его основной циркулирующий метаболит М1 не индуцировали CYP1A2, 2B6, 2C8, 2C9, 2C19 и 3A4. In vitro рисдиплам и М1 не угнетали (обратимое или зависящее от времени угнетение) ни один из исследуемых изоферментов CYP (CYP1A2, 2B6, 2C8, 2C9, 2C19, 2D6), за исключением CYP3A.
Препарат Еврисди® является слабым ингибитором CYP3A. У здоровых взрослых лиц приём препарата Еврисди® один раз в сутки в течение 2 недель несколько повышал экспозицию мидазолама, высокочувствительного субстрата CYP3A (AUC на 11 %; Cmax на 16 %). Такое взаимодействие не считается клинически значимым, поэтому коррекция дозы субстратов CYP3A не требуется.
С учётом результатов применения фармакокинетической модели, основанной на физиологии (PBPK), подобный эффект ожидается у детей и младенцев в возрасте от 2 месяцев.
Исследования in vitro показали, что рисдиплам и его основной метаболит не являются существенными ингибиторами MDR1 человека, полипептида-транспортера органических анионов (OATP)1B1, OATP1B3, транспортера органических анионов 1 и 3 (OAT 1 и 3). Однако рисдиплам и его основной метаболит in vitro являются ингибиторами полипептида-транспортера органических катионов 2 (OCT2) человека и транспортеров белков множественной резистентности и выведения токсинов (MATE)1 и транспортеров MATE2-K. При применении в терапевтических концентрациях активного вещества взаимодействие лекарственного средства с субстратами OCT2 не ожидается. Влияние сопутствующего применения рисдиплама на фармакокинетику субстратов MATE1 и MATE2-K у человека неизвестно. На основании данных in vitro препарат Еврисди® может увеличивать плазменную концентрацию активных веществ, выводящихся с помощью MATE1 или MATE2-K, например метформина (см. раздел «Фармакокинетика»). Если невозможно избежать сопутствующего применения, следует осуществлять мониторинг на предмет токсичности, связанной с лекарственным средством, и рассмотреть вопрос о снижении дозы другого одновременно применяемого препарата при необходимости.
Влияние других лекарственных средств на Еврисди®
Рисдиплам метаболизируется в основном с участием флавинсодержащих монооксигеназ 1 и 3 (FMO1 и 3), а также с помощью CYP 1A1, 2J2, 3A4 и 3A7. Рисдиплам не является субстратом белка множественной резистентности 1 человека (MDR1).
При одновременном применении сильного ингибитора CYP3A итраконазола в дозе 200 мг дважды в сутки и рисдиплама в дозе 6 мг однократно перорально не наблюдалось клинически значимого влияния на фармакокинетику (ФК) рисдиплама (увеличение AUC на 11 %, снижение Cmax на 9 %). При одновременном применении с ингибитором CYP3A коррекция дозы препарата Еврисди® не требуется.
Не ожидается взаимодействия с другими лекарственными средствами через опосредованные FMO1 и FMO3 пути передачи сигнала.
Особенности применения.
Общая информация
В исследованиях на животных отмечались изменения со стороны сетчатки, эпителия, особенно со стороны кожи и желудочно-кишечного тракта, а также признаки токсичности со стороны костного мозга (изменения в клиническом анализе крови). На сегодняшний день риск таких изменений у людей нельзя окончательно оценить из-за ограниченных долгосрочных данных о безопасности.
Эмбриофетальная токсичность
В исследованиях на животных наблюдалась эмбриофетальная токсичность. Пациентов репродуктивного возраста следует информировать о рисках. Необходимо применять высокоэффективные методы контрацепции во время лечения и по меньшей мере в течение 1 месяца после приема последней дозы препарата Эврисди® женщинами и в течение 4 месяцев после приема последней дозы препарата Эврисди® мужчинами (см. раздел «Способ применения и дозы»).
Потенциальное влияние на фертильность мужчин
С учетом обратимых эффектов препарата Эврисди® на мужскую фертильность, наблюдавшихся в исследованиях на животных, пациентам-мужчинам не следует быть донорами спермы во время лечения и в течение 4 месяцев после приема последней дозы препарата Эврисди® (см. раздел «Фармакокинетика»).
Следует избегать контакта порошка и восстановленного перорального раствора с кожей. При попадании лекарственного средства (порошка или раствора) на кожу пораженный участок необходимо промыть водой с мылом.
Вспомогательные вещества.
Этот лекарственный препарат содержит 0,38 мг бензоата натрия в 1 мл. Данный препарат содержит менее 1 ммоль натрия (23 мг) на дозу, то есть считается без содержания натрия.
Этот лекарственный препарат содержит изомальт. Пациентам с редкой врожденной непереносимостью фруктозы не следует применять данный лекарственный препарат.
Применение в период беременности или лактации.
С учетом результатов доклинических исследований, фертильность мужчин может нарушаться во время лечения препаратом Эврисди®. В репродуктивных органах крыс и обезьян отмечались дегенерация сперматозоидов и снижение количества сперматозоидов.
С пациентами мужского пола перед началом лечения препаратом Эврисди® необходимо обсудить стратегии сохранения фертильности. Пациенты мужского пола могут рассмотреть возможность сохранения спермы до начала лечения или после периода без лечения в течение не менее 4 месяцев (см. раздел «Особенности применения»).
С учетом результатов доклинических исследований, влияние препарата Эврисди® на фертильность женщин не ожидается.
Женщин репродуктивного возраста следует обследовать на беременность до начала лечения препаратом Эврисди®.
Пациентам мужского и женского пола репродуктивного возраста следует соблюдать следующие требования в отношении контрацепции:
- Пациентам мужского пола и их партнершам репродуктивного возраста следует применять высокоэффективную контрацепцию во время лечения препаратом Эврисди® и в течение по меньшей мере 4 месяцев после приема последней дозы препарата.
- Пациентам женского пола репродуктивного возраста следует применять высокоэффективную контрацепцию во время лечения препаратом Эврисди® и в течение по меньшей мере 1 месяца после приема последней дозы препарата.
Беременность
Отсутствуют клинические данные по применению препарата Эврисди® у беременных. Рисдиплам продемонстрировал эмбриофетотоксическое и тератогенное действие у животных. С учетом данных, полученных в исследованиях на животных, рисдиплам проникает через плацентарный барьер и может вызывать поражение плода.
Препарат Эврисди® не следует применять во время беременности, если только в этом нет четкой необходимости. Если беременной женщине требуется лечение препаратом Эврисди®, ей необходимо четко объяснить потенциальный риск для плода.
Лактация
Неизвестно, выделяется ли препарат Эврисди® в грудное молоко человека. Исследования на крысах показали, что рисдиплам выделяется в грудное молоко. Поскольку потенциальный вред для новорожденного, находящегося на грудном вскармливании, неизвестен, лечащий врач должен принять решение о дальнейшей терапии пациента. Во время лечения препаратом Эврисди® грудное вскармливание не рекомендуется.
Способность влиять на скорость реакции при управлении автотранспортом или другими механизмами.
Влияние препарата Эврисди® на скорость реакции при управлении автотранспортом или другими механизмами не изучалось в соответствующих исследованиях.
Способ применения и дозы.
Оральный раствор препарата Еврисди® должен готовить медицинский специалист (то есть врач или фармацевт) перед выдачей пациенту.
Перед применением первой дозы медицинскому работнику необходимо обсудить с пациентом или лицом, ухаживающим за пациентом, как приготовить и принять назначенную суточную дозу (см. ниже «Инструкции по обращению»).
Начинать и контролировать лечение препаратом Еврисди® должны врачи, имеющие опыт диагностики и лечения пациентов со спинальной мышечной атрофией.
Программа клинической разработки не включала пациентов с СМА типа IV.
Рекомендуемое дозирование
Лекарственное средство Еврисди® принимают перорально один раз в сутки, примерно в одно и то же время ежедневно, с использованием оральных шприцев для повторного применения, предоставляемых в упаковке. Рекомендуемая доза препарата Еврисди® для лечения СМА определяется в зависимости от возраста и массы тела пациента (см. таблицу 5).
Таблица 5
Режим дозирования в зависимости от возраста и массы тела
| Возраст и масса тела |
Рекомендуемая суточная доза |
| < 2 месяцев |
0,15 мг/кг |
| от 2 месяцев до < 2 лет |
0,20 мг/кг |
| ≥ 2 лет и масса тела < 20 кг |
0,25 мг/кг |
| ≥ 2 лет и масса тела ≥ 20 кг |
5 мг |
а На основании скорректированного возраста для недоношенных новорождённых.
Изменение дозы должно проводиться под наблюдением медицинского работника. Лечение дозой выше 5 мг в сутки до настоящего времени не изучалось. Имеются лишь очень ограниченные данные о безопасности, полученные в пострегистрационный период у новорождённых в возрасте до 16 дней, которым применяли препарат Эврисди® в рекомендованной дозе (см. подразделы «Фармакокинетика», «Дети»).
Пациенты с нарушением функции печени
Пациентам с лёгкими или умеренными нарушениями функции печени коррекция дозы не требуется. Применение препарата Эврисди® у пациентов с тяжёлыми нарушениями функции печени не изучалось (см. раздел «Фармакокинетика»).
Пациенты с нарушением функции почек
Безопасность и эффективность применения препарата Эврисди® у пациентов с нарушением функции почек не изучались. Не ожидается необходимость коррекции дозы у пациентов с нарушением функции почек (см. раздел «Фармакокинетика»).
Пациенты пожилого возраста
Клинические исследования препарата Эврисди® не включали пациентов в возрасте 65 лет и старше, поэтому не установлено, могут ли они реагировать на лечение иначе, чем более молодые пациенты.
Дети
Применение препарата Эврисди® у пациентов с СМА в возрасте до 2 месяцев подтверждается данными о фармакокинетике и безопасности у детей в возрасте от 16 дней (см. разделы «Побочные реакции» и подразделы «Клиническая эффективность» и «Фармакокинетика»). Отсутствуют клинические или фармакокинетические данные об использовании препарата у недоношенных новорождённых или новорождённых в возрасте до 16 дней (см. подраздел «Клиническая эффективность» выше).
Пропущенный приём
Лекарственное средство Эврисди® принимают перорально один раз в сутки примерно в одно и то же время ежедневно. Если приём дозы препарата Эврисди® был пропущен, препарат следует принять как можно скорее, если задержка составляет не более 6 часов по сравнению с запланированным временем приёма, после чего обычный режим дозирования можно восстановить со следующего дня. В противном случае пропущенную дозу принимать не следует, а следующую дозу нужно принять в запланированное время следующего дня.
Если доза была проглочена не полностью или возникла рвота после приёма препарата Эврисди®, не следует принимать другую дозу для компенсации утерянной. Необходимо дождаться следующего дня и принять следующую дозу в запланированное время.
Способ применения
Для введения суточной дозы препарата Эврисди® необходимо использовать многоразовый оральный шприц, поставляемый в картонной упаковке вместе с лекарственным средством (см. таблицу 6).
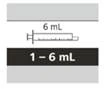
Таблица 6
Выбор соответствующего многоразового орального шприца для приёма назначенной суточной дозы Эврисди®
| Размер шприца |
Объем дозирования |
Цена деления шприца |
| 1 мл |
от 0,3 до 1 мл |
0,01 мл |
| 6 мл |
от 1 до 6 мл |
0,1 мл |
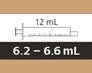
| 12 мл |
от 6,2 до 6,6 мл |
0,2 мл |
Для расчета объема дозировки необходимо также учитывать цену деления шприца. Округляйте объем дозы в большую или меньшую сторону до ближайшего значения деления, указанного на выбранном оральном шприце (например, с 6,3 мл до 6,4 мл, с 3,03 мл до 3 мл и с 1,05 до 1,1 мл).
Лекарственное средство Еврисди® в виде раствора необходимо принимать сразу после набора в оральный шприц для многократного использования. Если содержимое шприца не было принято в течение 5 минут, следует освободить оральный шприц от препарата (см. ниже «Утилизация неиспользованного лекарственного средства/лекарственного средства с истекшим сроком годности») и приготовить новую дозу.
Лекарственное средство Еврисди® следует принимать после еды. Пациенту необходимо выпить воды после приема Еврисди®, чтобы убедиться, что препарат полностью проглочен. Если пациент не может глотать и у него установлен назогастральный зонд или гастростомическая трубка, Еврисди® можно вводить через зонд/трубку. После введения препарата зонд/трубку следует промыть водой (см. ниже «Инструкции по обращению»).
Инструкции по обращению
Инструкции, которым необходимо следовать до, во время и после приготовления орального раствора:
- Раствор всегда должен готовить медицинский специалист (то есть врач или фармацевт).
- Следует избегать вдыхания порошка препарата Еврисди®. Обратите внимание на местные нормы и используйте соответствующее оборудование для приготовления раствора Еврисди®.
- Надевайте перчатки.
- Не используйте порошок, если истек срок годности. Срок годности порошка указан на этикетке флакона и картонной коробке.
- Не выдавайте восстановленный раствор, если срок годности готового к употреблению орального раствора — дата, указанная на этикетке флакона и картонной коробке — превышает первоначальный срок годности порошка.
- Избегайте контакта лекарственного средства с кожей. Если лекарственное средство (порошок или раствор) попало на кожу, промойте участок водой с мылом.
- Не используйте лекарственное средство, если какое-либо содержимое упаковки повреждено или отсутствует.
- Для приготовления раствора используйте очищенную воду или воду для инъекций.
- Не используйте оральные шприцы, кроме тех, которые вложены в картонную коробку вместе с лекарственным средством.
- Не смешивайте Еврисди® с пищей или жидкостями (например, с молоком или молочной смесью).
- Не смешивайте Еврисди® из новой бутылки с препаратом из бутылки, которую вы в данный момент используете.
Пациент или лицо, ухаживающее за пациентом, должны быть проинструктированы медицинским работником о том, как правильно готовить и принимать назначенную суточную дозу до выдачи приготовленного раствора.
Приготовление орального раствора
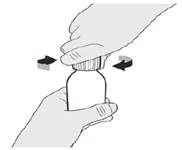
Наливают 79 мл очищенной воды или воды для инъекций во флакон с лекарственным средством.
Вставляют вдавливаемый адаптер для флакона в горлышко флакона, проталкивая его вниз.
После полного закрытия флакона взбалтывайте в течение 15 секунд.
Через 10 минут должен быть получен прозрачный раствор. Если раствор не стал прозрачным, его следует взбалтывать еще в течение 15 секунд.
Необходимо рассчитать 64 дня с момента приготовления раствора. День приготовления раствора считается днем 0. Рассчитанную дату следует указать на этикетке флакона и картонной коробке в поле: Восстановленный оральный раствор УТИЛИЗИРОВАТЬ ПОСЛЕ: (день/месяц/год).
Утилизация неиспользованного лекарственного средства/лекарственного средства с истекшим сроком годности
Попадание лекарственных средств в окружающую среду должно быть сведено к минимуму. Нельзя утилизировать лекарственные средства через канализацию, а также следует избегать утилизации с бытовыми отходами.
Неиспользованный препарат/препарат со сроком действия, который истек, должен быть утилизирован профессионально в месте выдачи лекарственного средства (врачом или фармацевтом).
Инструкция по приготовлению раствора (ТОЛЬКО ДЛЯ МЕДИЦИНСКИХ РАБОТНИКОВ, В ТОМ ЧИСЛЕ ВРАЧЕЙ И ФАРМАЦЕВТОВ)
Оральный раствор Еврисди® должен быть приготовлен медицинским работником перед выдачей пациенту.
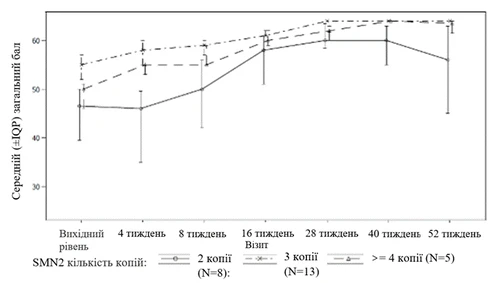
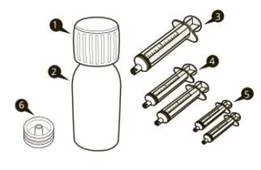
| Одна упаковка препарата Еврисди® содержит (см. рисунок A):
|
Рисунок A |
Важная информация о препарате Эврисди®
- Раствор всегда должен готовиться медицинским работником (врачом или фармацевтом).
- Избегать вдыхания порошка препарата Эврисди®. Соблюдать местные требования и использовать соответствующее оборудование для приготовления раствора Эврисди®.
- Надевать перчатки.
- Не использовать этот препарат после истечения срока годности. Дата окончания срока годности указана на этикетке флакона и картонной коробке.
- Избегать любого контакта этого препарата с кожей. При случайном контакте (с порошком или раствором) необходимо промыть место контакта водой с мылом.
- Не использовать этот препарат, если любая часть упаковки повреждена или отсутствует.
- Для приготовления раствора использовать очищенную воду или воду для инъекций.
- Не использовать многоразовые оральные шприцы, кроме тех, которые входят в упаковку.
- Не выдавать приготовленный раствор, если срок годности готового к употреблению орального раствора — дата, указанная на этикетке флакона и картонной коробке, — превышает дату окончания срока годности порошка.
- Перед выдачей приготовленного раствора медицинский работник должен проинструктировать пациента или ухаживающего лица о порядке приготовления и приема суточной дозы.
Как хранить препарат Эврисди®
- Хранить порошок (лекарственное средство, еще не приготовленное в виде раствора) в картонной коробке при комнатной температуре не выше 25 °C с целью защиты от света и влаги.
- Хранить раствор (лекарственное средство, приготовленное в виде раствора) в вертикальном положении в холодильнике при температуре 2–8 °C в картонной коробке для защиты содержимого от света.
- При необходимости оральный раствор можно хранить при комнатной температуре (ниже 40 °C) суммарно не более 5 дней. Не хранить оральный раствор при температуре выше 40 °C.
- Хранить оральный раствор во флаконе оригинальной упаковки в вертикальном положении и с плотно закрытой крышкой.
Приготовление раствора
|
Рисунок В |
Шаг 1 Осторожно постучите по дну флакона, чтобы разрыхлить порошок (см. рисунок B). |
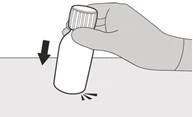
|
Рисунок С |
Шаг 2 Нажмите на крышку вниз, а затем поверните её влево (против часовой стрелки), чтобы снять (см. рисунок С). Крышку не выбрасывайте. |
|
Рисунок D |
Шаг 3 Осторожно добавьте 79 мл очищенной воды или воды для инъекций во флакон с препаратом (см. рисунок D). |
|
Рисунок Е |
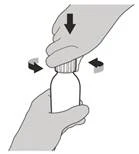
Шаг 4 Удерживайте флакон с препаратом одной рукой на столе. Вставьте уплотнительный адаптер для флакона в его горловину, нажимая на него сверху другой рукой, пока адаптер полностью не прижмётся к краю горловины флакона (см. рисунок Е). |
|
Рисунок F |
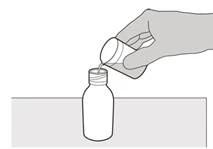
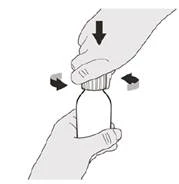
Шаг 5 Наденьте крышку обратно на флакон. Поверните крышку вправо (по часовой стрелке), чтобы закрыть флакон. Убедитесь, что флакон плотно закрыт, затем тщательно взбалтывайте в течение 15 секунд (см. рисунок F). |
|
Рисунок G |
Шаг 6 Определите дату «ВЫБРОСИТЬ ПОСЛЕ: (день/месяц/год)», отсчитав 64 дня с момента приготовления раствора (примечание: день приготовления считается днём 0. Например, если вы приготовили раствор 1 апреля, дата «ВЫБРОСИТЬ ПОСЛЕ» будет 4 июня). Укажите эту дату в соответствующем поле на этикетке флакона (см. рисунок G) и на картонной коробке: Восстановленный пероральный раствор ВЫБРОСИТЬ ПОСЛЕ: (день/месяц/год). Поместите флакон обратно в картонную коробку вместе со шприцами (в индивидуальных пакетиках) и инструкцией по медицинскому применению. Храните всё в холодильнике, держа флакон в вертикальном положении. |
ИНСТРУКЦИЯ ПО ПРИМЕНЕНИЮ ОРАЛЬНОГО РАСТВОРА
Перед началом приёма препарата Еврисди® необходимо внимательно прочитать и понять инструкцию по медицинскому применению. В ней содержится информация о том, как приготовить и ввести дозу препарата Еврисди® с помощью многоразового орального шприца через рот, чрезвычайный гастростомический зонд (PEG-зонд) или назогастральный зонд (гастростомическую трубку). Если у вас возникли вопросы по применению препарата Еврисди®, обратитесь к лечащему врачу или фармацевту. Перед выдачей оральный раствор Еврисди® должен быть приготовлен медицинским работником, таким как врач или фармацевт.
Вы должны получить препарат Еврисди® в виде жидкости в бутылке. Не используйте препарат, если в бутылке находится порошок, и в этом случае обратитесь к лечащему врачу или фармацевту.
Перед тем как принимать/вводить первую дозу, медицинский работник должен объяснить вам, как правильно приготовить и принимать/вводить назначенную суточную дозу.
- Попросите лечащего врача или фармацевта показать вам, какой из оральных шприцов, входящих в упаковку, следует использовать для введения, и как определить суточную дозу.
- Если нужный оральный шприц(ы) был(и) потерян(ы) или повреждён(ы), обратитесь к лечащему врачу или фармацевту. Он(а) подскажет вам, как продолжить приём препарата.
- Для измерения назначенной суточной дозы всегда используйте оральный шприц подходящего размера, который можно использовать повторно.
- Описание подходящего многоразового орального шприца также приведено в таблице 6: «Выбор подходящего многоразового орального шприца для введения назначенной суточной дозы Еврисди®». Обратитесь к лечащему врачу или фармацевту, если у вас есть вопросы по правильному выбору шприца.
- Не используйте препарат Еврисди®, если в бутылке отсутствует адаптер для бутылки. В этом случае обратитесь к лечащему врачу или фармацевту.
- Не используйте препарат Еврисди® после даты, указанной на этикетке бутылки и картонной коробке, как «Восстановленный оральный раствор УТИЛИЗИРОВАТЬ ПОСЛЕ: (день/месяц/год)». Если на этикетке бутылки или на картонной коробке отсутствует дата «Восстановленный оральный раствор УТИЛИЗИРОВАТЬ ПОСЛЕ: (день/месяц/год)», уточните её у лечащего врача или фармацевта.
- Не смешивайте Еврисди® с пищей или другими жидкостями (например, с молоком или молочной смесью).
- Не используйте Еврисди®, если бутылка или оральный шприц повреждены.
- Избегайте контакта Еврисди® с кожей. Если препарат попал на кожу, промойте участок водой с мылом.
- Если вы пролили Еврисди®, сначала промокните место сухой бумажной салфеткой, затем очистите его водой с мылом. Утилизируйте бумажную салфетку вместе с бытовыми отходами и тщательно вымойте руки водой с мылом.
- Если в бутылке недостаточно препарата Еврисди® для назначенной дозы, верните бутылку и использованные оральные шприцы в место выдачи (лечащему врачу или фармацевту) для надлежащей утилизации. Используйте новую бутылку Еврисди® для отбора назначенной суточной дозы. Не смешивайте Еврисди® из новой бутылки с препаратом из бутылки, которую вы использовали ранее.
Каждая коробка препарата Еврисди® содержит (см. рисунок A):
|
|
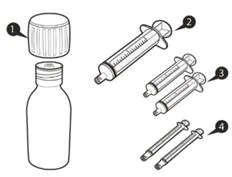
Рисунок A
A) Подготовка и набор правильного объема дозировки
Как выбрать соответствующий оральный шприц для назначенной дозы лекарственного средства Еврисди®
|
|
|
|
|
|

Как приготовить суточную дозу препарата Еврісді®
|
Рисунок В |
Шаг A1 Нажмите на крышку вниз, а затем поверните её влево (против часовой стрелки), чтобы снять (см. рисунок В). Крышку не выбрасывайте. |
|||||
|
Рисунок С |
Шаг A2 Полностью нажмите поршень перорального шприца вниз, чтобы удалить воздух из перорального шприца (см. рисунок С). |
|||||
|
Рисунок D |
Шаг А3 Удерживая флакон в вертикальном положении, вставьте наконечник шприца в адаптер флакона (см. рисунок D). |
|||||
|
Рисунок Е |
Шаг A4 Осторожно переверните флакон вверх дном вместе с плотно вставленным наконечником шприца в адаптер флакона (см. рисунок Е). |
|||||
|
Рисунок F |
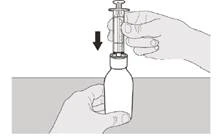
Шаг A5 Медленно потяните поршень назад, чтобы набрать назначенную вам суточную дозу препарата Еврісді® из флакона. Верхнюю часть чёрного ограничителя хода поршня необходимо выровнять с отметкой миллилитров на пероральном шприце, соответствующей назначенной вам суточной дозе (см. рисунок F). После набора правильной дозы удерживайте поршень на месте, чтобы он не двигался. |
|||||
|
Рисунок G |
Шаг A6 Продолжайте удерживать поршень на месте, чтобы он не двигался. Оставьте пероральный шприц в адаптере флакона и верните флакон в вертикальное положение. Поставьте флакон на ровную поверхность. Извлеките пероральный шприц из адаптера флакона, аккуратно потянув его вверх, удерживая поршень на месте (см. рисунок G). |
|||||
|
Рисунок Н |
Шаг A7 Держите пероральный шприц так, чтобы наконечник шприца был направлен вверх. Проверьте препарат Еврісді® в пероральном шприце. Если в пероральном шприце наблюдаются крупные пузырьки воздуха (см. рисунок H) или если вы набрали неправильную суточную дозу препарата Еврісді®, плотно вставьте наконечник шприца обратно в адаптер флакона. Нажмите поршень вниз, чтобы препарат Еврісді® перетёк обратно во флакон, и повторите шаги A4–A7. Принимайте или давайте препарат Еврісді® сразу после набора в пероральный шприц. Если препарат не был принят в течение 5 минут, удалите и утилизируйте препарат Еврісді® из перорального шприца и наберите новую дозу. |
|||||
|
Рисунок І |
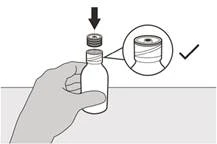
Шаг A8 Оставьте адаптер флакона на флаконе. Наденьте крышку обратно на флакон. Поверните крышку вправо (по часовой стрелке), чтобы плотно закрыть флакон (см. рисунок І). |
|||||
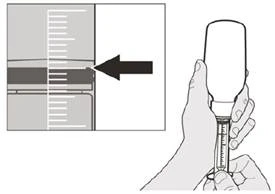
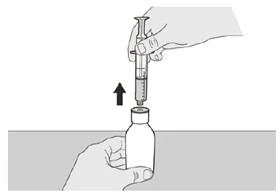
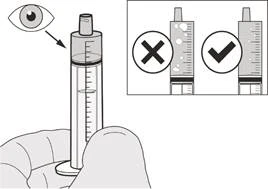
Если вы принимаете суточную дозу препарата Еврісді® перорально, соблюдайте инструкции в разделе «B) Как принимать дозу препарата Еврісді® перорально».
Если вы принимаете суточную дозу препарата Еврісді® через гастростомическую трубку, соблюдайте инструкции в разделе «C) Как вводить дозу препарата Еврісді® через гастростомическую трубку».
Если вы принимаете суточную дозу препарата Еврісді® через назогастральный зонд, соблюдайте инструкции в разделе «D) Как вводить дозу препарата Еврісді® через назогастральный зонд».
Оральные шприцы для препарата Еврісді® были специально разработаны для совместимости с системой ENFit®. Если имеющийся зонд для питания не совместим с системой ENFit®, может понадобиться переходной адаптер ENFit для подключения шприца препарата Еврісді® к гастростомической трубке или назогастральному зонду.
B) Как принимать суточную дозу препарата Еврісді® перорально
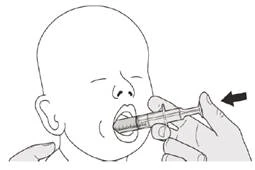
Находитесь в вертикальном положении (предпочтительно сидя), принимая суточную дозу Еврісді® через рот.
|
Рисунок J |
Шаг B1 Положите оральный шприц в рот наконечником вдоль любой щеки. Медленно нажмите на поршень до конца, чтобы ввести полную дозу препарата Еврісді® (см. рисунок J). Попадание препарата Еврісді® в горло или слишком быстрое введение может вызвать удушье. |
|
Рисунок К |
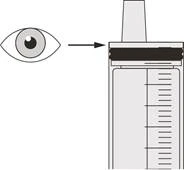
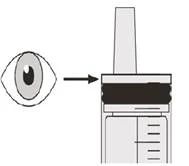
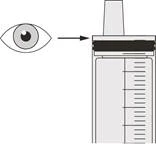
Шаг B2 Проверьте, остался ли препарат Эврисди® в оральном шприце (см. рисунок К). |
|
Рисунок L |
Шаг B3 Выпейте немного воды сразу после приёма назначенной дозы препарата Эврисди® (см. рисунок L). См. шаг E для получения информации об очистке шприца. |
C) Как вводить суточную дозу препарата Еврісді® через гастростомическую трубку
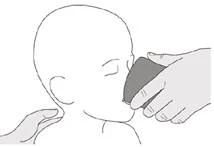
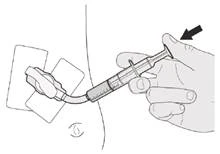
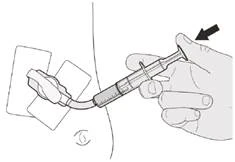
Если вы вводите препарат Еврісді® через гастростомическую трубку, попросите вашего врача показать вам, каким образом осматривать гастростомическую трубку перед введением препарата Еврісді®.
|
Рисунок М |
Шаг C1 Вставьте наконечник орального шприца в гастростомическую трубку. Медленно до конца нажмите на поршень, чтобы ввести полную дозу препарата Еврісді® (см. рисунок M). |
|
Рисунок N |
Шаг C2 Проверьте, остался ли препарат Еврісді® в оральном шприце (см. рисунок N). |
|
Рисунок О |
Шаг C3 Промойте гастростомическую трубку 10–20 мл воды сразу после введения назначенной дозы препарата Еврісді® (см. рисунок O). См. шаг E для получения информации об очистке шприца. |
D) Как вводить суточную дозу препарата Еврисди® через назогастральный зонд
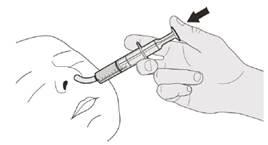
Если вы вводите препарат Еврисди® через назогастральный зонд, попросите вашего врача показать вам, каким образом проверять назогастральный зонд перед введением препарата Еврисди®.
|
Рисунок P |
Шаг D1 Вставьте наконечник орального шприца в назогастральный зонд. Медленно до конца нажмите на поршень, чтобы ввести полную дозу препарата Еврісді® (см. рисунок P). |
|
Рисунок Q |
Шаг D2 Проверьте, остался ли препарат Еврісді® в оральном шприце (см. рисунок Q). |
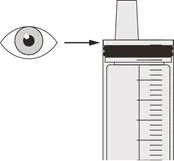
| Рисунок R |
Шаг D3 Промойте назогастральный зонд 10–20 мл воды сразу после введения назначенной дозы препарата Евриді® (см. рисунок R). См. шаг E для информации об очистке шприца. |
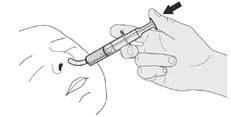
Е) Как очистить шприц для перорального применения после использования
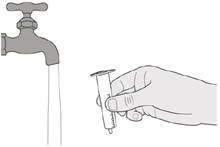
|
Рисунок S |
Шаг E1 Вытяните поршень (против сопротивления) из шприца. Хорошо промойте цилиндр орального шприца под чистой проточной водой (см. рисунок S). |
|
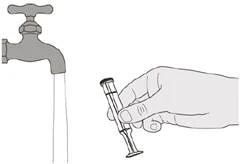
Рисунок Т |
Шаг E2 Хорошо промойте поршень под чистой проточной водой (см. рисунок T). |
|
Рисунок U |
Шаг E3 Проверьте, что цилиндр и поршень орального шприца чистые. Положите цилиндр и поршень орального шприца на чистую поверхность в безопасное место для просушивания (см. рисунок U). Вымойте руки. После того как цилиндр и поршень орального шприца просохнут, вставьте поршень обратно в цилиндр орального шприца и храните шприц до следующего применения. |
Дети.
Безопасность и эффективность лекарственного средства Еврисди® у пациентов в возрасте до 16 дней не установлена (см. раздел «Фармакокинетика»).
Передозировка.
Отсутствует опыт передозировки препарата Еврисди® в клинических исследованиях. Антидот при передозировке препаратом Еврисди® неизвестен. При передозировке следует тщательно наблюдать за состоянием пациента и начать поддерживающую терапию.
Побочные реакции.
Классификация частоты возникновения побочных реакций лекарственного средства: очень часто (≥ 1/10), часто (от ≥ 1/100 до < 1/10), нечасто (от ≥ 1/1 000 до < 1/100), редко (от ≥ 1/10 000 до < 1/1000), очень редко (< 1/10 000); частота неизвестна (невозможно рассчитать на основании имеющихся данных).
Краткая характеристика профиля безопасности
У пациентов с СМА с инфантильным началом наиболее часто наблюдавшимися побочными реакциями в клинических исследованиях лекарственного средства Еврисди® были повышение температуры тела (54,8 %), высыпания (29 %) и диарея (19,4 %). У пациентов с СМА с более поздним началом наиболее часто наблюдавшимися побочными реакциями в клинических исследованиях лекарственного средства Еврисди® были повышение температуры тела (21,7 %), головная боль (20 %), диарея (16,7 %) и высыпания (16,7 %).
Указанные выше побочные реакции возникали без идентифицированных клинических особенностей или характерной временной связи и в целом исчезали без прекращения лечения лекарственным средством Еврисди® у пациентов с СМА с инфантильным началом и поздним началом.
Таблица 7
Сводка побочных реакций у пациентов с СМА с инфантильным началом и поздним началом, наблюдавшихся в клинических исследованиях препарата Еврисди®
| Класс системы органов |
СМА с инфантильным началом2 (тип 1) |
СМА с поздним началом3 (тип 2 и 3) |
|
| Желудочно-кишечные расстройства |
|||
| Диарея |
Очень часто |
Очень часто |
|
| Тошнота |
Не применимо |
Часто |
|
| Язвы слизистой оболочки полости рта и афтозные язвы |
Часто |
Часто |
|
| Нарушения со стороны кожи и подкожных тканей |
|||
| Высыпания1 |
Очень часто |
Очень часто |
|
| Расстройства со стороны нервной системы |
|||
| Головная боль |
Не применимо |
Очень часто |
|
| Общие нарушения и реакции в месте введения |
|||
| Повышение температуры тела (включая гиперпирексию) |
Очень часто |
Очень часто |
|
| Инфекции и инвазии |
|||
| Инфекция мочевыводящих путей (включая цистит) |
Часто |
Часто |
|
| Нарушения со стороны костно-мышечной системы и соединительной ткани |
|||
| Артралгия |
Не применимо |
Часто |
|
1 В том числе дерматит, угревидный дерматит, аллергический дерматит, эритема, фолликулит, сыпь, эритематозная сыпь, макулопапулезная сыпь, папулезная сыпь.
2 У пациентов с СМА с младенческим началом (исследование FIREFISH, части 1 и 2) побочные реакции определены как явления, возникшие у ≥ 2 % пациентов и более, и возможная причинная связь которых с препаратом Эвризди®.
3 У пациентов с СМА с поздним началом (исследование SUNFISH, часть 2) побочные реакции определены как явления, возникшие у ≥ 2 % пациентов, получавших лечение препаратом Эвризди®, по сравнению с группой плацебо в период двойного слепого контролируемого лечения, и возможная причинная связь которых с препаратом Эвризди®.
Имеющиеся данные о безопасности ограничены по числу пациентов, получавших препарат Эвризди®, и по продолжительности экспозиции. Могут наблюдаться потенциально редкие и потенциально серьезные побочные реакции (ПР), которые не были выявлены в ходе исследовательской программы.
В исследовании RAINBOWFISH получены ограниченные данные о безопасности применения лекарственного средства Эвризди® у новорождённых и младенцев с пресимптоматической СМА. Исследование RAINBOWFISH — это открытое исследование с одной группой, включавшее 26 пациентов с пресимптоматической СМА в возрасте от 16 до 41 дня (диапазон массы тела от 3,1 до 5,7 кг) на момент введения первой дозы. В первичном анализе средняя продолжительность терапии составила 20,4 месяца (диапазон: 10,6 до 41,9 месяцев) (см. раздел «Фармакологические свойства», подраздел «Клиническая эффективность»).
Профиль безопасности препарата Эвризди® у пациентов с пресимптоматическим состоянием в исследовании RAINBOWFISH можно сравнить с профилем безопасности у пациентов с симптоматической СМА, получавших лечение препаратом Эвризди® в ходе клинических исследований. Долгосрочные данные на данный момент отсутствуют.
Профиль безопасности у пациентов с СМА, ранее получавших другую модифицирующую терапию
На основании первичного анализа участников исследования JEWELFISH профиль безопасности препарата Эвризди® у ранее леченных пациентов, получавших лечение препаратом Эвризди® в течение периода до 59 месяцев в исследовании JEWELFISH, согласуется с профилем безопасности при лечении ранее не леченных пациентов с СМА, получавших препарат Эвризди® в исследованиях FIREFISH (части 1 и 2), SUNFISH (части 1 и 2) и RAINBOWFISH.
В исследование JEWELFISH были включены пациенты, ранее получавшие нусинерсен (n = 76) или онасемноген абепарвовек (n = 14) (см. подраздел «Клиническая эффективность»).
Доклинические эффекты
Доклинические эффекты, касающиеся структуры сетчатки, эпителиальной ткани и гематологических параметров, описанные в разделе «Доклинические данные», на сегодняшний день не наблюдались в проведённых клинических исследованиях препарата Эвризди® при СМА.
Удлинение интервала QT
Анализ фармакокинетики/фармакодинамики показал отсутствие признаков удлинения QTc при применении препарата Эвризди® в терапевтическом диапазоне экспозиции, однако отсутствуют соответствующие данные при применении препарата Эвризди® при экспозиции, превышающей терапевтический диапазон.
Опыт применения в пострегистрационный период
В пострегистрационный период наблюдался кожный васкулит, симптомы которого исчезли после окончательного отмены лекарственного средства Эвризди®. На основании имеющихся данных невозможно определить категорию заболеваемости и частоту возникновения данной побочной реакции. Имеются лишь ограниченные данные о применении в пострегистрационный период у новорождённых в возрасте до 16 дней.
Срок годности.
2 года.
После восстановления готовый к применению оральный раствор стабилен в течение 64 дней при хранении в холодильнике при температуре 2–8 ºС. При необходимости пациент или лицо, осуществляющее уход, могут хранить оральный раствор при комнатной температуре (ниже 40 °C) в общей сложности не более 5 дней.
Условия хранения.
Хранить порошок для орального раствора при температуре не выше 25 ºС в оригинальной упаковке для защиты от света и влаги. Хранить в недоступном для детей месте.
Хранить готовый к применению оральный раствор в холодильнике при температуре 2–8 ºС в оригинальной упаковке для защиты от света. Держать флакон плотно закрытым и всегда хранить в вертикальном положении.
Несовместимость.
Не наблюдалось несовместимости между препаратом Эвризди® и рекомендованными многоразовыми шприцами для орального введения препарата.
Упаковка.
Стеклянный флакон объёмом 100 мл коричневого цвета (класс III в соответствии с USP и Ph. Eur.) с белой крышкой с функцией защиты от вскрытия детьми (внешняя оболочка из полиэтилена высокой плотности; внутренняя оболочка из гомополимера полипропилена) с кольцом контроля первого вскрытия и прокладкой из полиэтилена и поливинилиденхлорида, в комплекте с 1 вдавливаемым адаптером для флакона, 2 многоразовыми оральными шприцами объёмом 1 мл (каждый в полиэтиленовом пакетике), 2 многоразовыми оральными шприцами объёмом 6 мл (каждый в полиэтиленовом пакетике) и 1 многоразовым оральным шприцем объёмом 12 мл (в полиэтиленовом пакетике), упакованными в полиэтиленовый пакет. По 1 флакону и 1 комплекту в картонной коробке.
Категория отпуска.
По рецепту.
Производитель.
Ф. Хоффманн-Ля Рош Лтд
Местонахождение производителя и адрес места осуществления его деятельности.
Вурмисвег, 4303 Кайзерсгауст, Швейцария