Eurisdii®
UcraniaContenido
INSTRUCCIONES PARA USO MÉDICO DEL MEDICAMENTO Evrysdi®
Composición:
Principio activo: risdiplam;
1 frasco contiene 60 mg de risdiplam;
1 ml de solución reconstituida contiene 0,75 mg de risdiplam;
Excipientes: manitol (E 421); isomalt (E 953); aroma de fresa; ácido tartárico; benzoato de sodio (E 211); polietilenglicol 6000; sucralosa; ácido ascórbico; edetato disódico dihidrato.
Forma farmacéutica. Polvo para solución oral.
Características fisicoquímicas principales: Polvo o polvo con grumos o masa pulverulenta de color amarillo claro, amarillo, amarillento, verdoso-amarillento o verde claro. La solución reconstituida es de color verde amarillento a amarillo.
Grupo farmacoterapéutico. Agentes que actúan sobre el sistema músculo-esquelético. Otros agentes utilizados en patologías del sistema músculo-esquelético.
Código ATC M09A X10.
Propiedades farmacológicas.
Farmacodinámica.
Mecanismo de acción
Risdiplam es un modificador del empalme del ARN mensajero precursor (pre-ARNm) del gen del factor de supervivencia de las neuronas motoras 2 (SMN2), diseñado para el tratamiento de la AME causada por mutaciones en el cromosoma 5q que conducen a una deficiencia de la proteína SMN. La deficiencia funcional de la proteína SMN es el mecanismo fisiopatológico de la AME de todos los tipos. Risdiplam corrige el empalme de SMN2, modificando el equilibrio desde la exclusión del exón 7 hacia la inclusión del exón 7 en el transcrito de ARNm, lo que resulta en un aumento de la producción de proteína SMN funcional y estable. De este modo, risdiplam trata la AME al aumentar y mantener niveles funcionales de la proteína SMN.
Electrofisiología cardíaca
El efecto de risdiplam sobre el intervalo QTc fue evaluado en un estudio realizado con 47 voluntarios adultos sanos. Dentro del rango terapéutico, risdiplam no prolongó el intervalo QTc.
Risdiplam se distribuye uniformemente por todas las partes del cuerpo, incluyendo el sistema nervioso central, al atravesar la barrera hematoencefálica, aumentando así los niveles de la proteína SMN en el SNC y en todo el organismo. La concentración de risdiplam en plasma sanguíneo y la concentración de la proteína SMN en sangre reflejan la distribución y los efectos farmacodinámicos de risdiplam en tejidos como el cerebro y los músculos.
En los estudios clínicos FIREFISH, SUNFISH y JEWELFISH, realizados con pacientes con atrofia muscular espinal (AME) de inicio infantil y con AME de inicio tardío, risdiplam provocó un aumento constante y sostenido del nivel de proteína SMN medido en sangre, con un cambio mediano superior al doble del valor basal durante las 4 semanas posteriores al inicio del tratamiento. Este aumento en el nivel de proteína SMN se mantuvo durante todo el período de tratamiento, al menos durante 24 meses (ver sección «Eficacia clínica»).
Eficacia clínica
La eficacia del medicamento Evrysdi® en el tratamiento de pacientes con forma infantil de AME (tipo 1) y con formas de AME de inicio tardío (tipos 2 y 3) fue evaluada en dos estudios clínicos principales, FIREFISH y SUNFISH, y confirmada con datos adicionales del estudio JEWELFISH. La eficacia de Evrysdi® en el tratamiento de pacientes con AME presintomática fue evaluada basándose en un análisis intermedio de los objetivos secundarios del estudio RAINBOWFISH, que continúa en curso.
Los pacientes con diagnóstico clínico de AME tipo 4 no participaron en los estudios clínicos.
En los estudios clínicos se demostró una eficacia a largo plazo durante al menos 24 meses de tratamiento. Existen datos limitados sobre el uso de Evrysdi® por más de 2 años.
Forma infantil de AME
El estudio BP39056 (FIREFISH) fue un estudio abierto de dos partes, para evaluar la eficacia, seguridad, farmacocinética y farmacodinámica de Evrysdi® en pacientes con AME tipo 1 sintomáticos (todos los pacientes tenían confirmación genética de la enfermedad con dos copias del gen SMN2). La parte 1 del estudio FIREFISH fue diseñada como un estudio de búsqueda de dosis. En la parte confirmatoria 2 del estudio FIREFISH se evaluó la eficacia de Evrysdi® a las dosis terapéuticas seleccionadas según los resultados de la parte 1 (ver sección «Instrucciones de uso y dosis»). Los pacientes de la parte 1 no participaron en la parte 2.
En total, 62 pacientes con AME tipo 1 sintomáticos fueron incluidos en la parte 1 (n = 21) y la parte 2 (n = 41) del estudio FIREFISH, de los cuales 58 pacientes recibieron la dosis terapéutica de Evrysdi®. La edad media al inicio de los síntomas clínicos fue de 1,5 meses (0,9–3 meses). La edad media al ingreso en el estudio fue de 5,6 meses (2,2–6,9 meses), y el tiempo medio entre el inicio de los síntomas y la primera dosis fue de 3,7 meses (1–6 meses). El 60 % de los pacientes eran mujeres, el 57 % eran de raza caucásica y el 29 % eran asiáticos. Al inicio del estudio, la puntuación mediana en la escala CHOP-INTEND fue de 23 (8–37) y la puntuación mediana en HINE-2 fue de 1 (0–5). Las características demográficas y clínicas basales de los pacientes incluidos en la parte 1 del estudio fueron comparables a las de la parte 2.
El objetivo primario del estudio fue la proporción de pacientes capaces de sentarse sin soporte durante al menos 5 segundos, definido como el ítem 22 de la escala de desarrollo infantil y temprano de Bayley — tercera edición (BSID-III) para evaluar la motricidad global, tras 12 meses de tratamiento con Evrysdi® en la parte 2 del estudio; este resultado se alcanzó en el 29 % de los pacientes (n = 12/41, IC del 90 %: 17,8 %, 43,1 %, p <0,0001).
Los resultados clave de eficacia en pacientes que recibieron tratamiento con Evrysdi® en el estudio FIREFISH (datos combinados de la parte 1 y la parte 2) se presentan en la tabla 1.
Tabla 1
Resumen de los resultados clave de eficacia a los 12 y 24 meses (FIREFISH, parte 1 y parte 2)
| Puntos finales de eficacia |
A los 12 meses |
A los 24 meses |
| Proporción de pacientes (IC del 90 %) |
||
| Hitos del desarrollo motor y función motora |
N = 58a |
|
| BSID-III: sentarse sin apoyo durante al menos 5 segundos |
32,8 % |
60,3 % |
| CHOP-INTEND: puntuación de 40 o más puntos |
56,9 % |
74,1 % |
| Aumento de la puntuación CHOP-INTEND en ≥ 4 puntos desde el valor basal |
89,7 % |
87,9 % |
| HINE-2: pacientes que respondieron según los criterios de desarrollo de la función motorab |
77,6 % |
82,8 % |
| Supervivencia y supervivencia libre de eventos |
N=62a |
|
| Supervivencia libre de eventosc |
87,1 % |
|
| Supervivencia |
91,9 % |
90,3 % |
| Alimentación |
N = 58a |
|
| Capacidad para recibir alimentación por vía orald |
84,5 % |
82,8 % |
BSID-III — Escala de Desarrollo Infantil y de la Primera Infancia de Bayley — Tercera Edición;
CHOP-INTEND — Prueba del Hospital Infantil de Filadelfia para evaluar las funciones motoras en enfermedades neuromusculares en lactantes;
HINE-2 — Módulo 2 del Examen Neurológico Infantil de Hammersmith.
a Los datos sobre supervivencia y supervivencia sin ventilación se combinaron para todos los pacientes que recibieron cualquier dosis de risdiplam en la parte 1 y parte 2 del estudio (n = 62). Para los hitos motores, función motora y alimentación, los puntos finales de eficacia, los datos se combinaron para todos los pacientes que recibieron la dosis terapéutica de risdiplam (todos los pacientes en la parte 2 del estudio y los pacientes en el grupo de dosis alta en la parte 1; n = 58).
b Definición de respuesta según HINE-2: la respuesta en este análisis se definió como un aumento ≥ 2 puntos (o el mayor incremento posible) en la capacidad de mover las piernas, O un aumento ≥ 1 punto en hitos motores como control del sostén de la cabeza, volteo, sentado, arrastre, de pie o marcha, Y una mejora en un mayor número de categorías del desarrollo motor que empeoramiento.
c El fenómeno correspondiente al punto final de ventilación permanente se definió como traqueostomía o ≥ 16 horas diarias de ventilación no invasiva, o intubación durante > 21 días consecutivos en ausencia o tras la resolución de un episodio agudo reversible. Cuatro pacientes cumplieron los criterios del punto final de ventilación permanente antes de los 24 meses. Estos cuatro pacientes alcanzaron un incremento de al menos 4 puntos en la escala CHOP-INTEND respecto al valor basal.
d Incluye pacientes que recibieron exclusivamente alimentación oral (41 pacientes a los 12 y 24 meses) y aquellos pacientes que recibieron alimentación oral combinada con sonda de alimentación (8 pacientes a los 12 meses y 7 pacientes a los 24 meses).
A los 24 meses, el 40 % (23/58) de los pacientes que recibieron la dosis terapéutica del medicamento Evrysdi® pudieron sentarse sin apoyo durante 30 segundos (BSID-III, ítem 26). Además, los pacientes continuaron alcanzando hitos motores adicionales según el parámetro HINE-2 a los 24 meses; el 78 % de los pacientes pudieron voltear (31 % pudieron voltear de lado, 7 % pudieron voltear desde decúbito prono a decúbito supino y 40 % pudieron voltear desde decúbito supino a decúbito prono) y el 28 % pudieron alcanzar la posición sentada (16 % soportaron peso y 12 % estuvieron de pie con apoyo).
La proporción de pacientes vivos sin necesidad de ventilación permanente (supervivencia libre de eventos) fue del 84 % entre todos los pacientes a los 24 meses. Seis lactantes fallecieron (4 durante los primeros 3 meses tras la inclusión en el estudio) y un paciente tuvo la terapia interrumpida prematuramente y falleció 3,5 meses después. A cuatro pacientes se les requirió ventilación permanente antes de los 24 meses.
AMC con inicio tardío
Estudio BP39055 (SUNFISH) — estudio multicéntrico en dos partes para evaluar la eficacia, seguridad, farmacocinética y farmacodinámica del medicamento Evrysdi® en pacientes con diagnóstico de AMC tipo 2 o 3, con edades comprendidas entre 2 y 25 años. La parte 1 fue una parte exploratoria de determinación de dosis y la parte 2 fue una parte confirmatoria aleatorizada, doble ciego y controlada con placebo. Los pacientes de la parte 1 del estudio no participaron en la parte 2.
El punto final primario se evaluó como el cambio en la puntuación de función motora (MFM32) a los 12 meses en comparación con el valor basal. El MFM32 permite evaluar un amplio rango de funciones motoras en una amplia variedad de pacientes con AMC. La puntuación total del MFM32 se expresa en porcentaje (rango: 0 a 100) del valor máximo posible, donde una puntuación más alta indica una mejor función motora. El MFM32 evalúa la capacidad de función motora relacionada con funciones diarias importantes. Pequeños cambios en la función motora pueden traducirse en mejoras o pérdidas significativas en la función diaria.
SUNFISH, parte 2
La parte 2 de SUNFISH es una parte aleatorizada, doble ciego y controlada con placebo, que incluyó a 180 pacientes no deambulantes con AMC tipo 2 (71 %) o tipo 3 (29 %). Los pacientes fueron aleatorizados en una proporción 2:1 para recibir el medicamento Evrysdi® en dosis terapéutica (véase la sección «Modo de administración y dosis») o placebo. La aleatorización se estratificó según la edad (de 2 a 5 años, de 6 a 11 años, de 12 a 17 años, de 18 a 25 años).
La edad media de los pacientes al inicio del tratamiento fue de 9 años (rango: 2–25 años), y la mediana del tiempo desde la aparición de los síntomas de AMC hasta el primer tratamiento fue de 102,6 (1–275) meses. Entre los participantes del estudio, el 51 % eran mujeres, el 67 % eran de raza caucásica y el 19 % eran de origen asiático. Al inicio del estudio, el 67 % de los pacientes tenían escoliosis (32 % de ellos con escoliosis grave). Los pacientes tenían una puntuación media basal del MFM32 de 46,1 y una puntuación RULM de 20,1. En general, las características demográficas basales fueron bien equilibradas entre los grupos que recibieron Evrysdi® y placebo, excepto por una diferencia en la proporción de pacientes con escoliosis (63,3 % en el grupo de Evrysdi® frente al 73,3 % en el grupo control con placebo).
Los resultados del análisis primario de la parte 2 del estudio SUNFISH sobre el cambio en la puntuación total del MFM32 respecto al valor basal a los 12 meses mostraron una diferencia clínicamente significativa y estadísticamente significativa entre los grupos de pacientes que recibieron tratamiento con Evrysdi® y placebo. Los resultados del análisis primario y los principales puntos finales secundarios se presentan en la Tabla 2 y en la Figura 1.
Tabla 2
Resumen de los resultados de eficacia en pacientes con AMC de inicio tardío tras 12 meses de tratamiento (parte 2 del estudio SUNFISH)
| Punto final |
Evrusdi ® (N = 120) |
Placebo (N = 60) |
| Punto final primario: |
||
| Cambio en el puntaje total MFM321 a los 12 meses respecto al valor basal, media cuadrática media (95 % IC) |
1,36 (0,61; 2,11) |
-0,19 (-1,22; 0,84) |
| Diferencia respecto al placebo (95 % IC), valor p2 |
1,55 (0,30; 2,81) 0,0156 |
|
| Puntos finales secundarios |
||
| Proporción de pacientes con un cambio en el puntaje total MFM321 de 3 o más a los 12 meses respecto al valor basal (95 % IC) |
38,3 % (28,9; 47,6) |
23,7 % (12,0; 35,4) |
| Relación de probabilidades de respuesta general (95 % IC), valor p ajustado (no ajustado)3,4 |
2,35 (1,01; 5,44) 0,0469 (0,0469) |
|
| Cambio en el puntaje total RULM5 a los 12 meses respecto al valor basal, media cuadrática media (95 % IC) |
1,61 (1,00; 2,22) |
0,02 (-0,8; 0,87) |
| Diferencia respecto al placebo (95 % IC), valor p ajustado (no ajustado)2,4 |
1,59 (0,55; 2,62) 0,0469 (0,0028) |
|
1 Sobre la base de la regla de datos faltantes respecto a MFM32, se excluyeron 6 pacientes del análisis (Evyresdi®, n = 115; grupo control con placebo, n = 59).
2 Los datos se analizaron mediante un modelo mixto con medidas repetidas, que incluyó como factores el valor basal total, el tratamiento, la visita, el grupo de edad, la interacción tratamiento por visita y la interacción basal por visita.
3 Los datos se analizaron mediante regresión logística con el valor basal total, el grupo de tratamiento y el grupo de edad como factores.
4 El valor p ajustado se obtuvo para los puntos finales incluidos en la prueba jerárquica y se calculó en base a todos los valores p de los puntos finales en el orden jerárquico hasta el punto final actual. El valor p sin ajustar se evaluó con un nivel de significación del 5 %.
5 Sobre la base de la regla de datos faltantes respecto a RULM, se excluyeron 3 pacientes del análisis (Evyresdi®, n = 119; grupo control con placebo, n = 58).
Después de completar 12 meses de tratamiento, 117 pacientes continuaron recibiendo Evyresdi®. En el momento del análisis a los 24 meses, los pacientes que recibieron tratamiento durante 24 meses mostraron una mejora adicional de la función motora durante el período comprendido entre los 12 y los 24 meses de terapia. El cambio medio respecto al valor basal en el MFM32 fue de 1,83 (IC: 0,74–2,92) y en el RULM de 2,79 (IC: 1,94–3,64).
| Cambio en el valor medio cuadrático medio del índice MFM32 total |
|
| Meses |
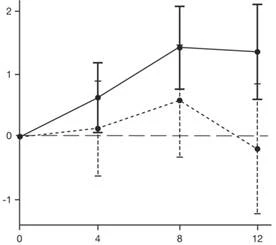
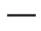
Evyrsdi® Placebo
* La barra de error representa un intervalo de confianza del 95 %.
† El puntaje total MFM se calculó según la guía del usuario, expresado como porcentaje del puntaje máximo posible en la escala (es decir, la suma de los puntos de los 32 ítems se dividió entre 96 y se multiplicó por 100).
Fig. 1. Cambio medio (LS) en el puntaje total MFM32 desde el valor basal a los 12 meses en la parte 2 del estudio SUNFISH.
SUNFISH, parte 1
La eficacia de Evyrsdi® en pacientes con AME de inicio tardío también se confirmó mediante los resultados de la parte 1 del estudio SUNFISH, un estudio de determinación de dosis. En la parte 1 se incluyeron 51 pacientes con AME tipos 2 y 3 (incluyendo 7 pacientes capaces de caminar) con edades entre 2 y 25 años. Tras un año de tratamiento con la dosis terapéutica (la dosis seleccionada para la parte 2), se observó una mejora clínicamente significativa en la función motora según los resultados del MFM32, con un cambio medio desde el valor basal de 2,7 puntos (IC del 95 %: 1,5; 3,8). La mejora en el MFM32 también se mantuvo durante un periodo de hasta 2 años de tratamiento con Evyrsdi® (cambio medio de 2,7 puntos (IC del 95 %: 1,2; 4,2)).
En un análisis comparativo, la función motora evaluada mediante el MFM se comparó entre la parte 1 del estudio SUNFISH y un control histórico con evolución natural de la enfermedad (ajustado por factores pronósticos clave). El cambio en el puntaje total MFM desde el valor basal a los 1 y 2 años fue mayor en los pacientes que recibieron Evyrsdi® en comparación con la cohorte de evolución natural (a 1 año: diferencia de 2,7 puntos; p < 0,0001; a 2 años: diferencia de 4 puntos; p < 0,0001). En la cohorte de evolución natural se observó una disminución de la función motora, como era esperado dada la progresión natural de la AME (cambio medio a 1 año: –0,6 puntos; a 2 años: –2 puntos).
AME presintomática
El estudio BN40703 (RAINBOWFISH) es un estudio unicéntrico, abierto y en curso, multicéntrico, para evaluar la eficacia, seguridad, farmacocinética y farmacodinámica de Evyrsdi® en recién nacidos desde el nacimiento hasta las 6 semanas de edad (al momento de recibir la primera dosis), con diagnóstico genético confirmado de AME, pero sin síntomas aún.
La eficacia del tratamiento con Evyrsdi® en AME presintomática se evaluó a los 12 meses en 26 pacientes [población de intención de tratar (ITT)]. La mediana de edad al recibir la primera dosis fue de 25 días (rango: 16 a 41 días), el 62 % eran mujeres y el 85 % eran de raza caucásica. Ocho, 13 y 5 pacientes tenían 2, 3 y ≥ 4 copias del gen SMN2, respectivamente. Al inicio del estudio, la mediana del puntaje CHOP-INTEND fue de 51,5 (rango: 35,0 a 62,0), la mediana del puntaje HINE-2 fue de 2,5 (rango: 0 a 6,0) y la mediana de la amplitud del potencial de acción del músculo del nervio cubital (CMAP) fue de 3,6 mV (rango: 0,5 a 6,7 mV).
La población primaria de análisis de eficacia (N = 5) incluyó pacientes con 2 copias del gen SMN2 y amplitud basal de CMAP ≥ 1,5 mV. En estos pacientes, la mediana del puntaje CHOP-INTEND fue de 48,0 (rango: 36,0–52,0), la mediana del puntaje HINE-2 fue de 2,0 (rango: 1,0 a 3,0) y la mediana de la amplitud de CMAP fue de 2,6 mV (rango: 1,6 a 3,8 mV) al inicio.
El punto final primario fue la proporción de pacientes en la población primaria de análisis de eficacia que fueron capaces de sentarse sin soporte durante al menos 5 segundos (escala BSID-III, ítem 22) a los 12 meses; una proporción estadísticamente significativa y clínicamente relevante alcanzó este hito en comparación con el criterio previamente establecido de eficacia del 5 %.
Los puntos finales clave de eficacia en pacientes tratados con Evyrsdi® se presentan en las tablas 3 y 4 y en la figura 2.
Tabla 3
Capacidad de sentarse según el ítem 22 de la escala BSID-III en pacientes presintomáticos a los 12 meses
| Punto final de evaluación de la eficacia |
Población de pacientes |
||
| Análisis primario de eficacia (N = 5) |
Pacientes con 2 copias del gen SMN2a (N = 8) |
ITT (N = 26) |
|
| Proporción de pacientes capaces de sentarse sin soporte durante al menos 5 segundos (escala BSID-III, ítem 22); (IC del 90 %) |
80 % (34,3 %; 99,0 %) |
87,5 % (52,9 %; 99,4 %) |
96,2 % (83,0 %; 99,8 %) |
Abreviaturas: BSID-III – Escalas de Desarrollo Bayley para Lactantes y Niños Pequeños, tercera edición; IC – intervalo de confianza; ITT – población con intención de tratar.
a Los pacientes con 2 copias del gen SMN2 tenían una amplitud media del PAMC de 2,0 (rango: 0,5–3,8) al inicio del estudio.
b El valor de p se basa en la prueba binomial exacta unilateral. El resultado se compara con un umbral del 5 %.
Además, el 80 % (4/5) de la población primaria de análisis de eficacia, el 87,5 % (7/8) de los pacientes con 2 copias del gen SMN2 y el 80,8 % (21/26) de los pacientes de la población ITT lograron la capacidad de sentarse sin soporte durante 30 segundos (BSID-III, ítem 26). Los pacientes de la población ITT también alcanzaron hitos motores medidos mediante el módulo HINE-2 en el mes 12 (N = 25). En esta población, el 96,0 % de los pacientes pudieron sentarse [1 paciente (1/8 pacientes con 2 copias del gen SMN2) logró sentarse establemente, y 23 pacientes (6/8, 13/13, 4/4 pacientes con 2, 3 y ≥ 4 copias del gen SMN2, respectivamente) pudieron girarse o voltearse]. Además, el 84 % de los pacientes pudieron ponerse de pie; el 32 % (N = 8) pudieron ponerse de pie con ayuda (3/8, 3/13 y 2/4 pacientes con 2, 3 y ≥ 4 copias del gen SMN2, respectivamente) y el 52 % (N = 13) pudieron ponerse de pie sin ayuda externa (1/8, 10/13 y 2/4 pacientes con 2, 3 y ≥ 4 copias del gen SMN2, respectivamente). Asimismo, el 72 % de los pacientes pudieron dar saltitos, caminar con o sin ayuda; el 8 % (N = 2) pudieron dar saltitos (2/8 pacientes con 2 copias del gen SMN2), el 16 % (N = 4) pudieron caminar con ayuda (3/13 y 1/4 pacientes con 3 y ≥ 4 copias del gen SMN2, respectivamente) y el 48 % (N = 12) pudieron caminar independientemente (1/8, 9/13 y 2/4 pacientes con 2, 3 y ≥ 4 copias del gen SMN2, respectivamente). Siete pacientes no fueron evaluados en el mes 12.
Tabla 4
Resumen de los puntos finales clave de eficacia en pacientes presintomáticos en el mes 12
| Puntos finales de eficacia |
Población ITT (N = 26) |
| Función motora |
|
| Proporción de pacientes que alcanzaron una puntuación total de 50 o más en la escala CHOP-INTEND (IC del 90 %) |
92 %a (76,9 %; 98,6 %) |
| Proporción de pacientes que alcanzaron una puntuación total de 60 o más en la escala CHOP-INTEND (IC del 90 %) |
80 %a (62,5 %; 91,8 %) |
| Alimentación |
|
| Proporción de pacientes capaces de alimentarse por vía oral (IC del 90 %) |
96,2 %b (83,0 %; 99,8 %) |
| Uso de los recursos del sistema sanitario |
|
| Proporción de pacientes que no requirieron hospitalización (IC del 90 %) |
92,3 % (77,7 %; 98,6 %) |
| Supervivencia libre de eventosd Proporción de pacientes con supervivencia libre de eventos (IC del 90 %) |
100 % (100 %; 100 %) |
Abreviaturas: CHOP INTEND – prueba del Hospital Infantil de Filadelfia para evaluar las funciones motoras en enfermedades neuromusculares en lactantes; IC – intervalo de confianza; ITT – población con intención de tratar.
a Basado en un número N = 25.
b La evaluación no se realizó en un paciente.
c Las hospitalizaciones incluían todos los casos de estancia hospitalaria de al menos dos días que no fueran debidos a requisitos del estudio.
d El evento en este caso significa muerte o necesidad de ventilación permanente; la ventilación permanente se define como traqueostomía o ≥ 16 horas de ventilación no invasiva al día, o intubación durante > 21 días consecutivos en ausencia o tras la desaparición de un episodio agudo reversible.
Abreviaturas: IQR – rango intercuartílico; SMN2 – gen de supervivencia de motoneuronas 2.
Fig. 2. Mediana de las puntuaciones totales CHOP-INTEND según visitas y número de copias del gen SMN2 (población ITT)
Uso en pacientes con AME que previamente recibieron otros tratamientos modificadores
Estudio BP39054 (JEWELFISH): estudio abierto, de un solo grupo, para evaluar la seguridad, tolerabilidad, farmacocinética y farmacodinámica del medicamento Evrysdi® en pacientes de 6 meses a 60 años de edad con AME infantil o con inicio tardío que previamente habían recibido tratamiento para la AME (incluyendo nusinersén y onasemnogén abeparvovec). De los 173 pacientes que recibieron Evrysdi®, 76 habían recibido previamente nusinersén (9 pacientes con AME tipo 1, 43 pacientes con AME tipo 2 y 24 pacientes con AME tipo 3) y 14 habían recibido previamente onasemnogén abeparvovec (4 pacientes con AME tipo 1 y 10 pacientes con AME tipo 2). La edad media de los pacientes al inicio del tratamiento con Evrysdi® fue de 14 años (rango de 1 a 60 años).
Al momento de la inclusión en el estudio, del grupo de 168 pacientes con edades entre 2 y 60 años, el 83 % presentaba escoliosis (39 % escoliosis grave) y el 63 % tenía una puntuación en la Escala Expandida de Evaluación de la Función Motora de Hammersmith (HFMSE) < 10 puntos. También se incluyeron en el estudio 15 pacientes capaces de caminar de forma independiente (de 5 a 46 años de edad).
Los resultados exploratorios de eficacia se evaluaron mediante la medición de la función motora según la edad, incluyendo escalas MFM-32 y RULM para pacientes de 2 a 60 años, BSID-III y HINE-2 para pacientes menores de 2 años y la prueba de la marcha de 6 minutos (6MWT) para pacientes ≥ 6 años capaces de caminar de forma independiente. Tras el análisis primario a los 24 meses de tratamiento, en pacientes de 2 a 60 años se observó una estabilización general de la función motora según las escalas MFM-32 y RULM (n = 137 y n = 133, respectivamente). Los pacientes menores de 2 años (n = 6) mantuvieron o mejoraron el desarrollo de la función motora, especialmente en la retención de la cabeza, volteo y sentarse sin apoyo. Los resultados de la 6MWT mostraron una mejora media de 30,88 metros (IC 95 %: -5,54; 67,29, n = 8). Todos los pacientes capaces de caminar de forma independiente mantuvieron esta capacidad.
Farmacocinética.
Los parámetros farmacocinéticos de risdiplam fueron caracterizados en adultos sanos y en pacientes con AME.
Tras la administración oral del medicamento Evrysdi® en dosis de 0,6 a 18 mg, la farmacocinética de risdiplam fue aproximadamente lineal. La farmacocinética de risdiplam fue mejor descrita mediante un modelo poblacional de farmacocinética con absorción mediante tránsito trifásico, distribución bicompartimental y eliminación de primer orden. Se determinó que la masa corporal y la edad del paciente tienen un impacto significativo en la farmacocinética del medicamento.
La exposición calculada (AUC0–24h media) en pacientes con AME infantil (de 2 a 7 meses al momento de la inclusión en el estudio) con la dosis recomendada de 0,2 mg/kg una vez al día fue de 1930 ng·h/mL. La exposición media calculada en lactantes de 16 días a < 2 meses con AME presintomática en el estudio RAINBOWFISH fue de 2020 ng·h/mL tras 2 semanas de administración diaria a una dosis de 0,15 mg/kg.
La exposición calculada en pacientes con AME de inicio tardío (de 2 a 25 años al momento de la inclusión en el estudio) en el estudio SUNFISH (parte 2) con la dosis terapéutica (0,25 mg/kg una vez al día para pacientes con peso corporal < 20 kg; 5 mg una vez al día para pacientes con peso corporal ≥ 20 kg) fue de 2010 ng·h/mL. La concentración máxima observada (Cmax media) fue de 194 ng/mL con una dosis de 0,2 mg/kg en el estudio FIREFISH y de 120 ng/mL en la parte 2 del estudio SUNFISH. La concentración máxima media calculada con una dosis de 0,15 mg/kg en el estudio RAINBOWFISH fue de 111 ng/mL.
Absorción
Risdiplam se absorbe rápidamente tras la administración oral en ayunas, con un tmax en plasma entre 1 y 4 horas. En los estudios clínicos, risdiplam se administró por la mañana junto con la comida o tras la alimentación con leche materna.
Disposición
Los parámetros farmacocinéticos poblacionales estimados fueron: 98 L – volumen de distribución central aparente, 93 L – volumen periférico y 0,68 L/hora – aclaramiento intercompartimental.
Risdiplam se une principalmente a la albúmina sérica, sin unión a la glicoproteína ácida alfa-1, con una fracción libre del 11 %.
Metabolismo
Risdiplam se metaboliza principalmente mediante las monooxigenasas flavin-dependientes 1 y 3 (FMO1 y FMO3), así como por las isoformas CYP 1A1, 2J2, 3A4 y 3A7. El fármaco original fue el componente principal detectado en plasma, representando el 83 % de la sustancia asociada al fármaco en circulación. El metabolito farmacológicamente inactivo M1 fue identificado como el principal metabolito circulante.
Eliminación
El parámetro poblacional de farmacocinética del aclaramiento aparente (CL/F) de risdiplam fue de 2,6 L/hora. La semivida efectiva de risdiplam fue de aproximadamente 50 horas en pacientes con AME.
Aproximadamente el 53 % de la dosis (14 % como risdiplam inalterado) se excretó por heces y el 28 % por orina (8 % como risdiplam inalterado).
Grupos de pacientes especiales
Alteración de la función hepática
La alteración hepática de grado leve o moderado no tuvo impacto en la FC de risdiplam. Tras la administración de 5 mg de risdiplam, las relaciones medias de Cmax y AUC fueron 0,95 y 0,80 en sujetos con alteración leve (n = 8) y 1,20 y 1,08 en pacientes con alteración moderada (n = 8), en comparación con pacientes con función hepática normal (grupo control, n = 10). La seguridad y la FC en pacientes con alteración hepática grave no han sido estudiadas hasta la fecha.
Alteración de la función renal
No se han realizado estudios de FC de risdiplam en pacientes con alteración de la función renal. La excreción renal de risdiplam en forma inalterada es mínima (8 %).
Pacientes de edad avanzada
No se han realizado estudios especiales de farmacocinética de Evrysdi® en pacientes con AME mayores de 60 años. Pacientes con AME hasta 60 años fueron incluidos en el estudio JEWELFISH. Pacientes sin AME hasta 69 años fueron incluidos en estudios clínicos de FC.
Pacientes pediátricos
El peso corporal y la edad del paciente fueron identificados como covariables en el análisis poblacional de FC. Por ello, la dosis se ajusta según la edad (menor o mayor de 2 meses y 2 años) y el peso corporal (hasta 20 kg) con el fin de lograr una exposición similar en diferentes grupos de edad y categorías de peso. No existen datos de farmacocinética disponibles para lactantes menores de 16 días.
Etnia
La farmacocinética de risdiplam no difiere entre pacientes japoneses y de raza caucásica.
Características clínicas.
Indicaciones.
Tratamiento de la atrofia muscular espinal (AME) asociada al 5q en pacientes pediátricos y adultos.
Contraindicaciones.
Hipersensibilidad conocida al risdiplam o a cualquiera de los excipientes mencionados en la sección «Composición».
Interacción con otros medicamentos y otras formas de interacción.
Efecto de Evrysdi® sobre otros medicamentos
In vitro, el risdiplam y su metabolito circulante principal M1 no indujeron las isoformas CYP1A2, 2B6, 2C8, 2C9, 2C19 y 3A4. In vitro, el risdiplam y M1 no inhibieron (inhibición reversible ni dependiente del tiempo) ninguna de las isoformas CYP estudiadas (CYP1A2, 2B6, 2C8, 2C9, 2C19, 2D6), excepto CYP3A.
Evrysdi® es un inhibidor débil de CYP3A. En adultos sanos, la administración de Evrysdi® una vez al día durante 2 semanas aumentó ligeramente la exposición al midazolam, un sustrato altamente sensible de CYP3A (AUC 11 %; Cmax 16 %). Esta interacción no se considera clínicamente significativa, por lo que no es necesario ajustar la dosis de los sustratos de CYP3A.
Debido a los resultados obtenidos con un modelo farmacocinético basado en la fisiología (PBPK), se espera un efecto similar en niños y lactantes a partir de los 2 meses de edad.
Los estudios in vitro demostraron que el risdiplam y su metabolito principal no son inhibidores significativos del transportador MDR1 humano, del polipéptido transportador de aniones orgánicos (OATP)1B1, OATP1B3, ni de los transportadores de aniones orgánicos 1 y 3 (OAT1 y OAT3). Sin embargo, el risdiplam y su metabolito principal son, in vitro, inhibidores del polipéptido transportador de cationes orgánicos 2 (OCT2) humano y de los transportadores de proteínas de resistencia múltiple y de excreción de toxinas (MATE)1 y MATE2-K. Cuando se utiliza en concentraciones terapéuticas, no se espera una interacción del medicamento con los sustratos de OCT2. El impacto del uso concomitante de risdiplam sobre la farmacocinética de los sustratos de MATE1 y MATE2-K en humanos no es conocido. Según datos in vitro, Evrysdi® podría aumentar la concentración plasmática de principios activos que se eliminan mediante MATE1 o MATE2-K, como por ejemplo la metformina (ver sección «Farmacocinética»). Si no es posible evitar la administración concomitante, se debe realizar un monitoreo de la toxicidad relacionada con el medicamento y considerar la posibilidad de reducir la dosis del otro medicamento administrado simultáneamente, si fuera necesario.
Efecto de otros medicamentos sobre Evrysdi®
El risdiplam se metaboliza principalmente por las flavin-monooxigenasas 1 y 3 (FMO1 y FMO3), así como por las CYP1A1, 2J2, 3A4 y 3A7. El risdiplam no es sustrato de la proteína de resistencia múltiple 1 humana (MDR1).
Cuando se administró concomitantemente itraconazol, un inhibidor potente de CYP3A, a una dosis de 200 mg dos veces al día, junto con risdiplam a una dosis única oral de 6 mg, no se observó un efecto clínicamente significativo sobre la farmacocinética (FK) del risdiplam (aumento del AUC en 11 %, disminución del Cmax en 9 %). No es necesario ajustar la dosis de Evrysdi® cuando se administra junto con un inhibidor de CYP3A.
No se espera interacción con otros medicamentos a través de las vías de señalización mediadas por FMO1 y FMO3.
Características de uso.
Información general
En estudios realizados en animales se observaron alteraciones en la retina, en el epitelio, especialmente en la piel y en el tracto gastrointestinal, así como signos de toxicidad en la médula ósea (cambios en el análisis sanguíneo clínico). Actualmente, el riesgo de tales alteraciones en humanos no puede evaluarse definitivamente debido a la limitada disponibilidad de datos de seguridad a largo plazo.
Toxicidad embrio-fetal
En estudios realizados en animales se observó toxicidad embrio-fetal. Se debe informar a los pacientes en edad reproductiva sobre los riesgos. Debe utilizarse un método anticonceptivo de alta eficacia durante el tratamiento y al menos durante 1 mes después de la administración de la última dosis de Evrysdi® en mujeres, y durante al menos 4 meses después de la administración de la última dosis de Evrysdi® en hombres (ver sección «Instrucciones de uso y dosis»).
Posible efecto sobre la fertilidad en hombres
Debido a los efectos reversibles del medicamento Evrysdi® sobre la fertilidad masculina observados en estudios en animales, los pacientes hombres no deben donar esperma durante el tratamiento ni durante los 4 meses posteriores a la administración de la última dosis de Evrysdi® (ver sección «Farmacocinética»).
Debe evitarse el contacto del polvo y de la solución oral reconstituida con la piel. Si el medicamento (polvo o solución) entra en contacto con la piel, la zona afectada debe lavarse inmediatamente con agua y jabón.
Sustancias auxiliares
Este medicamento contiene 0,38 mg de benzoato de sodio por 1 ml. Este medicamento contiene menos de 1 mmol de sodio (23 mg) por dosis, por lo tanto se considera prácticamente libre de sodio.
Este medicamento contiene isomalta. No debe administrarse a pacientes con intolerancia hereditaria rara a la fructosa.
Uso durante el embarazo o la lactancia
Debido a los resultados de estudios preclínicos, la fertilidad en hombres puede verse afectada durante el tratamiento con Evrysdi®. En órganos reproductivos de ratas y monos se observaron degeneración espermática y reducción del número de espermatozoides.
Antes de iniciar el tratamiento con Evrysdi®, se debe discutir con los pacientes de sexo masculino estrategias para preservar la fertilidad. Los pacientes de sexo masculino podrían considerar la posibilidad de preservar su esperma antes del inicio del tratamiento o tras un período sin tratamiento de al menos 4 meses (ver sección «Características de uso»).
Debido a los resultados de estudios preclínicos, no se espera que Evrysdi® tenga un efecto sobre la fertilidad en mujeres.
Las mujeres en edad fértil deben ser evaluadas para descartar embarazo antes de iniciar el tratamiento con Evrysdi®.
Los pacientes de sexo masculino y femenino en edad reproductiva deben cumplir con los siguientes requisitos en cuanto a anticoncepción:
- Los pacientes de sexo masculino y sus parejas en edad fértil deben utilizar un método anticonceptivo de alta eficacia durante el tratamiento con Evrysdi® y al menos durante 4 meses después de la administración de la última dosis del medicamento.
- Las pacientes de sexo femenino en edad fértil deben utilizar un método anticonceptivo de alta eficacia durante el tratamiento con Evrysdi® y al menos durante 1 mes después de la administración de la última dosis del medicamento.
Embarazo
No existen datos clínicos sobre el uso de Evrysdi® en mujeres embarazadas. Risdiplam ha demostrado tener efectos embrio-fetotóxicos y teratogénicos en animales. Debido a los datos obtenidos en estudios en animales, risdiplam atraviesa la barrera placentaria y puede causar daño fetal.
Evrysdi® no debe administrarse durante el embarazo a menos que sea estrictamente necesario. Si una mujer embarazada requiere tratamiento con Evrysdi®, debe informársele claramente sobre el riesgo potencial para el feto.
Lactancia
No se sabe si Evrysdi® se excreta en la leche materna humana. Estudios en ratas han demostrado que risdiplam se excreta en la leche materna. Dado que el potencial de daño para el lactante es desconocido, el médico tratante deberá tomar una decisión sobre la continuación del tratamiento del paciente. No se recomienda la lactancia durante el tratamiento con Evrysdi®.
Capacidad para afectar la rapidez de reacción al conducir vehículos o manejar maquinaria
No se han realizado estudios específicos para evaluar el efecto de Evrysdi® sobre la capacidad de reacción al conducir vehículos o manejar maquinaria.
Vía de administración y dosis.
La solución oral del medicamento Evrysdi® debe ser preparada por un profesional médico (es decir, un médico o farmacéutico) antes de entregársela al paciente.
Antes de la administración de la primera dosis, el profesional sanitario debe discutir con el paciente o con la persona encargada de su cuidado cómo preparar y tomar la dosis diaria prescrita (ver más abajo «Instrucciones de manipulación»).
El tratamiento con Evrysdi® debe ser iniciado y supervisado por médicos con experiencia en el diagnóstico y tratamiento de pacientes con atrofia muscular espinal.
El programa de desarrollo clínico no incluyó pacientes con AME tipo IV.
Dosificación recomendada
El medicamento Evrysdi® se administra por vía oral una vez al día, aproximadamente a la misma hora cada día, utilizando las jeringas orales reutilizables proporcionadas en el envase. La dosis recomendada de Evrysdi® para el tratamiento de la AME se determina según la edad y el peso corporal del paciente (ver tabla 5).
Tabla 5
Régimen de dosificación según edad y peso corporal
| Edad y peso corporal |
Dosis diaria recomendada |
| < 2 meses |
0,15 mg/kg |
| de 2 meses a < 2 años |
0,20 mg/kg |
| ≥ 2 años y peso corporal < 20 kg |
0,25 mg/kg |
| ≥ 2 años y peso corporal ≥ 20 kg |
5 mg |
a En base a la edad corregida para neonatos prematuros.
La modificación de la dosis debe realizarse bajo supervisión médica. Hasta la fecha, no se ha estudiado el tratamiento con una dosis diaria superior a 5 mg. Solo existen datos muy limitados sobre la seguridad obtenidos durante el período poscomercialización en recién nacidos menores de 16 días de edad que recibieron el medicamento Evrysdi® en la dosis recomendada (ver secciones «Farmacocinética», «Pediátricos»).
Pacientes con alteración de la función hepática
No se requiere ajuste de la dosis en pacientes con alteración hepática leve o moderada. No se ha estudiado la administración del medicamento Evrysdi® en pacientes con alteración hepática grave (ver sección «Farmacocinética»).
Pacientes con alteración de la función renal
No se han estudiado la seguridad y eficacia del medicamento Evrysdi® en pacientes con alteración de la función renal. No se espera que sea necesaria la corrección de la dosis en pacientes con alteración de la función renal (ver sección «Farmacocinética»).
Pacientes de edad avanzada
Los estudios clínicos con el medicamento Evrysdi® no incluyeron pacientes de 65 años o más, por lo que no se ha determinado si podrían responder de manera diferente al tratamiento en comparación con pacientes más jóvenes.
Pediátricos
El uso del medicamento Evrysdi® en pacientes pediátricos con AME desde los 2 meses de edad está respaldado por datos de farmacocinética y seguridad en niños desde los 16 días de edad (ver secciones «Reacciones adversas», y los apartados «Eficacia clínica» y «Farmacocinética»). No existen datos clínicos ni farmacocinéticos sobre el uso del medicamento en recién nacidos prematuros o neonatos menores de 16 días de edad (ver el apartado «Eficacia clínica» anterior).
Olvido de la dosis
El medicamento Evrysdi® se toma por vía oral una vez al día, aproximadamente a la misma hora todos los días. Si se olvida tomar la dosis de Evrysdi®, debe tomarse tan pronto como sea posible, siempre que la demora no supere las 6 horas respecto al horario programado, y al día siguiente se puede reanudar el régimen habitual de dosificación. En caso contrario, no debe tomarse la dosis olvidada y se debe tomar la siguiente dosis en el momento programado del día siguiente.
Si la dosis no se traga completamente o si se produce vómito tras la ingestión del medicamento Evrysdi®, no debe tomarse otra dosis para compensar la dosis perdida. Debe esperarse hasta el día siguiente para tomar la siguiente dosis en el momento programado.
Vía de administración
Para la administración de la dosis diaria del medicamento Evrysdi®, debe utilizarse la jeringa oral reutilizable suministrada en el envase de cartón junto con el medicamento (ver tabla 6).
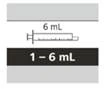
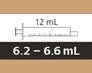
Tabla 6
Selección de la jeringa oral reutilizable adecuada para la administración de la dosis diaria prescrita de Evrysdi®
| Tamaño de la jeringa |
Volumen de dosificación |
Precisión de división de la jeringa |
| 1 ml |
de 0,3 a 1 ml |
0,01 ml |
| 6 ml |
de 1 a 6 ml |
0,1 ml |
| 12 ml |
de 6,2 a 6,6 ml |
0,2 ml |
Para calcular el volumen de la dosis, también debe tenerse en cuenta el valor de la división de la jeringa. Redondee el volumen de la dosis hacia arriba o hacia abajo al valor más cercano marcado en la jeringa oral seleccionada (por ejemplo, de 6,3 ml a 6,4 ml, de 3,03 ml a 3 ml y de 1,05 a 1,1 ml).
El medicamento Evrysdi® en forma de solución debe administrarse inmediatamente después de aspirarlo en la jeringa oral para uso múltiple. Si el contenido de la jeringa no se administra dentro de los 5 minutos siguientes, debe vaciarse la jeringa oral del medicamento (véase más adelante «Eliminación del medicamento no utilizado/medicamento caducado») y debe prepararse una nueva dosis.
El medicamento Evrysdi® debe tomarse después de las comidas. El paciente debe beber agua después de tomar Evrysdi® para asegurarse de que el medicamento se ha tragado completamente. Si el paciente no puede tragar y tiene una sonda nasogástrica o una sonda de gastrostomía colocada, Evrysdi® puede administrarse a través de la sonda/tubo. Tras la administración del medicamento, la sonda/tubo debe enjuagarse con agua (véase más adelante «Instrucciones de manipulación»).
Instrucciones de manipulación
Instrucciones que deben seguirse antes, durante y después de la preparación de la solución oral:
- La solución siempre debe prepararla un profesional sanitario (es decir, un médico o un farmacéutico).
- Debe evitarse la inhalación del polvo del medicamento Evrysdi®. Tenga en cuenta las normas locales y utilice el equipo adecuado para la preparación de la solución de Evrysdi®.
- Lleve guantes.
- No utilice el polvo si ha caducado. La fecha de caducidad del polvo está impresa en la etiqueta del frasco y en la caja de cartón.
- No distribuya la solución reconstituida si la fecha de caducidad de la solución oral lista para usar —indicada en la etiqueta del frasco y en la caja de cartón— supera la fecha de caducidad original del polvo.
- Evite el contacto del medicamento con la piel. Si el medicamento (polvo o solución) entra en contacto con la piel, lávese la zona con agua y jabón.
- No utilice el medicamento si el contenido del envase está dañado o falta alguna parte.
- Para la preparación de la solución, utilice agua purificada o agua para inyección.
- No utilice jeringas orales distintas de las incluidas en la caja de cartón junto con el medicamento.
- No mezcle Evrysdi® con alimentos ni con otros líquidos (por ejemplo, leche o fórmula láctea).
- No mezcle Evrysdi® de un frasco nuevo con el contenido de un frasco que esté utilizando actualmente.
El paciente o la persona encargada de su cuidado deben recibir instrucciones de un profesional sanitario sobre cómo preparar y tomar la dosis diaria prescrita antes de que se entregue la solución preparada.
Preparación de la solución oral
Añada 79 ml de agua purificada o agua para inyección al frasco con el medicamento.
Introduzca el adaptador de perforación en la abertura del frasco, empujándolo hacia abajo.
Una vez que el frasco esté completamente cerrado, agítelo durante 15 segundos.
Al cabo de 10 minutos, se obtendrá una solución transparente. Si la solución no se ha vuelto transparente, agítela durante otros 15 segundos.
Debe calcularse un periodo de 64 días a partir de la fecha de preparación de la solución. El día de la preparación se considera el día 0. La fecha calculada debe anotarse en la etiqueta del frasco y en la caja de cartón en el campo: SOLUCIÓN ORAL RECONSTITUIDA DEBE DISTRIBUIRSE ANTES DEL: (día/mes/año).
Eliminación del medicamento no utilizado/medicamento caducado
La entrada de medicamentos en el medio ambiente debe reducirse al mínimo. No elimine los medicamentos por las aguas residuales ni mediante la eliminación con la basura doméstica.
El medicamento no utilizado o caducado debe eliminarse de forma profesional en el lugar de dispensación del medicamento (por el médico o el farmacéutico).
Instrucciones para la preparación de la solución (SOLO PARA PROFESIONALES SANITARIOS, ESPECIALMENTE MÉDICOS O FARMACÉUTICOS)
La solución oral de Evrysdi® debe prepararse por un profesional sanitario antes de entregársela al paciente.
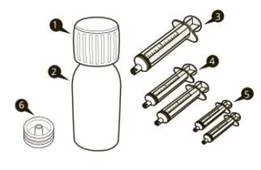
| Un envase del medicamento Evrysdi® contiene (véase la figura A):
|
Figura A |
Información importante sobre el medicamento Evrysdi®
- La solución siempre debe prepararse por un profesional sanitario (médico o farmacéutico).
- Evitar inhalar el polvo del medicamento Evrysdi®. Cumplir con los requisitos locales y utilizar el equipo adecuado para la preparación de la solución de Evrysdi®.
- Usar guantes.
- No utilizar este medicamento después de la fecha de caducidad. La fecha de caducidad se indica en la etiqueta del frasco y en la caja de cartón.
- Evitar cualquier contacto de este medicamento con la piel. Si ocurre contacto (con el polvo o con la solución), lavar inmediatamente la zona afectada con agua y jabón.
- No utilizar este medicamento si alguno de los componentes del envase está dañado o falta.
- Para la preparación de la solución, utilizar agua purificada o agua para preparaciones inyectables.
- No utilizar jeringas orales reutilizables, salvo aquellas suministradas en el envase.
- No dispensar la solución preparada si la fecha de caducidad de la solución oral lista para usar —indicada en la etiqueta del frasco y en la caja de cartón— excede la fecha de caducidad del polvo.
- Antes de dispensar la solución preparada, el profesional sanitario debe instruir al paciente o cuidador sobre cómo preparar y tomar la dosis diaria.
Cómo conservar el medicamento Evrysdi®
- Conservar el polvo (medicamento aún no preparado como solución) en su caja de cartón a temperatura ambiente no superior a 25 °C, para protegerlo de la luz y la humedad.
- Conservar la solución (medicamento ya preparado como solución) en posición vertical, en el refrigerador a una temperatura de 2–8 °C, dentro de la caja de cartón para proteger su contenido de la luz.
- Si fuera necesario, la solución oral puede mantenerse a temperatura ambiente (por debajo de 40 °C) durante un período acumulado no superior a 5 días. No conservar la solución oral a temperaturas superiores a 40 °C.
- Conservar la solución oral en el frasco original en posición vertical y con la tapa firmemente cerrada.
Preparación de la solución
|
Figura B |
Paso 1 Golpee suavemente el fondo del frasco para aflojar el polvo (véase la Figura B). |
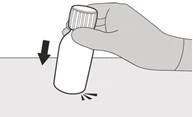
|
Figura C |
Paso 2 Retire la tapa presionándola hacia abajo y luego girando hacia la izquierda (en sentido antihorario) (ver Figura C). No deseche la tapa. |
|
Figura D |
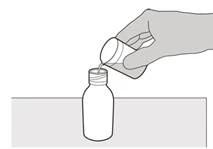
Paso 3 Agregue cuidadosamente 79 ml de agua purificada o agua para inyección al frasco con el medicamento (ver Figura D). |
|
Figura E |
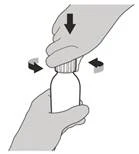
Paso 4 Sujete el frasco con el medicamento con una mano sobre una superficie plana. Inserte el adaptador de perforación en la abertura del frasco, presionando hacia abajo con la otra mano hasta que el adaptador esté completamente asentado en el borde de la abertura del frasco (ver Figura E). |
|
Figura F |
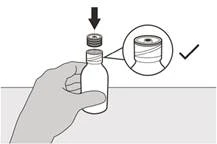
Paso 5 Vuelva a colocar la tapa sobre el frasco. Gire la tapa hacia la derecha (en sentido horario) para cerrar el frasco. Asegúrese de que el frasco esté completamente cerrado y agite bien durante 15 segundos (ver Figura F). |
|
Figura G |
Paso 6 Determine la fecha de «DESPUÉS DE ESTA FECHA, DÉJELO DE USAR: (día/mes/año)», contando 64 días a partir de la fecha de preparación de la solución (nota: el día de preparación se considera el día 0. Por ejemplo, si preparó la solución el 1 de abril, la fecha de «DESPUÉS DE ESTA FECHA, DÉJELO DE USAR» será el 4 de junio). Anote esta fecha en el campo correspondiente de la etiqueta del frasco (ver Figura G) y en la caja de cartón: Solución oral reconstituida, DÉJELO DE USAR DESPUÉS DE: (día/mes/año). Guarde el frasco nuevamente en la caja de cartón junto con las jeringas (en sus sobres) y el prospecto. Guarde todo en el refrigerador, manteniendo el frasco en posición vertical. |
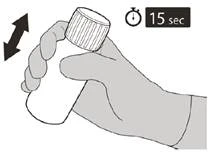
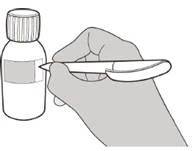
INSTRUCCIONES PARA EL USO DE LA SOLUCIÓN ORAL
Debe leer y comprender las instrucciones para la administración médica antes de comenzar a usar Euresid®. Estas instrucciones contienen información sobre cómo preparar y administrar la dosis de Euresid® utilizando una jeringa oral reutilizable, por vía oral, a través de una sonda de gastrostomía (sonda PEG) o una sonda nasogástrica. Si tiene alguna pregunta sobre el uso de Euresid®, consulte a su médico o farmacéutico. Antes de dispensarse, la solución oral de Euresid® debe ser preparada por un profesional sanitario, como un médico o un farmacéutico.
Usted recibirá Euresid® en forma de líquido en un frasco. No utilice el medicamento si en el frasco se encuentra en forma de polvo; en tal caso, comuníqueselo a su médico o farmacéutico.
Antes de administrar o tomar la primera dosis, un profesional sanitario debe instruirle sobre cómo preparar y administrar la dosis diaria.
- Pida a su médico o farmacéutico que le muestre cuál de las jeringas orales incluidas en el envase debe utilizar y cómo determinar la dosis diaria.
- Si se ha perdido o dañado la jeringa oral necesaria, comuníqueselo a su médico o farmacéutico. Ellos le indicarán cómo continuar con el tratamiento.
- Para medir la dosis diaria prescrita, utilice siempre la jeringa del tamaño adecuado, que puede reutilizarse.
- La descripción de la jeringa oral reutilizable adecuada para usted también se encuentra en la tabla 6: «Selección de la jeringa oral reutilizable adecuada para la dosis diaria prescrita de Euresid®». Consulte a su médico o farmacéutico si tiene dudas sobre cómo elegir correctamente la jeringa.
- No utilice Euresid® si no encuentra el adaptador para el frasco dentro del envase. En tal caso, comuníqueselo a su médico o farmacéutico.
- No utilice Euresid® después de la fecha indicada como «Solución oral reconstituida DESCARTAR DESPUÉS DEL: (día/mes/año)» en la etiqueta del frasco y en la caja de cartón. Si en la etiqueta del frasco o en la caja de cartón no aparece la fecha «Solución oral reconstituida DESCARTAR DESPUÉS DEL: (día/mes/año)», consulte dicha fecha a su médico o farmacéutico.
- No mezcle Euresid® con alimentos ni con otros líquidos (por ejemplo, leche o fórmula láctea).
- No utilice Euresid® si el frasco o la jeringa oral están dañados.
- Evite el contacto de Euresid® con la piel. Si Euresid® entra en contacto con su piel, lávese la zona con agua y jabón.
- Si derrama Euresid®, limpie la zona con una toalla de papel seca y luego lávela con agua y jabón. Deseche la toalla de papel en la basura y lávese bien las manos con agua y jabón.
- Si no hay suficiente cantidad de Euresid® en el frasco para la dosis prescrita, devuelva el frasco y las jeringas orales usadas al lugar de dispensación (médico o farmacéutico) para su correcta eliminación. Use un nuevo frasco de Euresid® para extraer la dosis diaria prescrita. No mezcle Euresid® del nuevo frasco con el medicamento del frasco previamente utilizado.
Cada caja de Euresid® contiene (ver figura A):
|
|
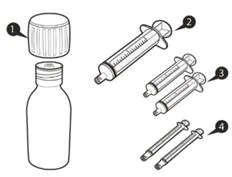
Figura A
A) Preparación y medición del volumen de dosis correcto
Cómo elegir la jeringa oral adecuada para la dosis prescrita del medicamento Eurisdí®
|
|
|
|
|
|

Cómo preparar la dosis diaria del medicamento Evrysdi®
|
Figura B |
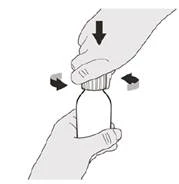
Paso A1 Presione la tapa hacia abajo y luego gírela hacia la izquierda (en sentido antihorario) para retirarla (véase la figura B). No deseche la tapa. |
|||||
|
Figura C |
Paso A2 Presione completamente hacia abajo el émbolo de la jeringa oral para expulsar todo el aire de la jeringa (véase la figura C). |
|||||
|
Figura D |
Paso A3 Sosteniendo el frasco en posición vertical, inserte la boquilla de la jeringa en el adaptador del frasco (véase la figura D). |
|||||
|
Figura E |
Paso A4 Gire cuidadosamente el frasco boca abajo, manteniendo firmemente insertada la boquilla de la jeringa en el adaptador del frasco (véase la figura E). |
|||||
|
Figura F |
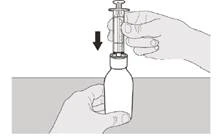
Paso A5 Extraiga lentamente el émbolo para aspirar la dosis diaria recetada del medicamento Eureisdí® del frasco. Debe alinear la parte superior del tope negro del émbolo con la marca de mililitros en la jeringa oral que corresponda a su dosis diaria recetada (véase la figura F). Una vez extraída la dosis correcta, sostenga firmemente el émbolo para evitar que se mueva. |
|||||
|
Figura G |
Paso A6 Continúe sosteniendo el émbolo sin moverlo. Deje la jeringa oral en el adaptador del frasco y coloque el frasco en posición vertical sobre una superficie plana. Retire cuidadosamente la jeringa del adaptador del frasco tirando suavemente hacia arriba, manteniendo el émbolo inmóvil (véase la figura G). |
|||||
|
Figura H |
Paso A7 Sostenga la jeringa oral con la punta orientada hacia arriba. Revise el medicamento Eureisdí® en la jeringa. Si observa grandes burbujas de aire en la jeringa oral (véase la figura H) o si ha extraído una dosis diaria incorrecta del medicamento Eureisdí®, inserte firmemente la boquilla de la jeringa en el adaptador del frasco. Presione el émbolo hacia abajo para devolver el medicamento Eureisdí® al frasco y repita los pasos A4–A7. Administre o tome el medicamento Eureisdí® inmediatamente después de cargarlo en la jeringa oral. Si el medicamento no se administra dentro de los 5 minutos, deseche el medicamento Eureisdí® de la jeringa oral y prepare una nueva dosis. |
|||||
|
Figura I |
Paso A8 Deje el adaptador del frasco colocado en el frasco. Vuelva a colocar la tapa sobre el frasco. Gire la tapa hacia la derecha (en sentido horario) para cerrar herméticamente el frasco (véase la figura I). |
|||||
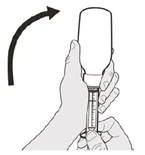
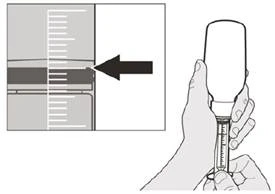
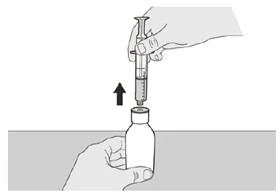
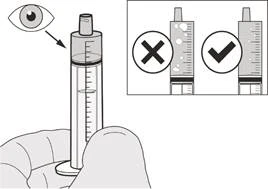
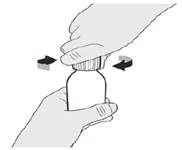
Si toma la dosis diaria del medicamento Evrysdi® por vía oral, siga las instrucciones en la sección «B) Cómo tomar la dosis del medicamento Evrysdi® por vía oral».
Si toma la dosis diaria del medicamento Evrysdi® a través de una sonda de gastrostomía, siga las instrucciones en la sección «C) Cómo administrar la dosis del medicamento Evrysdi® a través de una sonda de gastrostomía».
Si toma la dosis diaria del medicamento Evrysdi® a través de una sonda nasogástrica, siga las instrucciones en la sección «D) Cómo administrar la dosis del medicamento Evrysdi® a través de una sonda nasogástrica».
Las jeringas orales para el medicamento Evrysdi® han sido diseñadas especialmente para ser compatibles con el sistema ENFit®. Si la sonda de alimentación disponible no es compatible con el sistema ENFit®, podría necesitar un adaptador transicional ENFit para conectar la jeringa del medicamento Evrysdi® a la sonda de gastrostomía o a la sonda nasogástrica.
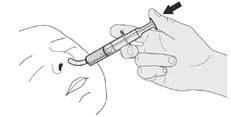
B) Cómo tomar la dosis diaria del medicamento Evrysdi® por vía oral
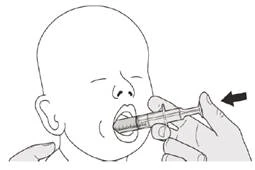
Manténgase en posición vertical (preferiblemente sentado) al tomar la dosis diaria de Evrysdi® por vía oral.
|
Figura J |
Paso B1 Coloque la jeringa oral en la boca con la boquilla a lo largo de cualquier mejilla. Lentamente, presione completamente el émbolo para administrar la dosis completa de Evrysdi® (véase la Figura J). El paso del medicamento Evrysdi® a la garganta o su administración demasiado rápida puede provocar dificultad para respirar. |
|
Figura K |
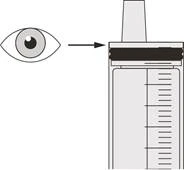
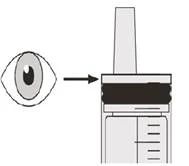
Paso B2 Compruebe si queda algún resto del medicamento Evrysdi® en la jeringa oral (véase la figura K). |
|
Figura L |
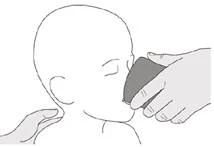
Paso B3 Beber un poco de agua inmediatamente después de tomar la dosis prescrita del medicamento Evrysdi® (véase la figura L). Véase el paso E para obtener información sobre la limpieza de la jeringa. |
C) Cómo administrar la dosis diaria del medicamento Evrysdi® a través de una sonda de gastrostomía
Si está administrando el medicamento Evrysdi® a través de una sonda de gastrostomía, pida a su médico que le muestre cómo inspeccionar la sonda de gastrostomía antes de administrar el medicamento Evrysdi®.
|
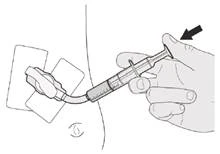
Figura M |
Paso C1 Coloque la punta de la jeringa oral en el tubo de gastrostomía. Presione lentamente el émbolo hasta el fondo para administrar la dosis completa del medicamento Evrysdi® (véase la figura M). |
|
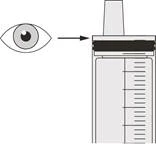
Figura N |
Paso C2 Verifique si queda medicamento Evrysdi® en la jeringa oral (véase la figura N). |
|
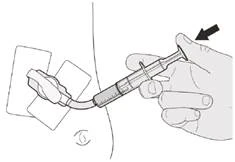
Figura O |
Paso C3 Lave el tubo de gastrostomía con 10-20 ml de agua inmediatamente después de administrar la dosis prescrita del medicamento Evrysdi® (véase la figura O). |
D) Cómo administrar la dosis diaria del medicamento Evrysdi® a través de una sonda nasogástrica
Si está administrando el medicamento Evrysdi® a través de una sonda nasogástrica, pida a su médico que le muestre cómo comprobar la colocación de la sonda nasogástrica antes de administrar el medicamento Evrysdi®.
|
Figura P |
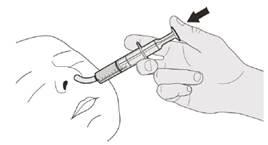
Paso D1 Coloque la punta de la jeringa oral en la sonda nasogástrica. Presione lentamente el émbolo hasta el fondo para administrar la dosis completa del medicamento Evrysdi® (véase la figura P). |
|
Figura Q |
Paso D2 Verifique si queda algún medicamento Evrysdi® en la jeringa oral (véase la figura Q). |
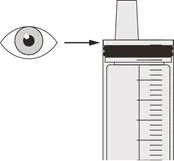
| Figura R |
Paso D3 Lave el sonda nasogástrica con 10-20 ml de agua inmediatamente después de administrar la dosis prescrita del medicamento Eurisdí® (ver Figura R). Ver Paso E para información sobre la limpieza de la jeringa. |
E) Cómo limpiar la jeringa oral después de su uso
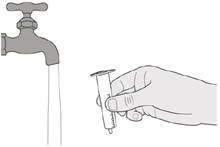
|
Figura S |
Paso E1 Saque el émbolo (contra resistencia) de la jeringa. Lave bien el cilindro de la jeringa oral con agua corriente limpia (ver Figura S). |
|
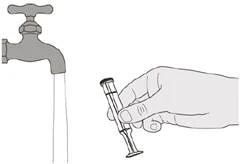
Figura T |
Paso E2 Lave bien el émbolo con agua corriente limpia (véase la figura T). |
|
Figura U |
Paso E3 Compruebe que el cilindro y el émbolo de la jeringa oral estén limpios. Coloque el cilindro y el émbolo de la jeringa oral sobre una superficie limpia en un lugar seguro para que se sequen (véase la Figura U). Lávese las manos. Una vez que el cilindro y el émbolo de la jeringa oral estén secos, coloque el émbolo nuevamente dentro del cilindro de la jeringa oral y guarde la jeringa para su próximo uso. |
Niños.
No se ha establecido la seguridad y eficacia del medicamento Evrisdi® en pacientes menores de 16 días de edad (ver sección «Farmacocinética»).
Sobredosis.
No existe experiencia clínica con sobredosis del medicamento Evrisdi® en estudios clínicos. No existe un antídoto conocido para la sobredosis con Evrisdi®. En caso de sobredosis, se debe observar cuidadosamente al paciente y comenzar un tratamiento de soporte.
Reacciones adversas
Clasificación de la frecuencia de aparición de reacciones adversas del medicamento: muy frecuentes (≥ 1/10), frecuentes (de ≥ 1/100 a < 1/10), poco frecuentes (de ≥ 1/1 000 a < 1/100), raras (de ≥ 1/10 000 a < 1/1 000), muy raras (< 1/10 000); frecuencia desconocida (no puede determinarse a partir de los datos disponibles).
Caracterización breve del perfil de seguridad
En pacientes con AME de inicio infantil, las reacciones adversas más frecuentes observadas en los estudios clínicos con el medicamento Evrysdi® fueron fiebre (54,8 %), erupción cutánea (29 %) y diarrea (19,4 %). En pacientes con AME de inicio tardío, las reacciones adversas más frecuentes observadas en los estudios clínicos con el medicamento Evrysdi® fueron fiebre (21,7 %), cefalea (20 %), diarrea (16,7 %) y erupción cutánea (16,7 %).
Las reacciones adversas mencionadas anteriormente ocurrieron sin características clínicas identificadas ni patrón temporal definido y, en general, desaparecieron sin necesidad de interrumpir el tratamiento con el medicamento Evrysdi® en pacientes con AME de inicio infantil y de inicio tardío.
Tabla 7
Resumen de reacciones adversas en pacientes con AME de inicio infantil y de inicio tardío, observadas en estudios clínicos con el medicamento Evrysdi®
| Clase de sistema orgánico |
CMA con inicio en la infancia2 (tipo 1) |
CMA con inicio tardío3 (tipo 2 y 3) |
|
| Alteraciones gastrointestinales |
|||
| Diárea |
Muy frecuente |
Muy frecuente |
|
| Náuseas |
No aplicable |
Frecuente |
|
| Úlceras en la mucosa oral y úlceras aftosas |
Frecuente |
Frecuente |
|
| Alteraciones de la piel y del tejido subcutáneo |
|||
| Erupción cutánea1 |
Muy frecuente |
Muy frecuente |
|
| Alteraciones del sistema nervioso |
|||
| Cefalea |
No aplicable |
Muy frecuente |
|
| Alteraciones generales y en el sitio de administración |
|||
| Aumento de la temperatura corporal (incluyendo hiperpirexia) |
Muy frecuente |
Muy frecuente |
|
| Infecciones e infestaciones |
|||
| Infección del tracto urinario (incluyendo cistitis) |
Frecuente |
Frecuente |
|
| Alteraciones del sistema osteomuscular y del tejido conectivo |
|||
| Artalgia |
No aplicable |
Frecuente |
|
1 Incluyendo dermatitis, dermatitis acneiforme, dermatitis alérgica, eritema, foliculitis, erupción cutánea, erupción eritematosa, erupción maculopapular, erupción papular.
2 En pacientes con AME de inicio infantil (FIREFISH, partes 1 y 2), las reacciones adversas se definieron como eventos que ocurrieron en ≥ 2 % de los pacientes o más, y cuya relación causal con el medicamento Evrysdi® es posible.
3 En pacientes con AME de inicio tardío (SUNFISH, parte 2), las reacciones adversas se definieron como eventos que ocurrieron en ≥ 2 % más frecuentemente en pacientes que recibieron tratamiento con Evrysdi® en comparación con el grupo placebo durante el período de tratamiento doble ciego controlado con placebo, y cuya relación causal con el medicamento Evrysdi® es posible.
Los datos disponibles sobre la seguridad son limitados en cuanto al número de pacientes que han recibido el medicamento Evrysdi® y a la duración de la exposición. Pueden observarse reacciones adversas potencialmente raras y potencialmente graves (RA) que no se identificaron durante el programa de estudios.
En el estudio RAINBOWFISH se obtuvieron datos limitados sobre la seguridad del uso del medicamento Evrysdi® en recién nacidos y lactantes con AME presintomática. El estudio RAINBOWFISH es un estudio abierto de grupo único que incluyó a 26 pacientes con AME presintomática con edades comprendidas entre los 16 y 41 días (rango de peso corporal entre 3,1 y 5,7 kg) en el momento de la administración de la primera dosis. En el análisis primario, la duración media del tratamiento fue de 20,4 meses (rango: 10,6 a 41,9 meses) (ver sección «Propiedades farmacológicas», subsección «Eficacia clínica»).
El perfil de seguridad del medicamento Evrysdi® en pacientes con estado presintomático en el estudio RAINBOWFISH es comparable al perfil de seguridad en pacientes con AME sintomática que recibieron tratamiento con Evrysdi® en los estudios clínicos. Actualmente no existen datos a largo plazo.
Perfil de seguridad en pacientes con AME que previamente recibieron otra terapia modificadora
Basado en el análisis primario de los participantes del estudio JEWELFISH, el perfil de seguridad de Evrysdi® en pacientes previamente tratados que recibieron tratamiento con Evrysdi® durante un período de hasta 59 meses en el estudio JEWELFISH, es coherente con el perfil de seguridad en pacientes con AME no previamente tratados que recibieron Evrysdi® en los estudios FIREFISH (partes 1 y 2), SUNFISH (partes 1 y 2) y RAINBOWFISH.
Los pacientes que previamente habían recibido nusinersén (n = 76) u onasemnogén abeparvovec (n = 14) fueron incluidos en el estudio JEWELFISH (ver subsección «Eficacia clínica»).
Efectos preclínicos
Los efectos preclínicos relacionados con la estructura de la retina, el tejido epitelial y los parámetros hematológicos, descritos en la sección «Datos preclínicos», hasta la fecha no se han observado en los estudios clínicos realizados con el medicamento Evrysdi® en AME.
Prolongación del QT
Un análisis de farmacocinética/farmacodinámica mostró ausencia de signos de prolongación del QTc tras la administración de Evrysdi® con exposición dentro del rango terapéutico; sin embargo, no existen datos adecuados tras la administración de Evrysdi® con exposición por encima del rango terapéutico.
Experiencia poscomercialización
Durante la fase poscomercialización se ha observado vasculitis cutánea, cuyos síntomas desaparecieron tras la interrupción definitiva del medicamento Evrysdi®. Con base en los datos disponibles, no es posible determinar la categoría de frecuencia ni la incidencia de esta reacción adversa. Solo existen datos limitados sobre su uso en poscomercialización en recién nacidos menores de 16 días de edad.
Duración del período de validez
2 años.
Tras la reconstitución, la solución oral lista para usar es estable durante 64 días si se conserva en nevera a una temperatura de 2-8 °C. Si fuera necesario, el paciente o la persona encargada de su cuidado pueden conservar la solución oral a temperatura ambiente (inferior a 40 °C) durante un período acumulado no superior a 5 días.
Condiciones de conservación
Conservar el polvo para solución oral a una temperatura no superior a 25 °C, en su envase original para protegerlo de la luz y la humedad. Mantener fuera del alcance de los niños.
Conservar la solución oral lista para usar en nevera a una temperatura de 2-8 °C, en su envase original para protegerla de la luz. Mantener el frasco bien cerrado y siempre en posición vertical.
Incompatibilidades
No se ha observado incompatibilidad entre el medicamento Evrysdi® y las jeringas orales reutilizables recomendadas para la administración oral del medicamento.
Envase
Frasco de vidrio ámbar de 100 ml (clase III según Farmacopea de EE. UU. y Farmacopea Europea), con tapón blanco con función de protección contra apertura por niños (envoltura externa de polietileno de alta densidad; envoltura interna de homopolímero de polipropileno), con anillo de control de primera apertura y revestimiento de polietileno y policloruro de vinilideno, acompañado de 1 adaptador de compresión para frasco, 2 jeringas orales reutilizables de 1 ml (cada una en bolsita de polietileno), 2 jeringas orales reutilizables de 6 ml (cada una en bolsita de polietileno), 1 jeringa oral reutilizable de 12 ml (en bolsita de polietileno), todo ello contenido en una bolsa de polietileno. Un frasco y un juego por caja de cartón.
Categoría de dispensación
Medicamento sujeto a prescripción médica.
Fabricante
F. Hoffmann-La Roche Ltd
Dirección del fabricante y lugar de ejercicio de su actividad
Wurmisweg, 4303 Kaiseraugst, Suiza