Evrisdi®
UkrainaSpis treści
INSTRUKCJA DOTYCZĄCA STOSOWANIA LEKU Evrisdi® (Evrysdi®)
Skład:
substancja czynna: risdiplam;
jedna butelka zawiera 60 mg risdiplamu;
1 ml odtworzonego roztworu zawiera 0,75 mg risdiplamu;
substancje pomocnicze: mannit (E 421); izomalit (E 953); aromat truskawkowy; kwas winny; benzoesan sodu (E 211); polietylenoglikol 6000; sukraloza; kwas askorbinowy; kwas etylenodiaminotetraacetynowy disodu, dwuwodny.
Postać farmaceutyczna. Proszek do sporządzenia roztworu doustnego.
Główne właściwości fizykochemiczne: proszek lub proszek z grudkami lub masą proszkową jasnożółtego, żółtego, szarawożółtego, zielonkawożółtego lub jasnozielonego koloru. Odtworzony roztwór o barwie od zielonkawożółtej do żółtej.
Grupa farmakoterapeutyczna. Leki wpływające na układ mięśniowo-szkieletowy. Inne leki stosowane w chorobach układu mięśniowo-szkieletowego.
Kod ATC M09A X10.
Właściwości farmakologiczne.
Farmakodynamika.
Mechanizm działania
Risdiplam jest modyfikatorem procesu splicingu pre-mRNA genu przeżycia neuronów ruchowych 2 (SMN2), zaprojektowanym do leczenia SMA spowodowanego mutacjami chromosomu 5q, prowadzącymi do niedoboru białka SMN. Funkcjonalny niedobór białka SMN jest mechanizmem patofizjologicznym SMA wszystkich typów. Risdiplam koryguje splicing SMN2, zmieniając równowagę z pomijania egzonu 7 na jego włączenie do transkryptu mRNA, co prowadzi do zwiększenia produkcji funkcjonalnego i stabilnego białka SMN. W ten sposób risdiplam leczy SMA poprzez podnoszenie i utrzymywanie funkcjonalnego poziomu białka SMN.
Elektrofizjologia serca
Wpływ risdiplamu na interwał QTc oceniano w badaniu przeprowadzonym u 47 zdrowych ochotników dorosłych. W zakresie działania terapeutycznego risdiplam nie wydłużał interwału QTc.
Risdiplam równomiernie rozkłada się w całym organizmie, w tym w ośrodkowym układzie nerwowym, przenikając przez barierę krew–mózg, zwiększając w ten sposób poziom białka SMN w OUN i całym organizmie. Stężenie risdiplamu w osoczu krwi oraz poziom białka SMN we krwi odzwierciedlają rozkład i efekty farmakodynamiczne risdiplamu w tkankach takich jak mózg i mięśnie.
W badaniach klinicznych FIREFISH, SUNFISH oraz JEWELFISH przeprowadzonych u pacjentów z wczesną postacią SMA (SMA typu 1) oraz pacjentów z późną postacią SMA, risdiplam powodował spójne i długotrwałe zwiększenie stężenia białka SMN we krwi, przy zmianie mediany o więcej niż podwójną wartość od punktu wyjściowego w ciągu 4 tygodni od rozpoczęcia leczenia. Takie zwiększenie poziomu białka SMN utrzymywało się przez cały okres leczenia trwający co najmniej 24 miesiące (patrz podrozdział „Skuteczność kliniczna”).
Kliniczna skuteczność
Skuteczność leku Evrisdi® w leczeniu pacjentów z wczesną postacią SMA (SMA typu 1) oraz z późną postacią SMA (SMA typu 2 i 3) została oceniona w dwóch podstawowych badaniach klinicznych FIREFISH i SUNFISH oraz potwierdzona dodatkowymi danymi z badania JEWELFISH. Skuteczność leku Evrisdi® w leczeniu pacjentów z przedobjawową postacią SMA została oceniona na podstawie pośredniej analizy wtórnych punktów końcowych trwającego badania RAINBOWFISH.
Pacjenci z klinicznym rozpoznaniem SMA typu 4 nie uczestniczyli w badaniach klinicznych.
W badaniach klinicznych wykazano długotrwałą skuteczność przez co najmniej 24 miesiące leczenia. Dane dotyczące zastosowania leku Evrisdi® przez okres dłuższy niż 2 lata są ograniczone.
Wczesna postać SMA
Badanie BP39056 (FIREFISH) – otwarte badanie dwuczęściowe oceniające skuteczność, bezpieczeństwo, farmakokinetykę i farmakodynamikę leku Evrisdi® u pacjentów z SMA typu 1 z objawami (wszyscy pacjenci mieli genetycznie potwierdzoną chorobę z obecnością dwóch kopii genu SMN2). Część 1 badania FIREFISH została zaprojektowana jako badanie doboru dawki. W części potwierdzającej (część 2) badania FIREFISH oceniano skuteczność leku Evrisdi® w dawkach terapeutycznych wybranych na podstawie wyników części 1 (patrz sekcja „Sposób stosowania i dawki”). Pacjenci z części 1 nie uczestniczyli w części 2.
Łącznie 62 pacjentów z SMA typu 1 z objawami zostało włączone do części 1 (n = 21) i części 2 (n = 41) badania FIREFISH, z których 58 pacjentów otrzymywało dawkę terapeutyczną leku Evrisdi®. Średni wiek pojawienia się objawów klinicznych wynosił 1,5 miesiąca (0,9–3 miesiąca). Średni wiek w momencie włączenia do badania wynosił 5,6 miesiąca (2,2–6,9 miesiąca), a średni czas między pojawieniem się objawów a podaniem pierwszej dawki wynosił 3,7 miesiąca (1–6 miesięcy). 60% pacjentów stanowiły kobiety, 57% – rasa kaukaska, 29% – Azjaci. Na początku badania mediana wyników testu CHOP-INTEND wynosiła 23 (8–37), a mediana wyników HINE-2 wynosiła 1 (0–5). Wyjściowe dane demograficzne i charakterystyka choroby pacjentów włączonego do części 1 badania były porównywalne z danymi pacjentów w części 2 badania.
Pierwszym punktem końcowym badania była frakcja pacjentów zdolnych do siadania bez wsparcia przez co najmniej 5 sekund, zgodnie z punktem 22 skali rozwoju niemowląt i małych dzieci Bayley’a – III wydanie (BSID-III) służącej ocenie motoryki ogólnej, po 12 miesiącach leczenia lekiem Evrisdi® w części 2 badania; osiągnięto ten wynik u 29% pacjentów (n = 12/41, 90% przedział ufności: 17,8%, 43,1%, p <0,0001).
Kluczowe wyniki dotyczące skuteczności u pacjentów leczonych lekiem Evrisdi® w badaniu FIREFISH (dane zestawione z części 1 i części 2) przedstawiono w tabeli 1.
Tabela 1
Streszczenie kluczowych wyników skuteczności po 12 i 24 miesiącach (FIREFISH, część 1 i część 2)
| Punkty końcowe skuteczności |
Po 12 miesiącach |
Po 24 miesiącach |
| Częstość u pacjentów (90 % CI) |
||
| Etapowanie rozwoju motorycznego i funkcja ruchowa |
N = 58a |
|
| BSID-III: siedzenie bez wsparcia przez co najmniej 5 sekund |
32,8 % |
60,3 % |
| CHOP-INTEND: liczba punktów 40 lub więcej |
56,9 % |
74,1 % |
| Zwiększenie punktów CHOP-INTEND o ≥ 4 od wartości wyjściowej |
89,7 % |
87,9 % |
| HINE-2: pacjenci odpowiadający kryteriom rozwoju funkcji motorycznejb |
77,6 % |
82,8 % |
| Przeżycie i przeżycie bez zdarzeń |
N=62a |
|
| Przeżycie bez zdarzeńc |
87,1 % |
|
| Przeżycie |
91,9 % |
90,3 % |
| Odżywianie |
N = 58a |
|
| Sposóbność do otrzymywania odżywiania doustnegod |
84,5 % |
82,8 % |
BSID-III — Skala rozwoju niemowląt i małych dzieci Bayleya — wydanie III;
CHOP-INTEND — Test Szpitala Dziecięcego w Filadelfii do oceny funkcji ruchowych u niemowląt z chorobami neuropatyczno-mięśniowymi;
HINE-2 — Moduł 2 badania neurologicznego Hammermitha u niemowląt.
a Dane dotyczące przeżycia i przeżycia bez wentylacji zostały połączone dla wszystkich pacjentów, którzy otrzymali dowolną dawkę risdiplamu w części 1 i części 2 badania (n = 62). W przypadku etapów rozwoju motorycznego, funkcji ruchowych oraz odżywiania dane dotyczące skuteczności zostały połączone dla wszystkich pacjentów, którzy otrzymali dawkę terapeutyczną risdiplamu (wszyscy pacjenci w części 2 badania oraz pacjenci z kohorty otrzymywania wysokiej dawki w części 1; n = 58).
b Definicja odpowiedzi według kryterium HINE-2: odpowiedź w tej analizie została zdefiniowana jako wzrost o ≥ 2 punkty (lub maksymalny możliwy wynik) w zdolności do kopania nogami LUB wzrost o ≥ 1 punkt w etapach rozwoju funkcji motorycznych, takich jak kontrola trzymania głowy, przewracanie się, siadanie, półzanie, stanie lub chodzenie, ORAZ poprawa w większej liczbie kategorii rozwoju funkcji motorycznych niż pogorszenie.
c Zjawisko spełniające kryterium punktu końcowego stałej wentylacji zdefiniowano jako tchawicę lub ≥ 16 godzin nieinwazyjnej wentylacji płuc na dobę, lub intubację trwającą > 21 dni z rzędu bez obecności lub po ustąpieniu ostrego, odwracalnego zjawiska. Czterem pacjentom spełniającym kryteria punktu końcowego stałej wentylacji do 24 miesięcy. Te cztery pacjentki osiągnęły wzrost wyniku o co najmniej 4 punkty w skali CHOP-INTEND w porównaniu z wynikiem wyjściowym.
d W tym pacjentów, którzy otrzymywali wyłącznie doustne odżywianie (41 pacjentów po 12 i 24 miesiącach), oraz pacjentów, którzy otrzymywali doustne odżywianie w połączeniu z sondą do żywienia (8 pacjentów po 12 miesiącach i 7 pacjentów po 24 miesiącach).
Po 24 miesiącach 40 % (23/58) pacjentów, którzy otrzymywali dawkę terapeutyczną leku Evrisdi®, mogło siadać bez wsparcia przez 30 sekund (BSID-III, punkt 26). Ponadto pacjenci kontynuowali osiąganie dodatkowych etapów rozwoju motorycznego według wskaźnika HINE-2 po 24 miesiącach; 78 % pacjentów potrafiło się przewracać (31 % pacjentów potrafiło przewracać się na bok, 7 % pacjentów potrafiło przewracać się z pozycji leżącej przodem na pozycję leżącą tyłem i 40 % pacjentów potrafiło przewracać się z pozycji leżącej tyłem na pozycję leżącą przodem) oraz 28 % pacjentów osiągnęło siadanie (16 % utrzymywało ciężar ciała i 12 % stało z wsparciem).
Udział pacjentów żyjących bez potrzeby stałej wentylacji (przeżycie bez zdarzeń) wynosił 84 % wśród wszystkich pacjentów po 24 miesiącach. Zmarło sześć niemowląt (4 w ciągu pierwszych 3 miesięcy po włączeniu do badania) i jednemu pacjentowi przedwcześnie przerwano leczenie, a 3,5 miesiąca po tym pacjent zmarł. Czterem pacjentom wymagana była stała wentylacja do 24 miesięcy.
MA z późnym przebiegiem
Badanie BP39055 (SUNFISH) – wieloośrodkowe badanie składające się z dwóch części, oceniające skuteczność, bezpieczeństwo, farmakokinetykę i farmakodynamikę leku Evrisdi® u pacjentów z rozpoznaniem MA typu 2 lub 3 w wieku od 2 do 25 lat. Część 1 była częścią badawczą ustalającą dawkę, a część 2 – randomizowaną, podwójnie ślepą, placebo-kontrolowaną częścią potwierdzającą. Pacjenci z części 1 badania nie brali udziału w części 2.
Pierwotnym punktem końcowym była ocena zmiany wyniku funkcji ruchowych (MFM32) po 12 miesiącach w porównaniu z wynikiem wyjściowym. MFM32 pozwala ocenić szeroki zakres funkcji ruchowych u szerokiej grupy pacjentów z MA. Ogólny wynik MFM32 wyrażany jest w procentach (zakres: od 0 do 100) maksymalnego możliwego wyniku, przy czym wyższy wynik wskazuje na lepszą funkcję ruchową. MFM32 określa zdolność do funkcji ruchowych związanych z ważnymi codziennymi czynnościami. Niewielkie zmiany funkcji ruchowych mogą prowadzić do istotnego poprawienia lub utraty codziennych funkcji.
SUNFISH, część 2
Część 2 badania SUNFISH to randomizowana, podwójnie ślepa, placebo-kontrolowana część badania, w której wzięło udział 180 pacjentów niezdolnych do chodzenia, z MA typu 2 (71 %) lub typu 3 (29 %). Pacjenci zostali losowo przydzieleni w stosunku 2:1 do otrzymywania leku Evrisdi® w dawce terapeutycznej (patrz sekcja „Sposób stosowania i dawki”) lub placebo. Randomizacja była stratyfikowana według wieku (od 2 do 5 lat, od 6 do 11 lat, od 12 do 17 lat, od 18 do 25 lat).
Średni wiek pacjentów w momencie rozpoczęcia leczenia wynosił 9 lat (zakres 2–25 lat), mediana czasu od wystąpienia objawów MA do pierwszego leczenia wynosiła 102,6 (1–275) miesiąca. Wśród uczestników badania 51 % pacjentów stanowiły kobiety, 67 % to rasa europejska, 19 % pochodzenie azjatyckie. W momencie rozpoczęcia badania 67 % pacjentów miało skoliozę (32 % z nich miało ciężką skoliozę). Pacjenci mieli średni wynik wyjściowy MFM32 wynoszący 46,1 i wynik RULM 20,1. Ogólnie początkowe dane demograficzne były dobrze zrównoważone między grupami przyjmującymi lek Evrisdi® i placebo, z wyjątkiem nierówności w liczbie pacjentów ze skoliozą (63,3 % pacjentów w grupie leku Evrisdi® i 73,3 % pacjentów w grupie placebo-kontrolnej).
Wyniki analizy pierwotnej części 2 badania SUNFISH dotyczące zmiany ogólnego wyniku MFM32 po 12 miesiącach wykazały klinicznie istotną i statystycznie istotną różnicę między grupami pacjentów otrzymujących leczenie lekiem Evrisdi® i placebo. Wyniki analizy pierwotnej oraz główne wtórne punkty końcowe przedstawiono w tabeli 2 i na rysunku 1.
Tabela 2
Podsumowanie wyników skuteczności u pacjentów z MA z późnym początkiem po 12 miesiącach leczenia (część 2 badania SUNFISH)
| Punkt końcowy |
Evrisdi® (N = 120) |
Placebo (N = 60) |
| Pierwotny punkt końcowy: |
||
| Zmiana ogólnego wyniku MFM321 po 12 miesiącach w porównaniu do stanu wyjściowego, średni wynik średniokwadratowy (95 %, przedział ufności) |
1,36 (0,61; 2,11) |
-0,19 (-1,22; 0,84) |
| Różnica w porównaniu do placebo (95 % przedział ufności), wartość p2 |
1,55 (0,30; 2,81) 0,0156 |
|
| Pośrednie punkty końcowe |
||
| Częstość pacjentów z poprawą ogólnego wyniku MFM321 o co najmniej 3 punkty po 12 miesiącach w porównaniu do stanu wyjściowego (95 % przedział ufności) |
38,3 % (28,9; 47,6) |
23,7 % (12,0; 35,4) |
| Stosunek szans ogólnej odpowiedzi (95 % przedział ufności), skorygowana (nieskorygowana) wartość p3,4 |
2,35 (1,01; 5,44) 0,0469 (0,0469) |
|
| Zmiana ogólnego wyniku RULM5 po 12 miesiącach w porównaniu do stanu wyjściowego, średni wynik średniokwadratowy (95 % przedział ufności) |
1,61 (1,00; 2,22) |
0,02 (-0,8; 0,87) |
| Różnica w porównaniu do placebo (95 % przedział ufności), skorygowana (nieskorygowana) wartość p2,4 |
1,59 (0,55; 2,62) 0,0469 (0,0028) |
|
1 Na podstawie zasady brakujących danych dotyczących MFM32 z analizy wykluczono 6 pacjentów (Evrisdi®, n = 115; grupa placebo, n = 59).
2 Dane analizowano za pomocą mieszanego modelu z powtarzanymi pomiarami z uwzględnieniem ogólnego wyniku wyjściowego, leczenia, wizyty, grupy wiekowej, efektu leczenia w zależności od wizyty oraz wyniku wyjściowego w zależności od wizyty.
3 Dane analizowano za pomocą regresji logistycznej dla ogólnego wyniku wyjściowego, grupy leczenia oraz grupy wiekowej.
4 Skorygowaną wartość p uzyskano dla punktów końcowych objętych testowaniem hierarchicznym i obliczono na podstawie wszystkich wartości p dla punktów końcowych w kolejności hierarchii aż do bieżącego punktu końcowego. Nieskorygowaną wartość p testowano przy poziomie istotności 5%.
5 Na podstawie zasady brakujących danych dotyczących RULM z analizy wykluczono 3 pacjentów (Evrisdi®, n = 119; grupa placebo, n = 58).
Po zakończeniu 12 miesięcy leczenia 117 pacjentów kontynuowało przyjmowanie leku Evrisdi®. W chwili analizy po 24 miesiącach pacjenci, którzy otrzymywali leczenie przez 24 miesiące, osiągnęli dalszą poprawę funkcji motorycznych w okresie pomiędzy 12. a 24. miesiącem terapii. Średnia zmiana wyniku wyjściowego MFM32 w porównaniu z poziomem wyjściowym wyniosła 1,83 (CI: 0,74–2,92), a dla RULM – 2,79 (CI: 1,94–3,64).
| Zmiana średniego kwadratowego średniego wyniku ogólnego wskaźnika MFM32 |
|
| Miesiące |
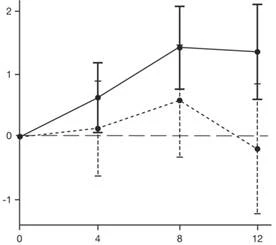
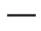
Evrisdi® Płacebo
* Słupki błędów oznaczają 95 % przedział ufności.
† Ogólny wynik MFM został obliczony zgodnie z instrukcją dla użytkownika i wyrażony jako procent maksymalnej możliwej liczby punktów dla skali (tj. suma punktów za 32 pozycje podzielona przez 96 i pomnożona przez 100).
Rys. 1. Średnia zmiana (LS) ogólnego wyniku MFM32 od poziomu wyjściowego po 12 miesiącach w części 2 badania SUNFISH.
SUNFISH, część 1
Skuteczność leku Evrisdi® u pacjentów z SMA z późnym początkiem potwierdzają również wyniki części 1 badania SUNFISH dotyczącego doboru dawki. Do części 1 zakwalifikowano 51 pacjentów z typem SMA 2 i 3 (w tym 7 pacjentów chodzących) w wieku od 2 do 25 lat. Po jednym roku leczenia dawką terapeutyczną (dawką wybraną do części 2) zaobserwowano klinicznie istotne poprawy funkcji motorycznej według wyników MFM32, ze średnią zmianą od poziomu wyjściowego wynoszącą 2,7 punktu (95 % PU: 1,5; 3,8). Poprawa wyników MFM32 utrzymywała się również w okresie do 2 lat leczenia lekiem Evrisdi® (średnia zmiana 2,7 punktu (95 % PU: 1,2; 4,2)).
W analizie poszukiwania danych funkcja motoryczna oceniana za pomocą skali MFM była porównywana w części 1 badania SUNFISH oraz w historycznej kohorcie z naturalnym przebiegiem choroby (ważonej według głównych czynników prognostycznych). Zmiana ogólnego wyniku MFM w porównaniu z poziomem wyjściowym po 1 i 2 latach była większa u pacjentów leczonych Evrisdi® w porównaniu z kohortą naturalnego przebiegu (po 1 roku: różnica 2,7 punktu; p < 0,0001; po 2 latach: różnica 4 punkty; p < 0,0001). W kohorcie naturalnego przebiegu zaobserwowano spadek funkcji motorycznej, zgodny z oczekiwanym postępem SMA (średnia zmiana po 1 roku: –0,6 punktu; po 2 latach: –2 punkty).
SMA przedobjawowa
Badanie BN40703 (RAINBOWFISH) — trwające jednogrupowe, otwarte, wieloośrodkowe badanie oceniające skuteczność, bezpieczeństwo, farmakokinetykę i farmakodynamikę leku Evrisdi® u niemowląt w wieku od urodzenia do 6 tygodni (w momencie podania pierwszej dawki), u których stwierdzono genetyczne rozpoznanie SMA, ale bez jeszcze występujących objawów.
Skuteczność leku Evrisdi® w przypadku SMA przedobjawowej oceniano po 12 miesiącach u 26 pacjentów [populacja ITT (intencja do leczenia)]. Mediana wieku tych pacjentów w momencie podania pierwszej dawki wynosiła 25 dni (zakres od 16 do 41 dni), 62 % stanowiły pacjentki płci żeńskiej, a 85 % — rasa kaukaska. Odpowiednio 8, 13 i 5 pacjentów posiadało 2, 3 i ≥ 4 kopie genu SMN2. Na początku badania mediana wyniku CHOP-INTEND wynosiła 51,5 (zakres od 35,0 do 62,0), mediana wyniku HINE-2 wynosiła 2,5 (zakres od 0 do 6,0), a mediana amplitudy potencjału działania mięśni nerwu łokciowego (CMAP) wynosiła 3,6 mV (zakres od 0,5 do 6,7 mV).
Pierwotna populacja analizy skuteczności (N = 5) obejmowała pacjentów z 2 kopiemi genu SMN2 i wyjściową amplitudą CMAP ≥ 1,5 mV. U tych pacjentów mediana wyniku CHOP-INTEND wynosiła 48,0 (zakres 36,0–52,0), mediana wyniku HINE-2 wynosiła 2,0 (zakres od 1,0 do 3,0), a mediana amplitudy CMAP wynosiła 2,6 mV (zakres od 1,6 do 3,8 mV) na poziomie wyjściowym.
Pierwszym punktem końcowym była frakcja pacjentów w pierwotnej populacji analizy skuteczności, którzy byli w stanie siadać bez wsparcia przez co najmniej 5 sekund (skala motoryki ogólnej BSID-III, pozycja 22) po 12 miesiącach; statystycznie istotna i klinicznie istotna frakcja pacjentów osiągnęła ten etap w porównaniu z wcześniej ustalonym kryterium skuteczności wynoszącym 5 %.
Kluczowe punkty końcowe skuteczności u pacjentów leczonych lekiem Evrisdi® przedstawiono w tabelach 3 i 4 oraz na rysunku 2.
Tabela 3
Umiejętność siadania zgodnie z definicją pozycji 22 skali BSID-III u pacjentów przedobjawowych po 12 miesiącach
| Punkt końcowy oceny skuteczności |
Populacja pacjentów |
||
| Pierwotna analiza skuteczności (N = 5) |
Pacjenci z dwiema kopiami genu SMN2a (N = 8) |
ITT (N = 26) |
|
| Częstość pacjentów, którzy byli w stanie siedzieć bez podpórki co najmniej 5 sekund (skala BSID-III, punkt 22); (90 % CI) |
80 % (34,3 %; 99,0 %) |
87,5 % (52,9 %; 99,4 %) |
96,2 % (83,0 %; 99,8 %) |
Skróty: BSID-III – Skala Bayleya rozwoju niemowląt i małych dzieci, trzecie wydanie; CI – przedział ufności; ITT – populacja intencyjna do leczenia.
a Pacjenci z dwoma kopiami genu SMN2 mieli średnią amplitudę CMAP wynoszącą 2,0 (zakres 0,5–3,8) na początku badania.
b Wartość p oparta na jednostronnym dokładnym teście dwumianowym. Wynik porównywany z progiem 5 %.
Ponadto 80 % (4/5) podstawowej populacji analizy skuteczności, 87,5 % (7/8) pacjentów z dwiema kopiami genu SMN2 oraz 80,8 % (21/26) pacjentów z populacji ITT osiągnęło zdolność siadania bez wsparcia przez 30 sekund (BSID-III, punkt 26). Pacjenci z populacji ITT osiągnęli również etapy ruchowe mierzone za pomocą modułu HINE-2 w 12. miesiącu (N = 25). W tej populacji 96,0 % pacjentów potrafiło siadać [1 pacjent (1/8 pacjentów z dwiema kopiami genu SMN2) osiągnął stabilne siadanie, a 23 pacjentów (6/8, 13/13, 4/4 pacjentów z 2, 3 i ≥ 4 kopiami genu SMN2 odpowiednio) potrafiło się obracać/przewracać]. Ponadto, 84 % pacjentów mogło stać; 32 % (N = 8) pacjentów mogło stać z pomocą (3/8, 3/13 i 2/4 pacjentów z 2, 3 i ≥ 4 kopiemi genu SMN2 odpowiednio) oraz 52 % (N = 13) pacjentów mogło stać bez pomocy zewnętrznej (1/8, 10/13 i 2/4 pacjentów z 2, 3 i ≥ 4 kopiemi genu SMN2 odpowiednio). Ponadto, 72 % pacjentów mogło podskakiwać, chodzić z pomocą lub bez; 8 % (N = 2) pacjentów mogło podskakiwać (2/8 pacjentów z dwiema kopiami genu SMN2), 16 % (N = 4) mogło chodzić z pomocą (3/13 i 1/4 pacjentów z 3 i ≥ 4 kopiemi genu SMN2 odpowiednio) oraz 48 % (N = 12) mogło chodzić samodzielnie (1/8, 9/13 i 2/4 pacjentów z 2, 3 i ≥ 4 kopiemi genu SMN2 odpowiednio). Siedmioro pacjentów nie poddano analizie w 12. miesiącu.
Tabela 4
Podsumowanie kluczowych punktów końcowych skuteczności dla pacjentów przedobjawowych w 12. miesiącu
| Punkty końcowe skuteczności |
Populacja ITT (N = 26) |
| Funkcja motoryczna |
|
| Częstość pacjentów osiągających wynik ogólny 50 lub wyższy w skali CHOP-INTEND (90% CI) |
92 %a (76,9 %; 98,6 %) |
| Częstość pacjentów osiągających wynik ogólny 60 lub wyższy w skali CHOP-INTEND (90% CI) |
80 %a (62,5 %; 91,8 %) |
| Odżywianie |
|
| Częstość pacjentów zdolnych do doustnego odżywiania (90% CI) |
96,2 %b (83,0 %; 99,8 %) |
| Wykorzystanie zasobów systemu opieki zdrowotnej |
|
| Częstość pacjentów, którzy nie wymagali hospitalizacji (90% CI) |
92,3 % (77,7 %; 98,6 %) |
| Przeżycie bez zdarzeńd Częstość pacjentów charakteryzujących się przeżyciem bez zdarzeń (90% CI) |
100 % (100 %; 100 %) |
Skróty: CHOP INTEND – test dziecięcego szpitala w Filadelfii do oceny funkcji ruchowych u niemowląt z chorobami neurologicznymi i mięśniowymi; CI – przedział ufności; ITT – populacja leczona.
a Na podstawie liczby N = 25.
b U jednego pacjenta nie przeprowadzono oceny.
c Hospitalizacje obejmowały wszystkie przypadki pobytu w szpitalu trwające co najmniej dwa dni, nie spowodowane wymaganiami badania.
d Zdarzenie w tym przypadku oznacza śmierć lub potrzebę stałej wentylacji; stała wentylacja definiowana jest jako traheostomia lub ≥ 16 godzin nieinwazyjnej wentylacji płuc na dobę, lub intubacja trwająca > 21 dni z rzędu w przypadku braku lub po ustąpieniu ostrego, odwracalnego stanu.
Skróty: IQR – rozstęp międzykwartylowy; SMN2 – gen przeżycia neuronów ruchowych 2.
Rys. 2. Mediana całkowitych punktów CHOP-INTEND według wizyt i liczby kopii genu SMN2 (populacja ITT)
Stosowanie u pacjentów z SMA, którzy wcześniej otrzymywali inne metody leczenia modyfikujące przebieg choroby
Badanie BP39054 (JEWELFISH) – jednogrupowe, otwarte badanie oceniające bezpieczeństwo, skuteczność, farmakokinetykę i farmakodynamikę leku Evrisdi® u pacjentów w wieku od 6 miesięcy do 60 lat z postacią niemowlęcą SMA lub SMA z późnym początkiem, którzy wcześniej otrzymywali leczenie SMA (w tym nusinersenem i onasemnogenem abeparvovek). Spośród 173 pacjentów, którzy otrzymali lek Evrisdi®, 76 pacjentów wcześniej otrzymywało leczenie nusinersenem (9 pacjentów z SMA typu 1, 43 pacjentów z SMA typu 2 i 24 pacjentów z SMA typu 3) oraz 14 pacjentów wcześniej otrzymało leczenie onasemnogenem abeparvovek (4 pacjenci z SMA typu 1 i 10 pacjentów z SMA typu 2). Średni wiek pacjentów na początku leczenia lekiem Evrisdi® wynosił 14 lat (zakres 1–60 lat).
Na moment włączenia do badania wśród 168 pacjentów w wieku 2–60 lat, 83 % miało skoliozę (39 % – ciężką skoliozę), a u 63 % wynik w rozszerzonej skali Hammerstona (HFMSE) wynosił < 10 punktów. Do badania włączono również 15 pacjentów, którzy byli w stanie samodzielnie chodzić (w wieku 5–46 lat).
Oceny wyników wyszukiwania skuteczności przeprowadzono poprzez pomiar funkcji ruchowych odpowiednich dla wieku, w tym za pomocą skal MFM-32 i RULM u pacjentów w wieku 2–60 lat, BSID-III i HINE-2 u pacjentów w wieku do 2 lat oraz testu sześciominutowego chodzenia (6MWT) u pacjentów w wieku ≥ 6 lat, którzy byli w stanie samodzielnie chodzić. Wyniki analizy pierwotnej po 24 miesiącach leczenia u pacjentów w wieku 2–60 lat wykazały ogólną stabilizację funkcji ruchowych według skal MFM-32 i RULM (n = 137 i n = 133 odpowiednio). Pacjenci w wieku do 2 lat (n = 6) zachowali lub poprawili rozwój funkcji ruchowych, w szczególności trzymanie głowy, przewracanie się i siadanie bez wsparcia. Wyniki 6MWT wykazały średnią poprawę o 30,88 metra (95 % CI: -5,54, 67,29, n = 8). Wszyscy pacjenci, którzy byli w stanie samodzielnie chodzić, zachowali zdolność chodzenia.
Farmakokinetyka.
Parametry farmakokinetyczne risdiplamu zostały scharakteryzowane u zdrowych dorosłych oraz u pacjentów z SMA.
Po podaniu doustnego roztworu leku Evrisdi® w dawkach od 0,6 do 18 mg farmakokinetyka risdiplamu była w przybliżeniu liniowa. Farmakokinetykę risdiplamu najlepiej opisano za pomocą modelu populacyjnego PK z wchłanianiem z trójkomorowym przejściem, dwukomorowym rozdziałem i eliminacją pierwszego rzędu. Stwierdzono, że masa ciała i wiek pacjenta mają istotny wpływ na farmakokinetykę leku.
Szacowana ekspozycja (średnie AUC0–24h) u pacjentów z postacią niemowlęcą SMA (w wieku 2–7 miesięcy w momencie włączenia do badania) przy zalecanej dawce 0,2 mg/kg raz dziennie wynosiła 1930 ng·h/ml. Średnia szacowana ekspozycja u niemowląt w wieku od 16 dni do < 2 miesięcy z przedobjawową SMA w badaniu RAINBOWFISH wynosiła 2020 ng·h/ml po 2 tygodniach codziennego przyjmowania w dawce 0,15 mg/kg.
Szacowana ekspozycja u pacjentów z SMA z późnym początkiem (w wieku 2–25 lat w momencie włączenia do badania) w badaniu SUNFISH (część 2) przy dawce terapeutycznej (0,25 mg/kg raz dziennie u pacjentów z masą ciała < 20 kg; 5 mg raz dziennie u pacjentów z masą ciała ≥ 20 kg) wynosiła 2010 ng·h/ml. Maksymalna stężenie obserwowane (średnie Cmax) wynosiło 194 ng/ml przy dawkowaniu 0,2 mg/kg w badaniu FIREFISH i 120 ng/ml w części 2 badania SUNFISH. Średnie szacowane maksymalne stężenie przy dawkowaniu 0,15 mg/kg w badaniu RAINBOWFISH wynosi 111 ng/ml.
Wchłanianie
Risdiplam był szybko wchłaniany po doustnym podaniu na czczo, przy czym tmax we krwi osoczu wahał się od 1 do 4 godzin. W badaniach klinicznych risdiplam podawano rano z posiłkiem lub po karmieniu piersią.
Rozkład
Szacowane parametry populacyjnej farmakokinetyki wynosiły: 98 l – pozorny objętościowy rozkład centralny, 93 l – objętość obwodowa i 0,68 l/godz. – klirens międzykompartmentowy.
Risdiplam wiąże się głównie z albuminami osocza, nie wiążąc się z alfa-1-kwasowymi glikoproteinami, przy czym frakcja wolna wynosi 11 %.
Metabolizm
Risdiplam jest metabolizowany głównie za pomocą zawierających flawinę monooksygenaz 1 i 3 (FMO1 i FMO3), a także izoenzymami CYP 1A1, 2J2, 3A4 i 3A7. Lek w formie pierwotnej był głównym składnikiem wykrytym w osoczu i stanowił 83 % substancji związanej z lekiem w krążeniu. Farmakologicznie nieaktywny metabolit M1 został zidentyfikowany jako główny metabolit krążący.
Eliminacja
Parametr populacyjnej farmakokinetyki pozornego klirensu (CL/F) risdiplamu wynosił 2,6 l/godz. Efektywny okres półtrwania risdiplamu wynosił około 50 godzin u pacjentów z SMA.
Około 53 % dawki (14 % w formie niezmienionego risdiplamu) wydalało się z kałem, a 28 % z moczem (8 % w formie niezmienionego risdiplamu).
Osobne grupy pacjentów
Zaburzenia funkcji wątroby
Lekkie i umiarkowane zaburzenia funkcji wątroby nie miały wpływu na PK risdiplamu. Po podaniu 5 mg risdiplamu średnie stosunki Cmax i AUC wynosiły 0,95 i 0,80 u osób z lekkim (n = 8) oraz 1,20 i 1,08 u pacjentów z umiarkowanym zaburzeniem funkcji wątroby (n = 8) w porównaniu do odpowiednich wartości u pacjentów z normalną funkcją wątroby (grupa kontrolna, n = 10). Bezpieczeństwo i PK u pacjentów z ciężkim zaburzeniem funkcji wątroby nie zostały dotychczas zbadane.
Zaburzenia funkcji nerek
Badania PK risdiplamu u pacjentów z zaburzeniem funkcji nerek nie były prowadzone. Wydalanie risdiplamu w niezmienionej formie przez nerki jest niewielkie (8 %).
Pacjenci starsi
Specjalistyczne badania farmakokinetyki leku Evrisdi® u pacjentów z SMA w wieku powyżej 60 lat nie były prowadzone. Pacjenci z SMA w wieku do 60 lat byli włączani do badania JEWELFISH. Pacjenci bez SMA w wieku do 69 lat byli włączani do klinicznych badań PK.
Dzieci
Masa ciała i wiek pacjenta zostały zidentyfikowane jako kowariate w analizie populacyjnej PK. Dlatego dawkę dostosowuje się w zależności od wieku (poniżej i powyżej 2 miesięcy oraz 2 lat) i masy ciała (do 20 kg), aby osiągnąć podobną ekspozycję w różnych grupach wiekowych i kategoriach wagowych. Brak danych farmakokinetycznych dotyczących niemowląt w wieku do 16 dni.
Pochodzenie etniczne
Farmakokinetyka risdiplamu nie różni się u Japończyków i pacjentów rasy europejskiej.
Charakterystyki kliniczne.
Wskazania.
Leczenie 5q-skojarzonej rdzeniowej atrofii mięśni (SMA) u dzieci i dorosłych pacjentów.
Przeciwwskazania.
Znana nadwrażliwość na risdiplam lub którykolwiek ze składników pomocniczych wymienionych w sekcji „Skład”.
Interakcje z innymi lekami i inne rodzaje interakcji.
Wpływ Evrisdi® na inne leki
In vitro risdiplam i jego główny krążący metabolit M1 nie indukowały CYP1A2, 2B6, 2C8, 2C9, 2C19 oraz 3A4. In vitro risdiplam i M1 nie hamowały (hamowania odwracalnego ani zależnego od czasu) żadnego z badanych izoenzymów CYP (CYP1A2, 2B6, 2C8, 2C9, 2C19, 2D6), z wyjątkiem CYP3A.
Lek Evrisdi® jest słabym inhibitorem CYP3A. U zdrowych dorosłych osób przyjmowanie leku Evrisdi® raz dziennie przez 2 tygodnie nieznacznie zwiększało ekspozycję na midazolam, wysoce wrażliwy substrat CYP3A (AUC o 11 %; Cmax o 16 %). Takiej interakcji nie uważa się za klinicznie istotną, dlatego nie jest wymagana korekta dawki substratów CYP3A.
Ze względu na wyniki zastosowania modelu farmakokinetycznego opartego na fizjologii (PBPK), podobny efekt można oczekiwać u dzieci i niemowląt w wieku od 2 miesięcy.
Badania in vitro wykazały, że risdiplam i jego główny metabolit nie są istotnymi inhibitorami MDR1 człowieka, organicznego polipeptydu-transportera anionów (OATP)1B1, OATP1B3, organicznego transportera anionów 1 i 3 (OAT 1 i 3). Jednakże risdiplam i jego główny metabolit in vitro są inhibitorami organicznego kationowego transportera 2 (OCT2) człowieka oraz transporterów białek oporności wielolekowej i wydalania toksyn (MATE)1 i transporterów MATE2-K. W przypadku stosowania w stężeniach terapeutycznych aktywnej substancji nie przewiduje się interakcji leku z substratami OCT2. Wpływ równoczesnego stosowania risdiplamu na farmakokinetykę substratów MATE1 i MATE2-K u człowieka jest nieznany. Na podstawie danych in vitro, lek Evrisdi® może zwiększać stężenie w osoczu aktywnych substancji wydzielanych za pomocą MATE1 lub MATE2-K, np. metformyny (patrz sekcja „Farmakokinetyka”). Jeśli nie można uniknąć równoczesnego stosowania, należy prowadzić monitorowanie pod kątem toksyczności związanej z lekiem oraz rozważyć możliwość zmniejszenia dawki innego równocześnie stosowanego leku w razie potrzeby.
Wpływ innych leków na Evrisdi®
Risdiplam metabolizowany jest głównie przy udziale flawinowych monooksygenaz 1 i 3 (FMO1 i 3), a także przy pomocy CYP 1A1, 2J2, 3A4 i 3A7. Risdiplam nie jest substratem białka oporności wielolekowej 1 człowieka (MDR1).
Podczas jednoczesnego stosowania silnego inhibitorem CYP3A itrakonazolu w dawce 200 mg dwa razy dziennie oraz risdiplamu w dawce 6 mg jednorazowo doustnie nie zaobserwowano klinicznie istotnego wpływu na farmakokinetykę (FK) risdiplamu (zwiększenie AUC o 11 %, zmniejszenie Cmax o 9 %). Podczas jednoczesnego stosowania z inhibitorem CYP3A nie jest wymagana korekta dawki leku Evrisdi®.
Nie przewiduje się interakcji z innymi lekami poprzez ścieżki sygnałowe pośredniczone przez FMO1 i FMO3.
Szczególne wytyczne dotyczące stosowania.
Ogólne informacje
W badaniach na zwierzętach obserwowano zmiany w siatkówce, nabłonku, szczególnie w skórze i przewodzie pokarmowym, oraz objawy toksyczności szpiku kostnego (zmiany w morfologii krwi). Obecnie nie można ostatecznie ocenić ryzyka takich zmian u ludzi ze względu na ograniczone dane długoterminowe dotyczące bezpieczeństwa.
Toksykoembriofetalna
W badaniach na zwierzętach zaobserwowano działanie toksyczne na embrion i płód. Pacjentów w wieku rozrodczym należy poinformować o istniejących ryzykach. Należy stosować skuteczne metody antykoncepcji podczas leczenia oraz co najmniej przez 1 miesiąc po podaniu ostatniej dawki leku Evrisdi® u kobiet i przez 4 miesiące po podaniu ostatniej dawki leku Evrisdi® u mężczyzn (patrz sekcja „Sposób i dawki stosowania”).
Potencjalny wpływ na płodność mężczyzn
Ze względu na odwracalne działanie leku Evrisdi® na płodność mężczyzn, zaobserwowane w badaniach na zwierzętach, mężczyźni nie powinni być dawcami nasienia podczas leczenia oraz przez 4 miesiące po podaniu ostatniej dawki leku Evrisdi® (patrz sekcja „Farmakokinetyka”).
Należy unikać kontaktu proszku oraz odtworzonego roztworu doustnego ze skórą. W przypadku kontaktu leku (proszku lub roztworu) ze skórą, miejsce narażone należy przemyć wodą z mydłem.
Substancje pomocnicze.
Ten lek zawiera 0,38 mg benzoesanu sodu w 1 ml. Ten lek zawiera mniej niż 1 mmol sodu (23 mg) na dawkę, co oznacza, że jest uznawany za bez sodu.
Ten lek zawiera izomaltozę. Pacjenci z rzadką wrodzoną nietolerancją fruktozy nie powinni stosować tego leku.
Stosowanie w okresie ciąży lub karmienia piersią.
Na podstawie wyników badań przedklinicznych, płodność mężczyzn może być zaburzona podczas leczenia lekiem Evrisdi®. U szczurów i małp obserwowano degenerację plemników oraz zmniejszenie liczby plemników.
U mężczyzn przed rozpoczęciem leczenia lekiem Evrisdi® należy omówić strategie zachowania płodności. Mężczyźni mogą rozważyć możliwość zabezpieczenia nasienia przed rozpoczęciem leczenia lub po okresie bez leczenia trwającym co najmniej 4 miesiące (patrz sekcja „Szczególne wytyczne dotyczące stosowania”).
Na podstawie wyników badań przedklinicznych, nie oczekuje się wpływu leku Evrisdi® na płodność kobiet.
Kobiety w wieku rozrodczym należy przebadać pod kątem ciąży przed rozpoczęciem leczenia lekiem Evrisdi®.
Pacjentom płci męskiej i żeńskiej w wieku rozrodczym należy przestrzegać następujących wymagań dotyczących antykoncepcji:
- Pacjenci płci męskiej i ich partnerki w wieku rozrodczym powinni stosować skuteczną antykoncepcję podczas leczenia lekiem Evrisdi® oraz przez co najmniej 4 miesiące po podaniu ostatniej dawki leku.
- Pacjentki płci żeńskiej w wieku rozrodczym powinny stosować skuteczną antykoncepcję podczas leczenia lekiem Evrisdi® oraz przez co najmniej 1 miesiąc po podaniu ostatniej dawki leku.
Ciąża
Brak danych klinicznych dotyczących stosowania leku Evrisdi® u kobiet w ciąży. Risdiplam wykazał działanie toksyczne na embrion i płód oraz działanie teratogenne u zwierząt. Na podstawie danych z badań na zwierzętach, risdiplam przenika przez barierę łożyskową i może powodować uszkodzenia płodu.
Leku Evrisdi® nie należy stosować w czasie ciąży, chyba że istnieje wyraźna konieczność. Jeżeli kobieta w ciąży wymaga leczenia lekiem Evrisdi®, należy jasno wyjaśnić potencjalne ryzyko dla płodu.
Karmienie piersią
Nie wiadomo, czy lek Evrisdi® wydzielany jest w mleko ludzkie. Badania na szczurach wykazały, że risdiplam wydzielany jest w mleko matki. Ze względu na nieznaną możliwość szkodzenia niemowlęciu karmionemu piersią, lekarz leczący powinien podjąć decyzję dotyczącą dalszej terapii pacjenta. W czasie leczenia lekiem Evrisdi® nie zaleca się karmienia piersią.
Wpływ na zdolność prowadzenia pojazdów lub obsługiwania maszyn.
W odpowiednich badaniach nie badano wpływu leku Evrisdi® na szybkość reakcji podczas prowadzenia pojazdów lub obsługi innych urządzeń.
Sposób stosowania i dawki
Roztwór doustny leku Evrisdi® powinien być przygotowany przez personel medyczny (tj. lekarza lub farmaceutę) przed wydaniem go pacjentowi.
Przed podaniem pierwszej dawki personel medyczny musi omówić z pacjentem lub osobą opiekującą się pacjentem, sposób przygotowania i przyjęcia przepisanej dawki dobowej (patrz poniżej „Instrukcje dotyczące postępowania”).
Leczenie lekiem Evrisdi® należy rozpoczynać i nadzorować lekarzom posiadającym doświadczenie w rozpoznawaniu i leczeniu pacjentów ze spinalną atrofią mięśniową.
Program klinicznego rozwoju nie obejmował pacjentów z SMA typu IV.
Zalecane dawkowanie
Lek Evrisdi® przyjmuje się doustnie raz dziennie, w mniej więcej tym samym czasie każdego dnia, za pomocą dołączonych do opakowania wielokrotnego użytku strzykawek do dawkowania doustnego. Zalecaną dawkę Evrisdi® w leczeniu SMA ustala się w zależności od wieku i masy ciała pacjenta (patrz tabela 5).
Tabela 5
Schemat dawkowania zależny od wieku i masy ciała
| Wiek i masa ciała |
Zalecana dawka dobową |
| < 2 miesięcy |
0,15 mg/kg |
| od 2 miesięcy do < 2 lat |
0,20 mg/kg |
| ≥ 2 lat i masa ciała < 20 kg |
0,25 mg/kg |
| ≥ 2 lat i masa ciała ≥ 20 kg |
5 mg |
a Na podstawie skorygowanego wieku dla niemowląt urodzonych przed terminem.
Dawkowanie należy dostosować pod nadzorem pracownika opieki zdrowia. Dotychczas nie badano dawki dziennych wyższych niż 5 mg. Dostępne są jedynie bardzo ograniczone dane dotyczące bezpieczeństwa u niemowląt w wieku do 16 dni, którym podawano Evrisdi® w dawce zalecanej w okresie po rejestrowym (patrz punkty „Farmakokinetyka”, „Dzieci”).
Pacjenci z zaburzeniem funkcji wątroby
U pacjentów z łagodnym lub umiarkowanym zaburzeniem funkcji wątroby nie jest wymagana korekta dawki. Nie badano stosowania Evrisdi® u pacjentów z ciężkim zaburzeniem funkcji wątroby (patrz sekcja „Farmakokinetyka”).
Pacjenci z zaburzeniem funkcji nerek
Nie badano bezpieczeństwa i skuteczności stosowania Evrisdi® u pacjentów z zaburzeniem funkcji nerek. Nie oczekuje się potrzeby korekty dawki u pacjentów z zaburzeniem funkcji nerek (patrz sekcja „Farmakokinetyka”).
Pacjenci w wieku podeszłym
Badania kliniczne Evrisdi® nie obejmowały pacjentów w wieku 65 lat i starszych, dlatego nie ustalono, czy odpowiedź na leczenie może być u nich inna niż u młodszych pacjentów.
Dzieci
Zastosowanie Evrisdi® u pacjentów z SMA w wieku do 2 miesięcy jest potwierdzone danymi farmakokinetycznymi i bezpieczeństwa u dzieci w wieku od 16 dni (patrz sekcje „Działania niepożądane” oraz podpunkty „Skuteczność kliniczna” i „Farmakokinetyka”). Nie ma danych klinicznych ani farmakokinetycznych dotyczących stosowania leku u niemowląt urodzonych przed terminem ani u noworodków w wieku do 16 dni (patrz powyższy podpunkt „Skuteczność kliniczna”).
Opóźniony przyjmowanie leku
Evrisdi® należy przyjmować doustnie raz dziennie, w przybliżeniu o tej samej porze każdego dnia. Jeżeli dawkę Evrisdi® pominięto, należy ją przyjąć tak szybko jak to możliwe, jeśli od zaplanowanego czasu minęło nie więcej niż 6 godzin, a następnie od następnego dnia powrócić do normalnego harmonogramu dawkowania. W przeciwnym razie nie należy uzupełniać pominiętej dawki, a następną dawkę należy przyjąć w zaplanowanym czasie następnego dnia.
Jeśli dawka nie została całkowicie połknięta lub wystąpiła wymiana po przyjęciu Evrisdi®, nie należy przyjmować kolejnej dawki w celu uzupełnienia utraconej dawki. Należy poczekać do następnego dnia, aby przyjąć następną dawkę w zaplanowanym czasie.
Sposób podania
Do podania dziennej dawki Evrisdi® należy użyć doustnej strzykawki wielokrotnego użytku dostarczanej w opakowaniu tekturowym razem z lekiem (patrz tabela 6).
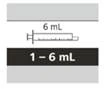
Tabela 6
Dobór odpowiedniej doustnej strzykawki wielokrotnego użytku do przyjęcia przepisanej dziennej dawki Evrisdi®
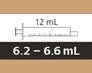
| Wielkość strzykawki |
Objętość dawkowania |
Cena podziału strzykawki |
| 1 ml |
od 0,3 do 1 ml |
0,01 ml |
| 6 ml |
od 1 do 6 ml |
0,1 ml |
| 12 ml |
od 6,2 do 6,6 ml |
0,2 ml |
Przy obliczaniu objętości dawki należy również uwzględnić wartość podziałki strzykawki. Zaokrąglij objętość dawki w górę lub w dół do najbliższej wartości podziałki oznaczonej na wybranej strzykawce dożylniej wielokrotnego użytku (np. z 6,3 ml do 6,4 ml, z 3,03 ml do 3 ml i z 1,05 do 1,1 ml).
Lek Evrisdi® w postaci roztworu należy przyjąć natychmiast po nabraniu go do strzykawki dożylniej wielokrotnego użytku. Jeśli zawartość strzykawki nie zostanie przyjęta w ciągu 5 minut, należy opróżnić strzykawkę dożylną z leku (patrz niżej „Unieszkodliwienie niewykorzystanego leku/skończonego leku”) i przygotować nową dawkę.
Lek Evrisdi® należy przyjmować po posiłku. Pacjent powinien wypić wodę po przyjęciu Evrisdi®, aby upewnić się, że lek został całkowicie połknięty. Jeśli pacjent nie może połykać i ma założoną sondę nosowo-żołądkową lub przetokę gastrostomijną, Evrisdi® można podawać przez sondę/przetokę. Po podaniu leku sondę/przetokę należy przemyć wodą (patrz niżej „Instrukcje dotyczące postępowania”).
Instrukcje dotyczące postępowania
Zasady, których należy przestrzegać przed, podczas i po przygotowaniu doustnego roztworu:
- Roztwór zawsze powinien być przygotowywany przez personel medyczny (czyli lekarza lub farmaceutę).
- Należy unikać wdychania proszku leku Evrisdi®. Należy zwrócić uwagę na lokalne przepisy i stosować odpowiednie wyposażenie do przygotowania roztworu Evrisdi®.
- Należy zakładać rękawiczki.
- Nie należy stosować proszku, jeśli upłynął jego termin ważności. Termin ważności proszku jest wydrukowany na etykiecie butelki i na tekturowej puszce.
- Nie należy wydawać odtworzonego roztworu, jeśli termin ważności gotowego do spożycia roztworu doustnego – data podana na etykiecie butelki i na tekturowej puszce – przekracza pierwotny termin ważności proszku.
- Unikaj kontaktu leku z powierzchnią skóry. Jeśli lek (proszek lub roztwór) dostanie się na skórę, należy przemyć miejsce wodą z mydłem.
- Nie należy stosować leku, jeśli zawartość opakowania jest uszkodzona lub brakuje jej.
- Do przygotowania roztworu należy używać wody oczyszczonej lub wody do wstrzykiwań.
- Nie należy używać strzykawek dożylnych innych niż te dołączone do tekturowej puszki razem z lekiem.
- Nie należy mieszać Evrisdi® z pożywieniem ani płynami (np. mlekiem lub mleczną mieszanką).
- Nie należy mieszać Evrisdi® z nowej butelki z lekiem z butelką, której aktualnie używasz.
Pacjent lub osoba opiekująca się pacjentem powinna zostać poinstruowana przez personel medyczny, jak należy przygotować i przyjąć przepisaną dawkę dobową przed dostarczeniem przygotowanego roztworu.
Przygotowanie doustnego roztworu
Nalej 79 ml wody oczyszczonej lub wody do wstrzykiwań do butelki z lekiem.
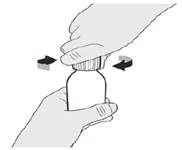
Włóż adapter wciskany do butelki do otworu butelki, wciskając go w dół.
Po całkowitym zamknięciu butelki wstrząśnij przez 15 sekund.
Po 10 minutach powinien powstać przezroczysty roztwór. Jeśli roztwór nie stanie się przezroczysty, należy go wstrząsnąć ponownie przez kolejne 15 sekund.
Należy obliczyć 64 dni od daty przygotowania roztworu. Dzień przygotowania roztworu uznaje się za dzień 0. Obliczoną datę należy wpisać na etykiecie butelki i na tekturowej puszce w polu: Odtworzony roztwór doustny WYRZUCIĆ PO: (dzień/miesiąc/rok).
Unieszkodliwienie niewykorzystanego leku/skończonego leku
Emisję leków do środowiska należy ograniczyć do minimum. Nie wolno unieszkodliwiać leków poprzez ścieki, należy również unikać unieszkodliwiania razem z odpadami komunalnymi.
Niewykorzystany lek/lek z upłyniętym okresem ważności należy profesjonalnie unieszkodliwić w miejscu wydawania leku (przez lekarza lub farmaceutę).
Instrukcja przygotowania roztworu (TYLKO DLA PERSONELU MEDYCZNEGO, W TYM LEKARZY LUB FARMACEUTÓW)
Roztwór doustny Evrisdi® powinien być przygotowany przez personel medyczny przed wydaniem pacjentowi.
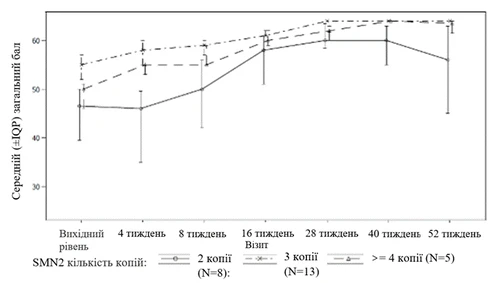
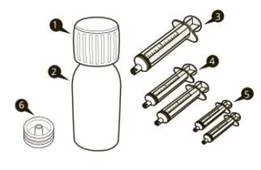
| Jedno opakowanie Evrisdi® zawiera (patrz rysunek A):
|
Rysunek A |
Ważne informacje dotyczące leku Evrisdi®
- Roztwór zawsze należy przygotowywać przez personel medyczny (lekarza lub farmaceutę).
- Unikać wdychania proszku leku Evrisdi®. Należy przestrzegać lokalnych wymogów i stosować odpowiednie wyposażenie podczas przygotowywania roztworu Evrisdi®.
- Należy nosić rękawice.
- Nie stosować tego leku po upływie terminu ważności. Data upływu terminu ważności jest podana na etykiecie butelki i na tekturowym opakowaniu.
- Unikać wszelkiego kontaktu tego leku z powierzchnią skóry. W przypadku kontaktu (z proszkiem lub roztworem) należy przemyć miejsce kontaktu wodą z mydłem.
- Nie stosować tego leku, jeśli którykolwiek element opakowania jest uszkodzony lub brakuje.
- Do przygotowania roztworu należy używać wody oczyszczonej lub wody do wstrzykiwań.
- Nie należy używać wielokrotnego dozownika doustnego oprócz dozownika dostarczonego w opakowaniu.
- Nie należy wydawać przygotowanego roztworu, jeśli termin ważności gotowego do użycia roztworu doustnego – data podana na etykiecie butelki i na tekturowym opakowaniu – przekracza datę upływu terminu ważności proszku.
- Przed wydaniem przygotowanego roztworu personelowi medycznemu należy poinstruować pacjenta lub opiekuna, w jaki sposób przygotować i przyjąć dobową dawkę.
Jak przechowywać lek Evrisdi®
- Proszek (środek leczniczy nieprzygotowany jeszcze jako roztwór) należy przechowywać w tekturowym opakowaniu w temperaturze pokojowej nie wyższej niż 25 °C w celu ochrony przed światłem i wilgocią.
- Roztwór (środek leczniczy przygotowany jako roztwór) należy przechowywać w położeniu pionowym w lodówce w temperaturze od 2 do 8 °C w tekturowym opakowaniu w celu ochrony zawartości przed światłem.
- W razie potrzeby roztwór doustny można przechowywać w temperaturze pokojowej (poniżej 40 °C) łącznie nie dłużej niż przez 5 dni. Nie należy przechowywać roztworu doustnego w temperaturze wyższej niż 40 °C.
- Roztwór doustny należy przechowywać w oryginalnej butelce w położeniu pionowym i z dobrze zamkniętym korkiem.
Przygotowanie roztworu
|
Rysunek B |
Krok 1 Delikatnie postukaj w dno fiolki, aby rozluźnić proszek (patrz rysunek B). |
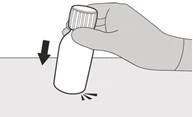
|
Rysunek C |
Krok 2 Zdejmij pokrywkę, naciskając ją w dół, a następnie skręcając w lewo (przeciwnie do ruchu wskazówek zegara) (patrz rysunek C). Nie wyrzucaj pokrywki. |
|
Rysunek D |
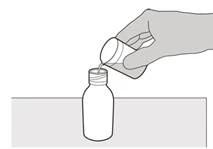
Krok 3 Delikatnie dodaj 79 ml wody do wody oczyszczonej lub wody do wstrzykiwań do butelki z lekiem (patrz rysunek D). |
|
Rysunek E |
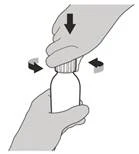
Krok 4 Trzymaj butelkę z lekiem jedną ręką na stole. Włóż adapter do naciskania butelki do otworu butelki, naciskając go w dół drugą ręką, aż adapter będzie całkowicie przylegał do brzegu otworu butelki (patrz rysunek E). |
|
Rysunek F |
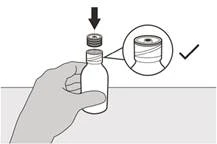
Krok 5 Należy ponownie założyć pokrywkę na butelkę. Obróć pokrywkę w prawo (zgodnie z ruchem wskazówek zegara), aby zamknąć butelkę. Upewnij się, że butelka jest całkowicie zamknięta, a następnie dokładnie wstrząśnij przez 15 sekund (patrz rysunek F). Poczekaj 10 minut. Powinieneś otrzymać klarowny roztwór. Jeśli nie, ponownie dokładnie wstrząśnij przez 15 sekund. |
|
Rysunek G |
Krok 6 Określ datę „WYRZUCIĆ PO: (dzień/miesiąc/rok)”, licząc 64 dni od dnia przygotowania roztworu (uwaga: dzień przygotowania liczy się jako dzień 0. Na przykład, jeśli przygotowałeś roztwór 1 kwietnia, datą „WYRZUCIĆ PO” będzie 4 czerwca). Wpisz tę datę w odpowiednie pole na etykiecie butelki (patrz rysunek G) oraz na tekturowej puszce: Odrodzony roztwór do doustnego stosowania WYRZUCIĆ PO: (dzień/miesiąc/rok). Umieść butelkę z powrotem do tekturowej puszki razem z strzykawkami (w torebkach) i instrukcją do użytkowania medycznego. Wszystko przechowuj w lodówce, trzymając butelkę w pionowym położeniu. |
INSTRUKCJA STOSOWANIA ROZTWORU DO PRZEDŁUŻNEGO
Należy uważnie przeczytać i zrozumieć instrukcję do stosowania medycznego przed rozpoczęciem przyjmowania Evrisdi®. Zawiera ona informacje na temat przygotowania i podania dawki Evrisdi® za pomocą wielokrotnego dożylnego strzykawki dożylnego doustnie, przez zgłębnik gastrostomii (sondę PEG) lub przez zgłębnik nosowożołądkowy (sondę do żołądka). W przypadku pytań dotyczących stosowania Evrisdi® należy skontaktować się z lekarzem lub farmaceutą. Przed wydaniem roztwór doustny Evrisdi® musi być przygotowany przez personel medyczny, takiego jak lekarz lub farmaceuta.
Lek Evrisdi® powinien być dostarczony w postaci płynu w butelce. Nie należy stosować, jeśli lek w fiolce jest w postaci proszku, i należy skontaktować się z lekarzem lub farmaceutą w takim przypadku.
Przed przyjęciem/podaniem pierwszej dawki personel medyczny musi poinstruować, jak dokładnie przygotować i przyjmować/podać dawkę dobową.
- Poproś lekarza lub farmaceutę o pokazanie, który z dostępnych w opakowaniu strzykawek doustnych należy użyć do podania i jak ustalić dawkę dobową.
- W przypadku zaginięcia lub uszkodzenia strzykawki doustnej należy skontaktować się z lekarzem lub farmaceutą, który doradzi, jak kontynuować przyjmowanie leku.
- Do dokładnego odmierzenia ustalonej dawki dobowej należy zawsze używać strzykawki odpowiedniego rozmiaru, przeznaczonej do wielokrotnego użytku.
- Opis odpowiedniej dla Ciebie strzykawki doustnej do ponownego użytku znajduje się również w Tabeli 6: „Wybór odpowiedniej strzykawki doustnej do ponownego użytku do podania przepisanej dawki dobowej Evrisdi®”. W razie pytań dotyczących właściwego wyboru strzykawki należy skonsultować się z lekarzem lub farmaceutą.
- Jeśli nie ma adaptera butelkowego w butelce, nie należy stosować Evrisdi® i należy skontaktować się z lekarzem lub farmaceutą.
- Nie należy stosować Evrisdi® po dacie „Roztwór doustny po przygotowaniu WYRZUĆ PO: (dzień/miesiąc/rok)”, podanej na etykiecie butelki i opakowaniu tekturowym. Jeśli na etykiecie butelki lub opakowaniu tekturowym nie ma daty „Roztwór doustny po przygotowaniu WYRZUĆ PO: (dzień/miesiąc/rok)”, należy zapytać lekarza lub farmaceuty o datę.
- Nie należy mieszać Evrisdi® z pożywieniem ani płynami (np. mlekiem lub mleczną mieszanką).
- Nie należy stosować Evrisdi®, jeśli butelka lub strzykawka doustna są uszkodzone.
- Należy unikać kontaktu Evrisdi® z powierzchnią skóry. W przypadku kontaktu Evrisdi® z powierzchnią skóry należy przemyć miejsce wodą z mydłem.
- W przypadku rozlania Evrisdi® należy przetrzeć miejsce suchą chusteczką papierową, a następnie oczyścić wodą z mydłem. Wyrzucić chusteczkę papierową do śmieci i dokładnie umyć ręce wodą z mydłem.
- Jeśli w butelce nie ma wystarczającej ilości leku Evrisdi® na przepisaną dawkę, należy zwrócić butelkę i używane strzykawki doustne do miejsca wydania (lekarzowi lub farmaceucie) w celu odpowiedniego utylizacji. Należy użyć nowej butelki Evrisdi® do odmierzenia przepisanej dawki dobowej. Nie wolno mieszać Evrisdi® z nowej butelki z lekiem z butelki, z której wcześniej korzystano.
Każde opakowanie leku Evrisdi® zawiera (patrz rysunek A):
|
|
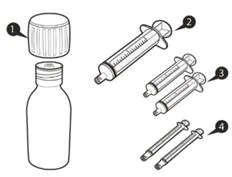
Rysunek A
A) Przygotowanie i pobranie właściwej objętości dawki
Jak wybrać odpowiedni strzykawkę doustną do przepisanego dawkowania leku Evrisdi®
|
|
|
|
|
|

Jak przygotować dobową dawkę leku Evrisdi®
|
Rysunek B |
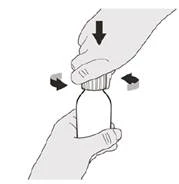
Krok A1 Zdejmij pokrywkę, naciskając ją w dół, a następnie odkręcając w lewo (przeciwnie do ruchu wskazówek zegara) (patrz rysunek B). Nie wyrzucaj pokrywki. |
|||||
|
Rysunek C |
Krok A2 Naciśnij tłok strzykawki do doustnego podania całkowicie w dół, aby usunąć powietrze ze strzykawki (patrz rysunek C). |
|||||
|
Rysunek D |
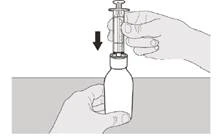
Krok A3 Trzymając butelkę w pionowej pozycji, wsuń końcówkę strzykawki do adaptera butelki (patrz rysunek D). |
|||||
|
Rysunek E |
Krok A4 Delikatnie odwróć butelkę do góry dnem, z końcówką strzykawki mocno włożoną w adapter butelki (patrz rysunek E). |
|||||
|
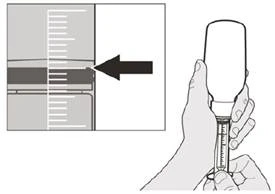
Rysunek F |
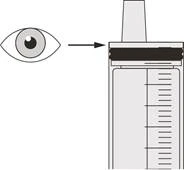
Krok A5 Powoli wyciągnij tłok, aby odmierzyć przepisaną dawkę dzienną leku Evrisdi® z butelki. Górna część czarnego ogranicznika ruchu tłoka powinna być wyrównana z oznaczeniem mililitrów na strzykawce do doustnego podania odpowiadającym przepisanej dawce dziennej (patrz rysunek F). Po odmierzeniu właściwej dawki trzymaj tłok nieruchomo w miejscu, aby się nie przesuwał. |
|||||
|
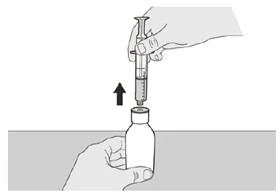
Rysunek G |
Krok A6 Trzymaj tłok nieruchomo w miejscu, aby się nie przesuwał. Pozostaw strzykawkę doustną w adapterze butelki i obróć butelkę do pozycji pionowej. Postaw butelkę na płaskiej powierzchni. Wyjmij strzykawkę doustną z adaptera butelki, delikatnie wyciągając ją do góry, trzymając tłok nieruchomo (patrz rysunek G). |
|||||
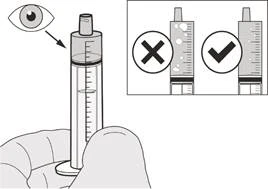
|
Rysunek H |
Krok A7 Trzymaj strzykawkę doustną tak, aby jej końcówka była skierowana do góry. Sprawdź lek Evrisdi® w strzykawce doustnej. Jeśli w strzykawce doustnej pojawią się duże pęcherzyki powietrza (patrz rysunek H) lub jeśli odmierzono niewłaściwą dawkę dzienną leku Evrisdi®, wsuń końcówkę strzykawki mocno do adaptera butelki. Naciśnij tłok w dół, aż lek Evrisdi® przepłynie z powrotem do butelki i powtórz kroki A4–A7. Podaj lub przyjmij lek Evrisdi® natychmiast po odmierzeniu go w strzykawce doustnej. Jeśli lek nie zostanie przyjęty w ciągu 5 minut, usuń i zutylizuj lek Evrisdi® ze strzykawki doustnej i odmierz nową dawkę. |
|||||
|
Rysunek I |
Krok A8 Pozostaw adapter butelki na butelce. Załóż ponownie pokrywkę na butelkę. Obróć pokrywkę w prawo (zgodnie z ruchem wskazówek zegara), aby szczelnie zamknąć butelkę (patrz rysunek I). |
|||||
Jeśli przyjmujesz dobową dawkę leku Evrisdi® doustnie, postępuj zgodnie z instrukcjami w punkcie «B) Jak przyjmować dawkę leku Evrisdi® doustnie».
Jeśli przyjmujesz dobową dawkę leku Evrisdi® przez rurkę gastrostomijną, postępuj zgodnie z instrukcjami w punkcie «C) Jak podawać dawkę leku Evrisdi® przez rurkę gastrostomijną».
Jeśli przyjmujesz dobową dawkę leku Evrisdi® przez sondę nosowo-żołądkową, postępuj zgodnie z instrukcjami w punkcie «D) Jak podawać dawkę leku Evrisdi® przez sondę nosowo-żołądkową».
Doustne strzykawki do leku Evrisdi® zostały specjalnie zaprojektowane pod kątem kompatybilności z systemem ENFit®. Jeśli posiada sonda do żywienia nie jest kompatybilna z systemem ENFit®, może być potrzebny przejściowy łącznik ENFit®, aby podłączyć strzykawkę do leku Evrisdi® do rurki gastrostomijnej lub sondy nosowo-żołądkowej.
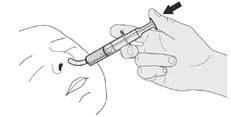
B) Jak przyjmować dawkę dobową leku Evrisdi® doustnie
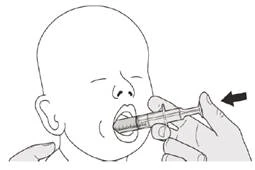
Podczas przyjmowania dawki dobowej Evrisdi® doustnie pozostawaj w pozycji pionowej (najlepiej w siadzie).
|
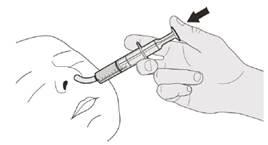
Rysunek J |
Krok B1 Umieść strzykawkę doustną w ustach końcówką wzdłuż dowolnej policzki. Wolno naciśnij do końca tłoczek, aby podać pełną dawkę leku Evrisdi® (patrz rysunek J). Wpłynięcie leku Evrisdi® do gardła lub zbyt szybkie podanie może spowodować duszność. |
|
Rysunek K |
Krok B2 Sprawdź, czy w strzykawce dożylniej pozostał jeszcze lek Evrisdi® (patrz rysunek K). |
|
Rysunek L |
Krok B3 Wypij trochę wody bezpośrednio po podaniu przepisanej dawki leku Evrisdi® (patrz rysunek L). Zobacz krok E w celu uzyskania informacji o czyszczeniu strzykawki. |
C) Jak podawać dobową dawkę leku Evrisdi® przez rurkę gastrostomijną
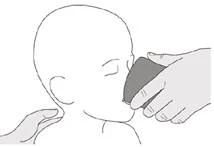
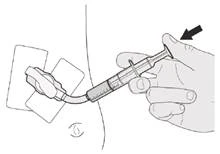
Jeśli podajesz lek Evrisdi® przez rurkę gastrostomijną, poproś lekarza o pokazanie, jak należy sprawdzić rurkę gastrostomijną przed podaniem leku Evrisdi®.
|
Rysunek M |
Krok C1 Umieścić końcówkę doustną strzykawki w rurce gastrostomijnej. Delikatnie nacisnąć tłoczek do końca, aby wprowadzić pełną dawkę leku Evrisdi® (patrz rysunek M). |
|
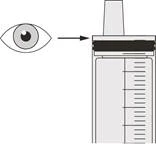
Rysunek N |
Krok C2 Sprawdzić, czy w strzykawce doustnej nie pozostał lek Evrisdi® (patrz rysunek N). |
|
Rysunek O |
Krok C3 Przemyć rurkę gastrostomijną 10–20 ml wody bezpośrednio po podaniu przepisanej dawki leku Evrisdi® (patrz rysunek O). Patrz krok E w celu uzyskania informacji na temat czyszczenia strzykawki. |
D) Jak podawać dobową dawkę leku Evrisdi® przez zgłębnik nosowo-żołądkowy
Jeśli podajesz lek Evrisdi® przez zgłębnik nosowo-żołądkowy, poproś lekarza o pokazanie, w jaki sposób sprawdzić zgłębnik nosowo-żołądkowy przed podaniem leku Evrisdi®.
|
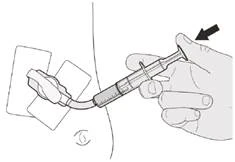
Rysunek P |
Krok D1 Umieść końcówkę strzykawki do doustnego podania w sondzie nosowo-żołądkowej. Naciskaj powoli tłoczek do końca, aby podać pełną dawkę leku Evrisdi® (patrz rysunek P). |
|
Rysunek Q |
Krok D2 Sprawdź, czy w strzykawce do doustnego podania nie pozostał żaden lek Evrisdi® (patrz rysunek Q). |
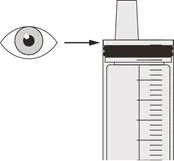
| Rycunek R |
Krok D3 Przepłucz sondę nosowo-żołądkową 10–20 ml wody bezpośrednio po podaniu przepisanej dawki leku Evrisdi® (patrz rycunek R). Informacje dotyczące czyszczenia strzykawki znajdują się w kroku E. |
E) Jak wyczyścić strzykawkę doustną po zastosowaniu
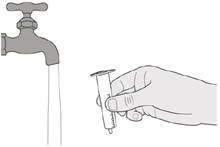
|
Rysunek S |
Krok E1 Wyciągnij strzykawka tłok (przeciwko oporowi) ze strzykawki. Dobrze wypłucz cylindryczny pojemnik strzykawki do doustnego podawania czystą bieżącą wodą (patrz rysunek S). |
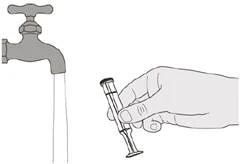
|
Rysunek T |
Krok E2 Dobrze przemyj tłok pod bieżącą czystą wodą (patrz rysunek T). |
|
Rysunek U |
Krok E3 Sprawdź, czy cylinder i tłoczek strzykawki do doustnego podawania są czyste. Połóż cylinder i tłoczek strzykawki do doustnego podawania na czystej powierzchni w bezpiecznym miejscu, aby wyschnęły (patrz rysunek U). Wymyj ręce. Po wyschnięciu cylindra i tłoczka strzykawki do doustnego podawania włóż tłoczek z powrotem do cylindra strzykawki i przechowuj strzykawkę do następnego użycia. |
Dzieci.
Bezpieczeństwo i skuteczność leku Evrisdi® u pacjentów w wieku do 16. dnia życia nie zostały ustalone (patrz sekcja „Farmakokinetyka”).
Przedawkowanie.
Brak doświadczenia z przedawkowaniem leku Evrisdi® w badaniach klinicznych. Nie istnieje znany antydotum w przypadku przedawkowania leku Evrisdi®. W przypadku przedawkowania należy dokładnie obserwować stan pacjenta i rozpocząć leczenie wspierające.
Efekty uboczne
Klasyfikacja częstości występowania działań niepożądanych leku: bardzo często (≥ 1/10), często (od ≥ 1/100 do < 1/10), rzadko (od ≥ 1/1 000 do < 1/100), rzadko (od ≥ 1/10 000 do < 1/1000), bardzo rzadko (< 1/10 000); częstość nieznana (nie można oszacować na podstawie dostępnych danych).
Krótki opis profilu bezpieczeństwa
U pacjentów z SMA o początku w okresie niemowlęcym najczęściej obserwowane działania niepożądane stwierdzone w badaniach klinicznych leku Evrisdi® to podwyższenie temperatury ciała (54,8%), wysypka (29%) oraz biegunka (19,4%). U pacjentów z SMA o późniejszym początku najczęściej obserwowane działania niepożądane stwierdzone w badaniach klinicznych leku Evrisdi® to podwyższenie temperatury ciała (21,7%), ból głowy (20%), biegunka (16,7%) oraz wysypka (16,7%).
Wymienione powyżej działania niepożądane występowały bez zidentyfikowanych cech klinicznych lub charakterystycznego związku z czasem i ogólnie ustępowały bez przerywania leczenia lekiem Evrisdi® u pacjentów z SMA o początku w okresie niemowlęcym oraz późnym początku.
Tabela 7
Streszczenie działań niepożądanych u pacjentów z SMA o początku w okresie niemowlęcym i późnym początku, obserwowanych w badaniach klinicznych leku Evrisdi®
| Klasa układu narządów |
MA zaczynająca się w okresie niemowlęcym2 (typ 1) |
MA zaczynająca się później3 (typ 2 i 3) |
|
| Zaburzenia żołądkowo-jelitowe |
|||
| Diareia |
Bardzo często |
Bardzo często |
|
| Nudności |
Nie dotyczy |
Często |
|
| Ulczki błony śluzowej jamy ustnej i aftowe |
Często |
Często |
|
| Zaburzenia ze strony skóry i tkanki podskórnej |
|||
| Wysypka1 |
Bardzo często |
Bardzo często |
|
| Zaburzenia ze strony układu nerwowego |
|||
| Ból głowy |
Nie dotyczy |
Bardzo często |
|
| Zaburzenia ogólne i reakcje w miejscu wstrzyknięcia |
|||
| Zwiększenie temperatury ciała (w tym hiperpirksję) |
Bardzo często |
Bardzo często |
|
| Infekcje i inwazje |
|||
| Infekcja dróg moczowych (w tym zapalenie pęcherza) |
Często |
Często |
|
| Zaburzenia ze strony układu kostno-mięśniowego i tkanki łącznej |
|||
| Artrologia |
Nie dotyczy |
Często |
|
1 W tym przypadku do objawów zaliczane są zapalenie skóry, zapalenie skóry typu trądzikowatego, zapalenie alergiczne skóry, rumień, zapalenie mieszków włosowych, wysypka, wysypka rumieniowa, wysypka plamista i grudkowa, wysypka grudkowa.
2 U pacjentów z SMA o wczesnym początku (badanie FIREFISH, części 1 i 2) reakcje niepożądane zdefiniowano jako zdarzenia występujące u ≥ 2% pacjentów lub więcej, których związek przyczynowy z lekiem Evrisdi® jest możliwy.
3 U pacjentów z SMA o późnym początku (badanie SUNFISH, część 2) reakcje niepożądane zdefiniowano jako zdarzenia występujące częściej o ≥ 2% u pacjentów leczonych lekiem Evrisdi® w porównaniu z grupą placebo w trakcie podwójnie ślepej, kontrolowanej fazy leczenia, oraz których związek przyczynowy z lekiem Evrisdi® jest możliwy.
Dostępne dane dotyczące bezpieczeństwa są ograniczone pod względem liczby pacjentów, którzy otrzymywali lek Evrisdi®, oraz długości ekspozycji. Możliwe są potencjalne rzadkie i potencjalnie poważne reakcje niepożądane (RN), które nie zostały wykryte w trakcie programu badań.
W badaniu RAINBOWFISH uzyskano ograniczone dane dotyczące bezpieczeństwa stosowania leku Evrisdi® u noworodków i niemowląt z przedobjawową SMA. Badanie RAINBOWFISH to otwarte badanie z jedną grupą, w którym wzięło udział 26 pacjentów z przedobjawową SMA w wieku od 16 do 41 dni (zakres masy ciała od 3,1 do 5,7 kg) w momencie podania pierwszej dawki. W analizie pierwotnej średnia długość trwania terapii wynosiła 20,4 miesiąca (zakres: 10,6 do 41,9 miesiąca) (patrz sekcja „Właściwości farmakologiczne”, podsekcja „Skuteczność kliniczna”).
Profil bezpieczeństwa leku Evrisdi® u pacjentów z przedobjawową SMA w badaniu RAINBOWFISH można porównać z profilem bezpieczeństwa u pacjentów z objawową SMA, którzy otrzymywali leczenie lekiem Evrisdi® w trakcie badań klinicznych. Dane długoterminowe są obecnie niedostępne.
Profil bezpieczeństwa u pacjentów z SMA, którzy wcześniej otrzymywali inną terapię modyfikującą chorobę
Na podstawie analizy pierwotnej uczestników badania JEWELFISH profil bezpieczeństwa leku Evrisdi® u wcześniej leczonych pacjentów, którzy otrzymywali leczenie lekiem Evrisdi® przez okres do 59 miesięcy w badaniu JEWELFISH, jest zgodny z profilem bezpieczeństwa u pacjentów z SMA wcześniej nieleczonych, którzy otrzymywali lek Evrisdi® w badaniach FIREFISH (części 1 i 2), SUNFISH (części 1 i 2) oraz RAINBOWFISH.
Do badania JEWELFISH zakwalifikowano pacjentów, którzy wcześniej otrzymywali nusinersen (n = 76) lub onasemnogen abeparvovek (n = 14) (patrz podsekcja „Skuteczność kliniczna”).
Efekty przedkliniczne
Efekty przedkliniczne dotyczące struktury siatkówki, tkanki nabłonkowej oraz parametrów hematologicznych, opisane w sekcji „Dane przedkliniczne”, do chwili obecnej nie zostały zaobserwowane w przeprowadzonych badaniach klinicznych leku Evrisdi® w SMA.
Wydłużenie QT
Analiza farmakokinetyki/farmakodynamiki wykazała brak objawów wydłużenia QTc w wyniku stosowania leku Evrisdi® w zakresie ekspozycji terapeutycznej; jednak nie ma odpowiednich danych dotyczących stosowania leku Evrisdi® przy ekspozycji przekraczającej zakres terapeutyczny.
Doświadczenie z okresu po rejestracji
W okresie po rejestracji zaobserwowano zapalenie naczyń skóry, którego objawy ustąpiły po całkowitym odstawieniu leku Evrisdi®. Na podstawie dostępnych danych niemożliwe jest określenie kategorii częstości występowania tej reakcji niepożądanej. Dostępne są jedynie ograniczone dane dotyczące stosowania u noworodków w wieku poniżej 16 dni w okresie po rejestracji.
Okres ważności.
2 lata.
Po odtworzeniu gotowy do użycia roztwór doustny jest stabilny przez 64 dni, gdy przechowywany jest w lodówce w temperaturze 2–8 ºC. W razie potrzeby pacjent lub osoba opiekująca się pacjentem może przechowywać roztwór doustny w temperaturze pokojowej (poniżej 40 °C) łącznie nie dłużej niż przez 5 dni.
Warunki przechowywania.
Przechowuj proszek do sporządzenia roztworu doustnego w temperaturze nie wyższej niż 25 ºC, w oryginalnym opakowaniu, aby chronić przed światłem i wilgocią. Przechowuj w miejscu niedostępnym dla dzieci.
Gotowy do użycia roztwór doustny należy przechowywać w lodówce w temperaturze 2–8 ºC, w oryginalnym opakowaniu, aby chronić przed światłem. Butelkę należy trzymać szczelnie zamkniętą i zawsze przechowywać w pozycji pionowej.
Niezgodność.
Nie zaobserwowano niezgodności między lekiem Evrisdi® a zalecanymi strzykawkami wielokrotnego użytku do doustnego podawania leku.
Opakowanie.
Szkło butelka o pojemności 100 ml w kolorze bursztynowym (klasa III zgodnie z Farm. USA i Farm. Eur.) z białą pokrywką z funkcją zabezpieczenia przed otwarciem przez dzieci (zewnętrzna warstwa z polietylenu o wysokiej gęstości; wewnętrzna warstwa z homopolimeru polipropylenu), z pierścieniem kontrolującym pierwsze otwarcie i wkładką z polietylenu i poli(chlorku winylidenu) w zestawie z 1 adapterem do butelki do wciskania, 2 strzykawkami doustnymi wielokrotnego użytku o pojemności 1 ml (każda w foliowej torebce polietylenowej), 2 strzykawkami doustnymi wielokrotnego użytku o pojemności 6 ml (każda w foliowej torebce polietylenowej) oraz 1 strzykawką doustną wielokrotnego użytku o pojemności 12 ml (w foliowej torebce polietylenowej), umieszczonymi w foliowej torebce polietylenowej. Po 1 butelce i 1 zestawie w tekturowym pudełku.
Kategoria wydawania.
Na receptę.
Producent.
F. Hoffmann-La Roche Ltd
Miejsce produkcji i adres siedziby producenta.
Wurmisweg, 4303 Kaiseraugst, Szwajcaria