Eurisdi®
UcrainaIndice
ISTRUZIONE per l'uso del medicinale Evrysdi® (Evrysdi®)
Composizione:
sostanza attiva: risdiplam;
1 flacone contiene 60 mg di risdiplam;
1 ml di soluzione ricostituita contiene 0,75 mg di risdiplam;
sostanze ausiliarie: mannitolo (E 421); isomalto (E 953); aroma di fragola; acido tartarico; benzoato di sodio (E 211); polietilenglicole 6000; sucralosio; acido ascorbico; edetato disodico diidrato.
Forma farmaceutica. Polvere per soluzione orale.
Principali caratteristiche fisico-chimiche: Polvere o polvere granulare o massa polverulenta di colore giallo chiaro o giallo, grigiastro-giallo, verdastro-giallo o verde chiaro. La soluzione ricostituita è di colore verdastro-giallo fino a giallo.
Categoria farmacoterapeutica. Medicinali che agiscono sull'apparato muscolo-scheletrico. Altri agenti utilizzati nelle patologie dell'apparato muscolo-scheletrico.
Codice ATC M09A X10.
Proprietà farmacologiche.
Farmacodinamica.
Meccanismo d'azione
Risdiplam è un modulatore dello splicing dell'RNA messaggero precursorio (pre-mRNA) del gene di sopravvivenza dei motoneuroni 2 (SMN2), sviluppato per il trattamento della SMA causata da mutazioni del cromosoma 5q che portano a una carenza della proteina SMN. La carenza funzionale della proteina SMN rappresenta il meccanismo patofisiologico della SMA di tutti i tipi. Risdiplam corregge lo splicing di SMN2, modificando l'equilibrio dall'esclusione all'inclusione dell'esone 7 nel trascritto mRNA, aumentando così la produzione di proteina SMN funzionale e stabile. Pertanto, risdiplam tratta la SMA aumentando e mantenendo livelli funzionali di proteina SMN.
Elettrofisiologia cardiaca
L'effetto di risdiplam sull'intervallo QTc è stato valutato in uno studio condotto su 47 volontari sani adulti. Entro il range terapeutico, risdiplam non ha prolungato l'intervallo QTc.
Risdiplam si distribuisce uniformemente in tutte le parti del corpo, compreso il sistema nervoso centrale, attraversando la barriera emato-encefalica, aumentando così i livelli di proteina SMN nel SNC e nell'intero organismo. La concentrazione plasmatica di risdiplam e i livelli di proteina SMN nel sangue riflettono la distribuzione e gli effetti farmacodinamici di risdiplam in tessuti come il cervello e i muscoli.
Negli studi clinici FIREFISH, SUNFISH e JEWELFISH, condotti su pazienti con SMA ad esordio infantile e con SMA ad esordio tardivo, risdiplam ha determinato un aumento costante e duraturo del livello di proteina SMN misurato nel sangue, con una variazione mediana superiore al doppio rispetto ai livelli basali entro 4 settimane dall'inizio del trattamento. Tale aumento dei livelli di proteina SMN è stato mantenuto per tutta la durata del trattamento, per almeno 24 mesi (vedi sezione «Efficacia clinica»).
Efficacia clinica
L'efficacia del medicinale Eurisdi® nel trattamento di pazienti con SMA di tipo infantile (SMA tipo 1) e con SMA ad esordio tardivo (SMA tipo 2 e 3) è stata valutata in due studi clinici principali, FIREFISH e SUNFISH, e ulteriormente confermata da dati aggiuntivi dello studio JEWELFISH. L'efficacia di Eurisdi® nel trattamento di pazienti con SMA presintomatica è stata valutata in base all'analisi intermedia di endpoint secondari dello studio RAINBOWFISH in corso.
Pazienti con diagnosi clinica di SMA tipo 4 non hanno partecipato agli studi clinici.
Negli studi clinici è stata dimostrata un'efficacia a lungo termine per almeno 24 mesi di trattamento. I dati sull'uso di Eurisdi® oltre i 2 anni sono limitati.
SMA di tipo infantile
Lo studio BP39056 (FIREFISH) – uno studio aperto in due parti, per valutare efficacia, sicurezza, farmacocinetica e farmacodinamica di Eurisdi® in pazienti con SMA tipo 1 sintomatici (tutti i pazienti avevano una conferma genetica della malattia con due copie del gene SMN2). La parte 1 dello studio FIREFISH è stata progettata come studio di ricerca della dose. Nella parte confermatoria 2 dello studio FIREFISH è stata valutata l'efficacia di Eurisdi® alle dosi terapeutiche selezionate sulla base dei risultati della parte 1 (vedi sezione «Modalità di somministrazione e posologia»). I pazienti della parte 1 non hanno partecipato alla parte 2.
Complessivamente, 62 pazienti con SMA tipo 1 sintomatico sono stati inclusi nella parte 1 (n = 21) e nella parte 2 (n = 41) dello studio FIREFISH, di cui 58 hanno ricevuto la dose terapeutica di Eurisdi®. L'età media all'esordio dei sintomi clinici era di 1,5 mesi (0,9–3 mesi). L'età media al momento dell'inclusione nello studio era di 5,6 mesi (2,2–6,9 mesi), mentre il tempo medio tra l'esordio dei sintomi e la prima dose era di 3,7 mesi (1–6 mesi). Il 60% dei pazienti era di sesso femminile, il 57% di razza caucasica e il 29% asiatici. All'inizio dello studio, il punteggio mediano CHOP-INTEND era 23 (8–37) e il punteggio mediano HINE-2 era 1 (0–5). Le caratteristiche demografiche e cliniche basali dei pazienti inclusi nella parte 1 erano sovrapponibili a quelle della parte 2.
Il principale endpoint dello studio era la percentuale di pazienti in grado di sedersi senza supporto per almeno 5 secondi, come definito dal punto 22 della scala di sviluppo per neonati e bambini in età prescolare di Bayley – III edizione (BSID-III) per la valutazione della motricità generale, dopo 12 mesi di trattamento con Eurisdi® nella parte 2 dello studio; tale risultato è stato raggiunto nel 29% dei pazienti (n = 12/41, IC 90%: 17,8%, 43,1%, p <0,0001).
I risultati chiave sull'efficacia nei pazienti trattati con Eurisdi® nello studio FIREFISH (dati combinati delle parti 1 e 2) sono riportati nella Tabella 1.
Tabella 1
Riepilogo dei risultati chiave sull'efficacia a 12 e 24 mesi (FIREFISH, parte 1 e parte 2)
| Endpoint di efficacia |
Dopo 12 mesi |
Dopo 24 mesi |
| Frazione di pazienti (IC 90 %) |
||
| Sviluppo motorio e funzione motoria |
N = 58a |
|
| BSID-III: sedersi senza supporto per almeno 5 secondi |
32,8 % |
60,3 % |
| CHOP-INTEND: punteggio pari o superiore a 40 |
56,9 % |
74,1 % |
| Aumento del punteggio CHOP-INTEND di ≥ 4 punti rispetto al basale |
89,7 % |
87,9 % |
| HINE-2: pazienti che rispondono secondo i criteri di sviluppo della funzione motoriab |
77,6 % |
82,8 % |
| Sopravvivenza e sopravvivenza libera da eventi |
N=62a |
|
| Sopravvivenza libera da eventic |
87,1 % |
|
| Sopravvivenza |
91,9 % |
90,3 % |
| Alimentazione |
N = 58a |
|
| Capacità di ricevere nutrizione oraled |
84,5 % |
82,8 % |
BSID-III — Scala di sviluppo Bayley per lattanti e bambini in età precoce — III edizione;
CHOP-INTEND — Test dell’Ospedale Pediatrico di Filadelfia per la valutazione delle funzioni motorie nelle malattie neuromuscolari nei neonati;
HINE-2 — Modulo 2 dell’esame neurologico Hammermith per lattanti.
a I dati sulla sopravvivenza e sulla sopravvivenza senza ventilazione sono stati combinati per tutti i pazienti che hanno ricevuto qualsiasi dose di risdiplam nella parte 1 e nella parte 2 dello studio (n = 62). Per i traguardi dello sviluppo motorio, la funzione motoria e l’alimentazione, i dati sugli endpoint di efficacia sono stati combinati per tutti i pazienti che hanno ricevuto la dose terapeutica di risdiplam (tutti i pazienti nella parte 2 dello studio e i pazienti del gruppo ad alta dose nella parte 1; n = 58).
b Definizione di risposta secondo il criterio HINE-2: la risposta in quest’analisi è definita come un aumento ≥ 2 punti (o il punteggio massimo possibile) nella capacità di pedalare con le gambe OPPURE un aumento ≥ 1 punto in uno dei seguenti traguardi dello sviluppo motorio: controllo del capo, rotolamento, seduta, strisciamento, stazione eretta o deambulazione, E un miglioramento in un numero maggiore di categorie dello sviluppo motorio rispetto al peggioramento.
c L’evento corrispondente all’endpoint della ventilazione permanente è definito come tracheostomia o ≥ 16 ore di ventilazione non invasiva al giorno, oppure intubazione per > 21 giorni consecutivi in assenza o dopo la risoluzione di un episodio acuto reversibile. Quattro pazienti soddisfacevano i criteri per l’endpoint della ventilazione permanente entro 24 mesi. Questi quattro pazienti hanno mostrato un aumento del punteggio CHOP-INTEND di almeno 4 punti rispetto al basale.
d Comprende i pazienti che ricevevano esclusivamente alimentazione orale (41 pazienti a 12 e 24 mesi) e quei pazienti che ricevevano alimentazione orale in combinazione con un sondino per nutrizione artificiale (8 pazienti a 12 mesi e 7 pazienti a 24 mesi).
A 24 mesi, il 40 % (23/58) dei pazienti che ricevevano la dose terapeutica del medicinale Eurisdi® era in grado di stare seduto senza supporto per 30 secondi (BSID-III, punto 26). Inoltre, i pazienti hanno continuato a raggiungere ulteriori traguardi dello sviluppo motorio secondo il punteggio HINE-2 a 24 mesi; il 78 % dei pazienti era in grado di rotolare (il 31 % dei pazienti riusciva a rotolare su un fianco, il 7 % riusciva a rotolare dalla posizione prona a supina e il 40 % riusciva a rotolare dalla posizione supina a prona) e il 28 % dei pazienti era riuscito a raggiungere la seduta (il 16 % sosteneva il peso corporeo e il 12 % stava in piedi con supporto).
La percentuale di pazienti vivi senza necessità di ventilazione permanente (sopravvivenza libera da eventi) era dell’84 % tra tutti i pazienti a 24 mesi. Sei neonati sono deceduti (4 nei primi 3 mesi dall’inclusione nello studio) e un paziente ha interrotto prematuramente il trattamento, morendo 3,5 mesi dopo. Quattro pazienti hanno richiesto ventilazione permanente entro 24 mesi.
SMN di tipo tardivo
Studio BP39055 (SUNFISH) – studio multicentrico in due parti per valutare l’efficacia, la sicurezza, la farmacocinetica e la farmacodinamica del medicinale Eurisdi® in pazienti con diagnosi di SMN di tipo 2 o 3, di età compresa tra 2 e 25 anni. La parte 1 era una fase esplorativa per la determinazione della dose e la parte 2 era una fase confermatoria randomizzata, in doppio cieco, controllata con placebo. I pazienti della parte 1 dello studio non hanno partecipato alla parte 2.
L’endpoint primario è stato valutato come il cambiamento nel punteggio della funzione motoria (MFM32) a 12 mesi rispetto al basale. L'MFM32 consente di valutare un’ampia gamma di funzioni motorie in una vasta popolazione di pazienti con SMN. Il punteggio totale MFM32 è espresso in percentuale (intervallo: da 0 a 100) del punteggio massimo possibile, dove un punteggio più alto indica una maggiore funzione motoria. L'MFM32 misura la capacità di eseguire funzioni motorie correlate ad attività quotidiane importanti. Piccoli cambiamenti nella funzione motoria possono portare a significativi miglioramenti o perdite nelle funzioni quotidiane.
SUNFISH, parte 2
La parte 2 di SUNFISH è una fase randomizzata, in doppio cieco, controllata con placebo, che ha coinvolto 180 pazienti non deambulanti con SMN di tipo 2 (71 %) o di tipo 3 (29 %). I pazienti sono stati randomizzati in rapporto 2:1 per ricevere il medicinale Eurisdi® alla dose terapeutica (vedi sezione «Modalità di somministrazione e posologia») o placebo. La randomizzazione è stata stratificata in base all’età (2-5 anni, 6-11 anni, 12-17 anni, 18-25 anni).
L’età media dei pazienti al momento dell’inizio del trattamento era di 9 anni (intervallo 2-25 anni), la mediana del tempo tra l’insorgenza dei sintomi di SMN e il primo trattamento era di 102,6 (1-275) mesi. Tra i partecipanti allo studio, il 51 % dei pazienti era di sesso femminile, il 67 % era di razza caucasica, il 19 % di origine asiatica. Al momento dell’inizio dello studio, il 67 % dei pazienti aveva scoliosi (il 32 % di questi aveva scoliosi grave). I pazienti avevano un punteggio medio basale MFM32 di 46,1 e un punteggio RULM di 20,1. Nel complesso, le caratteristiche demografiche basali erano ben bilanciate tra i gruppi che assumevano Eurisdi® e placebo, ad eccezione di una differenza nel numero di pazienti con scoliosi (63,3 % nel gruppo Eurisdi® e 73,3 % nel gruppo placebo).
I risultati dell’analisi primaria della parte 2 dello studio SUNFISH riguardo al cambiamento del punteggio totale MFM32 rispetto al basale a 12 mesi hanno dimostrato una differenza clinicamente significativa e statisticamente significativa tra i gruppi di pazienti trattati con Eurisdi® e quelli trattati con placebo. I risultati dell’analisi primaria e i principali endpoint secondari sono riportati nella Tabella 2 e nella Figura 1.
Tabella 2
Riassunto dei risultati di efficacia nei pazienti con SMN ad insorgenza tardiva dopo 12 mesi di trattamento (parte 2 dello studio SUNFISH)
| Punto finale |
Eurisdi® (N = 120) |
Placebo (N = 60) |
| Punto finale primario: |
||
| Variazione del punteggio totale MFM321 a 12 mesi rispetto al basale, media quadratica media (95 % IC) |
1,36 (0,61; 2,11) |
-0,19 (-1,22; 0,84) |
| Differenza rispetto al placebo (95 % IC), valore p2 |
1,55 (0,30; 2,81) 0,0156 |
|
| Punti finali secondari |
||
| Frazione di pazienti con variazione del punteggio totale MFM321 di 3 o più a 12 mesi rispetto al basale (95 % IC) |
38,3 % (28,9; 47,6) |
23,7 % (12,0; 35,4) |
| Rapporto di probabilità di risposta globale (95 % IC), valore p corretto (non corretto)3,4 |
2,35 (1,01; 5,44) 0,0469 (0,0469) |
|
| Variazione del punteggio totale RULM5 a 12 mesi rispetto al basale, media quadratica media (95 % IC) |
1,61 (1,00; 2,22) |
0,02 (-0,8; 0,87) |
| Differenza rispetto al placebo (95 % IC), valore p corretto (non corretto)2,4 |
1,59 (0,55; 2,62) 0,0469 (0,0028) |
|
1 Sulla base della regola di esclusione per dati mancanti relativi a MFM32, 6 pazienti sono stati esclusi dall’analisi (Eurisdi®, n = 115; gruppo placebo, n = 59).
2 I dati sono stati analizzati mediante un modello misto con misure ripetute, includendo come covariate il valore basale, il trattamento, la visita, il gruppo d’età, l’effetto dell’interazione trattamento per visita e l’interazione basale per visita.
3 I dati sono stati analizzati mediante regressione logistica per il punteggio basale totale, il gruppo di trattamento e il gruppo d’età.
4 Il valore p aggiustato è stato ottenuto per gli endpoint inclusi nel testing gerarchico ed è stato calcolato sulla base di tutti i valori p degli endpoint nell’ordine gerarchico fino all’endpoint corrente. Il valore p non aggiustato è stato testato con un livello di significatività del 5%.
5 Sulla base della regola di esclusione per dati mancanti relativi a RULM, 3 pazienti sono stati esclusi dall’analisi (Eurisdi®, n = 119; gruppo placebo, n = 58).
Dopo 12 mesi di trattamento, 117 pazienti hanno continuato a ricevere Eurisdi®. Al momento dell’analisi a 24 mesi, i pazienti che avevano ricevuto il trattamento per 24 mesi hanno mostrato un ulteriore miglioramento della funzione motoria tra i mesi 12 e 24. La variazione media rispetto al basale nel punteggio MFM32 è stata di 1,83 (IC: 0,74–2,92), mentre per RULM è stata di 2,79 (IC: 1,94–3,64).
| Variazione del valore medio quadratico medio dell'indice totale MFM32 |
|
| Mesi |
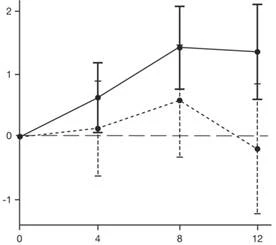
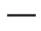
Eurisdi® Placebo
* The error bar represents the 95 % confidence interval.
† The total MFM score was calculated according to the user manual and expressed as a percentage of the maximum possible score for the scale (i.e., the sum of scores for 32 items divided by 96 and multiplied by 100).
Fig. 1. Mean (LS) change in total MFM32 score from baseline at 12 months in part 2 of the SUNFISH study.
SUNFISH, part 1
The efficacy of Eurisdi® in patients with later-onset SMA is also supported by results from part 1 of the SUNFISH dose-finding study. Part 1 included 51 patients with type 2 and type 3 SMA (including 7 ambulant patients) aged 2–25 years. After one year of treatment with the therapeutic dose (the dose selected for part 2), a clinically meaningful improvement in motor function was observed based on MFM32, with a mean change from baseline of 2.7 points (95 % CI: 1.5; 3.8). Improvement in MFM32 was maintained over up to 2 years of treatment with Eurisdi® (mean change of 2.7 points (95 % CI: 1.2; 4.2)).
In an exploratory analysis, motor function assessed by MFM was compared between part 1 of SUNFISH and a historical natural history control cohort (weighted based on key prognostic factors). Change in total MFM score from baseline at 1 and 2 years was greater in patients treated with Eurisdi® compared to the natural history cohort (at 1 year: difference of 2.7 points; p < 0.0001; at 2 years: difference of 4 points; p < 0.0001). In the natural history cohort, a decline in motor function was observed, as expected due to the natural progression of SMA (mean change at 1 year: –0.6 points; at 2 years: –2 points).
Presymptomatic SMA
Study BN40703 (RAINBOWFISH) is an ongoing, single-arm, open-label, multicenter study evaluating the efficacy, safety, pharmacokinetics, and pharmacodynamics of Eurisdi® in infants aged from birth to 6 weeks (at first dose administration) with a genetic diagnosis of SMA but who are asymptomatic.
Efficacy of Eurisdi® in presymptomatic SMA was evaluated at month 12 in 26 patients [intention-to-treat (ITT) population]. The median age at first dose was 25 days (range: 16 to 41 days), 62 % were female, and 85 % were of Caucasian race. Eight patients had 2 copies of the SMN2 gene, 13 had 3 copies, and 5 had ≥ 4 copies. At baseline, the median CHOP-INTEND score was 51.5 (range: 35.0 to 62.0), the median HINE-2 score was 2.5 (range: 0 to 6.0), and the median compound muscle action potential amplitude of the ulnar nerve (CMAP) was 3.6 mV (range: 0.5 to 6.7 mV).
The primary efficacy analysis population (N = 5) included patients with 2 copies of the SMN2 gene and baseline CMAP amplitude ≥ 1.5 mV. In these patients, the median CHOP-INTEND score was 48.0 (range: 36.0–52.0), the median HINE-2 score was 2.0 (range: 1.0 to 3.0), and the median CMAP amplitude was 2.6 mV (range: 1.6 to 3.8 mV) at baseline.
The primary endpoint was the proportion of patients in the primary efficacy analysis population who were able to sit without support for at least 5 seconds (BSID-III gross motor scale item 22) at month 12; a statistically significant and clinically meaningful proportion of patients achieved this milestone compared to the predefined efficacy criterion of 5 %.
Key efficacy endpoints in patients treated with Eurisdi® are presented in Tables 3 and 4 and Figure 2.
Table 3
Ability to sit according to BSID-III item 22 definition in presymptomatic patients at month 12
| Punto finale di valutazione dell'efficacia |
Popolazione dei pazienti |
||
| Analisi primaria dell'efficacia (N = 5) |
Pazienti con due copie del gene SMN2a (N = 8) |
ITT (N = 26) |
|
| Proporzione di pazienti in grado di sedersi senza supporto per almeno 5 secondi (punto 22 della scala BSID-III); (IC 90 %) |
80 % (34,3 %; 99,0 %) |
87,5 % (52,9 %; 99,4 %) |
96,2 % (83,0 %; 99,8 %) |
Abbreviations: BSID-III – Bayley Scales of Infant and Toddler Development, Third Edition; CI – confidence interval; ITT – intention-to-treat population.
a Patients with two copies of the SMN2 gene had a mean CMAP amplitude of 2.0 (range 0.5–3.8) at baseline.
b The p-value is based on a one-sided exact binomial test. The result is compared to a 5% threshold.
Additionally, 80% (4/5) of the primary efficacy analysis population, 87.5% (7/8) of patients with two copies of the SMN2 gene, and 80.8% (21/26) of the ITT population achieved the ability to sit without support for 30 seconds (BSID-III, item 26). The ITT population also achieved motor milestones assessed by the HINE-2 at month 12 (N = 25). In this population, 96.0% of patients were able to sit [1 patient (1/8 patients with two copies of the SMN2 gene) achieved stable sitting, and 23 patients (6/8, 13/13, 4/4 patients with 2, 3, and ≥4 copies of the SMN2 gene, respectively) were able to roll over/turn]. Furthermore, 84% of patients were able to stand; 32% (N = 8) were able to stand with support (3/8, 3/13, and 2/4 patients with 2, 3, and ≥4 copies of the SMN2 gene, respectively), and 52% (N = 13) were able to stand independently (1/8, 10/13, and 2/4 patients with 2, 3, and ≥4 copies of the SMN2 gene, respectively). Additionally, 72% of patients were able to bounce, walk with or without support; 8% (N = 2) were able to bounce (2/8 patients with two copies of the SMN2 gene), 16% (N = 4) were able to walk with support (3/13 and 1/4 patients with 3 and ≥4 copies of the SMN2 gene, respectively), and 48% (N = 12) were able to walk independently (1/8, 9/13, and 2/4 patients with 2, 3, and ≥4 copies of the SMN2 gene, respectively). Seven patients were not assessed at month 12.
Table 4
Summary of key efficacy endpoints for presymptomatic patients at month 12
| Punti finali di efficacia |
Popolazione ITT (N = 26) |
| Funzione motoria |
|
| Frazione di pazienti che hanno raggiunto un punteggio totale di 50 o superiore nella scala CHOP-INTEND (IC 90%) |
92 %a (76,9 %; 98,6 %) |
| Frazione di pazienti che hanno raggiunto un punteggio totale di 60 o superiore nella scala CHOP-INTEND (IC 90%) |
80 %a (62,5 %; 91,8 %) |
| Alimentazione |
|
| Frazione di pazienti in grado di alimentarsi per via orale (IC 90%) |
96,2 %b (83,0 %; 99,8 %) |
| Utilizzo delle risorse del sistema sanitario |
|
| Frazione di pazienti che non hanno richiesto ricovero (IC 90%) |
92,3 % (77,7 %; 98,6 %) |
| Sopravvivenza senza eventid Frazione di pazienti caratterizzati da sopravvivenza senza eventi (IC 90%) |
100 % (100 %; 100 %) |
Abbreviazioni: CHOP INTEND – test dell’Ospedale dei Bambini di Filadelfia per la valutazione delle funzioni motorie nei neonati con malattie neuromuscolari; IC – intervallo di confidenza; TTP – popolazione trattata.
a Basato su un numero N = 25.
b La valutazione non è stata effettuata per un paziente.
c Le ospedalizzazioni includevano tutti i ricoveri ospedalieri di almeno due giorni di durata non motivati dalle esigenze dello studio.
d L’evento in questo caso indica decesso o necessità di ventilazione permanente; la ventilazione permanente è definita come tracheostomia o ≥ 16 ore di ventilazione non invasiva al giorno, oppure intubazione per > 21 giorni consecutivi in assenza o dopo la scomparsa di un evento acuto reversibile.
Abbreviazioni: IQR – intervallo interquartile; SMN2 – gene di sopravvivenza dei motoneuroni 2.
Fig. 2. Mediana dei punteggi totali CHOP-INTEND in base alle visite e al numero di copie del gene SMN2 (popolazione TTP)
Uso in pazienti con SMA precedentemente trattati con altre terapie modulanti
Lo studio BP39054 (JEWELFISH) è uno studio aperto, a gruppo singolo, per valutare la sicurezza, tollerabilità, farmacocinetica e farmacodinamica di Eurisdi® in pazienti con SMA infantile o con SMA ad esordio tardivo, di età compresa tra 6 mesi e 60 anni, precedentemente trattati per SMA (inclusi nusinersen e onasemnogene abeparvovec). Dei 173 pazienti trattati con Eurisdi®, 76 avevano ricevuto un trattamento precedente con nusinersen (9 pazienti con SMA di tipo 1, 43 con SMA di tipo 2 e 24 con SMA di tipo 3) e 14 avevano ricevuto in precedenza onasemnogene abeparvovec (4 pazienti con SMA di tipo 1 e 10 con SMA di tipo 2). L’età media dei pazienti all’inizio del trattamento con Eurisdi® era di 14 anni (range: 1–60 anni).
Al momento dell’inclusione nello studio, tra i 168 pazienti di età compresa tra 2 e 60 anni, l’83% presentava scoliosi (39% scoliosi grave) e il 63% aveva un punteggio sulla scala estesa di valutazione della funzione motoria di Hammersmith (HFMSE) inferiore a 10 punti. Nello studio sono stati inclusi anche 15 pazienti in grado di deambulare autonomamente (età compresa tra 5 e 46 anni).
Gli endpoint di efficacia sono stati valutati mediante misurazione della funzione motoria in base all’età, inclusi gli strumenti MFM-32 e RULM per pazienti di età compresa tra 2 e 60 anni, BSID-III e HINE-2 per pazienti di età inferiore a 2 anni e il test del cammino di 6 minuti (6MWT) per pazienti di età ≥ 6 anni in grado di deambulare autonomamente. I risultati dell’analisi primaria dopo 24 mesi di trattamento hanno mostrato una stabilità generale della funzione motoria nei pazienti di età compresa tra 2 e 60 anni, secondo le scale MFM-32 e RULM (n = 137 e n = 133 rispettivamente). I pazienti di età inferiore a 2 anni (n = 6) hanno mantenuto o migliorato lo sviluppo della funzione motoria, in particolare il controllo della testa, il rotolamento e la capacità di sedersi senza supporto. I risultati del 6MWT mostrano un miglioramento medio di 30,88 metri (IC 95%: -5,54; 67,29; n = 8). Tutti i pazienti in grado di deambulare autonomamente hanno mantenuto tale capacità.
Farmacocinetica
I parametri farmacocinetici di risdiplam sono stati caratterizzati in soggetti sani adulti e in pazienti con SMA.
Dopo somministrazione orale della soluzione di Eurisdi® in dosi comprese tra 0,6 e 18 mg, la farmacocinetica di risdiplam è risultata approssimativamente lineare. La farmacocinetica di risdiplam è stata meglio descritta mediante un modello popolazionale di PK con assorbimento tramite transito a tre camere, distribuzione bicompartimentale ed eliminazione di primo ordine. È stato osservato che il peso corporeo e l’età del paziente hanno un impatto significativo sulla farmacocinetica del farmaco.
L’esposizione stimata (AUC media 0–24h) nei pazienti con SMA infantile (età compresa tra 2 e 7 mesi al momento dell’inclusione nello studio) alla dose raccomandata di 0,2 mg/kg una volta al giorno è risultata pari a 1930 ng·h/mL. L’esposizione media stimata nei neonati di età compresa tra 16 giorni e < 2 mesi con SMA presintomatica nello studio RAINBOWFISH è stata di 2020 ng·h/mL dopo 2 settimane di somministrazione giornaliera alla dose di 0,15 mg/kg.
L’esposizione stimata nei pazienti con SMA ad esordio tardivo (età compresa tra 2 e 25 anni al momento dell’inclusione nello studio) nello studio SUNFISH (parte 2) con la dose terapeutica (0,25 mg/kg una volta al giorno per pazienti con peso corporeo < 20 kg; 5 mg una volta al giorno per pazienti con peso corporeo ≥ 20 kg) è risultata pari a 2010 ng·h/mL. La concentrazione massima osservata (Cmax media) è stata di 194 ng/mL con una dose di 0,2 mg/kg nello studio FIREFISH e di 120 ng/mL nella parte 2 dello studio SUNFISH. La concentrazione massima media stimata con una dose di 0,15 mg/kg nello studio RAINBOWFISH è di 111 ng/mL.
Assorbimento
Risdiplam viene rapidamente assorbito dopo somministrazione orale a digiuno, con un tmax plasmatico compreso tra 1 e 4 ore. Negli studi clinici, risdiplam è stato somministrato al mattino con il pasto o dopo l’allattamento al seno.
Distribuzione
I parametri farmacocinetici popolazionali stimati sono: volume apparente di distribuzione centrale pari a 98 L, volume periferico pari a 93 L e clearance intercompartimentale pari a 0,68 L/ora.
Risdiplam si lega principalmente all’albumina sierica, senza legame con la glicoproteina acida alfa-1; la frazione libera è pari all’11%.
Metabolismo
Risdiplam è principalmente metabolizzato dalle monoossigenasi flaviniche 1 e 3 (FMO1 e FMO3), nonché dagli isoenzimi CYP 1A1, 2J2, 3A4 e 3A7. Il principio attivo invariato è risultato il componente principale rilevato nel plasma, rappresentando l’83% della sostanza associata al farmaco in circolo. Il metabolita farmacologicamente inattivo M1 è stato identificato come il principale metabolita circolante.
Eliminazione
Il parametro farmacocinetico popolazionale della clearance apparente (CL/F) di risdiplam è pari a 2,6 L/ora. L’emivita effettiva di eliminazione di risdiplam è di circa 50 ore nei pazienti con SMA.
Circa il 53% della dose (14% come risdiplam invariato) è stato eliminato attraverso le feci e il 28% attraverso le urine (8% come risdiplam invariato).
Popolazioni speciali
Alterazioni della funzione epatica
Le alterazioni epatiche di grado lieve e moderato non hanno influenzato la PK di risdiplam. Dopo somministrazione di 5 mg di risdiplam, i rapporti medi di Cmax e AUC sono stati rispettivamente 0,95 e 0,80 in soggetti con compromissione epatica lieve (n = 8) e 1,20 e 1,08 in pazienti con compromissione epatica moderata (n = 8), rispetto ai valori nei pazienti con funzione epatica normale (gruppo di controllo, n = 10). La sicurezza e la PK nei pazienti con grave compromissione epatica non sono state ancora studiate.
Alterazioni della funzione renale
Studi di PK di risdiplam in pazienti con compromissione renale non sono stati condotti. L’eliminazione renale di risdiplam in forma invariata è trascurabile (8%).
Pazienti anziani
Studi specifici di farmacocinetica di Eurisdi® in pazienti con SMA di età superiore a 60 anni non sono stati condotti. Pazienti con SMA di età fino a 60 anni sono stati inclusi nello studio JEWELFISH. Pazienti senza SMA di età fino a 69 anni sono stati inclusi negli studi clinici di PK.
Bambini
Il peso corporeo e l’età del paziente sono stati identificati come covariate nell’analisi PK popolazionale. Pertanto, la dose viene aggiustata in base all’età (sotto e sopra 2 mesi e 2 anni) e al peso corporeo (fino a 20 kg) al fine di ottenere un’esposizione simile in diverse fasce d’età e categorie di peso. Non sono disponibili dati farmacocinetici nei neonati di età inferiore a 16 giorni.
Appartenenza etnica
La farmacocinetica di risdiplam non differisce tra pazienti giapponesi e pazienti di razza caucasica.
Caratteristiche cliniche.
Indicazioni.
Trattamento della atrofia muscolare spinale (SMA) associata a 5q in pazienti pediatrici e adulti.
Controindicazioni.
Ipersensibilità nota al risdiplam o ad uno qualsiasi degli eccipienti elencati nella sezione «Composizione».
Interazioni con altri medicinali e altre forme di interazione.
Influenza di Eurisdi® su altri medicinali
In vitro, il risdiplam e il suo metabolita circolante principale M1 non inducono CYP1A2, 2B6, 2C8, 2C9, 2C19 e 3A4. In vitro, il risdiplam e M1 non inibiscono (inibizione reversibile o dipendente dal tempo) nessuno degli isoenzimi CYP studiati (CYP1A2, 2B6, 2C8, 2C9, 2C19, 2D6), ad eccezione del CYP3A.
Eurisdi® è un inibitore debole del CYP3A. In adulti sani, l’assunzione di Eurisdi® una volta al giorno per 2 settimane ha leggermente aumentato l’esposizione al midazolam, un substrato altamente sensibile del CYP3A (AUC +11 %; Cmax +16 %). Tale interazione non è considerata clinicamente rilevante, pertanto non è necessaria alcuna correzione della dose dei substrati del CYP3A.
Sulla base dei risultati ottenuti con modelli farmacocinetici basati sulla fisiologia (PBPK), un effetto simile è atteso nei bambini e nei neonati a partire dai 2 mesi di età.
Studi in vitro hanno dimostrato che il risdiplam e il suo metabolita principale non sono inibitori significativi del MDR1 umano, del polipeptide trasportatore di anioni organici (OATP)1B1, OATP1B3, né dei trasportatori di anioni organici 1 e 3 (OAT 1 e 3). Tuttavia, in vitro, il risdiplam e il suo metabolita principale sono inibitori del polipeptide trasportatore di cationi organici 2 (OCT2) umano e dei trasportatori di proteine di resistenza multipla ed escrezione di tossine (MATE)1 e dei trasportatori MATE2-K. Quando somministrati alle concentrazioni terapeutiche, non è prevista un’interazione del medicinale con i substrati di OCT2. L’impatto dell’assunzione concomitante di risdiplam sulla farmacocinetica dei substrati di MATE1 e MATE2-K nell’uomo non è noto. Sulla base di dati in vitro, Eurisdi® potrebbe aumentare la concentrazione plasmatica di principi attivi eliminati tramite MATE1 o MATE2-K, ad esempio la metformina (vedere sezione «Farmacocinetica»). Se non è possibile evitare la somministrazione concomitante, si raccomanda il monitoraggio per la tossicità correlata al farmaco e si dovrebbe considerare la riduzione della dose dell’altro medicinale somministrato contemporaneamente, se necessario.
Influenza di altri medicinali su Eurisdi®
Il risdiplam è metabolizzato principalmente dalle monoossigenasi flaviniche 1 e 3 (FMO1 e 3), nonché dai CYP 1A1, 2J2, 3A4 e 3A7. Il risdiplam non è un substrato della proteina di resistenza multipla 1 umana (MDR1).
La somministrazione concomitante di itraconazolo, un inibitore potente del CYP3A, alla dose di 200 mg due volte al giorno, e di risdiplam alla dose di 6 mg in singola dose orale, non ha mostrato alcun effetto clinicamente significativo sulla farmacocinetica (PK) del risdiplam (aumento dell’AUC del 11 %, riduzione del Cmax del 9 %). Pertanto, non è necessaria alcuna correzione della dose di Eurisdi® quando somministrato concomitantemente con inibitori del CYP3A.
Non sono previste interazioni con altri medicinali attraverso i percorsi di segnalazione mediati da FMO1 e FMO3.
Caratteristiche particolari di impiego.
Informazioni generali
Negli studi sugli animali sono state osservate alterazioni della retina, dell'epitelio, in particolare della cute e del tratto gastrointestinale, e segni di tossicità sul midollo osseo (modifiche negli esami ematici clinici). Attualmente, il rischio di tali alterazioni nell'uomo non può essere valutato definitivamente a causa della limitata disponibilità di dati a lungo termine sulla sicurezza.
Tossicità embriofetale
Negli studi sugli animali è stata osservata tossicità embriofetale. I pazienti in età fertile devono essere informati dei rischi. Durante il trattamento e almeno per 1 mese dopo l’assunzione dell’ultima dose del medicinale Eurisdi®, le donne devono utilizzare metodi contraccettivi altamente efficaci, mentre gli uomini devono utilizzare metodi contraccettivi altamente efficaci per almeno 4 mesi dopo l’assunzione dell’ultima dose del medicinale Eurisdi® (vedere la sezione «Modalità di somministrazione e posologia»).
Possibile effetto sulla fertilità negli uomini
A causa degli effetti reversibili del medicinale Eurisdi® sulla fertilità maschile osservati negli studi sugli animali, gli uomini in trattamento non devono donare sperma durante il trattamento e per almeno 4 mesi dopo l’assunzione dell’ultima dose del medicinale Eurisdi® (vedere la sezione «Farmacocinetica»).
È necessario evitare il contatto tra la polvere e la soluzione orale ricostituita e la pelle. In caso di contatto del medicinale (polvere o soluzione) con la pelle, la zona interessata deve essere lavata con acqua e sapone.
Sostanze eccipienti
Questo medicinale contiene 0,38 mg di benzoato di sodio per 1 ml. Questo medicinale contiene meno di 1 mmol di sodio (23 mg) per dose, pertanto è considerato "privo di sodio".
Questo medicinale contiene isomalto. I pazienti con rara intolleranza ereditaria al fruttosio non devono assumere questo medicinale.
Uso durante la gravidanza o l’allattamento
In base ai risultati degli studi preclinici, la fertilità negli uomini può essere compromessa durante il trattamento con il medicinale Eurisdi®. Negli organi riproduttivi di ratti e scimmie sono state osservate degenerazione degli spermatozoi e riduzione del numero di spermatozoi.
Prima dell’inizio del trattamento con il medicinale Eurisdi®, è necessario discutere con i pazienti di sesso maschile strategie per la conservazione della fertilità. I pazienti di sesso maschile possono prendere in considerazione la possibilità di conservare lo sperma prima dell’inizio del trattamento o dopo un periodo senza trattamento di almeno 4 mesi (vedere la sezione «Caratteristiche particolari di impiego»).
In base ai risultati degli studi preclinici, non ci si aspetta che il medicinale Eurisdi® influisca sulla fertilità femminile.
Le donne in età fertile devono essere sottoposte a test per escludere una gravidanza prima dell’inizio del trattamento con il medicinale Eurisdi®.
Ai pazienti di sesso maschile e femminile in età fertile devono essere raccomandate le seguenti misure contraccettive:
- Ai pazienti di sesso maschile e alle loro partner in età fertile deve essere richiesto di utilizzare un metodo contraccettivo altamente efficace durante il trattamento con il medicinale Eurisdi® e per almeno 4 mesi dopo l’assunzione dell’ultima dose del medicinale.
- Alle pazienti di sesso femminile in età fertile deve essere richiesto di utilizzare un metodo contraccettivo altamente efficace durante il trattamento con il medicinale Eurisdi® e per almeno 1 mese dopo l’assunzione dell’ultima dose del medicinale.
Gravidanza
Non esistono dati clinici sull’uso del medicinale Eurisdi® in donne in gravidanza. Il risdiplam ha dimostrato effetti embriofetotossici e teratogeni negli animali. Sulla base dei dati ottenuti negli studi sugli animali, il risdiplam attraversa la barriera placentare e può causare danni al feto.
Il medicinale Eurisdi® non deve essere utilizzato durante la gravidanza, a meno che non sia strettamente necessario. Se una donna in gravidanza necessita di trattamento con il medicinale Eurisdi®, deve essere chiaramente informata del rischio potenziale per il feto.
Allattamento
Non è noto se il medicinale Eurisdi® sia escreto nel latte materno umano. Studi su ratti hanno dimostrato che il risdiplam è escreto nel latte materno. Poiché il potenziale rischio per il neonato allattato al seno non è noto, il medico curante deve valutare attentamente la prosecuzione della terapia del paziente. L’allattamento al seno non è raccomandato durante il trattamento con il medicinale Eurisdi®.
Capacità di guidare veicoli o di usare macchinari
L’effetto del medicinale Eurisdi® sulla capacità di guidare veicoli o di usare macchinari non è stato studiato in studi clinici adeguati.
Modalità di somministrazione e dosi
La soluzione orale del medicinale Eurisdi® deve essere preparata da un professionista sanitario (ad esempio medico o farmacista) prima della consegna al paziente.
Prima della prima dose, il professionista sanitario deve discutere con il paziente o con la persona che si prende cura di lui come preparare e assumere la dose giornaliera prescritta (vedere di seguito «Istruzioni per l’uso»).
Il trattamento con Eurisdi® deve essere iniziato e monitorato da medici esperti nella diagnosi e nel trattamento dei pazienti con atrofia muscolare spinale.
Il programma di sviluppo clinico non include pazienti con SMA di tipo IV.
Dosaggio raccomandato
Il medicinale Eurisdi® va assunto per via orale una volta al giorno, approssimativamente alla stessa ora ogni giorno, utilizzando le siringhe orali riutilizzabili fornite nella confezione. La dose raccomandata di Eurisdi® per il trattamento della SMA è determinata in base all’età e al peso corporeo del paziente (vedere tabella 5).
Tabella 5
Regime posologico in base all’età e al peso corporeo
| Età e peso corporeo |
Dose giornaliera raccomandata |
| < 2 mesi |
0,15 mg/kg |
| da 2 mesi a < 2 anni |
0,20 mg/kg |
| ≥ 2 anni e peso corporeo < 20 kg |
0,25 mg/kg |
| ≥ 2 anni e peso corporeo ≥ 20 kg |
5 mg |
sulla base dell'età corretta per neonati prematuri.
La modifica della dose deve essere effettuata sotto la supervisione di un operatore sanitario. Il trattamento con una dose giornaliera superiore a 5 mg non è stato studiato. Sono disponibili solo dati molto limitati sulla sicurezza, ottenuti nel periodo post-commercializzazione, in neonati di età inferiore a 16 giorni ai quali è stato somministrato Eurisdi® nella dose raccomandata (vedi sottosezioni «Farmacocinetica», «Popolazione pediatrica»).
Pazienti con compromissione epatica
Nei pazienti con compromissione epatica lieve o moderata non è necessaria alcuna correzione della dose. L'uso di Eurisdi® nei pazienti con compromissione epatica grave non è stato studiato (vedi sezione «Farmacocinetica»).
Pazienti con compromissione renale
La sicurezza e l'efficacia di Eurisdi® nei pazienti con compromissione renale non sono state studiate. Non si prevede la necessità di aggiustamento della dose nei pazienti con compromissione renale (vedi sezione «Farmacocinetica»).
Pazienti anziani
Gli studi clinici con Eurisdi® non hanno incluso pazienti di età pari o superiore a 65 anni; pertanto, non è stato stabilito se questi pazienti possano rispondere al trattamento in modo diverso rispetto ai pazienti più giovani.
Popolazione pediatrica
L'uso di Eurisdi® in pazienti con SMA di età inferiore a 2 mesi è supportato da dati di farmacocinetica e sicurezza in bambini di età pari o superiore a 16 giorni (vedi sezioni «Effetti indesiderati» e sottosezioni «Efficacia clinica» e «Farmacocinetica»). Non sono disponibili dati clinici o farmacocinetici sull'uso del medicinale in neonati prematuri o neonati di età inferiore a 16 giorni (vedi sottosezione «Efficacia clinica» sopra).
Assunzione differita
Il medicinale Eurisdi® deve essere assunto per via orale una volta al giorno, approssimativamente allo stesso orario ogni giorno. Se si dimentica di assumere una dose di Eurisdi®, il medicinale deve essere assunto il prima possibile, purché il ritardo rispetto all’orario previsto non superi le 6 ore; quindi, il normale schema di somministrazione può essere ripreso il giorno successivo. In caso contrario, non si deve assumere la dose dimenticata e la dose successiva deve essere assunta all’orario previsto il giorno seguente.
Se la dose non viene completamente ingerita o se si verifica vomito dopo l’assunzione di Eurisdi®, non si deve assumere un’altra dose per compensare quella persa. Si deve attendere il giorno successivo per assumere la dose successiva all’orario previsto.
Modalità di somministrazione
Per somministrare la dose giornaliera di Eurisdi® è necessario utilizzare la siringa orale riutilizzabile fornita nella confezione di cartone insieme al medicinale (vedi tabella 6).
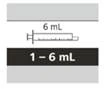
Tabella 6
Selezione della siringa orale riutilizzabile appropriata per assumere la dose giornaliera prescritta di Eurisdi®
| Dimensione della siringa |
Volume della dose |
Valore di divisione della siringa |
| 1 ml |
da 0,3 a 1 ml |
0,01 ml |
| 6 ml |
da 1 a 6 ml |
0,1 ml |
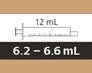
| 12 ml |
da 6,2 a 6,6 ml |
0,2 ml |
Per il calcolo del volume della dose è necessario considerare anche il valore della graduazione della siringa. Arrotondare il volume della dose per eccesso o per difetto al valore di graduazione più vicino indicato sulla siringa orale selezionata (ad esempio, da 6,3 ml a 6,4 ml, da 3,03 ml a 3 ml e da 1,05 a 1,1 ml).
Il medicinale Eurisdi® in forma di soluzione deve essere assunto immediatamente dopo essere stato aspirato nella siringa orale riutilizzabile. Se il contenuto della siringa non viene assunto entro 5 minuti, la siringa orale deve essere svuotata del medicinale (vedere sotto "Smaltimento del medicinale non utilizzato/medicinale scaduto") e deve essere preparata una nuova dose.
Il medicinale Eurisdi® deve essere assunto dopo i pasti. Al paziente deve essere somministrata acqua dopo l'assunzione di Eurisdi® per assicurarsi che il medicinale sia stato completamente deglutito. Se il paziente non è in grado di deglutire e ha un sondino nasogastrico o una sonda gastrostomica, Eurisdi® può essere somministrato attraverso il sondino/la sonda. Dopo la somministrazione del medicinale, il sondino/la sonda devono essere sciacquati con acqua (vedere sotto "Istruzioni per l'uso").
Istruzioni per l'uso
Istruzioni da seguire prima, durante e dopo la preparazione della soluzione orale:
- La soluzione deve sempre essere preparata da un operatore sanitario (ovvero medico o farmacista).
- Evitare l'inalazione della polvere del medicinale Eurisdi®. Prestare attenzione alle norme locali e utilizzare l'equipaggiamento adeguato per la preparazione della soluzione Eurisdi®.
- Indossare guanti.
- Non utilizzare la polvere se la data di scadenza è trascorsa. La data di scadenza della polvere è riportata sull'etichetta della fiala e sulla confezione di cartone.
- Non dispensare la soluzione ricostituita se la data di scadenza della soluzione orale pronta all'uso – indicata sull'etichetta della fiala e sulla confezione di cartone – supera la data di scadenza iniziale della polvere.
- Evitare il contatto del medicinale con la pelle. Se il medicinale (polvere o soluzione) entra in contatto con la pelle, lavare l'area con acqua e sapone.
- Non utilizzare il medicinale se qualsiasi componente della confezione è danneggiato o mancante.
- Per la preparazione della soluzione utilizzare acqua purificata o acqua per preparazioni iniettabili.
- Non utilizzare siringhe orali diverse da quelle incluse nella confezione di cartone insieme al medicinale.
- Non mescolare Eurisdi® con cibi o liquidi (ad esempio latte o formula lattea).
- Non mescolare Eurisdi® da una nuova fiala con il medicinale da una fiala precedentemente aperta in uso.
Il paziente o la persona che si prende cura del paziente devono essere istruiti da un operatore sanitario su come preparare e assumere la dose giornaliera prescritta prima della consegna della soluzione pronta.
Preparazione della soluzione orale
Aggiungere 79 ml di acqua purificata o acqua per preparazioni iniettabili nella fiala contenente il medicinale.
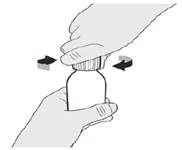
Inserire l'adattatore premunibile nel collo della fiala, spingendolo verso il basso.
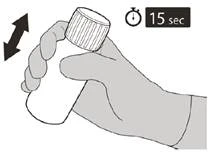
Dopo aver chiuso completamente la fiala, agitare per 15 secondi.
Dopo 10 minuti si dovrà ottenere una soluzione limpida. Se la soluzione non è ancora limpida, agitare ulteriormente per altri 15 secondi.
È necessario calcolare 64 giorni a partire dalla data di preparazione della soluzione. Il giorno di preparazione della soluzione è considerato il giorno 0. La data calcolata deve essere indicata sull'etichetta della fiala e sulla confezione di cartone nel campo: Soluzione orale ricostituita DA SMALTIRE DOPO: (giorno/mese/anno).
Smaltimento del medicinale non utilizzato/medicinale scaduto
L'immissione di medicinali nell'ambiente deve essere ridotta al minimo. Non smaltire i medicinali tramite acque reflue, né con i rifiuti domestici.
Il medicinale non utilizzato o scaduto deve essere smaltito professionalmente presso il luogo di dispensazione del medicinale (medico o farmacista).
Istruzioni per la preparazione della soluzione (SOLO PER OPERATORI SANITARI, IN PARTICOLARE MEDICI O FARMACISTI)
La soluzione orale di Eurisdi® deve essere preparata da un operatore sanitario prima della dispensazione al paziente.
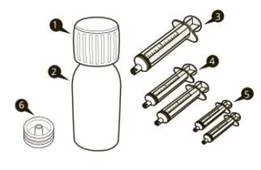
| Un imballaggio del medicinale Eurisdi® contiene (vedi figura A):
|
Figura A |
Informazioni importanti sul medicinale Eurisdi®
- La soluzione deve sempre essere preparata da un operatore sanitario (medico o farmacista).
- Evitare l'inalazione della polvere del medicinale Eurisdi®. Rispettare i requisiti locali e utilizzare l'equipaggiamento adeguato per la preparazione della soluzione di Eurisdi®.
- Indossare guanti.
- Non utilizzare questo medicinale dopo la data di scadenza indicata sull'etichetta del flacone e sulla confezione di cartone.
- Evitare qualsiasi contatto di questo medicinale con la pelle. In caso di contatto (con la polvere o con la soluzione), lavare l'area interessata con acqua e sapone.
- Non utilizzare questo medicinale se uno qualsiasi degli elementi dell'imballaggio è danneggiato o mancante.
- Per la preparazione della soluzione, utilizzare acqua purificata o acqua per preparazioni iniettabili.
- Non utilizzare siringhe orali riutilizzabili, eccetto quelle fornite nella confezione.
- Non distribuire la soluzione preparata se la data di scadenza della soluzione orale pronta all'uso – indicata sull'etichetta del flacone e sulla confezione di cartone – supera la data di scadenza della polvere.
- Prima di consegnare la soluzione preparata all'operatore sanitario, è necessario istruire il paziente o il caregiver su come preparare e assumere la dose giornaliera.
Come conservare il medicinale Eurisdi®
- Conservare la polvere (medicinale non ancora preparato come soluzione) nella confezione di cartone a temperatura ambiente non superiore a 25 °C, al riparo dalla luce e dall'umidità.
- Conservare la soluzione (medicinale preparato come soluzione) in posizione verticale in frigorifero a una temperatura compresa tra 2–8 °C, all'interno della confezione di cartone per proteggere il contenuto dalla luce.
- Se necessario, la soluzione orale può essere conservata a temperatura ambiente (inferiore a 40 °C) per un periodo totale non superiore a 5 giorni. Non conservare la soluzione orale a temperature superiori a 40 °C.
- Conservare la soluzione orale nel flacone originale in posizione verticale e con il tappo ben chiuso.
Preparazione della soluzione
|
Figura B |
Passo 1 Battere delicatamente sul fondo del flaconcino per allentare la polvere (vedere Figura B). |
|
Figura C |
Passo 2 Premere verso il basso sul tappo e poi svitarlo ruotando verso sinistra (in senso antiorario) (vedere Figura C). Non gettare via il tappo. |
|
Figura D |
Passo 3 Aggiungere con attenzione 79 ml di acqua purificata o di acqua per preparazioni iniettabili nel flacone del medicinale (vedere Figura D). |
|
Figura E |
Passo 4 Tenere il flacone del medicinale con una mano appoggiata sul tavolo. Inserire l’adattatore per la compressione nel collo del flacone, premendo dall’alto con l’altra mano finché l’adattatore non sarà completamente premuto fino al bordo dell’apertura del flacone (vedere Figura E). |
|
Figura F |
Passo 5 Rimettere il tappo sul flacone. Ruotare il tappo a destra (in senso orario) per chiudere il flacone. Assicurarsi che il flacone sia completamente chiuso, quindi agitare energicamente per 15 secondi (vedere Figura F). |
|
Figura G |
Passo 6 Determinare la data di «DA GETTARE DOPO: (giorno/mese/anno)», calcolando 64 giorni dopo la preparazione della soluzione (nota: il giorno di preparazione è considerato giorno 0. Ad esempio, se si prepara la soluzione il 1° aprile, la data «DA GETTARE DOPO» sarà il 4 giugno). Indicare questa data nello spazio previsto sull'etichetta del flacone (vedere Figura G) e sulla scatola di cartone: Soluzione orale ricostituita DA GETTARE DOPO: (giorno/mese/anno). |
ISTRUZIONI PER L'USO DELLA SOLUZIONE ORALE
È necessario leggere e comprendere le istruzioni per l'uso medico prima di iniziare a prendere Eurisdi®. Queste istruzioni contengono informazioni su come preparare e somministrare la dose di Eurisdi® utilizzando una siringa orale riutilizzabile per via orale, tramite sonda gastrostomica (sonda PEG) o sonda nasogastrica (sonda gastrica). Se ha domande riguardo all'uso di Eurisdi®, si rivolga al medico o al farmacista. Prima della dispensazione, la soluzione orale di Eurisdi® deve essere preparata da un operatore sanitario, come un medico o un farmacista.
Il farmaco Eurisdi® deve essere consegnato in forma liquida in una bottiglia. Non utilizzi il medicinale se si trova in flacone sotto forma di polvere; in tal caso, si rivolga al medico o al farmacista.
Prima di assumere/somministrare la prima dose, un operatore sanitario dovrà istruirla su come preparare e assumere/somministrare la dose giornaliera.
- Chieda al proprio medico o al farmacista di mostrarle quale delle siringhe orali contenute nella confezione deve utilizzare e come determinare la dose giornaliera.
- Se una siringa orale è andata persa o danneggiata, si rivolga al medico o al farmacista, che le indicherà come procedere con l'assunzione del farmaco.
- Per misurare la dose giornaliera prescritta, utilizzi sempre la siringa riutilizzabile della misura corretta.
- La descrizione della siringa orale riutilizzabile adatta al suo caso è riportata anche nella tabella 6: «Scelta della siringa orale riutilizzabile appropriata per la somministrazione della dose giornaliera prescritta di Eurisdi®». Si rivolga al medico o al farmacista se ha dubbi su come scegliere correttamente la siringa.
- Se l'adattatore per flacone non è presente nella bottiglia, non utilizzi Eurisdi® e si rivolga al medico o al farmacista.
- Non utilizzi Eurisdi® dopo la data indicata sull'etichetta della bottiglia e sulla confezione di cartone come «Soluzione orale ricostituita DA GETTARE DOPO: (giorno/mese/anno)». Se sulla etichetta della bottiglia o sulla confezione di cartone non è indicata la data «Soluzione orale ricostituita DA GETTARE DOPO: (giorno/mese/anno)», chieda la data al medico o al farmacista.
- Non mescoli Eurisdi® con cibo o liquidi (ad esempio latte o formula lattea).
- Non utilizzi Eurisdi® se la bottiglia o la siringa orale sono danneggiate.
- Eviti il contatto di Eurisdi® con la pelle. Se Eurisdi® dovesse entrare in contatto con la pelle, lavi l'area interessata con acqua e sapone.
- Se dovesse versare Eurisdi®, pulisca l'area con un tovagliolo di carta asciutto, quindi deterga con acqua e sapone. Getti il tovagliolo di carta nei rifiuti e si lavi bene le mani con acqua e sapone.
- Se nella bottiglia non è presente una quantità sufficiente di Eurisdi® per la dose prescritta, restituisca la bottiglia e le siringhe orali utilizzate al punto di distribuzione (medico o farmacista) per lo smaltimento appropriato. Utilizzi una nuova bottiglia di Eurisdi® per prelevare la dose giornaliera prescritta. Non mescoli Eurisdi® da una nuova bottiglia con il prodotto di una bottiglia già utilizzata in precedenza.
Ogni confezione di Eurisdi® contiene (vedi figura A):
|
|
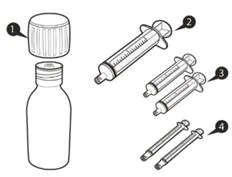
Figura A
A) Preparazione e prelievo del volume corretto della dose
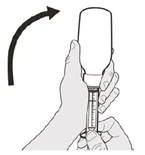
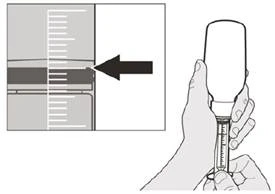
Come scegliere la siringa orale appropriata per la dose prescritta del medicinale Eurisdi®
|
|
|
|
|
|

Come preparare la dose giornaliera del medicinale Eurisdi®
|
Figura B |
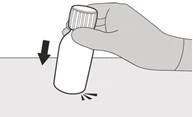
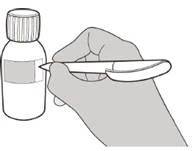
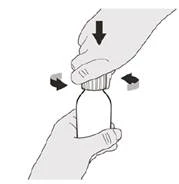
Passo A1 Premere verso il basso sul coperchio e ruotarlo verso sinistra (in senso antiorario) per rimuoverlo (vedere Figura B). Non gettare via il coperchio. |
|||||
|
Figura C |
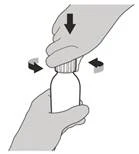
Passo A2 Premere completamente verso il basso lo stantuffo della siringa orale per espellere tutta l'aria dalla siringa orale (vedere Figura C). |
|||||
|
Figura D |
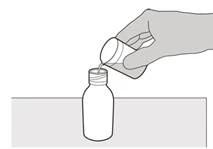
Passo A3 Tenendo la bottiglia in posizione verticale, inserire la punta della siringa nell'applicatore della bottiglia (vedere Figura D). |
|||||
|
Figura E |
Passo A4 Ruotare con attenzione la bottiglia capovolta, con la punta della siringa ben inserita nell'applicatore della bottiglia (vedere Figura E). |
|||||
|
Figura F |
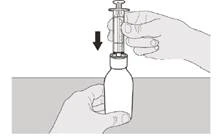
Passo A5 Tirare lentamente lo stantuffo per aspirare la dose giornaliera prescritta del medicinale Eurisdi® dalla bottiglia. Allineare la parte superiore del limitatore di corsa nero con la marcatura dei millilitri sulla siringa orale corrispondente alla dose giornaliera prescritta (vedere Figura F). Dopo aver aspirato la dose corretta, mantenere lo stantuffo fermo per evitare che si muova. |
|||||
|
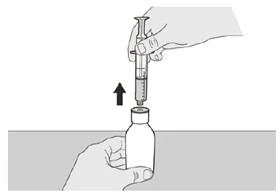
Figura G |
Passo A6 Continuare a mantenere lo stantuffo fermo per evitare che si muova. Lasciare la siringa orale nell'applicatore della bottiglia e riportare la bottiglia in posizione verticale. Appoggiare la bottiglia su una superficie piana. Estrarre con attenzione la siringa orale dall'applicatore della bottiglia tirandola verso l'alto, mantenendo lo stantuffo fermo (vedere Figura G). |
|||||
|
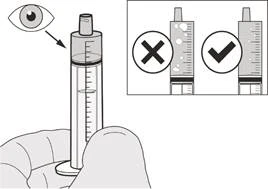
Figura H |
Passo A7 Tenere la siringa orale con la punta rivolta verso l'alto. Controllare il medicinale Eurisdi® all'interno della siringa orale. Se sono presenti bolle d'aria di grandi dimensioni nella siringa orale (vedere Figura H) oppure se è stata aspirata una dose giornaliera errata di Eurisdi®, reinserire saldamente la punta della siringa nell'applicatore della bottiglia. Premere lo stantuffo verso il basso in modo che il medicinale Eurisdi® ritorni nella bottiglia e ripetere i passi da A4 ad A7. Assumere o somministrare immediatamente il medicinale Eurisdi® dopo averlo aspirato nella siringa orale. Se il medicinale non viene assunto entro 5 minuti, eliminare ed eliminare il medicinale Eurisdi® dalla siringa orale e aspirare una nuova dose. |
|||||
|
Figura I |
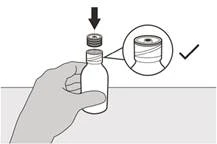
Passo A8 Lasciare l'applicatore sulla bottiglia. Rimettere il coperchio sulla bottiglia. Ruotare il coperchio verso destra (in senso orario) per chiudere ermeticamente la bottiglia (vedere Figura I). |
|||||
Se sta assumendo la dose giornaliera di Eurisdi® per via orale, seguire le istruzioni nella sezione «B) Come assumere la dose giornaliera di Eurisdi® per via orale».
Se sta assumendo la dose giornaliera di Eurisdi® tramite sonda gastrostomica, seguire le istruzioni nella sezione «C) Come somministrare la dose giornaliera di Eurisdi® tramite sonda gastrostomica».
Se sta assumendo la dose giornaliera di Eurisdi® tramite sonda nasogastrica, seguire le istruzioni nella sezione «D) Come somministrare la dose giornaliera di Eurisdi® tramite sonda nasogastrica».
Le siringhe orali per Eurisdi® sono state appositamente progettate per essere compatibili con il sistema ENFit®. Se la sonda per l'alimentazione disponibile non è compatibile con il sistema ENFit®, potrebbe essere necessario un connettore di transizione ENFit per collegare la siringa di Eurisdi® alla tubazione gastrostomica o alla sonda nasogastrica.
B) Come assumere la dose giornaliera di Eurisdi® per via orale
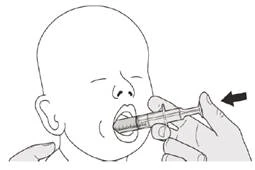
Assumere la dose giornaliera di Eurisdi® per via orale mantenendosi in posizione verticale (preferibilmente seduti).
|
Figura J |
Passaggio B1 Posizionare la siringa orale in bocca con la punta lungo una delle guance. Spingere lentamente lo stantuffo fino in fondo per somministrare l'intera dose di Eurisdi® (vedere Figura J). Se Eurisdi® raggiunge la gola o viene somministrato troppo rapidamente, potrebbe causare soffocamento. |
|
Figura K |
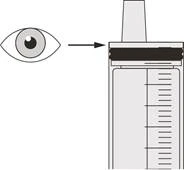
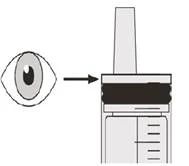
Passo B2 Verificare se nel siringa orale è rimasto del farmaco Eurisdi® (vedere Figura K). |
|
Figura L |
Passo B3 Assumere un po' d'acqua subito dopo aver assunto la dose prescritta di Eurisdi® (vedere Figura L). Per informazioni sulla pulizia della siringa, vedere il passo E. |
C) Come somministrare la dose giornaliera di Eurisdi® attraverso un tubo gastrostomico
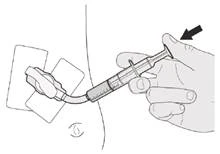
Se sta somministrando Eurisdi® attraverso un tubo gastrostomico, chieda al suo medico di mostrarle come ispezionare il tubo gastrostomico prima della somministrazione di Eurisdi®.
|
Figura M |
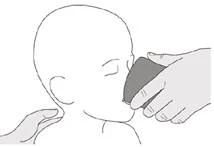
Passo C1 Inserire la punta della siringa orale nel tubo gastrostomico. Premere lentamente lo stantuffo fino in fondo per somministrare l'intera dose di Eurisdi® (vedere Figura M). |
|
Figura N |
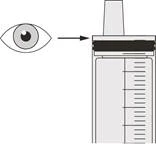
Passo C2 Verificare che non sia rimasto alcun farmaco Eurisdi® nella siringa orale (vedere Figura N). |
|
Figura O |
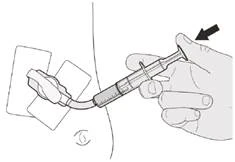
Passo C3 Sciacquare il tubo gastrostomico con 10-20 ml di acqua immediatamente dopo la somministrazione della dose prescritta di Eurisdi® (vedere Figura O). Vedere Passo E per informazioni sulla pulizia della siringa. |
D) Come somministrare la dose giornaliera di Eurisdi® tramite sonda nasogastrica
Se sta somministrando Eurisdi® tramite sonda nasogastrica, chieda al suo medico di mostrarle come verificare correttamente la posizione della sonda nasogastrica prima di somministrare Eurisdi®.
|
Figura P |
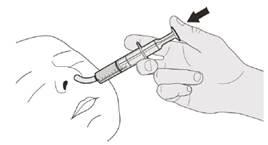
Passo D1 Inserire la punta della siringa orale nel sondino nasogastrico. Premere lentamente lo stantuffo fino in fondo per somministrare la dose completa di Eurisdi® (vedere la Figura P). |
|
Figura Q |
Passo D2 Verificare che non sia rimasto alcun medicinale Eurisdi® nella siringa orale (vedere la Figura Q). |
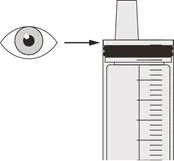
| Figura R |
Passaggio D3 Sciacquare il sondino nasogastrico con 10-20 ml di acqua immediatamente dopo la somministrazione della dose prescritta di Eurisdi® (vedere Figura R). Vedere il Passaggio E per informazioni sulla pulizia della siringa. |
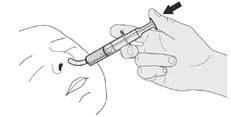
E) Come pulire la siringa orale dopo l'uso
|
Figura S |
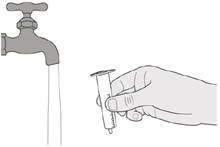
Passo E1 Estrarre lo stantuffo (contro resistenza) dalla siringa. Sciacquare accuratamente il cilindro della siringa orale con acqua corrente pulita (vedere Figura S). |
|
Figura T |
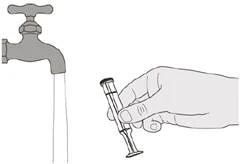
Passaggio E2 Sciacquare accuratamente lo stantuffo sotto acqua corrente pulita (vedere Figura T). |
|
Figura U |
Passo E3 Verificare che il cilindro e lo stantuffo della siringa orale siano puliti. Posizionare il cilindro e lo stantuffo della siringa orale su una superficie pulita in un luogo sicuro per farli asciugare (vedere Figura U). Lavarsi le mani. Dopo che il cilindro e lo stantuffo della siringa orale si sono asciugati, reinserire lo stantuffo nel cilindro della siringa orale e conservare la siringa per il successivo utilizzo. |
Neonati.
La sicurezza e l'efficacia del medicinale Eurisdi® nei pazienti di età inferiore a 16 giorni non sono state stabilite (vedere il paragrafo «Farmacocinetica»).
Sovradosaggio.
Non vi è esperienza di sovradosaggio con il medicinale Eurisdi® negli studi clinici. Non esiste un antidoto noto in caso di sovradosaggio con Eurisdi®. In caso di sovradosaggio, si raccomanda un attento monitoraggio del paziente e l'inizio di una terapia di supporto.
Reazioni avverse
Classificazione della frequenza di insorgenza delle reazioni avverse del medicinale: molto comune (≥ 1/10), comune (da ≥ 1/100 a < 1/10), non comune (da ≥ 1/1 000 a < 1/100), raro (da ≥ 1/10 000 a < 1/1000), molto raro (< 1/10 000); frequenza non nota (non può essere calcolata sulla base dei dati disponibili).
Breve descrizione del profilo di sicurezza
Nei pazienti con SMA ad esordio infantile, le reazioni avverse più comuni osservate negli studi clinici con il medicinale Eurisdi® sono state aumento della temperatura corporea (54,8%), eruzioni cutanee (29%) e diarrea (19,4%). Nei pazienti con SMA ad esordio tardivo, le reazioni avverse più comuni osservate negli studi clinici con il medicinale Eurisdi® sono state aumento della temperatura corporea (21,7%), cefalea (20%), diarrea (16,7%) ed eruzioni cutanee (16,7%).
Le reazioni avverse sopra indicate si sono verificate senza caratteristiche cliniche identificabili o relazione temporale specifica e in generale sono scomparse senza interruzione del trattamento con il medicinale Eurisdi® nei pazienti con SMA ad esordio infantile e tardivo.
Tabella 7
Riepilogo delle reazioni avverse osservate nei pazienti con SMA ad esordio infantile e tardivo negli studi clinici con il medicinale Eurisdi®
| Classe di sistema/organo |
SMA infantile2 (tipo 1) |
SMA tardiva3 (tipo 2 e 3) |
|
| Disturbi gastrointestinali |
|||
| Diaree |
Molto comune |
Molto comune |
|
| Nausea |
Non applicabile |
Comune |
|
| Lesioni della mucosa orale e ulcere aftose |
Comune |
Comune |
|
| Disturbi della cute e del tessuto sottocutaneo |
|||
| Eruzione cutanea1 |
Molto comune |
Molto comune |
|
| Disturbi del sistema nervoso |
|||
| Cefalea |
Non applicabile |
Molto comune |
|
| Alterazioni sistemiche e condizioni in sede di somministrazione |
|||
| Aumento della temperatura corporea (inclusa iperpiressia) |
Molto comune |
Molto comune |
|
| Infezioni e infestazioni |
|||
| Infezione delle vie urinarie (inclusa cistite) |
Comune |
Comune |
|
| Alterazioni del sistema muscoloscheletrico e del tessuto connettivo |
|||
| Artralgia |
Non applicabile |
Comune |
|
1 Compresi dermatite, dermatite acneiforme, dermatite allergica, eritema, follicolite, eruzione cutanea, eruzione eritematosa, eruzione maculopapulare, eruzione papulosa.
2 Negli pazienti con SMA ad esordio infantile (studio FIREFISH, parti 1 e 2), le reazioni avverse sono state definite come eventi verificatisi in ≥ 2% dei pazienti o più, con possibile relazione causale con il medicinale Eurisdi®.
3 Negli pazienti con SMA ad esordio tardivo (studio SUNFISH, parte 2), le reazioni avverse sono state definite come eventi che si sono verificati in almeno il 2% in più di pazienti trattati con Eurisdi® rispetto al gruppo placebo durante il periodo di trattamento in doppio cieco controllato con placebo, con possibile relazione causale con il medicinale Eurisdi®.
I dati disponibili sulla sicurezza sono limitati per quanto riguarda il numero di pazienti trattati con Eurisdi® e la durata dell’esposizione. Possono verificarsi potenziali reazioni avverse rare e potenzialmente gravi (RA) non identificate durante il programma di studi.
Sono disponibili dati limitati sulla sicurezza dell’uso di Eurisdi® nei neonati e nei lattanti con SMA presintomatica dallo studio RAINBOWFISH. Lo studio RAINBOWFISH è uno studio aperto a singolo braccio che ha coinvolto 26 pazienti con SMA presintomatica di età compresa tra 16 e 41 giorni (intervallo di peso corporeo da 3,1 a 5,7 kg) al momento della prima dose. Nell’analisi primaria, la durata media del trattamento è stata di 20,4 mesi (intervallo: da 10,6 a 41,9 mesi) (vedere la sezione «Proprietà farmacologiche», sottosezione «Efficacia clinica»).
Il profilo di sicurezza di Eurisdi® nei pazienti con SMA presintomatica nello studio RAINBOWFISH è paragonabile al profilo di sicurezza nei pazienti con SMA sintomatica trattati con Eurisdi® negli studi clinici. Attualmente non sono disponibili dati a lungo termine.
Profilo di sicurezza nei pazienti con SMA precedentemente trattati con un’altra terapia modificante la malattia
Sulla base dell’analisi primaria dei partecipanti allo studio JEWELFISH, il profilo di sicurezza di Eurisdi® nei pazienti precedentemente trattati, che hanno ricevuto Eurisdi® per un periodo fino a 59 mesi nello studio JEWELFISH, è coerente con il profilo di sicurezza osservato nei pazienti con SMA non precedentemente trattati che hanno ricevuto Eurisdi® negli studi FIREFISH (parti 1 e 2), SUNFISH (parti 1 e 2) e RAINBOWFISH.
I pazienti precedentemente trattati con nusinersen (n = 76) o onasemnogene abeparvovec (n = 14) sono stati inclusi nello studio JEWELFISH (vedere la sottosezione «Efficacia clinica»).
Effetti preclinici
Gli effetti preclinici riguardanti la struttura della retina, il tessuto epiteliale e i parametri ematologici, descritti nella sezione «Dati preclinici», non sono stati osservati negli studi clinici condotti con Eurisdi® per la SMA.
Prolungamento dell’intervallo QT
L’analisi farmacocinetica/farmacodinamica ha mostrato l’assenza di segni di prolungamento dell’intervallo QTc in seguito all’uso di Eurisdi® con esposizione nell’intervallo terapeutico; tuttavia, non sono disponibili dati adeguati per esposizioni superiori all’intervallo terapeutico.
Esperienza post-commercializzazione
Durante l’uso post-commercializzazione è stato osservato vasculite cutanea, i cui sintomi sono scomparsi dopo la sospensione definitiva di Eurisdi®. Sulla base dei dati disponibili, non è possibile determinare la categoria di frequenza di questa reazione avversa. Sono disponibili solo dati limitati sull’uso nei neonati di età inferiore a 16 giorni nel periodo post-commercializzazione.
Periodo di validità
2 anni.
Dopo la ricostituzione, la soluzione orale pronta all’uso è stabile per 64 giorni se conservata in frigorifero a una temperatura compresa tra 2–8 °C. Se necessario, il paziente o la persona che se ne prende cura possono conservare la soluzione orale a temperatura ambiente (inferiore a 40 °C) per un periodo totale non superiore a 5 giorni.
Condizioni di conservazione
Conservare la polvere per soluzione orale a una temperatura non superiore a 25 °C, nella confezione originale, per proteggerla dalla luce e dall’umidità. Conservare in un luogo inaccessibile ai bambini.
Conservare la soluzione orale pronta all’uso in frigorifero a una temperatura compresa tra 2–8 °C, nella confezione originale, per proteggerla dalla luce. Tenere il flacone ben chiuso e conservarlo sempre in posizione verticale.
Incompatibilità
Non sono state osservate incompatibilità tra Eurisdi® e le siringhe orali riutilizzabili raccomandate per la somministrazione orale del medicinale.
Confezionamento
Flacone di vetro ambrato da 100 ml (classe III secondo Farmacopea degli Stati Uniti e Farmacopea Europea) con tappo bianco dotato di sistema di sicurezza per la chiusura a prova di bambino (rivestimento esterno in polietilene ad alta densità; rivestimento interno in polipropilene omo-polimero), anello di controllo dell’apertura e disco interno in polietilene e polivinilidene cloruro, completo di 1 adattatore per flacone, 2 siringhe orali riutilizzabili da 1 ml (ognuna in un sacchetto di polietilene), 2 siringhe orali riutilizzabili da 6 ml (ognuna in un sacchetto di polietilene), 1 siringa orale riutilizzabile da 12 ml (in un sacchetto di polietilene), il tutto contenuto in un sacchetto di polietilene. Una fiala e un set completo sono confezionati in una scatola di cartone.
Categoria di vendita
Sotto prescrizione medica.
Produttore
F. Hoffmann-La Roche Ltd
Sede del produttore e indirizzo del luogo di attività
Wurmisweg, 4303 Kaiseraugst, Svizzera