Uman Complex 500 IU/20 ml
Ukraina
Spis treści
INSTRUKCJA stosowania leku Uman Complex 500 IU/20 ml
Skład:
1 fiolka zawiera:
substancje czynne: Human prothrombin complex
czynnik krzepnięcia krwi człowieka IX – 500 IU;
czynnik krzepnięcia krwi człowieka II – 500 IU;
czynnik krzepnięcia krwi człowieka X – 400 IU;
substancje pomocnicze:
cytrynian sodu – 51,6 mg;
chlorek sodu – 162,0 mg;
glicyna – 92,6 mg;
heparyna – nie więcej niż 250 IU (nie więcej niż 12,5 IU/ml);
antytrombina III – nie więcej niż 2,5 IU (nie więcej niż 0,125 IU/ml);
rozpuszczalnik:
woda do wstrzykiwań – 20 ml.
Czynnik IX jest kalibrowany zgodnie z międzynarodowym standardem.
Zawartość całkowitego białka w fiolce wynosi ≤ 300 mg. Specyficzna aktywność preparatu wynosi więcej niż 0,6 IU/mg, wyrażona jako aktywność czynnika IX.
Substancje pomocnicze o znanym działaniu: lek zawiera do 0,20 mmol (lub 4,6 mg) sodu na 1 ml rozcieńczonego roztworu (równoważnik 4 mmol lub 92 mg sodu na fiolkę). Należy to uwzględnić u pacjentów stosujących dietę kontrolowaną pod względem zawartości sodu.
Postać leku.
Proszek do sporządzenia roztworu do wlewu.
Główne właściwości fizykochemiczne: preparat stanowi biały lub lekko zabarwiony proszek lub substancję kruchą, silnie higroskopijną.
Grupa farmakoterapeutyczna. Środki przeciwkrwotoczne. Kombinacja czynników krzepnięcia krwi IX, II, VII i X. Kod ATC B02BD01.
Właściwości immunologiczne i biologiczne.
Farmakodynamika.
Czynniki krzepnięcia II, VII, IX i X, syntetyzowane w wątrobie za pomocą witaminy K, są zwykle nazywane kompleksem protrombinowym.
Czynnik VII – zymogen aktywnej proteazy serynowej czynnika VIIa, który inicjuje krzepnięcie krwi drogą zewnętrzną. Kompleks czynnik tkankowy – czynnik VIIa aktywuje czynniki krzepnięcia X oraz IX, w wyniku czego powstają czynniki IXa i Xa. W dalszym ciągu aktywacji kaskady krzepnięcia aktywowany jest protrombina (czynnik II), która przekształca się w trombinę. Pod wpływem trombiny fibrynogen przekształca się w fibrynę, w wyniku czego powstaje skrzeplina.
Normalna produkcja trombiny ma również kluczowe znaczenie dla funkcjonowania płytek krwi jako części hemostazy pierwotnej.
Odstępstwo ciężkiej niedoboru czynnika VII prowadzi do zmniejszenia tworzenia trombiny i skłonności do krwawień z powodu osłabionej funkcji tworzenia fibryny oraz osłabionej hemostazy pierwotnej. Odstępstwo niedoboru czynnika IX jest jedną z klasycznych hemofilii (hemofilia B). Odstępstwo niedoboru czynnika II lub czynnika X występuje bardzo rzadko, ale w ciężkiej postaci powoduje krwawienia podobne do krwawień w klasycznej hemofilii.
Nabyte niedobory czynników krzepnięcia zależnych od witaminy K obserwuje się podczas leczenia antagonistami witaminy K. Gdy niedobór staje się poważny, prowadzi to do skłonności do znaczących krwawień, głównie takich jak krwawienia do przestrzeni zaotrzewnowej lub mózgowe, a nie do mięśni i stawów. Ciężka niewydolność wątroby prowadzi również do wyraźnie obniżonych poziomów czynników krzepnięcia zależnych od witaminy K oraz skłonności do krwawień klinicznych, które jednak często mają charakter złożony z powodu jednoczesnego trwałego niewielkiego krzepnięcia wewnątrznaczyniowego, niskich poziomów płytek krwi, niedoboru inhibitorów krzepnięcia oraz zaburzonego fibrynolizy.
Podanie ludzkiego kompleksu protrombinowego zapewnia zwiększenie poziomu czynników krzepnięcia zależnych od witaminy K we krwi, a także może przez pewien czas korygować zaburzenia krzepnięcia u pacjentów z niedoborem jednego lub kilku czynników.
Farmakokinetyka.
| Czynnik krzepnięcia |
Okres półtrwania |
| Czynnik II |
40–60 godzin |
| Czynnik IX |
16–30 godzin |
| Czynnik X |
30–60 godzin |
Dane przedkliniczne dotyczące bezpieczeństwa.
Składnik czynników kompleksu protrombinowego jest naturalną częścią ludzkiej osocza i działa podobnie jak czynniki endogenne.
Badania toksyczności pojedynczej dawki nie są istotne, ponieważ wyższe dawki prowadzą do przeciążenia.
Badania toksyczności wielokrotnej dawki u zwierząt nie są możliwe ze względu na interferencję przeciwciał, które powstają przeciwko białku heterologicznemu.
Nawet dawki znacznie przekraczające zalecane dla człowieka na 1 kg masy ciała nie wykazują żadnego działania toksycznego u zwierząt poddanych badaniom.
Ponieważ doświadczenie kliniczne wskazuje na brak objawów działania kancerogennego i mutagennego czynników kompleksu protrombinowego ludzkiej osocza, badania eksperymentalne, szczególnie u gatunków heterologicznych, nie są uzasadnione ani obowiązkowe.
Właściwości kliniczne.
Wskazania.
- Leczenie krwawień i profilaktyka krwawień przed zabiegami operacyjnymi w przypadku nabytej niedostateczności czynników krzepnięcia związanego z kompleksem protrombinowym, np. niedostateczności wywołanej leczeniem antagonistami witaminy K lub przedawkowaniem antagonistów witaminy K, gdy konieczna jest szybka korekcja niedoboru.
- Leczenie krwawień i profilaktyka krwawień przed zabiegami operacyjnymi w przypadku wrodzonej niedostateczności dowolnego czynnika krzepnięcia zależnego od witaminy K, gdy nie ma dostępnego leku zawierającego oczyszczony specyficzny czynnik krzepnięcia.
Przeciwwskazania.
Nadwrażliwość na substancję czynną lub na którąkolwiek ze substancji pomocniczych.
Znana alergia na heparynę lub w wywiadzie heparynowe zakrzepowe małopłytkowość (HIT).
Interakcje z innymi lekami i inne rodzaje oddziaływań.
Leki zawierające ludzki kompleks protrombinowy niwelują działanie antagonistów witaminy K.
Interakcje z innymi lekami nie są znane.
Wpływ na wyniki badań biologicznych
Podczas wykonywania testów krzepnięcia wrażliwych na heparynę u pacjentów otrzymujących wysokie dawki ludzkiego kompleksu protrombinowego należy uwzględnić obecność heparyny jako składnika podawanego leku.
Dzieci.
Brak danych specyficznych dotyczących dzieci.
Właściwości stosowania.
Leczenie zaburzeń krzepnięcia powinno być prowadzone przez specjalistów z odpowiednim doświadczeniem.
U pacjentów z nabytym niedoborem czynników krzepnięcia zależnych od witaminy K (np. podczas leczenia antagonistami witaminy K) lek Uman Complex należy stosować wyłącznie w przypadku konieczności szybkiej korekty poziomu kompleksu protrombinowego, np. w przypadku nasilonych krwawień lub nagłych zabiegów chirurgicznych. W innych przypadkach zazwyczaj wystarczy zmniejszyć dawkę antagonisty witaminy K i/lub podać witaminę K.
Pacjenci otrzymujący antyagonisty witaminy K mogą mieć skłonność do hiperkoagulacji, a podanie ludzkiego kompleksu protrombinowego może ją sprowokować.
W przypadku wrodzonego niedoboru dowolnego czynnika krzepnięcia zależnego od witaminy K, należy, jeśli to możliwe, stosować specyficzny czynnik krzepnięcia.
W przypadku wystąpienia reakcji alergicznej lub reakcji anafilaktycznej należy natychmiast przerwać wstrzykiwanie/infuzję.
W przypadku szoku należy przeprowadzić standardowe leczenie medyczne.
Ochrona wirusowa
Standardowe środki zapobiegania infekcjom w wyniku stosowania leków wytworzonych z krwi lub osocza ludzkiego obejmują dobór dawców, badanie indywidualnych dawek lub pul osocza pod kątem specyficznych markerów infekcji oraz wprowadzenie skutecznych etapów technologicznych inaktywacji/usuwania wirusów.
Mimo to, przy stosowaniu leków wytworzonych z krwi lub osocza ludzkiego, nie można całkowicie wykluczyć możliwości przeniesienia czynników infekcyjnych. Dotyczy to również nieznanych wirusów lub innych patogenów.
Podjęte środki są uzasadnione jako skuteczne wobec wirusów otoczkowych, takich jak HIV, wirusów zapalenia wątroby B i C, a także wobec wirusa zapalenia wątroby A bez otoczki. Środki stosowane mogą mieć ograniczoną skuteczność wobec wirusów bez otoczki, takich jak parwowirus B19. Zakażenie parwowirusem B19 może być poważne u kobiet w ciąży (zakażenie płodu), a także u osób z niedoborem odporności lub zwiększonym erytropoezą (np. przy anemii hemolitycznej).
Należy rozważyć możliwość przeprowadzenia odpowiedniej szczepionki (przeciwko zapaleniu wątroby A i B) u pacjentów regularnie otrzymujących ludzki kompleks protrombinowy.
W celu zachowania powiązania między pacjentem a serią leku, zaleca się ściśle zapisywanie w karcie medycznej pacjenta numeru serii leku Uman Complex przy każdym jego podaniu.
Stosowanie ludzkiego kompleksu protrombinowego wiąże się ze zwiększonym ryzykiem rozsianej wewnątrznaczyniowej koagulacji, powikłań zakrzepowo-zatorowych oraz zawału mięśnia sercowego. Pacjentów otrzymujących ludzki kompleks protrombinowy należy dokładnie przebadać pod kątem objawów wewnątrznaczyniowej koagulacji lub zakrzepów.
Z uwagi na potencjalne ryzyko wystąpienia powikłań zakrzepowo-zatorowych, konieczne jest dokładne monitorowanie z odpowiednim badaniem biologicznym w celu wykrycia pierwszych objawów zakrzepowych i objawów koagulopatii podczas podawania tego leku pacjentom z chorobą wieńcową serca lub z przebytym zawałem mięśnia sercowego, pacjentom z chorobą wątroby, pacjentom po operacji, noworodkom lub pacjentom z ryzykiem wystąpienia powikłań zakrzepowo-zatorowych lub rozsianej wewnątrznaczyniowej koagulacji. W każdym z tych przypadków potencjalna korzyść z leczenia lekiem Uman Complex musi przewyższać ryzyko wystąpienia wskazanych powikłań.
Brak danych dotyczących stosowania leku Uman Complex w przypadku krwawienia okołoporodowego spowodowanego niedoborem witaminy K u noworodków.
Dzieci.
Brak specyficznych danych dotyczących dzieci.
Stosowanie w okresie ciąży lub karmienia piersią.
W trakcie kontrolowanych badań klinicznych nie ustalono bezpieczeństwa stosowania ludzkiego kompleksu protrombinowego u kobiet w ciąży.
Badania doświadczalne na zwierzętach nie są odpowiednie do oceny bezpieczeństwa wpływu leku na funkcję rozrodczą, rozwój embrionalny/fetalny, przebieg ciąży oraz rozwój okołoporodowy i poporodowy u człowieka.
Dlatego ludzki kompleks protrombinowy może być stosowany w czasie ciąży i laktacji wyłącznie w przypadku nagłej potrzeby.
Wpływ na zdolność do kierowania pojazdami i obsługiwanie maszyn.
Nie przeprowadzono badań dotyczących wpływu na zdolność kierowania pojazdami i obsługiwanie innych maszyn.
Sposób stosowania i dawki.
Poniżej przedstawiono jedynie ogólne zalecenia dotyczące dawkowania. Leczenie należy rozpoczynać wyłącznie pod nadzorem specjalisty z doświadczeniem w leczeniu zaburzeń krzepnięcia. Dawkowanie i długość trwania terapii zastępczej zależą od ciężkości zaburzenia, lokalizacji i stopnia krwawienia, a także od stanu klinicznego pacjenta.
Ilość i częstotliwość podania należy dobrać indywidualnie dla każdego pacjenta.
Interwały dawkowania powinny być dostosowane do różnych okresów półtrwania różnych czynników krzepnięcia w kompleksie protrombinowym (patrz „Farmakokinetyka”). Indywidualne wymagania dawkowe mogą być określone wyłącznie na podstawie regularnych pomiarów indywidualnych poziomów czynników krzepnięcia we krwi lub za pomocą ogólnych testów oceny poziomu kompleksu protrombinowego (czas protrombinowy), a także przy ciągłym monitorowaniu stanu klinicznego pacjenta.
W przypadku istotnych zabiegów chirurgicznych konieczny jest dokładny monitoring terapii zastępczej za pomocą badań koagulacyjnych (określenie specyficznego czynnika krzepnięcia i/lub ogólne testy oceny poziomu kompleksu protrombinowego).
Krwawienia i profilaktyka krwawień przedoperacyjnych podczas leczenia antagonistami witaminy K
Dawka będzie zależeć od wartości początkowej (przed leczeniem) i docelowej międzynarodowego stosunku znormalizowanego (INR). Korekcja zaburzeń hemostazy spowodowanych antagonistami witaminy K utrzymuje się około 6–8 godzin. Jednak działanie witaminy K, podawanej jednocześnie, osiąga zazwyczaj skuteczność w ciągu 4–6 godzin. W związku z tym po podaniu witaminy K ponowne leczenie kompleksem protrombinowym ludzkim zazwyczaj nie jest konieczne.
Ponieważ te zalecenia są empiryczne, a ponadto ponieważ czas odbudowy i trwania efektu mogą się różnić, monitorowanie INR podczas leczenia jest obowiązkowe.
Krwawienia i profilaktyka krwawień przedoperacyjnych przy wrodzonej niedostateczności dowolnego czynnika krzepnięcia zależnego od witaminy K, gdy brakuje leku zawierającego specyficzny czynnik krzepnięcia
Wymaganą dawkę do leczenia oblicza się na podstawie danych empirycznych, zgodnie z którymi około 1 IU czynnika IX na 1 kg masy ciała zwiększa aktywność czynnika IX o 0,01 IU/ml; 1 IU czynnika II lub czynnika X na 1 kg masy ciała zwiększa aktywność czynnika II lub czynnika X odpowiednio o 0,02 i 0,017 IU/ml.
Dawkę danego czynnika podaje się w jednostkach międzynarodowych (IU) zgodnie z obowiązującymi standardami WHO dla każdego czynnika. Aktywność danego czynnika krzepnięcia we krwi wyraża się w procentach (w stosunku do osocza normalnego) lub w jednostkach międzynarodowych (w stosunku do międzynarodowego standardu dla danego czynnika krzepnięcia).
Jedna jednostka międzynarodowa (IU) aktywności czynnika krzepnięcia odpowiada ilości znajdującej się w 1 ml normalnego osocza ludzkiego.
Na przykład, obliczenie wymaganej dawki czynnika X opiera się na danych empirycznych, że 1 jednostka międzynarodowa (IU) czynnika X na 1 kg masy ciała zwiększa aktywność czynnika X we krwi o 0,017 IU/ml.
Wymaganą dawkę oblicza się według następującego wzoru:
Wymagana liczba jednostek = masa ciała (kg) × pożądane zwiększenie aktywności czynnika X (IU/ml) × 60,
gdzie
60 (ml/kg) – wartość odwrotna oczekiwanego odnowienia.
Jeśli znana jest indywidualna wartość odnowienia, należy ją wykorzystać do obliczeń.
Dzieci.
Bezpieczeństwo i skuteczność leku Uman Complex 500 IU/20 ml u dzieci nie zostały ustalone.
Sposób podania
Rozpuścić lek zgodnie z poniższym opisem. Lek Uman Complex 500 IU/20 ml należy podawać dożylnie w formie wstrzyknięcia lub powolnej infuzji.
Zaleca się podawanie nie więcej niż 100 IU/kg masy ciała dziennie.
Odtwarzanie proszku za pomocą rozpuszczalnika:
- Przygotować fiolkę z proszkiem i fiolkę z rozpuszczalnikiem do temperatury pokojowej.
- Utrzymywać temperaturę pokojową przez cały czas trwania procesu odtwarzania (maksymalnie 10 minut).
- Usunąć ochronne nakładki z fiolki z proszkiem i fiolki z rozpuszczalnikiem.
- Przetrzeć powierzchnie przekładek obu fiolkek alkoholem etylowym.
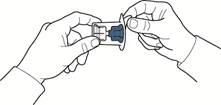
- Otworzyć opakowanie urządzenia, ostrożnie odklejając górną pokrywkę, nie dotykając wnętrza opakowania (rys. A).
- Nie wyjmować urządzenia z opakowania.
- Odwrócić opakowanie z urządzeniem i włożyć plastikowy kolec przez przekładkę fiolki z rozpuszczalnikiem, tak aby niebieska część urządzenia połączyła się z fiolką z rozpuszczalnikiem (rys. B).
- Trzymając za krawędź opakowania, usunąć je, nie dotykając samego urządzenia (rys. C).
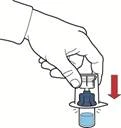
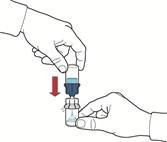
- Upewnić się, że fiolka z proszkiem znajduje się na stabilnej powierzchni; odwrócić układ tak, aby fiolka z rozpuszczalnikiem była nad urządzeniem; nacisnąć przezroczysty adapter na przekładce fiolki z proszkiem, aby włożyć plastikowy kolec przez przekładkę fiolki z proszkiem; rozpuszczalnik automatycznie przepłynie do fiolki z proszkiem (rys. D).
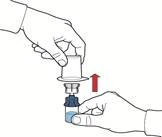
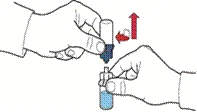
- Po przepłynięciu rozpuszczalnika odkręcić niebieską część układu, do której podłączona jest fiolka z rozpuszczalnikiem, i usunąć ją (rys. E).
- Delikatnie wstrząsać fiolką aż do całkowitego rozpuszczenia proszku. Nie wstrząsać zbyt energicznie, aby uniknąć pienienia się roztworu (rys. F).
| Rys. A |
Rys. B |
|
|
|
| Rys. C |
Rys. D |
|
|
|
| Rys. E |
Rys. F |
|
|
|
Wprowadzenie roztworu
Otrzymany roztwór powinien być przejrzysty lub lekko opalizujący.
Przed wstrzyknięciem roztwór należy wizualnie sprawdzić pod kątem obecności cząstek lub zmiany barwy. Nie należy stosować, jeśli roztwór jest mętny lub zawiera osad.
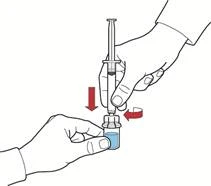
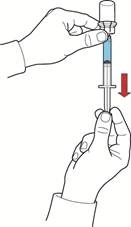
- Przesuwając tłok, napełnić strzykawkę powietrzem, podłączyć do urządzenia i wprowadzić powietrze do fiolki z odtworzonym roztworem (rys. E).
- Zachowując tłok w tym samym położeniu, odwrócić układ do góry nogami tak, aby fiolka z odtworzonym roztworem znajdowała się nad urządzeniem, i powoli przesuwając tłok, nabrać koncentrat do strzykawki (rys. Ż).
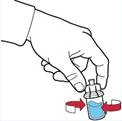
- Odłączyć strzykawkę, obracając ją przeciwnie do ruchu wskazówek zegara.
- Wizualnie sprawdzić roztwór w strzykawce – powinien być przejrzysty lub lekko opalizujący oraz nie zawierać cząstek.
- Podłączyć do strzykawki igłę-skośnik i podać lek dożylnie drogą infuzji lub powolnej iniekcji.
| Rys. E |
Rys. Ż |
|
|
|
Zawartość otwartych fiol należy użyć natychmiast.
Roztwór odnowiony, pobrany do strzykawki, należy użyć natychmiast.
Wszelkie niewykorzystane leki lub ich pozostałości należy zniszczyć zgodnie z lokalnymi wymaganiami.
Dzieci.
Nie ustalono bezpieczeństwa i skuteczności leku Uman Complex 500 IU/20 ml u dzieci.
Przedawkowanie.
Podawanie wysokich dawek ludzkiego kompleksu protrombinowego było związane z przypadkami zawału mięśnia sercowego, rozsianej wewnątrznaczyniowej koagulacji, zakrzepicy żył i zakrzembicy płucnej. W związku z tym w przypadku przedawkowania zwiększa się ryzyko wystąpienia powikłań tromboembolicznych lub rozsianej wewnątrznaczyniowej koagulacji.
Dzieci
Brak danych specyficznych dotyczących dzieci.
Efekty uboczne
Krótki opis profilu bezpieczeństwa
Rzadko obserwowano reakcje alergiczne lub reakcje anafilaktyczne.
Zastępcza terapia ludzkim kompleksem protrombinowym może rzadko prowadzić do powstawania przeciwciał cyrkulujących, które hamują jeden lub kilka czynników ludzkiego kompleksu protrombinowego. Jeśli takie inhibitory powstają, objawiają się słabą odpowiedzią kliniczną.
W rzadkich przypadkach występuje gorączka.
Istnieje potencjalne ryzyko rozwoju reakcji tromboembolicznych po podaniu ludzkiego kompleksu protrombinowego, takich jak embolia i zakrzepica, rozsiane wewnątrznaczyniowe krzepnięcie krwi (DIC) oraz zawał mięśnia sercowego.
Informacje dotyczące bezpieczeństwa w zakresie ryzyka przeniesienia czynników zakaźnych znajdują się w sekcji „Szczególne wskazania dotyczące stosowania”.
Efekty uboczne, które mogą wystąpić podczas stosowania ludzkiego kompleksu protrombinowego, przedstawiono w poniższej tabeli zgodnie z klasyfikacją układów narządów MedDRA (KSN i terminy preferowane).
Częstość została oszacowana według następujących umownych kategorii: bardzo często (≥1/10); często (≥1/100 do <1/10); rzadko (≥1/1 000 do <1/100); rzadko (≥1/10 000 do <1/1 000); bardzo rzadko (<1/10 000); częstość nieznana (nie można oszacować na podstawie dostępnych danych).
| Standardna klasa systemu organów MedDRA |
Reakcje niepożądane(MedDRA, termin preferowanego użycia) |
Częstotliwość |
| Zaburzenia układu immunologicznego |
Nadwrażliwość |
Nieznana |
| Reakcje anafilaktycznego typu |
Nieznana |
|
| Zaburzenia naczyniowe |
Embolia |
Nieznana |
| Tromboza |
Nieznana |
|
| Rozsiane wewnątrznaczyniowe krzepnięcie krwi (DIC) |
Nieznana |
|
| Infarkt mięśnia sercowego |
Nieznana |
|
| Zaburzenia ogólne i reakcje w miejscu podania |
Piroza |
Nieznana |
| Badania diagnostyczne |
Przeciwciała hamujące |
Nieznana |
Dzieci
Nie ma dostępnych specyficznych danych dotyczących dzieci.
Donoszenie o podejrzewanych reakcjach niepożądanych
Donoszenie o podejrzewanych reakcjach niepożądanych po rejestracji produktu leczniczego jest ważne, ponieważ pozwala na dalsze monitorowanie stosunku korzyści do ryzyka. Osoby pracujące w służbie zdrowia powinny zgłaszać wszelkie podejrzewane reakcje niepożądane za pośrednictwem krajowego systemu zgłaszania.
Termin ważności.
Proszek (lekkożylny produkt liofilizowany): 3 lata.
Roztwórnik (woda do wstrzykiwań): 5 lat.
Roztwór należy użyć natychmiast po przygotowaniu.
Nie należy stosować produktu, jeśli roztwór jest mętny lub zawiera osad po odtworzeniu.
Warunki przechowywania.
Przechowywać w lodówce w temperaturze od 2 do 8 ºC.
Przechowywać fiolkę w oryginalnym opakowaniu tekturowym w celu ochrony przed światłem.
Nie zamrażać.
Niezgodność.
Koncentrat ludzkiego kompleksu protrombinowego nie może być mieszany z innymi produktami leczniczymi.
Do stosowania należy używać wyłącznie zestawu do wstrzykiwań/wlewu dostarczanego wraz z lekiem, ponieważ nieskuteczne leczenie może być spowodowane adsorpcją czynników krzepnięcia na wewnętrznych powierzchniach niektórych zestawów do wstrzykiwań/wlewów.
Opakowanie.
500 IU w fiolce nr 1, w zestawie z rozcieńczalnikiem (woda do wstrzykiwań) 20 ml w fiolce nr 1 oraz zestawem do rozcieńczenia i podania, w opakowaniu tekturowym.
Kategoria wydawania.
Na receptę.
Producent.
Kedrion S.p.A.
Adres producenta oraz miejsce prowadzenia działalności.
Via Provinciale, Bolognana 55027, Gallicano, Lucca, Włochy.