Uman Complex 500 MO/20 ml/UMAN COMPLEX 500 IU/20 ml
Ucraina
Indice
ISTRUZIONI PER L'USO MEDICINALE DEL MEDICINALE UMAN COMPLEX 500 MO/20 ml/UMAN COMPLEX 500 IU/20 ml
Composizione:
1 flaconcino contiene:
principi attivi: Human prothrombin complex
fattore della coagulazione del sangue umano IX – 500 MO;
fattore della coagulazione del sangue umano II – 500 MO;
fattore della coagulazione del sangue umano X – 400 MO;
eccipienti:
citrato di sodio – 51,6 mg;
cloruro di sodio – 162,0 mg;
glicina – 92,6 mg;
eparina – non più di 250 MO (non più di 12,5 MO/ml);
antitrombina III – non più di 2,5 MO (non più di 0,125 MO/ml);
solvente:
acqua per preparazioni iniettabili – 20 ml.
Il fattore IX è titolato secondo lo standard internazionale.
Il contenuto di proteine totali nel flaconcino è ≤ 300 mg. L'attività specifica del medicinale è superiore a 0,6 MO/mg, espressa come attività del fattore IX.
Eccipienti con effetto noto: il medicinale contiene fino a 0,20 mmol (o 4,6 mg) di sodio per 1 ml di soluzione diluita (equivalente a 4 mmol, o 92 mg, di sodio per flaconcino). Ciò deve essere tenuto in considerazione nei pazienti sottoposti a dieta controllata per il sodio.
Forma farmaceutica.
Polvere per soluzione per infusione.
Principali proprietà fisico-chimiche: il medicinale è costituito da una polvere bianca o leggermente colorata o da una massa friabile, molto igroscopica.
Gruppo farmacoterapeutico. Agenti antiemorragici. Combinazione dei fattori della coagulazione II, VII, IX e X. Codice ATC B02B D01.
Proprietà immunologiche e biologiche.
Farmacodinamica.
I fattori della coagulazione II, VII, IX e X, sintetizzati nel fegato con l'aiuto della vitamina K, sono solitamente denominati complesso protrombinico.
Il fattore VII è uno zimogeno della serina proteasi attiva fattore VIIa, che inizia la coagulazione del sangue attraverso la via estrinseca. Il complesso fattore tessutale – fattore VIIa attiva i fattori della coagulazione X e IX, portando alla formazione dei fattori IXa e Xa. Con l'ulteriore attivazione della cascata della coagulazione, la protrombina (fattore II) viene attivata e trasformata in trombina. Sotto l'azione della trombina, il fibrinogeno si trasforma in fibrina, con conseguente formazione del coagulo.
La normale formazione della trombina è inoltre essenziale per la funzione piastrinica come parte dell'emostasi primaria.
La carenza grave isolata del fattore VII porta a una ridotta formazione di trombina e a una tendenza al sanguinamento per un'indebolita formazione del fibrinogeno e un'emostasi primaria compromessa. La carenza isolata del fattore IX è una delle emofilie classiche (emofilia B). La carenza isolata del fattore II o del fattore X è molto rara, ma nella forma grave causa emorragie simili a quelle dell'emofilia classica.
La carenza acquisita dei fattori della coagulazione dipendenti dalla vitamina K si osserva durante il trattamento con antagonisti della vitamina K. Se la carenza diventa grave, ciò porta a una tendenza a emorragie significative, prevalentemente retroperitoneali o cerebrali, piuttosto che muscolari o articolari. Anche una grave insufficienza epatica porta a livelli marcatamente ridotti dei fattori della coagulazione dipendenti dalla vitamina K e a una tendenza a emorragia clinica, che tuttavia è spesso complessa a causa della contemporanea attivazione intravascolare cronica, bassi livelli di piastrine, carenza di inibitori della coagulazione e alterato fibrinolisi.
L'amministrazione del complesso protrombinico umano determina un aumento dei livelli plasmatici dei fattori della coagulazione dipendenti dalla vitamina K e può temporaneamente correggere i disturbi della coagulazione in pazienti con carenza di uno o più fattori.
Farmacocinetica.
| Fattore della coagulazione |
Periodo di dimezzamento |
| Fattore II |
40 – 60 ore |
| Fattore IX |
16 – 30 ore |
| Fattore X |
30 – 60 ore |
Dati preclinici di sicurezza.
Il concentrato dei fattori del complesso protrombinico è un componente normale del plasma umano e agisce in modo analogo ai fattori endogeni.
Gli studi sulla tossicità di dose singola non sono significativi, poiché dosi più elevate portano a sovraccarico.
Gli studi sulla tossicità di dose ripetuta negli animali non sono possibili a causa dell'interferenza degli anticorpi formati contro la proteina eterologa.
Anche dosi notevolmente superiori a quelle raccomandate per l'uomo per kg di peso corporeo non mostrano alcun effetto tossico negli animali sottoposti a studio.
Poiché l'esperienza clinica indica l'assenza di segni di azione cancerogena e mutagena dei fattori del complesso protrombinico del plasma umano, studi sperimentali, specialmente su specie eterologhe, non sono giustificatamente obbligatori.
Caratteristiche cliniche.
Indicazioni.
- Trattamento delle emorragie e profilassi preoperatoria delle emorragie in caso di carenza acquisita dei fattori della coagulazione del complesso protrombinico, ad esempio carenza indotta da trattamento con antagonisti della vitamina K o sovradosaggio di antagonisti della vitamina K, quando è necessaria una rapida correzione della carenza.
- Trattamento delle emorragie e profilassi preoperatoria in caso di carenza ereditaria di qualsiasi fattore della coagulazione dipendente dalla vitamina K, quando non è disponibile un prodotto contenente il fattore specifico purificato.
Controindicazioni.
Ipersensibilità al principio attivo o a qualsiasi eccipiente.
Allergia nota all’eparina o anamnesi di trombocitopenia indotta da eparina.
Interazioni con altri medicinali e altre forme di interazione.
I preparati del complesso protrombinico umano neutralizzano l'azione degli antagonisti della vitamina K.
Non sono note interazioni con altri medicinali.
Interferenze con i test biologici
Nei test di coagulazione sensibili all’eparina, si deve tenere conto dell’eparina contenuta nel prodotto somministrato nei pazienti che ricevono alte dosi di complesso protrombinico umano.
Bambini.
Non sono disponibili dati specifici nei bambini.
Caratteristiche particolari di utilizzo.
Il trattamento dei disturbi della coagulazione deve essere effettuato da personale specializzato con esperienza specifica.
Nei pazienti con carenza acquisita dei fattori della coagulazione dipendenti dalla vitamina K (ad esempio, durante il trattamento con antagonisti della vitamina K), il medicinale Uman Complex 500 MO/20 ml/UMAN COMPLEX 500 IU/20 ml deve essere utilizzato solo in caso di necessità di correzione rapida dei livelli del complesso protrombinico, ad esempio in presenza di emorragie significative o interventi chirurgici urgenti. In altri casi, di solito è sufficiente ridurre il dosaggio dell'antagonista della vitamina K e/o somministrare vitamina K.
I pazienti che ricevono antagonisti della vitamina K possono essere predisposti all'ipercoagulabilità, e la somministrazione del complesso protrombinico umano potrebbe indurla.
In caso di carenza congenita di uno qualsiasi dei fattori della coagulazione dipendenti dalla vitamina K, se disponibile, si deve utilizzare il fattore specifico.
In caso di reazione allergica o di reazione anafilattica, l'iniezione/l'infusione deve essere immediatamente interrotta.
In caso di shock, deve essere praticato un trattamento medico standard.
Sicurezza virale
Le misure standard per prevenire infezioni derivanti dall'uso di medicinali ottenuti dal sangue o dal plasma umano comprendono la selezione dei donatori, lo screening delle singole donazioni o dei pool di plasma per specifici marcatori di infezione, nonché l'inserimento di fasi tecnologiche efficaci di inattivazione/rimozione dei virus.
Nonostante ciò, quando si utilizzano medicinali ottenuti dal sangue o dal plasma umano, non si può escludere completamente la possibilità di trasmissione di agenti infettivi. Ciò include anche la trasmissione di virus sconosciuti o di altri patogeni.
Le misure adottate sono considerate efficaci nei confronti dei virus a membrana, come HIV, epatite B ed epatite C, nonché nei confronti del virus senza membrana dell'epatite A. Tuttavia, le misure adottate potrebbero avere efficacia limitata nei confronti dei virus senza membrana, come il parvovirus B19. L'infezione da parvovirus B19 può essere grave per le donne in gravidanza (infezione del feto) e per persone con immunodeficienza o con aumentata eritropoiesi (ad esempio, in caso di anemia emolitica).
Si deve prendere in considerazione la possibilità di effettuare una vaccinazione adeguata (contro epatite A e B) per i pazienti che ricevono regolarmente il complesso protrombinico umano.
Per mantenere un collegamento tra il paziente e il lotto del prodotto, si raccomanda vivamente di registrare ogni volta, nella cartella clinica del paziente, il numero di lotto del medicinale Uman Complex 500 MO/20 ml/UMAN COMPLEX 500 IU/20 ml al momento della somministrazione.
L'uso del complesso protrombinico umano è associato a un aumento del rischio di coagulazione intravascolare disseminata, complicanze tromboemboliche e infarto del miocardio. I pazienti che ricevono il complesso protrombinico umano devono essere attentamente monitorati per la comparsa di sintomi di coagulazione intravascolare o di trombosi.
A causa del potenziale rischio di complicanze tromboemboliche, è necessario effettuare un rigoroso monitoraggio con adeguati test biologici per individuare i primi sintomi trombotici e di coagulopatia nei pazienti con cardiopatia coronarica o anamnesi di infarto del miocardio, nei pazienti con malattia epatica, nei pazienti post-operatori, nei neonati o nei pazienti a rischio di sviluppare eventi tromboembolici o coagulazione intravascolare disseminata. In ciascuno di questi casi, il potenziale beneficio del trattamento con il medicinale Uman Complex 500 MO/20 ml/UMAN COMPLEX 500 IU/20 ml deve superare il rischio delle complicanze sopra menzionate.
Non sono disponibili dati sull'uso del medicinale Uman Complex 500 MO/20 ml/UMAN COMPLEX 500 IU/20 ml in caso di emorragia perinatale dovuta a carenza di vitamina K nei neonati.
Bambini.
Non sono disponibili dati specifici nei bambini.
Uso durante la gravidanza o l'allattamento.
Negli studi clinici controllati non è stata stabilita la sicurezza dell'uso del complesso protrombinico umano in donne in gravidanza.
Gli studi sperimentali sugli animali non sono adatti per valutare la sicurezza dell'effetto del medicinale sulla funzione riproduttiva, lo sviluppo embrionale/fetale, l'andamento della gravidanza e lo sviluppo peri- e postnatale nell'uomo.
Pertanto, il complesso protrombinico umano può essere utilizzato durante la gravidanza e l'allattamento solo in caso di stretta necessità.
Capacità di influenzare la velocità di reazione nella guida di autoveicoli o nell'uso di macchinari.
Non sono stati effettuati studi sull'effetto del medicinale sulla capacità di guidare autoveicoli o di lavorare con macchinari.
Modalità e posologia
Di seguito sono riportate solo raccomandazioni generali relative alla posologia. Il trattamento deve essere iniziato esclusivamente sotto la supervisione di uno specialista esperto nel trattamento dei disturbi della coagulazione. La posologia e la durata della terapia sostitutiva dipendono dalla gravità del disturbo, dal sito e dal grado di emorragia, nonché dallo stato clinico del paziente.
La quantità e la frequenza di somministrazione devono essere calcolate singolarmente per ogni paziente.
Gli intervalli di somministrazione devono essere adattati ai diversi tempi di emivita dei vari fattori della coagulazione del complesso protrombinico (vedere "Farmacocinetica"). Le esigenze individuali di posologia possono essere determinate solo sulla base di misurazioni regolari dei livelli plasmatici individuali dei fattori della coagulazione o mediante test generali per la determinazione dei livelli del complesso protrombinico (tempo di protrombina), nonché con un costante monitoraggio dello stato clinico del paziente.
In caso di interventi chirurgici significativi, è estremamente necessario effettuare un accurato monitoraggio della terapia sostitutiva mediante esami coagulativi (determinazione del fattore di coagulazione specifico e/o test generali per i livelli del complesso protrombinico).
Emorragie e prevenzione preoperatoria delle emorragie durante il trattamento con antagonisti della vitamina K
La dose dipenderà dal valore iniziale (prima del trattamento) e dal valore obiettivo del rapporto normalizzato internazionale (INR). La correzione dei disturbi dell'emostasi indotti dagli antagonisti della vitamina K persiste per circa 6-8 ore. Tuttavia, l'effetto della vitamina K, quando somministrata contemporaneamente, si manifesta generalmente entro 4-6 ore. Pertanto, dopo la somministrazione di vitamina K, di solito non è necessario un ulteriore trattamento con complesso protrombinico umano.
Poiché queste raccomandazioni sono empiriche e poiché il recupero e la durata dell'effetto possono variare, il monitoraggio dell'INR durante il trattamento è obbligatorio.
Emorragie e prevenzione preoperatoria delle emorragie in caso di carenza congenita di un qualsiasi fattore della coagulazione dipendente dalla vitamina K, quando non è disponibile un prodotto contenente il fattore specifico
La dose necessaria per il trattamento viene calcolata sulla base di dati empirici, secondo cui circa 1 UI del fattore IX per 1 kg di peso corporeo aumenta l'attività del fattore IX di 0,01 UI/ml; 1 UI del fattore II o del fattore X per 1 kg di peso corporeo aumenta l'attività del fattore II o del fattore X rispettivamente di 0,02 e 0,017 UI/ml.
La dose del fattore somministrato è espressa in unità internazionali (UI) secondo gli standard attuali dell'OMS per ciascun fattore. L'attività plasmatica del fattore di coagulazione specifico è espressa in percentuale (rispetto al plasma normale) oppure in unità internazionali (rispetto allo standard internazionale per il fattore di coagulazione specifico).
Un'unità internazionale (UI) di attività di un fattore della coagulazione corrisponde alla quantità presente in 1 ml di plasma normale umano.
Ad esempio, il calcolo della dose necessaria del fattore X si basa su dati empirici secondo cui 1 unità internazionale (UI) di fattore X per 1 kg di peso corporeo aumenta l'attività plasmatica del fattore X di 0,017 UI/ml.
La dose necessaria viene calcolata con la seguente formula:
Quantità necessaria di unità = peso corporeo (kg) × aumento desiderato del fattore X (UI/ml) × 60,
dove
60 (ml/kg) è il valore reciproco del recupero atteso.
Se è noto il valore di recupero individuale, tale valore deve essere utilizzato per il calcolo.
Bambini.
La sicurezza e l'efficacia del medicinale Uman Complex 500 MO/20 ml/UMAN COMPLEX 500 IU/20 ml nei bambini non sono state stabilite.
Modalità di somministrazione
Ricostituire il medicinale come descritto di seguito. Il medicinale Uman Complex 500 MO/20 ml/UMAN COMPLEX 500 IU/20 ml deve essere somministrato per via endovenosa mediante iniezione o infusione lenta.
Si raccomanda di non somministrare più di 100 UI/kg di peso corporeo al giorno.
Ricostituzione della polvere con il solvente:
- Portare il flaconcino con la polvere e il flaconcino con il solvente alla temperatura ambiente.
- Mantenere la temperatura ambiente per tutta la durata del processo di ricostituzione (massimo 10 minuti).
- Rimuovere i tappi protettivi dal flaconcino con la polvere e dal flaconcino con il solvente.
- Disinfettare le superfici dei tappi di entrambi i flaconcini con alcol etilico.
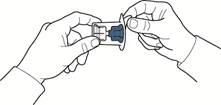
- Aprire la confezione del dispositivo, staccando con attenzione il coperchio superiore senza toccare la parte interna della confezione (fig. A).
- Non estrarre il dispositivo dalla confezione.
- Capovolgere la confezione con il dispositivo e inserire la punta di plastica attraverso il tappo del flaconcino con il solvente, in modo che la parte blu del dispositivo si connetta al flaconcino con il solvente (fig. B).
- Tenendo il bordo della confezione, rimuoverla senza toccare il dispositivo stesso (fig. C).
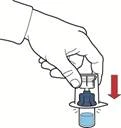
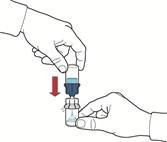
- Assicurarsi che il flaconcino con la polvere sia posizionato su una superficie stabile; capovolgere il sistema in modo che il flaconcino con il solvente si trovi sopra il dispositivo; premere sull'adattatore trasparente sul tappo del flaconcino con la polvere in modo da far penetrare la punta di plastica attraverso il tappo del flaconcino con la polvere; il solvente si sposterà automaticamente nel flaconcino con la polvere (fig. D).
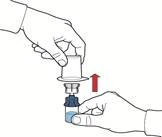
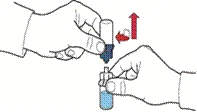
- Dopo lo spostamento del solvente, svitare la parte blu del sistema, a cui è collegato il flaconcino del solvente, e rimuoverla (fig. E).
- Agitare delicatamente il flaconcino fino a completo scioglimento della polvere. Non agitare energeticamente per evitare la formazione di schiuma (fig. F).
| Fig. A |
Fig. B |
|
|
|
| Fig. C |
Fig. D |
|
|
|
| Fig. E |
Fig. F |
|
|
|
Somministrazione della soluzione
La soluzione ottenuta deve essere limpida o leggermente opalescente.
Prima della somministrazione, la soluzione deve essere ispezionata visivamente per verificare la presenza di particelle o cambiamenti di colore. Non utilizzare se la soluzione è torbida o contiene sedimenti.
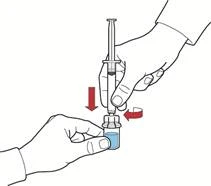
- Tirando indietro lo stantuffo, riempire la siringa con aria, collegarla al dispositivo e iniettare l'aria nel flacone contenente la soluzione ricostituita (fig. E).
- Mantenendo lo stantuffo nella stessa posizione, capovolgere il sistema in modo che il flacone con la soluzione ricostituita si trovi sopra il dispositivo e, tirando lentamente indietro lo stantuffo, aspirare il concentrato nella siringa (fig. F).
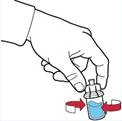
- Scollegare la siringa ruotandola in senso antiorario.
- Ispezionare visivamente la soluzione nella siringa: deve essere limpida o leggermente opalescente e non contenere particelle.
- Applicare all'estremità della siringa un ago a farfalla e somministrare il medicinale per via endovenosa mediante infusione o iniezione lenta.
| Fig. E |
Fig. F |
|
|
|
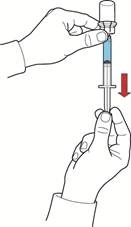
Il contenuto dei flaconi aperti deve essere utilizzato immediatamente.
La soluzione ricostituita aspirata in siringa deve essere utilizzata immediatamente.
Qualsiasi farmaco non utilizzato o i suoi residui devono essere eliminati in conformità ai requisiti locali.
Bambini.
La sicurezza e l'efficacia del medicinale Uman Complex 500 MO/20 ml/UMAN COMPLEX 500 IU/20 ml nei bambini non sono state stabilite.
Sovradosaggio.
L’infusione di alte dosi di complessi protrombinici umani è stata associata a casi di infarto miocardico, coagulazione intravascolare disseminata, trombosi venose e tromboembolia polmonare. Pertanto, in caso di sovradosaggio, aumenta il rischio di sviluppare complicanze tromboemboliche o coagulazione intravascolare disseminata.
Bambini
Non sono disponibili dati specifici nei bambini.
Effetti indesiderati.
Descrizione sintetica del profilo di sicurezza
Raramente sono state osservate reazioni allergiche o di tipo anafilattico.
La terapia sostitutiva con complesso protrombinico umano può raramente indurre la formazione di anticorpi circolanti in grado di inibire uno o più fattori del complesso protrombinico umano. Se tali inibitori si sviluppano, si manifestano con una risposta clinica scarsa.
In rari casi è stata osservata febbre.
Esiste un potenziale rischio di sviluppare reazioni tromboemboliche dopo l’infusione del complesso protrombinico umano, come embolia e trombosi, coagulazione intravascolare disseminata e infarto del miocardio.
Per informazioni sulla sicurezza riguardo al rischio di trasmissione di agenti infettivi, vedere la sezione «Particolari avvertenze e precauzioni per l’uso».
Gli effetti indesiderati che possono verificarsi durante l’impiego del complesso protrombinico umano sono riportati nella tabella sottostante secondo la classificazione per sistemi e organi MedDRA (SOC) e i termini preferenziali.
La frequenza è stata valutata secondo le seguenti categorie convenzionali: molto frequente (≥1/10); frequente (≥1/100 a <1/10); non frequente (≥1/1 000 a <1/100); raro (≥1/10 000 a <1/1 000); molto raro (<1/10 000), frequenza non nota (non può essere stimata sulla base dei dati disponibili).
| Classe sistemica standard MedDRA |
Reazioni avverse(MedDRA, termine di preferenza) |
Frequenza |
| Disturbi del sistema immunitario |
Ipersensibilità |
Sconosciuta |
| Reazioni di tipo anafilattico |
Sconosciuta |
|
| Disturbi vascolari |
Embolia |
Sconosciuta |
| Trombosi |
Sconosciuta |
|
| Coagulazione intravascolare disseminata |
Sconosciuta |
|
| Infarto del miocardio |
Sconosciuta |
|
| Disturbi generali e condizioni in relazione al sito di somministrazione |
Piressia |
Sconosciuta |
| Esami diagnostici |
Anticorpi inibitori |
Sconosciuta |
Popolazione pediatrica
Non sono disponibili dati specifici nei bambini.
Segnalazione delle reazioni avverse sospette
La segnalazione delle reazioni avverse sospette dopo l'autorizzazione del medicinale è importante, in quanto permette di continuare a monitorare il rapporto beneficio/rischio del medicinale. I professionisti del settore sanitario devono segnalare qualsiasi reazione avversa sospetta attraverso il sistema nazionale di segnalazione.
Periodo di validità.
Polvere (preparato liofilizzato): 3 anni.
Solvente (acqua per preparazioni iniettabili): 5 anni.
La soluzione ricostituita deve essere utilizzata immediatamente.
Non utilizzare il medicinale se, dopo la ricostituzione, la soluzione è torbida o contiene particelle in sospensione.
Condizioni di conservazione.
Conservare in frigorifero a una temperatura compresa tra 2 e 8 ºC.
Conservare il flaconcino nella confezione esterna originale per proteggerlo dalla luce.
Non congelare.
Incompatibilità.
Il concentrato del complesso protrombinico umano non deve essere miscelato con altri medicinali.
Deve essere utilizzato esclusivamente il set per iniezione/infusione fornito, poiché un trattamento inefficace potrebbe essere causato dall'adsorbimento dei fattori della coagulazione sulle superfici interne di alcuni set per iniezione/infusione.
Confezione.
1 flaconcino da 500 MO in un set con 1 flaconcino da 20 ml di solvente (acqua per preparazioni iniettabili) e un set per la ricostituzione e la somministrazione, in una confezione di cartone.
Categoria di prescrizione.
Sotto prescrizione medica.
Produttore.
Kedrion S.p.A.
Indirizzo del produttore e sede operativa.
Via Provinciale, Bolognana 55027, Gallicano (Lucca), Italia.