Trycor® 145 mg
Ukraina
Spis treści
INSTRUKCJA dotyczÄ cÄ stosowania leku Trycor® 145 mg (TRICOR® 145 MG)
SkÅ ad:
substancja czynna: fenofibrat;
1 tabletka zawiera 145 mg fenofibratu;
substancje pomocnicze: hipromeloza, sodok dokuzat, sacharoza, sodowa lawrzynian siarkowego, laktoza jednowodna, celuloza mikrokrysztaÅ‚yczna silikatyzowana, krospliwidon, stearynian magnezu, Opadry® OY-B-28920 (alkohol polowinylowy, dwutlenek tytanu (E 171), talk, lecytyna sojowa, żelina ksantanowa).
PostaÄ c leku. Tabletki powlekane.
GÅ ówne wÅ‚aÅ›ciwoÅ›ci fizykochemiczne: biaÅ‚e, owalne tabletki powlekane oznaczone kodem „145” z jednej strony i logo firmy „Fournier” z drugiej strony.
Grupa farmakoterapeutyczna. Åšrodki hipolipidemiczne. Leki obniżajÄ ce poziom cholesterolu i trójglicerydów w osoczu. Fibraty. Kod ATC C10AB05.
Właściwości farmakologiczne.
Farmakodynamika.
Dislipidemia
Fenofibrat jest pochodną kwasu fibrowego, którego efekty lipidomodyfikujące u człowieka są pośredniczone przez aktywację receptora aktywowanego przez proliferator peroksysomów typu alfa (PPARα).
Poprzez aktywację PPARα fenofibrat zwiększa lipolizę oraz eliminację z osocza aterogennych cząstek bogatych w trójglicerydy poprzez aktywację lipazy lipoproteinowej oraz zmniejszenie powstawania apoproteiny CIII. Aktywacja PPARα powoduje również zwiększenie syntezy apoproteiny AI i AII.
Wyżej wymienione efekty fenofibratu na lipoproteiny prowadzą do zmniejszenia frakcji lipoprotein o bardzo niskiej i niskiej gęstości (VLDL i LDL) zawierających apoproteiny B oraz do zwiększenia frakcji lipoprotein o wysokiej gęstości (HDL) zawierających apoproteiny AI i AII.
Ponadto, poprzez modulację syntezy i katabolizmu frakcji VLDL, fenofibrat zwiększa klirens LDL oraz zmniejsza ilość drobnych, gęstych cząstek LDL, których poziom jest podwyższony u osób z aterogennym fenotypem lipoproteinowym, co często występuje u pacjentów z ryzykiem rozwoju choroby niedokrwiennej serca.
W badaniach klinicznych stosowanie fenofibratu prowadziło do obniżenia poziomu cholesterolu ogólnego o 20–25%, trójglicerydów o 40–55% oraz wzrostu poziomu cholesterolu HDL o 10–30%.
U pacjentów z hipercholesterolemią, u których poziom cholesterolu LDL obniżał się o 20–35%, ogólny wpływ na poziom cholesterolu prowadził do zmniejszenia stosunków: cholesterol ogólny/chloresterol HDL, cholesterol LDL/chloresterol HDL lub apoproteina B/apoproteina AI, które są markerami ryzyka aterogennego.
Istnieją dowody, że leczenie fibratami może zmniejszyć częstość występowania zdarzeń w przebiegu choroby niedokrwiennej serca, jednak fibraty nie wykazały zmniejszenia ogólnej śmiertelności w pierwotnej ani wtórnej profilaktyce chorób układu sercowo-naczyniowego.
Badanie ACCORD lipid było randomizowanym, placebo-kontrolowanym badaniem z udziałem 5518 pacjentów z cukrzycą typu 2, którzy otrzymywali fenofibrat w połączeniu ze symwastatyną. Terapia fenofibratem w połączeniu ze symwastatyną w porównaniu do monoterapii symwastatyną nie wykazała istotnych różnic w oddziaływaniu na złożony punkt końcowy pierwotny – niezgonny zawał mięśnia sercowego, niezgonny udar mózgu i śmierć sercowo-naczyniowa (stosunek ryzyka [SR] 0,92, 95% CI 0,79-1,08, p = 0,32; redukcja ryzyka absolutnego: 0,74%). W uprzednio wybranej podgrupie pacjentów z dyslipidemią, którzy mieli najniższy tercyl cholesterolu HDL (≤ 34 mg/dl lub 0,88 mmol/l) oraz najwyższy tercyl TG (≥ 204 mg/dl lub 2,3 mmol/l) przed rozpoczęciem leczenia, terapia skojarzona fenofibratem i symwastatyną w porównaniu do monoterapii symwastatyną wykazała 31% względne zmniejszenie ryzyka złożonego punktu końcowego pierwotnego (stosunek ryzyka [SR] 0,69, 95% CI 0,49-0,97, p = 0,03; redukcja ryzyka absolutnego: 4,95%). Analiza innej uprzednio wybranej podgrupy wykazała istotną statystycznie interakcję między leczeniem a płcią (p = 0,01), wskazującą na możliwą korzyść terapii skojarzonej u mężczyzn (p = 0,037), ale potencjalnie wyższe ryzyko wystąpienia punktu końcowego pierwotnego u kobiet otrzymujących terapię skojarzoną w porównaniu do monoterapii symwastatyną (p = 0,069). Zjawisko to nie było obserwowane w wyżej wspomnianej podgrupie pacjentów z dyslipidemią, jednakże nie było również jednoznacznych dowodów na korzyść dla kobiet z dyslipidemią otrzymujących fenofibrat w połączeniu ze symwastatyną, a potencjalny szkodliwy efekt w tej podgrupie nie może być wykluczony.
Zewnętrzne nacieki cholesterolu (kserosty i kseroty zwojowe) mogą istotnie się zmniejszać lub całkowicie zanikać podczas leczenia fenofibratem.
U pacjentów z podwyższonym poziomem fibrynogenu, którzy leczono fenofibratem, zaobserwowano istotne obniżenie tego parametru, podobnie jak u pacjentów z podwyższonym poziomem lipoproteiny (a). Fenofibrat obniża poziomy innych markerów stanu zapalnego, takich jak białko C-reaktywne.
Działanie urkozuryczne fenofibratu, prowadzące do obniżenia poziomu kwasu moczowego o około 25%, należy uznać za dodatkowy korzystny efekt leku u pacjentów z dyslipidemią i hiperurykemią.
Antyagregacyjny wpływ fenofibratu na płytki krwi został wykazany w badaniach na zwierzętach oraz w badaniu klinicznym, które wykazały zmniejszenie agregacji płytek krwi wywołanej ADP, kwasem arachidonowym i adrenalina.
Retinopatia cukrzycowa
W kilku mechanizmów zaproponowano do wyjaśnienia efektów fenofibratu u pacjentów z proliferacyjną retinopatią cukrzycową (PRC) i cukrzycowym obrzękiem plamki (DME) in vitro oraz na modelach gryzoni. Wykazano, że fenofibrat zmniejsza ekspresję czynnika wzrostu śródbłonka naczyń (VEGF), który jest głównym czynnikiem angiogennym w PRC, zmniejsza przepuszczalność naczyń i apoptozę nabłonka barwnikowego siatkówki, które sprzyjają rozwojowi DME.
Badanie FIELD było wielonarodowym, randomizowanym badaniem z udziałem 9795 pacjentów z cukrzycą typu 2. Wybranych pacjentów losowano do grupy leczenia fenofibratem 200 mg na dobę (n = 4895) lub do grupy placebo (n = 4900). W podbadaniu okulistycznym z udziałem 1012 pacjentów wykonano standaryzowane fotografie siatkówki, które oceniano według kryteriów ETDRS (Early Treatment Diabetic Retinopathy Study) w celu określenia skumulowanej częstości wystąpienia retinopatii cukrzycowej i jej poszczególnych objawów. Analizy przeprowadzono u wszystkich pacjentów, którzy rozpoczęli leczenie. W podbadaniu okulistycznym pierwotny punkt końcowy – postęp 2-stopniowy w stadium retinopatii – istotnie się nie różnił między dwiema grupami ogółem (46 [9,6%] pacjentów w grupie fenofibratu vs 57 [12,3%] w grupie placebo, p = 0,19) ani w podgrupie pacjentów bez wcześniejszej retinopatii (43 [11,4%] vs 43 [11,7%], p = 0,87). Z drugiej strony, w grupie pacjentów z już istniejącą retinopatią, 2-stopniowy postęp obserwowano u istotnie mniejszej liczby pacjentów przy stosowaniu fenofibratu niż w grupie placebo (3 [3,1%] vs 14 [14,6%] pacjentów, p = 0,004).
Informacje dotyczące leczenia laserowego retinopatii cukrzycowej, które było z góry ustalonym trzeciorzędnym punktem końcowym w głównym badaniu, zbierano przy każdej wizycie pacjenta do kliniki. Potrzeba przeprowadzenia pierwszego leczenia laserowego wszystkich retinopatii była istotnie niższa przy leczeniu fenofibratem w porównaniu do placebo (164 [3,4%] pacjentów w grupie fenofibratu vs 238 [4,9%] w grupie placebo; stosunek ryzyka [SR] 0,69, 95% CI 0,56-0,84, p = 0,0002; redukcja ryzyka absolutnego 1,5% [0,7-2,3]). Potrzeba takiego leczenia nie zależała od stężenia lipidów w osoczu krwi.
W podgrupie 2856 uczestników badania ACCORD (ACCORD Eye) oceniano wpływ trzech strategii leczenia na postęp retinopatii cukrzycowej: intensywnej lub standardowej terapii glikemii (docelowy poziom HbA1c <6,0% lub odpowiednio 7,0–7,9%), dyslipidemii (terapia skojarzona fenofibrat 160 mg/dobę plus symwastatyna lub placebo plus symwastatyna) lub kontroli ciśnienia tętniczego skurczowego (docelowy wskaźnik <120 lub <140 mm Hg). Postęp retinopatii cukrzycowej określano po 4 latach na podstawie wzrostu wartości w skali ETDRS o co najmniej 3 punkty (na podstawie oceny stereoskopowych fotografii siedmiu pól dna oka) lub rozwoju retinopatii cukrzycowej wymagającej fotokoagulacji laserowej lub witrerektomii.
Częstość postępu retinopatii cukrzycowej wyniosła 6,5% w grupie intensywnej terapii dyslipidemii z zastosowaniem fenofibratu w porównaniu do 10,2% w grupie placebo (skorygowany stosunek szans 0,60; 95% CI 0,42-0,87, p = 0,006). Stwierdzono, że intensywna terapia skojarzona dyslipidemii zmniejszała częstość postępu retinopatii cukrzycowej.
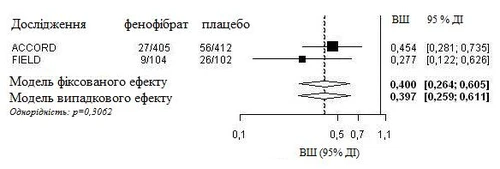
Przeprowadzono zintegrowaną analizę danych indywidualnych pacjentów z badania FIELD i opublikowanych informacji z badania ACCORD Eye. Złożony pierwotny punkt końcowy badania ACCORD Eye został zastosowany do badania FIELD, tj. nasilenie ciężkości w skali ETDRS o 3 punkty, fotokoagulacja lub witrerektomia w leczeniu proliferacyjnej retinopatii cukrzycowej. Oba badania były jednorodne (zastosowano model efektu stałego) i wykazały ogólne 60-procentowe zmniejszenie postępu retinopatii cukrzycowej (stosunek szans: 0,40, 95% CI 0,26-0,61).
Postęp retinopatii cukrzycowej (DR) u pacjentów z DR na początku badania: analiza złożona badań FIELD PSP-DR i ACCORD Eye z wykorzystaniem pierwotnego punktu końcowego badania ACCORD Eye.
Farmakokinetyka.
Wchłanianie.
Maksymalne stężenie leku w osoczu (Cmax) osiągane jest po 2–4 godzinach po podaniu doustnym. Stężenie w osoczu pozostaje stabilne przy długotrwałym stosowaniu u wszystkich pacjentów.
W przeciwieństwie do wcześniejszych preparatów fenofibratu, przy stosowaniu preparatu Trycor® 145 mg w formie nanocząstek jego maksymalne stężenie w osoczu oraz całkowite narażenie nie zależą od przyjęcia posiłku, dlatego Trycor® 145 mg można przyjmować niezależnie od posiłku.
W badaniu wpływu posiłku na wchłanianie leku po podaniu nowej formy tabletek 145 mg fenofibratu zdrowym mężczyznom i kobietom na czczo oraz z posiłkiem bogatym w tłuszcze wykazano, że przyjęcie posiłku nie wpływa na ekspozycję (AUC i Cmax) kwasu fenofibrowego.
Rozkład.
Kwas fenofibrowy w znacznym stopniu wiąże się z albuminą osocza krwi (powyżej 99%).
Metabolizm i wydalanie.
Po podaniu doustnym fenofibrat szybko ulega hydrolizie przez esterazy do aktywnego metabolitu – kwasu fenofibrowego. Niezmieniony fenofibrat nie jest wykrywany w osoczu krwi. Fenofibrat nie jest substratem CYP 3A4 i nie bierze udziału w wątrobowym metabolizmie mikrosomalnym.
Lek wydalany jest głównie z moczem. Prawie całkowicie wydala się w ciągu 6 dni. Fenofibrat wydalany jest głównie w postaci kwasu fenofibrowego i jego koniugatu glukuronidowego. U pacjentów w wieku starszym wyraźny całkowity klirens kwasu fenofibrowego z osocza krwi nie ulega zmianie.
Badania kinetyki po podaniu dawki pojedynczej i długotrwałym leczeniu wykazały, że lek nie gromadzi się w organizmie. Kwas fenofibrowy nie jest usuwany podczas hemodializy.
Okres półtrwania kwasu fenofibrowego wynosi około 20 godzin.
Właściwości kliniczne.
Wskazania.
Trycor® 145 mg jest wskazany jako uzupełnienie diety oraz innych nielików stosowanych metod leczenia (np. aktywności fizycznej, redukcji masy ciała) w następujących stanach:
- ciężka hipertrójglicerydemia z niskim poziomem cholesterolu lipoprotein o wysokiej gęstości lub bez niego;
- hiperlipidemia mieszana w przypadkach, gdy stosowanie statyn jest przeciwwskazane lub występuje nietolerancja statyn;
- hiperlipidemia mieszana u pacjentów z wysokim ryzykiem sercowo-naczyniowym, jako dodatek do terapii statyną, gdy poziom trójglicerydów i cholesterolu lipoprotein o wysokiej gęstości nie jest odpowiednio kontrolowany.
Retinopatia cukrzycowa: Trycor® 145 mg jest wskazany w celu zmniejszenia postępu retinopatii cukrzycowej u pacjentów z cukrzycą typu 2 i istniejącą retinopatią cukrzycową.
Przeciwwskazania.
Niewydolność wątroby (w tym zespół żółciowy wątroby i niejasne, utrzymujące się zaburzenia funkcji wątroby).
Stwierdzone choroby pęcherzyka żółciowego.
Ciężkie przewlekłe choroby nerek.
Choroba trzustki o charakterze przewlekłym lub ostrym, z wyjątkiem przypadków ostrego zapalenia trzustki wywołanego ciężką hipertrójglicerydemią.
Stwierdzona alergia światłoczuła lub reakcje fototoksyczne podczas leczenia fibratami lub ketoprofenem.
Nadwrażliwość na substancję czynną lub na którąkolwiek z substancji pomocniczych wymienionych w sekcji „Skład”.
Trycor® 145 mg nie powinien być stosowany u pacjentów z alergią na orzechy ziemne, olej z orzechów ziemnych lub lecytynę sojową, ani podobne produkty ze względu na ryzyko wystąpienia reakcji nadwrażliwości.
Interakcje z innymi lekami i inne rodzaje interakcji.
Leki przeciwkrzepliwe doustne.
Fenofibran potęguje działanie doustnych leków przeciwkrzepliwych i może zwiększać ryzyko krwawień. Zaleca się zmniejszenie dawki leku przeciwkrzepliwego o około 1/3 na początku leczenia, a następnie stopniowe dostosowanie dawki w zależności od wartości INR (międzynarodowego wskaźnika znormalizowanego).
Cyklosporyna.
W trakcie jednoczesnego stosowania fenofibranu i cyklosporyny odnotowano kilka ciężkich przypadków odwracalnych zaburzeń funkcji nerek. Dlatego należy dokładnie monitorować funkcję nerek u pacjentów stosujących tę kombinację, a w przypadku wystąpienia ciężkich zmian w wynikach badań laboratoryjnych należy przerwać stosowanie fenofibranu.
Inhibitory HMG-CoA reduktazy i inne fibraty.
Ryzyko poważnego toksycznego działania na mięśnie wzrasta przy jednoczesnym stosowaniu fibratu z inhibitorami HMG-CoA reduktazy lub innymi fibratami. Należy zachować ostrożność przy stosowaniu tej terapii kombinowanej i dokładnie monitorować pacjentów pod kątem objawów toksycznego działania na mięśnie (patrz sekcja „Szczególne ostrzeżenia i środki ostrożności”).
Glitazony.
Podczas jednoczesnego stosowania fenofibranu i glitazonów obserwowano przypadki odwracalnego paradoksalnego obniżenia poziomu cholesterolu LPWZ. Dlatego zaleca się kontrolowanie poziomu cholesterolu LPWZ podczas stosowania tej kombinacji leków i przerwanie ich stosowania, jeśli poziom cholesterolu LPWZ staje się zbyt niski.
Fermenty cytochromu P450.
Badania in vitro z wykorzystaniem mikrosom wątroby człowieka wykazały, że fenofibran i kwas fenofibrowy nie są inhibitorami izoform cytochromu (CYP) P450 CYP3A4, CYP2D6, CYP2E1 ani CYP1A2. W stężeniach terapeutycznych są słabymi inhibitorami CYP2C19 i CYP2A6 oraz słabymi lub umiarkowanymi inhibitorami CYP2C9.
U pacjentów stosujących jednocześnie fenofibran i leki metabolizowane przez CYP2C19, CYP2A6 oraz szczególnie przez CYP2C9, które mają wąski zakres terapeutyczny, należy dokładnie monitorować stan pacjenta i w razie potrzeby dostosować dawkę tych leków.
Szczególne ostrzeżenia i środki ostrożności.
Hyperlipidemia wtórna.
Przed rozpoczęciem leczenia fenofibranem należy odpowiednio leczyć stany, które są przyczyną hipercholesterolemii wtórnej, takie jak niekontrolowana cukrzyca typu 2, niedoczynność tarczycy, zespół nerczycowy, dysproteinemia, obturacyjna choroba wątroby lub alkoholizm. Hyperlipidemia wtórna może również występować u pacjentów przyjmujących leki, takie jak diuretyki, β-blokery, estrogeny, progestageny, doustne środki antykoncepcyjne, leki immunosupresyjne oraz inhibitory proteaz. W takich przypadkach należy ustalić, czy hiperlipidemia ma charakter pierwotny czy wtórny (może być spowodowana przez wyżej wymienione leki).
Funkcja wątroby.
Podobnie jak przy stosowaniu innych leków obniżających stężenie lipidów, u niektórych pacjentów obserwowano wzrost stężenia transaminaz. W większości przypadków wzrost ten był tymczasowy, niewielki i bezobjawowy. Zaleca się kontrolę stężenia transaminaz co 3 miesiące przez pierwsze 12 miesięcy leczenia oraz okresowo później. Należy zwrócić uwagę na wzrost stężenia transaminaz i przerwać leczenie, jeśli stężenia AST i ALT przekraczają trzykrotnie górną granicę normy. W przypadku wystąpienia objawów zapalenia wątroby (np. żółtaczki, świądu) oraz potwierdzenia rozpoznania wynikami badań laboratoryjnych należy przerwać leczenie fenofibranem.
Trzustka.
U pacjentów przyjmujących fenofibrany donoszono o wystąpieniu zapalenia trzustki (patrz sekcje „Przeciwwskazania” oraz „Efekty niepożądane”). Może to być spowodowane niewystarczającą skutecznością leczenia u pacjentów z ciężką hipertriglicerydemią, bezpośrednią toksycznością leku lub zjawiskiem wtórnym, spowodowanym kamieniami w dróg żółciowych lub powstawaniem błota prowadzącym do obturacji przewodu żółciowego wspólnego.
Mięśnie.
Podczas stosowania fibratów i innych leków obniżających stężenie lipidów zgłaszano toksyczne działanie na mięśnie, w tym rzadkie przypadki rabdomiolizy z niewydolnością nerek lub bez niej. Częstość występowania tego zaburzenia wzrasta przy hiperalbuminii i w przypadku wcześniejszej niewydolności nerek. Pacjentom z czynnikami zwiększającymi ryzyko miopatii i/lub rabdomiolizy, takimi jak wiek powyżej 70 roku życia, choroby mięśni dziedziczne w wywiadzie osobistym lub rodzinnym, zaburzenia funkcji nerek, niedoczynność tarczycy oraz nadużywanie alkoholu, może grozić zwiększone ryzyko rozwoju rabdomiolizy. U takich pacjentów należy dokładnie ocenić stosunek korzyści do ryzyka przed rozpoczęciem leczenia fenofibranem.
Toksyczne działanie na mięśnie należy podejrzewać u pacjentów z uogólnioną mialgia, mięsitisem, skurczami mięśniowymi i osłabieniem mięśni oraz/lub znacznym wzrostem stężenia CK (pięciokrotnie wyższym niż górna granica normy). W takich przypadkach należy przerwać leczenie fenofibranem.
Ryzyko toksycznego działania na mięśnie może wzrosnąć, jeśli lek jest stosowany jednocześnie z innym fibrynem lub inhibitem HMG-CoA reduktazy, szczególnie u pacjentów z chorobą mięśni. Dlatego jednoczesne stosowanie fenofibranu z inhibitem HMG-CoA reduktazy lub z innym fibrynem należy ograniczyć do terapii rezerwowej u pacjentów z ciężką, złożoną dyslipidemią i wysokim ryzykiem sercowo-naczyniowym, bez wywiadu chorób mięśni, przy jednoczesnym dokładnym monitorowaniu możliwego toksycznego działania na mięśnie.
Funkcja nerek.
Jeśli stężenie kreatyniny wzrośnie o więcej niż 50% w stosunku do GGN (górnej granicy normy), leczenie fenofibrany należy przerwać. Zaleca się kontrolę stężenia kreatyniny w ciągu pierwszych 3 miesięcy od rozpoczęcia leczenia oraz okresowo później (dotyczące dawkowania patrz sekcja „Sposób stosowania i dawki”).
Substancje pomocnicze.
Preparat zawiera laktozę, dlatego nie powinien być podawany pacjentom z rzadkimi, dziedzicznymi schorzeniami, takimi jak nietolerancja galaktozy, niedobór laktoazy lub zespół złego wchłaniania glukozy-galaktozy.
Preparat zawiera sacharozę, dlatego nie powinien być podawany pacjentom z rzadkimi, dziedzicznymi schorzeniami, takimi jak nietolerancja fruktozy, zespół złego wchłaniania glukozy-galaktozy lub niedobór sacharazy-izomalatazy.
Stosowanie w czasie ciąży lub karmienia piersią.
Ciąża. Brak wystarczających danych dotyczących stosowania fenofibranu u kobiet w ciąży. W badaniach na zwierzętach nie zaobserwowano żadnego działania teratogennego. Efekty embrionotoksyczne obserwowano po podawaniu leku w dawkach toksycznych dla organizmu matki. Potencjalne ryzyko dla człowieka jest nieznane. Dlatego Trycor® 145 mg w czasie ciąży należy stosować wyłącznie po dokładnej ocenie stosunku korzyści do ryzyka.
Karmienie piersią. Nie wiadomo, czy fenofibran i/lub jego metabolity przenikają do mleka matki. Nie można wykluczyć ryzyka dla niemowląt karmionych piersią, dlatego fenofibranu nie należy stosować w czasie karmienia piersią.
Plodność. W badaniach na zwierzętach obserwowano odwracalne działanie na płodność. Brak danych klinicznych dotyczących wpływu na płodność po stosowaniu preparatu Trycor® 145 mg.
Wpływ na zdolność prowadzenia pojazdów i obsługiwania maszyn.
Trycor® 145 mg nie wpływa lub wpływa nieznacznie na zdolność prowadzenia pojazdów i obsługiwanie maszyn.
Sposób stosowania i dawki
Trycor® 145 mg można przyjmować w dowolnym czasie doby niezależnie od posiłków (patrz sekcja „Właściwości farmakologiczne. Farmakokinetyka”). Tabletki należy połykać całkowicie, popijając szklanką wody.
Diety rozpoczętej przed włączeniem leku należy kontynuować.
W trakcie leczenia hiperlipidemii skuteczność terapii należy kontrolować poprzez oznaczanie poziomów lipidów we krwi. Jeśli po kilku miesiącach (np. po 3 miesiącach) nie uzyskuje się odpowiedniej odpowiedzi na leczenie, należy rozważyć dodatkowe lub inne środki terapeutyczne.
Dorośli
Zalecana dawka to 1 tabletka zawierająca 145 mg fenofibratu, 1 razy dziennie. Pacjentom, którzy przyjmują 1 kapsułkę zawierającą 200 mg fenofibratu lub 1 tabletę zawierającą 160 mg fenofibratu, można zastąpić te dawki 1 tabletką leku Trycor® 145 mg bez dodatkowego dopasowywania dawki.
Jeśli pacjent wymaga stosowania fenofibratu w dwóch wskazaniach (hiperlipidemia i retinopatia cukrzycowa), należy przyjmować tylko 1 tabletkę leku Trycor® 145 mg dziennie.
Pacjenci w wieku podeszłym
Pacjentom w wieku podeszłym, którzy nie mają zaburzeń funkcji nerek, zaleca się zwykłą dawkę dla dorosłych.
Zaburzenia funkcji nerek
Pacjentom z zaburzeniami funkcji nerek należy zmniejszyć dawkę. W przypadku przewlekłych chorób nerek o umiarkowanym nasileniu (klirens kreatyniny od 30 do 60 ml/min) stosowanie fenofibratu w dostępnej dawce 145 mg nie jest zalecane.
Pacjentom z ciężkimi przewlekłymi chorobami nerek (klirens kreatyniny < 30 ml/min) stosowanie fenofibratu jest przeciwwskazane.
Zaburzenia funkcji wątroby
Trycor® 145 mg nie powinien być stosowany u pacjentów z zaburzeniami funkcji wątroby ze względu na brak danych.
Dzieci
Bezpieczeństwo i skuteczność stosowania fenofibratu u dzieci i młodzieży (do 18. roku życia) nie zostały ustalone, a odpowiednie dane są niedostępne. Dlatego fenofibrat nie powinien być stosowany u dzieci i młodzieży (do 18. roku życia).
Przedawkowanie
Zgłaszano jedynie pojedyncze przypadki przedawkowania fenofibratu. W większości przypadków nie odnotowano żadnych objawów przedawkowania.
Nie zna się specyficznego antydota. W przypadku podejrzenia przedawkowania należy przeprowadzić leczenie objawowe i podjąć odpowiednie środki wspierające, jeśli to konieczne. Fenofibrat nie jest usuwany podczas hemodializy.
Niepożądane działania.
Najczęściej obserwowane niepożądane reakcje podczas terapii fenofibratem to zaburzenia trawienne, zaburzenia ze strony żołądka lub jelit.
Poniższe niepożądane zjawiska obserwowano w badaniach klinicznych kontrolowanych placebo (n=2344) z podaną częstością:
| Klasa układu narządów według MedDRA |
Bardzo często ≥ 1/10 |
Często ≥ 1/100, <1/10 |
Nieczęsto ≥ 1/1000, <1/100 |
Rzadko ≥ 1/10000, <1/1000 |
| Zaburzenia ze strony krwi i układu chłonnego |
Obniżenie poziomu hemoglobiny Obniżenie liczby białych krwinek |
|||
| Zaburzenia ze strony układu odpornościowego |
Nadwrażliwość (w tym reakcja anafilaktyczna) |
|||
| Zaburzenia ze strony układu nerwowego |
Ból głowy |
|||
| Zaburzenia ze strony układu naczyniowego |
Zatorowość tętnicza (zatorowość płucna, zakrzepica żył głębokich)** |
|||
| Zaburzenia ze strony przewodu pokarmowego |
Objawy i dolegliwości ze strony układu trawiennego (ból brzucha, nudności, wymioty, biegunka, wzdęcia) |
Choroba trzustki* |
||
| Zaburzenia ze strony układu wątrobowo-żółciowego |
Zwiększenie poziomu aminotransferaz (patrz punkt „Szczególne ostrzeżenia i środki ostrożności”) |
Choroba kamicy żółciowej (patrz punkt „Szczególne ostrzeżenia i środki ostrożności”) |
Zapalenie wątroby |
|
| Zaburzenia ze strony skóry i tkanki podskórnej |
Reakcje skórne nadwrażliwości (np. wysypka, świąd, pokrzywka) |
Łysienie Reakcje fotouczulenia |
||
| Zaburzenia ze strony mięśni szkieletowych, tkanki łącznej i kości |
Zaburzenia mięśniowe (np. mialgia, miązg, skurcze mięśni i osłabienie) |
|||
| Zaburzenia ze strony układu rozrodczego i gruczołów mlekowych |
Dysfunkcja seksualna |
|||
| Odchylenia od normy stwierdzone w wyniku badań laboratoryjnych |
Zwiększony poziom homocysteiny we krwi*** |
Zwiększony poziom kreatyniny we krwi |
Zwiększony poziom mocznika we krwi |
* W badaniu FIELD – randomizowanym, placebo kontrolowanym badaniu, w którym wzięło udział 9795 pacjentów z cukrzycą typu 2 – u pacjentów przyjmujących fenofibrat zaobserwowano istotne statystycznie zwiększenie częstości występowania zapalenia trzustki w porównaniu z grupą otrzymującą placebo (0,8 % i 0,5 % odpowiednio, p = 0,031).
** Zaobserwowano istotne statystycznie zwiększenie częstości występowania zakrzepicy płucnej (0,7 % w grupie otrzymującej placebo i 1,1 % w grupie przyjmującej fenofibrat; p = 0,022) oraz nieistotne statystycznie zwiększenie częstości występowania zakrzepicy żył głębokich (1,0 % w grupie otrzymującej placebo [48/4900 pacjentów] i 1,4 % w grupie przyjmującej fenofibrat [67/4895 pacjentów]; p = 0,074).
*** Średnie zwiększenie stężenia homocysteiny we krwi u pacjentów przyjmujących fenofibrat wynosiło 6,5 µmol/l i było odwracalne po zakończeniu leczenia fenofibratem. Podwyższone ryzyko wystąpienia zjawisk żylnej trombozy może być związane ze zwiększonym poziomem homocysteiny. Kliniczna istotność tego zjawiska nie jest ustalona.
Oprócz działań niepożądanych zaobserwowanych w badaniach klinicznych, w okresie postmarketingowym stosowania leku Trycor® 145 mg otrzymano spontaniczne zgłoszenia działań niepożądanych wymienionych poniżej; na podstawie dostępnych danych niemożliwe jest ustalenie dokładnej częstości ich występowania, dlatego sklasyfikowano ją jako „nieznana”.
Zaburzenia ze strony układu oddechowego, klatki piersiowej i jamy mostkowej: choroba płucna typu międzypłucia.
Zaburzenia ze strony tkanki mięśniowej, tkanki łącznej i kości: rabdomioliza.
Zaburzenia ze strony układu wątrobowo-żółciowego: żółtaczka, powikłania kamicy żółciowej (np. zapalenie pęcherzyka żółciowego, zapalenie dróg żółciowych, kolka żółciowa).
Zaburzenia ze strony skóry i tkanki podskórnej: ciężkie reakcje skórne (np. wielopostaciowe zaczerwienienie, zespół Stevensa-Johnsona, toksyczne martwicze odłuszczenie nabłonka).
Zaburzenia ze strony układu nerwowego: zmęczenie.
W przypadku wystąpienia jakichkolwiek działań niepożądanych podczas stosowania leku lub skarg dotyczących jakości leku, prosimy o powiadomienie firmę Abbott Ukraine LLC pod numerem telefonu: +38 044-498-60-80 lub e-mail: [email protected].
Okres ważności. 3 lata.
Warunki przechowywania. Przechowywać w oryginalnym opakowaniu w temperaturze nie wyższej niż 25 °C w miejscu niedostępnym dla dzieci.
Opakowanie. Po 10 tabletów w blisterze, po 2 lub 3 blisterach w tece kartonowej.
Kategoria wydawania. Na receptę.
Producenci. Fournier Laboratories Ireland Limited, Irlandia / Fournier Laboratories Ireland Limited, Ireland.
Astrea Fontaine, Francja / Astrea Fontaine, France.
Miejsce położenia producentów oraz adresy siedzib działalności. Anngrove, Carrigtwohill, Co. Cork, Irlandia / Anngrove, Carrigtwohill, Co. Cork, Ireland.
Rue Des Pres Potets, Fontaine Les Dijon, 21121, Francja / Rue Des Pres Potets, Fontaine Les Dijon, 21121, France.