Tricor® 145 mg
Ukraine
Table of Contents
INSTRUCTIONS FOR MEDICAL USE OF THE MEDICINAL PRODUCT TRICOR® 145 MG (TRICOR® 145 MG)
Composition:
Active ingredient: fenofibrate;
1 tablet contains 145 mg of fenofibrate;
Excipients: hypromellose, sodium docusate, sucrose, sodium lauryl sulfate, lactose monohydrate, microcrystalline silicified cellulose, crospovidone, magnesium stearate, Opadry**®** OY-B-28920 (polyvinyl alcohol, titanium dioxide (E 171), talc, soy lecithin, xanthan gum).
Pharmaceutical form. Film-coated tablets.
Main physicochemical properties: white, oval, film-coated tablets marked with "145" on one side and the company logo "Fournier" on the other.
Pharmacotherapeutic group. Hypolipidemic agents. Agents that reduce serum cholesterol and triglyceride levels. Fibrates. ATC code C10AB05.
Pharmacological Properties
Pharmacodynamics.
Dyslipidemia
Fenofibrate is a derivative of fibric acid, whose lipid-modifying effects in humans are mediated through activation of the peroxisome proliferator-activated receptor alpha (PPARα).
By activating PPARα, fenofibrate enhances lipolysis and clearance of triglyceride-rich atherogenic particles from plasma by stimulating lipoprotein lipase activity and reducing the production of apolipoprotein CIII. Activation of PPARα also increases the synthesis of apolipoproteins AI and AII.
These effects of fenofibrate on lipoproteins lead to a reduction in the fraction of very low-density lipoproteins (VLDL) and low-density lipoproteins (LDL) containing apolipoprotein B, and to an increase in the fraction of high-density lipoproteins (HDL) containing apolipoproteins AI and AII.
Additionally, by modulating the synthesis and catabolism of VLDL fractions, fenofibrate enhances LDL clearance and reduces the number of small, dense LDL particles, whose levels are elevated in individuals with an atherogenic lipoprotein phenotype commonly observed in patients at risk of developing ischemic heart disease.
In clinical studies, treatment with fenofibrate reduced total cholesterol levels by 20–25%, triglycerides by 40–55%, and increased HDL cholesterol levels by 10–30%.
In patients with hypercholesterolemia, LDL cholesterol levels decreased by 20–35%, and the overall effect on cholesterol levels led to a reduction in the ratios of total cholesterol/HDL cholesterol, LDL cholesterol/HDL cholesterol, or apolipoprotein B/apolipoprotein AI, which are markers of atherogenic risk.
Evidence suggests that fibrate therapy may reduce the incidence of ischemic heart disease events; however, fibrates have not demonstrated a reduction in overall mortality in primary or secondary prevention of cardiovascular diseases.
The ACCORD lipid study was a randomized, placebo-controlled trial involving 5,518 patients with type 2 diabetes who were treated with fenofibrate in addition to simvastatin. Combination therapy with fenofibrate and simvastatin, compared to simvastatin monotherapy, showed no significant differences in the effect on the composite primary endpoint—nonfatal myocardial infarction, nonfatal stroke, and cardiovascular death (risk ratio [RR] 0.92, 95% CI 0.79–1.08, p = 0.32; absolute risk reduction: 0.74%). In a pre-specified subgroup of patients with dyslipidemia who had the lowest tertile of HDL cholesterol (≤34 mg/dL or 0.88 mmol/L) and the highest tertile of triglycerides (≥204 mg/dL or 2.3 mmol/L) at baseline, combination therapy with fenofibrate and simvastatin, compared to simvastatin monotherapy, demonstrated a 31% relative risk reduction in the composite primary endpoint (risk ratio [RR] 0.69, 95% CI 0.49–0.97, p = 0.03; absolute risk reduction: 4.95%). Analysis of another pre-specified subgroup revealed a statistically significant interaction between treatment and sex (p = 0.01), suggesting potential benefit in men (p = 0.037), but a potentially higher risk of the primary endpoint in women receiving combination therapy compared to simvastatin monotherapy (p = 0.069). This phenomenon was not observed in the aforementioned subgroup of patients with dyslipidemia, but there were also no clear evidence of benefit for women with dyslipidemia receiving fenofibrate plus simvastatin, and a potential harmful effect in this subgroup cannot be excluded.
Extra-vascular cholesterol deposits (tendon and tuberous xanthomas) may significantly decrease or completely disappear during treatment with fenofibrate.
In patients with elevated fibrinogen levels, significant reductions were observed during fenofibrate treatment, as well as in patients with elevated lipoprotein(a) levels. Fenofibrate reduces levels of other inflammatory markers such as C-reactive protein.
The uricosuric effect of fenofibrate, which leads to a reduction in uric acid levels by approximately 25%, should be considered an additional beneficial effect of the drug in patients with dyslipidemia and hyperuricemia.
An anti-aggregatory effect of fenofibrate on platelets has been demonstrated in animal studies and in a clinical trial, which showed reduced platelet aggregation induced by ADP, arachidonic acid, and adrenaline.
Diabetic Retinopathy
Several mechanisms have been proposed to explain the effects of fenofibrate in patients with proliferative diabetic retinopathy (PDR) and diabetic macular edema (DME) in vitro and in rodent models. Fenofibrate has been shown to reduce retinal expression of vascular endothelial growth factor (VEGF), a key angiogenic factor in PDR, and to reduce vascular permeability and apoptosis of the retinal pigment epithelium, both of which contribute to the development of DME.
The FIELD study was a multinational randomized trial involving 9,795 patients with type 2 diabetes. Patients were randomized to receive either fenofibrate 200 mg daily (n = 4,895) or placebo (n = 4,900). In an ophthalmologic sub-study involving 1,012 patients, standardized retinal photographs were taken and assessed using ETDRS (Early Treatment Diabetic Retinopathy Study) criteria to determine the cumulative incidence of diabetic retinopathy and its individual manifestations. Analyses were performed in all patients who started treatment. In the ophthalmologic sub-study, the primary endpoint—two-step progression in retinopathy stage—did not differ significantly between the two groups overall (46 [9.6%] patients in the fenofibrate group vs. 57 [12.3%] in the placebo group, p = 0.19), or in the subgroup of patients without pre-existing retinopathy (43 [11.4%] vs. 43 [11.7%], p = 0.87). Conversely, in the subgroup of patients with existing retinopathy, two-step progression was significantly less frequent in the fenofibrate group compared to placebo (3 [3.1%] vs. 14 [14.6%] patients, p = 0.004).
Information on laser treatment for diabetic retinopathy, predefined as a tertiary endpoint in the main study, was collected at each patient visit. The need for first laser treatment for any retinopathy was significantly lower in the fenofibrate group compared to placebo (164 [3.4%] patients in the fenofibrate group vs. 238 [4.9%] in the placebo group; risk ratio [RR] 0.69, 95% CI 0.56–0.84, p = 0.0002; absolute risk reduction: 1.5% [0.7–2.3]). This benefit was independent of plasma lipid concentrations.
In a subgroup of 2,856 participants from the ACCORD study (ACCORD Eye), the impact of three treatment strategies on the progression of diabetic retinopathy was evaluated: intensive vs. standard glycemic control (target HbA1c <6.0% or 7.0–7.9%, respectively), dyslipidemia (combination therapy with fenofibrate 160 mg/day plus simvastatin vs. placebo plus simvastatin), or systolic blood pressure control (target <120 vs. <140 mm Hg). Progression of diabetic retinopathy was assessed after 4 years by an increase of 3 or more steps on the ETDRS scale (based on stereoscopic seven-field fundus photography) or by the development of diabetic retinopathy requiring laser photocoagulation or vitrectomy.
The rate of progression of diabetic retinopathy was 6.5% in the group receiving intensive dyslipidemia therapy with fenofibrate compared to 10.2% in the placebo group (adjusted odds ratio 0.60; 95% CI 0.42–0.87, p = 0.006). It was concluded that intensive combined dyslipidemia therapy reduced the rate of progression of diabetic retinopathy.
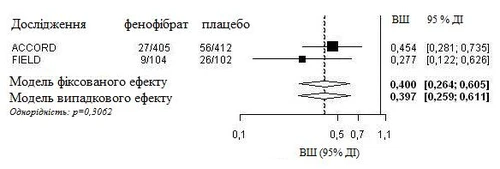
An integrated analysis of individual patient data from the FIELD study and published information from the ACCORD Eye study was conducted. The composite primary endpoint from the ACCORD Eye study was applied to the FIELD study, i.e., a 3-step worsening on the ETDRS scale, photocoagulation, or vitrectomy for treatment of proliferative diabetic retinopathy. Both studies were homogeneous (using a fixed-effects model) and showed an overall 60% reduction in the progression of diabetic retinopathy (odds ratio: 0.40, 95% CI 0.26–0.61).
Progression of diabetic retinopathy (DR) in patients with DR at baseline: integrated analysis of the FIELD PSP-DR and ACCORD Eye studies using the primary endpoint of the ACCORD Eye study.
Pharmacokinetics.
Absorption.
Maximum plasma concentration (Cmax) is reached within 2–4 hours after oral administration. Plasma concentrations remain stable during long-term use in all patients.
Unlike previous fenofibrate formulations, the maximum plasma concentration and overall exposure of fenofibrate in the form of nanoparticles (Tricor® 145 mg) are not affected by food intake; therefore, Tricor® 145 mg can be administered independently of meals.
In a food-effect study, healthy men and women received a single 145 mg dose of the new tablet formulation of fenofibrate either fasting or with a high-fat meal. The results showed that food intake did not affect the exposure (AUC and Cmax) of fenofibric acid.
Distribution.
Fenofibric acid is highly bound to plasma albumin (>99%).
Metabolism and Elimination.
After oral administration, fenofibrate is rapidly hydrolyzed by esterases to its active metabolite, fenofibric acid. Unchanged fenofibrate is not detectable in plasma. Fenofibrate is not a substrate for CYP3A4 and does not undergo hepatic microsomal metabolism.
The drug is primarily excreted in urine. It is almost completely eliminated within 6 days. Fenofibrate is excreted mainly as fenofibric acid and its glucuronide conjugate. In elderly patients, the overall plasma clearance of fenofibric acid is not significantly altered.
Pharmacokinetic studies after single-dose and long-term administration showed no accumulation of the drug in the body. Fenofibric acid is not removed by hemodialysis.
The elimination half-life of fenofibric acid is approximately 20 hours.
Clinical characteristics.
Indications.
Tricor® 145 mg is indicated as an adjunct to diet and other non-pharmacological treatment methods (e.g., physical exercise, weight reduction) in the following conditions:
- Severe hypertriglyceridemia with or without low HDL-cholesterol levels;
- Mixed dyslipidemia in cases where statin therapy is contraindicated or statin intolerance exists;
- Mixed dyslipidemia in patients with high cardiovascular risk, as an adjunct to statin therapy, when triglyceride and HDL-cholesterol levels are not adequately controlled.
Diabetic retinopathy: Tricor® 145 mg is indicated to reduce the progression of diabetic retinopathy in patients with type 2 diabetes mellitus and existing diabetic retinopathy.
Contraindications.
Hepatic insufficiency (including biliary cirrhosis and unexplained persistent liver function abnormalities).
Established gallbladder disease.
Severe chronic renal disease.
Chronic or acute pancreatitis, except in cases of acute pancreatitis caused by severe hypertriglyceridemia.
Established photoallergy or phototoxic reactions during treatment with fibrates or ketoprofen.
Hypersensitivity to the active substance or to any of the excipients listed in the section "Composition".
Tricor® 145 mg should also not be used in patients with allergy to peanuts, peanut oil, or soy lecithin, or similar products due to the risk of hypersensitivity reactions.
Interaction with other medicinal products and other forms of interaction.
Oral anticoagulants.
Fenofibrate enhances the effect of oral anticoagulants and may increase the risk of bleeding. It is recommended to reduce the anticoagulant dose by approximately one-third at the start of treatment and subsequently adjust it gradually according to the INR (International Normalized Ratio).
Cyclosporine.
Several severe cases of reversible renal function impairment have been reported during concomitant use of fenofibrate and cyclosporine. Therefore, renal function should be carefully monitored in patients receiving this combination, and fenofibrate should be discontinued if severe laboratory abnormalities occur.
HMG-CoA reductase inhibitors and other fibrates.
The risk of serious muscle toxicity increases with concomitant use of fenofibrate and HMG-CoA reductase inhibitors or other fibrates. Such combination therapy should be used with caution, and patients should be closely monitored for signs of muscle toxicity (see section "Special precautions").
Glitazones.
Cases of reversible paradoxical decrease in HDL-cholesterol levels have been observed during concomitant use of fenofibrate and glitazones. Therefore, monitoring of HDL-cholesterol levels is recommended during combination therapy, and treatment should be discontinued if HDL-cholesterol levels become too low.
Cytochrome P450 enzymes.
In vitro studies using human liver microsomes have shown that fenofibrate and fenofibric acid are not inhibitors of cytochrome P450 (CYP) isoforms CYP3A4, CYP2D6, CYP2E1, or CYP1A2. At therapeutic concentrations, they are weak inhibitors of CYP2C19 and CYP2A6, and weak to moderate inhibitors of CYP2C9.
Close monitoring is required in patients receiving fenofibrate concomitantly with drugs metabolized by CYP2C19, CYP2A6, and especially CYP2C9, particularly those with a narrow therapeutic index, and dose adjustments should be made if necessary.
Special precautions for use.
Secondary hyperlipidemia.
Prior to initiating fenofibrate therapy, appropriate treatment of underlying conditions causing secondary hypercholesterolemia should be undertaken, such as uncontrolled type 2 diabetes mellitus, hypothyroidism, nephrotic syndrome, dysproteinemia, obstructive liver disease, or alcoholism. Secondary hypercholesterolemia associated with pharmacological therapy may occur in patients receiving diuretics, β-blockers, estrogens, progestogens, combined oral contraceptives, immunosuppressants, and protease inhibitors. In such cases, it is necessary to determine whether hyperlipidemia is primary or secondary (elevated lipid levels possibly induced by the aforementioned therapeutic agents).
Liver function.
As with other lipid-lowering agents, elevations in transaminase levels have been reported in some patients receiving fenofibrate. In most cases, these increases were transient, mild, and asymptomatic. Transaminase levels should be monitored every 3 months during the first 12 months of therapy and periodically thereafter. Patients with increasing transaminase levels should be closely monitored, and treatment should be discontinued if AST and ALT levels exceed three times the upper limit of normal. Fenofibrate should be discontinued in case of symptoms suggestive of hepatitis (e.g., jaundice, pruritus) and confirmed by laboratory test results.
Pancreas.
Cases of pancreatitis have been reported in patients taking fenofibrate (see sections "Contraindications" and "Adverse reactions"). This may result from inadequate therapeutic response in patients with severe hypertriglyceridemia, a direct drug effect, or a secondary phenomenon mediated by gallstones or sludge formation causing obstruction of the common bile duct.
Muscles.
Myotoxicity, including rare cases of rhabdomyolysis with or without renal impairment, has been reported with fibrate and other lipid-lowering agents. The incidence of this adverse effect increases in the presence of hypoalbuminemia and a history of renal impairment. Patients with risk factors for myopathy and/or rhabdomyolysis—including age ≥70 years, personal or family history of hereditary muscle disorders, renal dysfunction, hypothyroidism, and alcohol abuse—are at increased risk of developing rhabdomyolysis. In such patients, the benefit-risk ratio of fenofibrate therapy should be carefully evaluated.
Myotoxicity should be suspected in patients presenting with diffuse myalgia, myositis, muscle cramps, weakness, and/or markedly elevated creatine kinase (CK) levels (more than 5 times the upper limit of normal). In such cases, fenofibrate treatment should be discontinued.
The risk of myotoxicity may be increased when fenofibrate is coadministered with another fibrate or a HMG-CoA reductase inhibitor, particularly in patients with pre-existing muscle disorders. Therefore, concomitant use of fenofibrate with a HMG-CoA reductase inhibitor or another fibrate should be reserved for patients with severe combined dyslipidemia and high cardiovascular risk, in the absence of any history of muscle disorders, and under strict monitoring for potential myotoxic effects.
Kidney function.
If creatinine levels increase by more than 50% above the upper limit of normal (ULN), fenofibrate therapy should be discontinued. Creatinine levels should be monitored within the first 3 months after initiation of treatment and periodically thereafter (see section "Dosage and administration" for dosing recommendations).
Excipients.
This medicinal product contains lactose; therefore, patients with rare hereditary conditions such as galactose intolerance, lactase deficiency, or glucose-galactose malabsorption should not take this product.
This medicinal product contains sucrose; therefore, patients with rare hereditary conditions such as fructose intolerance, glucose-galactose malabsorption, or sucrase-isomaltase deficiency should not take this product.
Use during pregnancy or breastfeeding.
Pregnancy. There are insufficient data on the use of fenofibrate in pregnant women. Animal studies have not shown any teratogenic effects. Embryotoxic effects were observed when the drug was administered at doses toxic to the maternal organism. The potential risk to humans is unknown. Therefore, Tricor® 145 mg should be used during pregnancy only after careful assessment of benefit versus risk.
Breastfeeding. It is unknown whether fenofibrate and/or its metabolites are excreted in human breast milk. Risk to breastfed infants cannot be ruled out; therefore, fenofibrate should not be used during breastfeeding.
Fertility. Reversible effects on fertility were observed in animal studies. There are no clinical data on the effect of Tricor® 145 mg on fertility in humans.
Effect on ability to drive and use machines.
Tricor® 145 mg has no effect or has a negligible effect on the ability to drive or operate machinery.
Dosage and Administration
Tricor® 145 mg can be taken at any time of the day, regardless of food intake (see section "Pharmacological Properties. Pharmacokinetics"). Tablets should be swallowed whole with a glass of water.
Dietary therapy initiated prior to administration of the drug should be continued.
During treatment of hyperlipidemia, treatment efficacy should be monitored by measuring serum lipid levels. If an adequate response is not achieved after several months (e.g., after 3 months), additional or alternative therapeutic measures should be considered.
Adults
The recommended dose is 1 tablet containing 145 mg of fenofibrate once daily. Patients receiving 1 capsule containing 200 mg of fenofibrate or 1 tablet containing 160 mg of fenofibrate may be switched directly to 1 tablet of Tricor® 145 mg without dose adjustment.
If fenofibrate is required for two indications (hyperlipidemia and diabetic retinopathy), only one tablet of Tricor® 145 mg once daily should be administered.
Elderly Patients
The usual adult dose is recommended for elderly patients without impaired renal function.
Renal Impairment
Dose reduction is required in patients with renal impairment. Fenofibrate at the current dosage of 145 mg is not recommended in patients with moderate chronic renal disease (creatinine clearance from 30 to 60 mL/min).
Fenofibrate is contraindicated in patients with severe chronic renal disease (creatinine clearance < 30 mL/min).
Hepatic Impairment
Tricor® 145 mg is not recommended for patients with hepatic impairment due to lack of data.
Children
The safety and efficacy of fenofibrate in children and adolescents (under 18 years of age) have not been established, and relevant data are lacking. Therefore, fenofibrate is not recommended for use in children and adolescents (under 18 years of age).
Overdose
Only isolated cases of fenofibrate overdose have been reported. In most cases, no symptoms of overdose were observed.
There is no specific antidote. In case of suspected overdose, symptomatic treatment and appropriate supportive measures should be initiated as necessary. Fenofibrate is not eliminated by hemodialysis.
Adverse reactions.
The most commonly reported adverse reactions during fenofibrate therapy are gastrointestinal disorders, disturbances related to the stomach or intestine.
The following adverse events were observed in placebo-controlled clinical studies (n=2344) with the specified frequency:
| MedDRA System Organ Class |
Very common ≥ 1/10 |
Common ≥ 1/100, <1/10 |
Uncommon ≥ 1/1000, <1/100 |
Rare ≥ 1/10000, <1/1000 |
| Blood and lymphatic system disorders |
Decreased hemoglobin levels Decreased white blood cell count |
|||
| Immune system disorders |
Hypersensitivity (including anaphylactic reaction) |
|||
| Nervous system disorders |
Headache |
|||
| Vascular disorders |
Thromboembolism (pulmonary embolism, deep vein thrombosis)** |
|||
| Gastrointestinal disorders |
Gastrointestinal symptoms (abdominal pain, nausea, vomiting, diarrhea, flatulence) |
Pancreatitis* |
||
| Hepatobiliary disorders |
Elevated transaminase levels (see section "Special precautions for use") |
Cholelithiasis (see section "Special precautions for use") |
Hepatitis |
|
| Skin and subcutaneous tissue disorders |
Hypersensitivity skin reactions (e.g., rash, pruritus, urticaria) |
Allopecia Photosensitivity reactions |
||
| Musculoskeletal and connective tissue disorders |
Muscle disorders (e.g., myalgia, myositis, muscle spasms and weakness) |
|||
| Reproductive system and breast disorders |
Sexual dysfunction |
|||
| Investigations |
Increased blood homocysteine level*** |
Increased blood creatinine level |
Increased blood urea level |
* In the FIELD study – a randomized, placebo-controlled study involving 9,795 patients with type 2 diabetes – a statistically significant increase in the incidence of pancreatitis was observed in patients receiving fenofibrate compared to those in the placebo group (0.8% vs. 0.5%, respectively; p = 0.031).
** A statistically significant increase in the incidence of pulmonary embolism was observed (0.7% in the placebo group vs. 1.1% in the fenofibrate group; p = 0.022), and a statistically non-significant increase in the incidence of deep vein thrombosis (1.0% in the placebo group [48/4900 patients] vs. 1.4% in the fenofibrate group [67/4895 patients]; p = 0.074).
*** The mean increase in blood homocysteine levels in patients treated with fenofibrate was 6.5 µmol/L and was reversible upon discontinuation of fenofibrate therapy. Elevated risk of venous thrombotic events may be associated with increased homocysteine levels. The clinical significance of this observation is not established.
In addition to adverse reactions observed in clinical trials, spontaneous reports of the following adverse reactions have been received during the post-marketing use of Tricor® 145 mg. The exact frequency cannot be determined based on available data; therefore, the frequency is classified as "unknown".
Respiratory, thoracic and mediastinal disorders: interstitial lung disease.
Musculoskeletal and connective tissue disorders: rhabdomyolysis.
Hepatobiliary disorders: jaundice, complications of gallstone disease (e.g. cholecystitis, cholangitis, biliary colic).
Skin and subcutaneous tissue disorders: severe skin reactions (e.g. erythema multiforme, Stevens-Johnson syndrome, toxic epidermal necrolysis).
Nervous system disorders: fatigue.
If any adverse reactions occur during use of the medicinal product or if there are any concerns regarding product quality, please report to Abbott Ukraine LLC at the following phone number: +38 044-498-60-80 or by e-mail: [email protected].
Shelf life. 3 years.
Storage conditions. Store in the original packaging at a temperature not exceeding 25 °C, in a place inaccessible to children.
Packaging. 10 tablets per blister, 2 or 3 blisters per cardboard box.
Prescription status. Prescription only.
Manufacturers. Fournier Laboratories Ireland Limited, Ireland / Fournier Laboratories Ireland Limited, Ireland.
Astrea Fontaine, France / Astrea Fontaine, France.
Manufacturers' addresses and locations of their operations. Anngrove, Carrigtwohill, Co. Cork, Ireland / Anngrove, Carrigtwohill, Co. Cork, Ireland.
Rue Des Pres Potets, Fontaine Les Dijon, 21121, France / Rue Des Pres Potets, Fontaine Les Dijon, 21121, France.