TriCor® 145 mg
Ucrania
Contenido
INSTRUCCIONES PARA USO MÉDICO DEL MEDICAMENTO TRAICOR® 145 MG (TRICOR® 145 MG)
Composición:
Principio activo: fenofibrato;
1 tableta contiene 145 mg de fenofibrato;
Sustancias auxiliares: hipromelosa, docosato de sodio, sacarosa, laurilsulfato de sodio, monohidrato de lactosa, celulosa microcristalina silicatada, crospovidona, estearato de magnesio, Opadry**®** OY-B-28920 (alcohol polivinílico, dióxido de titanio (E 171), talco, lecitina de soja, goma xantana).
Forma farmacéutica. Tabletas recubiertas con película.
Características físico-químicas principales: tabletas blancas, ovales, recubiertas con película, con la inscripción «145» en un lado y el logotipo de la compañía «Fournier» en el otro.
Grupo farmacoterapéutico. Agentes hipolipidémicos. Preparados que reducen los niveles de colesterol y triglicéridos en suero sanguíneo. Fibratos. Código ATC C10AB05.
Propiedades farmacodinámicas.
Dislipidemia
El fenofibrato es un derivado del ácido fíbrico cuyos efectos modificadores de lípidos en humanos están mediados por la activación del receptor activado por proliferadores de peroxisomas alfa (PPARα).
Mediante la activación de PPARα, el fenofibrato potencia la lipólisis y la eliminación de partículas aterogénicas ricas en triglicéridos de la sangre mediante la activación de la lipoproteína lipasa y la reducción de la producción de apolipoproteína CIII. La activación de PPARα también provoca un aumento en la síntesis de apolipoproteínas AI y AII.
Los efectos mencionados del fenofibrato sobre las lipoproteínas conducen a una disminución de las fracciones de lipoproteínas de muy baja y baja densidad (VLDL y LDL) que contienen apolipoproteína B, y a un aumento de la fracción de lipoproteínas de alta densidad (HDL) que contienen apolipoproteínas AI y AII.
Además, mediante la modulación de la síntesis y el catabolismo de las fracciones de VLDL, el fenofibrato potencia la depuración de LDL y reduce la cantidad de partículas pequeñas y densas de LDL, cuyos niveles están elevados en personas con fenotipo lipoproteínico aterogénico, común en pacientes con riesgo de enfermedad coronaria.
En estudios clínicos, con el uso de fenofibrato, el nivel de colesterol total disminuyó entre un 20-25 %, los triglicéridos entre un 40-55 %, y el nivel de colesterol HDL aumentó entre un 10-30 %.
En pacientes con hipercolesterolemia, en quienes el nivel de colesterol LDL disminuye entre un 20-35 %, el efecto global sobre el colesterol conduce a una reducción de las relaciones colesterol total/colesterol HDL, colesterol LDL/colesterol HDL o apolipoproteína B/apolipoproteína AI, que son marcadores de riesgo aterogénico.
Existe evidencia de que el tratamiento con fármacos fibratos puede reducir la frecuencia de eventos en enfermedad coronaria, pero los fibratos no han demostrado reducir la mortalidad total en prevención primaria o secundaria de enfermedades cardiovasculares.
El estudio ACCORD lipid fue un ensayo aleatorizado controlado con placebo que incluyó a 5518 pacientes con diabetes tipo 2 tratados con fenofibrato además de simvastatina. La terapia combinada con fenofibrato y simvastatina, en comparación con la monoterapia con simvastatina, no mostró diferencias significativas respecto al efecto sobre el punto final primario combinado: infarto de miocardio no fatal, accidente cerebrovascular no fatal y muerte cardiovascular (razón de riesgos [RR] 0,92, IC del 95 % 0,79-1,08, p = 0,32; reducción del riesgo absoluto: 0,74 %). En un subgrupo predefinido de pacientes con dislipidemia que tenían el tercil más bajo de colesterol HDL (≤ 34 mg/dl o 0,88 mmol/l) y el tercil más alto de triglicéridos (≥ 204 mg/dl o 2,3 mmol/l) antes del tratamiento, la terapia combinada con fenofibrato y simvastatina mostró una reducción relativa del 31 % en el riesgo del punto final primario combinado en comparación con la monoterapia con simvastatina (razón de riesgos [RR] 0,69, IC del 95 % 0,49-0,97, p = 0,03; reducción del riesgo absoluto: 4,95 %). Un análisis de otro subgrupo predefinido reveló una interacción estadísticamente significativa entre el tratamiento y el sexo (p = 0,01), lo que sugiere un posible beneficio de la terapia combinada en hombres (p = 0,037), pero un riesgo potencialmente mayor del punto final primario en mujeres que recibieron terapia combinada en comparación con la monoterapia con simvastatina (p = 0,069). Este fenómeno no se observó en el subgrupo mencionado anteriormente de pacientes con dislipidemia, pero tampoco hubo pruebas claras de beneficio en mujeres con dislipidemia que recibieron fenofibrato junto con simvastatina, y un posible efecto perjudicial en este subgrupo no puede descartarse.
Los depósitos extravasculares de colesterol (xantomas tendinosos y tuberosos) pueden reducirse significativamente o desaparecer completamente durante el tratamiento con fenofibrato.
En pacientes con niveles elevados de fibrinógeno, se observó una reducción significativa de este parámetro con el tratamiento con fenofibrato, al igual que en pacientes con niveles elevados de lipoproteína (a). El fenofibrato reduce también los niveles de otros marcadores inflamatorios, como la proteína C reactiva.
El efecto uricosúrico del fenofibrato, que conduce a una reducción del nivel de ácido úrico en aproximadamente un 25 %, debe considerarse un efecto favorable adicional del fármaco en pacientes con dislipidemia e hiperuricemia.
El efecto antiagregante del fenofibrato sobre las plaquetas se ha demostrado en estudios en animales y en un estudio clínico, que mostraron una reducción de la agregación plaquetaria inducida por ADP, ácido araquidónico y adrenalina.
Retinopatía diabética
Se han propuesto varios mecanismos para explicar los efectos del fenofibrato en pacientes con retinopatía diabética proliferativa (RDP) y edema macular diabético (EMD) in vitro y en modelos de roedores. Se ha demostrado que el fenofibrato reduce la expresión retiniana del factor de crecimiento endotelial vascular (VEGF), que es el principal factor angiogénico en la RDP, y disminuye la permeabilidad vascular y el apoptosis del epitelio pigmentario de la retina, que contribuyen al desarrollo del EMD.
El estudio FIELD fue un ensayo aleatorizado multinacional que incluyó a 9795 pacientes con diabetes tipo 2. Los pacientes seleccionados fueron aleatorizados a recibir tratamiento con fenofibrato 200 mg/día (n = 4895) o placebo (n = 4900). En un subestudio oftalmológico con 1012 pacientes se tomaron fotografías estandarizadas de la retina, que se evaluaron según los criterios del ETDRS (Early Treatment Diabetic Retinopathy Study) para determinar la frecuencia acumulada de desarrollo de retinopatía diabética y sus manifestaciones individuales. Los análisis se realizaron en todos los pacientes que iniciaron el tratamiento. En el subestudio oftalmológico, el punto final primario, el progresión de dos etapas en la escala de retinopatía, no mostró diferencias significativas entre los dos grupos en general (46 [9,6 %] pacientes en el grupo de fenofibrato frente a 57 [12,3 %] en el grupo placebo, p = 0,19), ni en el subgrupo de pacientes sin retinopatía previa (43 [11,4 %] frente a 43 [11,7 %], p = 0,87). Por el contrario, en el subgrupo de pacientes con retinopatía ya existente, la progresión de dos etapas se observó en un número significativamente menor de pacientes en el grupo de fenofibrato que en el grupo placebo (3 [3,1 %] frente a 14 [14,6 %], p = 0,004).
La información sobre el tratamiento láser de la retinopatía diabética, que fue una tercera variable principal predefinida en el estudio principal, se recogió en cada visita clínica del paciente. La necesidad de realizar el primer tratamiento láser para cualquier retinopatía fue significativamente menor con el tratamiento con fenofibrato en comparación con placebo (164 [3,4 %] pacientes en el grupo fenofibrato frente a 238 [4,9 %] en el grupo placebo; razón de riesgos [RR] 0,69, IC del 95 % 0,56-0,84, p = 0,0002; reducción del riesgo absoluto: 1,5 % [0,7-2,3]). La necesidad de este tratamiento no dependió de la concentración de lípidos en sangre.
En un subgrupo de 2856 participantes del estudio ACCORD (ACCORD Eye) se evaluó el impacto de tres estrategias terapéuticas sobre la progresión de la retinopatía diabética: tratamiento intensivo o estándar de la glucemia (nivel objetivo de HbA1c <6,0 % o entre 7,0 y 7,9 %, respectivamente), dislipidemia (terapia combinada con fenofibrato 160 mg/día más simvastatina o placebo más simvastatina) o control de la presión arterial sistólica (objetivo <120 o <140 mmHg). La progresión de la retinopatía diabética se definió a los 4 años como un aumento de 3 o más puntos en la escala ETDRS (según la evaluación de fotografías estereoscópicas de siete campos del fondo de ojo) o el desarrollo de retinopatía diabética que requirió fotocoagulación láser o vitrectomía.
La frecuencia de progresión de la retinopatía diabética fue del 6,5 % en el grupo de tratamiento intensivo de la dislipidemia con fenofibrato en comparación con el 10,2 % en el grupo placebo (razón de riesgos ajustada 0,60; IC del 95 % 0,42-0,87, p = 0,006). Se concluyó que la terapia combinada intensiva para la dislipidemia redujo la frecuencia de progresión de la retinopatía diabética.
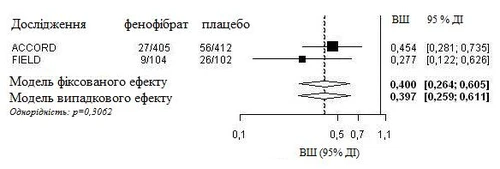
Se realizó un análisis integrado de datos individuales de pacientes del estudio FIELD y de la información publicada del estudio ACCORD Eye. El punto final primario combinado del estudio ACCORD Eye se aplicó al estudio FIELD, es decir, empeoramiento de la gravedad en la escala ETDRS en 3 puntos, fotocoagulación o vitrectomía para el tratamiento de la retinopatía diabética proliferativa. Ambos estudios fueron homogéneos (modelo de efectos fijos) y mostraron una reducción global del 60 % en la progresión de la retinopatía diabética (razón de riesgos: 0,40, IC del 95 % 0,26-0,61).
Progresión de la retinopatía diabética (DR) en pacientes con DR al inicio del estudio: análisis combinado de los estudios FIELD PSP-DR y ACCORD Eye utilizando el punto final primario del estudio ACCORD Eye.
Farmacocinética.
Absorción.
La concentración máxima del fármaco en plasma (Cmax) se alcanza entre 2 y 4 horas tras la administración oral. La concentración en plasma permanece estable durante el tratamiento prolongado en todos los pacientes.
A diferencia de los fármacos fenofibrato anteriores, al administrar Tricor® 145 mg en forma de nanopartículas, su concentración máxima en plasma y la exposición total no dependen de la ingesta de alimentos, por lo que Tricor® 145 mg puede administrarse independientemente de las comidas.
En un estudio sobre el efecto de los alimentos en la absorción del fármaco, tras la administración de la nueva formulación de comprimidos de 145 mg de fenofibrato a hombres y mujeres sanos en ayunas y con una comida rica en grasas, se demostró que la ingesta de alimentos no afecta a la exposición (AUC y Cmax) del ácido fenofibróico.
Distribución.
El ácido fenofibróico se une en gran medida a la albúmina plasmática (más del 99 %).
Metabolismo y eliminación.
Tras la administración oral, el fenofibrato se hidroliza rápidamente por las esterasas hasta su metabolito activo, el ácido fenofibróico. El fenofibrato sin modificar no se detecta en plasma. El fenofibrato no es sustrato del CYP 3A4 y no participa en el metabolismo microsomal hepático.
El fármaco se elimina principalmente por orina. Prácticamente todo el fármaco se elimina en 6 días. El fenofibrato se excreta principalmente como ácido fenofibróico y su conjugado glucurónido. En pacientes de edad avanzada, el aclaramiento plasmático total aparente del ácido fenofibróico no se modifica significativamente.
Los estudios de cinética tras una dosis única y tras tratamiento prolongado mostraron que el fármaco no se acumula en el organismo. El ácido fenofibróico no se elimina mediante hemodiálisis.
El periodo de semivida de eliminación del ácido fenofibróico es de aproximadamente 20 horas.
Características clínicas.
Indicaciones.
TriCor® 145 mg está indicado como complemento de la dieta y de otros métodos no farmacológicos de tratamiento (por ejemplo, ejercicio físico, reducción de peso) en las siguientes condiciones:
- hipertrigliceridemia grave con o sin niveles bajos de colesterol de lipoproteínas de alta densidad;
- hiperlipidemia mixta en casos en que el uso de estatinas está contraindicado o existe intolerancia a las estatinas;
- hiperlipidemia mixta en pacientes con alto riesgo cardiovascular, como complemento al tratamiento con estatinas, cuando los niveles de triglicéridos y colesterol de lipoproteínas de alta densidad no están adecuadamente controlados.
Retinopatía diabética: TriCor® 145 mg está indicado para reducir la progresión de la retinopatía diabética en pacientes con diabetes mellitus tipo 2 y retinopatía diabética existente.
Contraindicaciones.
Insuficiencia hepática (incluyendo cirrosis biliar y alteraciones persistentes de la función hepática de causa no aclarada).
Enfermedades establecidas de la vesícula biliar.
Enfermedades renales crónicas graves.
Pancreatitis crónica o aguda, excepto en casos de pancreatitis aguda provocada por hipertrigliceridemia grave.
Fotopatía o reacciones fototóxicas previamente establecidas durante el tratamiento con fármacos fibratos o con ketoprofeno.
Hipersensibilidad al principio activo o a cualquiera de los excipientes enumerados en la sección «Composición».
Además, TriCor® 145 mg no debe administrarse a pacientes con alergia al cacahuete, al aceite de cacahuete o al lecitina de soja, o productos similares, debido al riesgo de reacciones de hipersensibilidad.
Interacción con otros medicamentos y otros tipos de interacción.
Anticoagulantes orales.
El fenofibrato potencia el efecto de los anticoagulantes orales y puede aumentar el riesgo de hemorragia. Se recomienda reducir la dosis del anticoagulante aproximadamente en un tercio al inicio del tratamiento y, posteriormente, ajustarla progresivamente según el valor del INR (Índice Internacional Normalizado).
Ciclosporina.
Se han descrito varios casos graves, aunque reversibles, de alteración de la función renal con el uso concomitante de fenofibrato y ciclosporina. Por tanto, debe controlarse cuidadosamente la función renal en los pacientes que reciben esta combinación, y debe suspenderse el fenofibrato si se presentan alteraciones graves en los parámetros de laboratorio.
Inhibidores de la HMG-CoA reductasa y otros fármacos fibratos.
El riesgo de toxicidad grave sobre el músculo aumenta con la administración concomitante de un fármaco fibrato con inhibidores de la HMG-CoA reductasa u otros fármacos fibratos. Esta terapia combinada debe utilizarse con precaución y se debe vigilar estrechamente al paciente por signos de toxicidad muscular (ver sección «Precauciones de uso»).
Glitazonas.
Se han observado casos de disminución paradójica y reversible del colesterol de lipoproteínas de alta densidad (HDL) con el uso concomitante de fenofibrato y glitazonas. Por lo tanto, se recomienda controlar los niveles de colesterol HDL durante el tratamiento combinado y suspender los fármacos si los niveles de colesterol HDL se vuelven demasiado bajos.
Enzimas del citocromo P450.
Estudios in vitro con microsomas hepáticos humanos han demostrado que el fenofibrato y el ácido fenofibróico no son inhibidores de las isoformas del citocromo P450 (CYP) CYP3A4, CYP2D6, CYP2E1 ni CYP1A2. En concentraciones terapéuticas, son inhibidores débiles de CYP2C19 y CYP2A6, y débiles o moderados inhibidores de CYP2C9.
Debe vigilarse cuidadosamente a los pacientes que reciben simultáneamente fenofibrato y medicamentos metabolizados por CYP2C19, CYP2A6 y especialmente CYP2C9, y que tengan un índice terapéutico estrecho, y ajustar la dosis de estos medicamentos si es necesario.
Características de uso.
Hiperlipidemia secundaria.
Antes de iniciar la terapia con fenofibrato, se debe tratar adecuadamente las condiciones subyacentes que causan la hipercolesterolemia secundaria, tales como diabetes mellitus tipo 2 no controlada, hipotiroidismo, síndrome nefrótico, disproteinemia, enfermedad hepática obstructiva o alcoholismo. La hipercolesterolemia secundaria asociada al tratamiento farmacológico puede observarse en pacientes que reciben diuréticos, betabloqueantes, estrógenos, progestágenos, anticonceptivos orales combinados, inmunosupresores e inhibidores de la proteasa. En tales casos, se debe determinar si la hiperlipidemia es primaria o secundaria (posible aumento de los niveles de lípidos provocado por los agentes terapéuticos mencionados anteriormente).
Función hepática.
Como con otros fármacos hipolipemiantes, en algunos pacientes se han observado aumentos de los niveles de transaminasas. En la mayoría de los casos, este incremento fue transitorio, leve y asintomático. Se recomienda controlar los niveles de transaminasas cada 3 meses durante el primer año de tratamiento y periódicamente posteriormente. Se debe prestar atención a los pacientes con niveles crecientes de transaminasas y suspender el tratamiento si los niveles de AST y ALT superan más de 3 veces el límite superior de la normalidad. Si aparecen síntomas de hepatitis (por ejemplo, ictericia, prurito) y el diagnóstico se confirma mediante análisis de laboratorio, se debe interrumpir el uso de fenofibrato.
Páncreas.
En pacientes que han tomado fenofibrato se han notificado casos de pancreatitis (ver secciones «Contraindicaciones» y «Reacciones adversas»). Esto puede ser consecuencia de una eficacia insuficiente del tratamiento en pacientes con hipertigliceridemia grave, un efecto directo del fármaco o un fenómeno secundario mediado por cálculos biliares o formación de lodo biliar con obstrucción del colédoco.
Músculos.
Durante el uso de fibratos y otros fármacos hipolipemiantes, se han notificado efectos tóxicos sobre el músculo, incluyendo casos raros de rabdomiólisis con o sin insuficiencia renal. La frecuencia de este trastorno aumenta en caso de hipoalbuminemia y antecedentes de insuficiencia renal. Los pacientes con factores que predisponen al desarrollo de miopatía y/o rabdomiólisis, tales como edad ≥ 70 años, antecedentes personales o familiares de trastornos musculares hereditarios, alteración de la función renal, hipotiroidismo o abuso de alcohol, pueden tener un riesgo aumentado de desarrollar rabdomiólisis. En estos pacientes se debe evaluar cuidadosamente el beneficio frente al riesgo del tratamiento con fenofibrato.
Se debe sospechar toxicidad muscular en pacientes con mialgia difusa, miositis, calambres musculares y debilidad y/o aumento marcado de la CK (más de 5 veces el límite superior de la normalidad). En tales casos, se debe suspender el tratamiento con fenofibrato.
El riesgo de toxicidad muscular puede aumentar si el fármaco se administra junto con otro fibrato o un inhibidor de la HMG-CoA reductasa, especialmente en caso de enfermedad muscular preexistente. Por tanto, la administración concomitante de fenofibrato con un inhibidor de la HMG-CoA reductasa u otro fibrato debe reservarse como terapia de rescate en pacientes con dislipidemia combinada grave y alto riesgo cardiovascular, sin antecedentes de enfermedad muscular y bajo estricta vigilancia del posible efecto tóxico sobre el músculo.
Función renal.
Si el nivel de creatinina aumenta más del 50 % respecto al límite superior de la normalidad (LSN), se debe interrumpir el tratamiento con fenofibrato. Se recomienda controlar los niveles de creatinina durante los primeros 3 meses tras el inicio del tratamiento y periódicamente posteriormente (para la dosificación, ver la sección «Posología y forma de administración»).
Sustancias auxiliares.
El medicamento contiene lactosa, por lo que no debe administrarse a pacientes con enfermedades hereditarias raras como intolerancia a la galactosa, deficiencia de lactasa o malabsorción de glucosa-galactosa.
El medicamento contiene sacarosa, por lo que no debe administrarse a pacientes con enfermedades hereditarias raras como intolerancia a la fructosa, malabsorción de glucosa-galactosa o deficiencia de sacarasa-isomaltasa.
Uso durante el embarazo o la lactancia.
Embarazo. No existen datos suficientes sobre el uso de fenofibrato en mujeres embarazadas. En estudios en animales no se observó ningún efecto teratogénico. Se observaron efectos embriotóxicos con dosis del fármaco tóxicas para el organismo materno. El riesgo potencial en humanos es desconocido. Por tanto, Tricor® 145 mg solo debe usarse durante el embarazo tras una evaluación cuidadosa del balance beneficio/riesgo.
Lactancia. No se sabe si el fenofibrato y/o sus metabolitos se excretan en la leche materna humana. No puede descartarse el riesgo para los lactantes, por lo que el fenofibrato no debe usarse durante la lactancia.
Fertilidad. En estudios en animales se observaron efectos reversibles sobre la fertilidad. No existen datos clínicos sobre el efecto del uso de Tricor® 145 mg sobre la fertilidad.
Capacidad para afectar la velocidad de reacción al conducir vehículos de motor o manejar maquinaria.
Tricor® 145 mg no afecta o tiene un efecto insignificante sobre la capacidad para conducir vehículos de motor o manejar maquinaria.
Vía de administración y dosis.
Se puede tomar Tricor® 145 mg en cualquier momento del día, independientemente de la ingestión de alimentos (ver sección «Propiedades farmacológicas. Farmacocinética»). Las tabletas deben tragarse enteras, acompañadas de un vaso de agua.
Debe continuarse la terapia dietética que se haya iniciado antes de la prescripción del medicamento.
Durante el tratamiento de la hiperlipidemia, la eficacia del tratamiento debe controlarse mediante la determinación de los niveles de lípidos en suero sanguíneo. Si tras varios meses (por ejemplo, tras 3 meses) no se logra una respuesta adecuada al tratamiento, deben considerarse medidas terapéuticas adicionales u otras alternativas.
Adultos
La dosis recomendada es de 1 tableta que contiene 145 mg de fenofibrato, una vez al día. A los pacientes que toman 1 cápsula que contiene 200 mg de fenofibrato, o 1 tableta que contiene 160 mg de fenofibrato, se les puede sustituir por 1 tableta de Tricor® 145 mg sin necesidad de ajuste adicional de la dosis.
Si un paciente necesita tomar fenofibrato por dos indicaciones (hiperlipidemia y retinopatía diabética), debe tomarse solamente una tableta de Tricor® 145 mg al día.
Pacientes de edad avanzada
En pacientes de edad avanzada sin alteración de la función renal se recomienda la dosis habitual para adultos.
Alteración de la función renal
En pacientes con alteración de la función renal es necesario reducir la dosis. El uso de fenofibrato en la dosis actual de 145 mg no se recomienda en casos de enfermedad renal crónica de intensidad moderada (clearance de creatinina entre 30 y 60 ml/min).
El uso de fenofibrato está contraindicado en pacientes con enfermedad renal crónica grave (clearance de creatinina < 30 ml/min).
Alteración de la función hepática
Tricor® 145 mg no se recomienda en pacientes con alteraciones de la función hepática debido a la falta de datos disponibles.
Niños
La seguridad y eficacia del uso de fenofibrato en niños y adolescentes (menores de 18 años) no han sido establecidas y no existen datos al respecto. Por tanto, el fenofibrato no se recomienda en niños y adolescentes (menores de 18 años).
Sobredosis.
Solo se han notificado casos aislados de sobredosis con fenofibrato. En la mayoría de los casos no se indicaron síntomas de sobredosis.
No existe antídoto específico. En caso de sospecha de sobredosis, debe administrarse tratamiento sintomático y las medidas de soporte adecuadas según sea necesario. El fenofibrato no se elimina mediante hemodiálisis.
Efectos adversos
Las reacciones adversas más frecuentemente notificadas durante la terapia con fenofibrato son trastornos digestivos y alteraciones gastrointestinales.
Los siguientes efectos adversos se observaron en estudios clínicos controlados con placebo (n=2344), con la frecuencia indicada:
| Clasificación por órganos según MedDRA |
Muy frecuentes ≥ 1/10 |
Frecuentes ≥ 1/100, <1/10 |
No frecuentes ≥ 1/1000, <1/100 |
Raros ≥ 1/10000, <1/1000 |
| Alteraciones de la sangre y del sistema linfático |
Disminución del nivel de hemoglobina Disminución del número de glóbulos blancos en sangre |
|||
| Alteraciones del sistema inmunitario |
Hipersensibilidad (incluyendo reacción anafiláctica) |
|||
| Alteraciones del sistema nervioso |
Cefalea |
|||
| Alteraciones del sistema vascular |
Tromboembolias (embolia pulmonar, trombosis venosa profunda)** |
|||
| Alteraciones del tubo digestivo |
Signos y síntomas gastrointestinales (dolor abdominal, náuseas, vómitos, diarrea, flatulencia) |
Pancreatitis* |
||
| Alteraciones del sistema hepatobiliar |
Aumento de los niveles de transaminasas (ver sección «Instrucciones de uso») |
Enfermedad por cálculos biliares (ver sección «Instrucciones de uso») |
Hepatitis |
|
| Alteraciones de la piel y del tejido subcutáneo |
Reacciones cutáneas de hipersensibilidad (por ejemplo, erupción, prurito, urticaria) |
Alopecia Reacciones de fotosensibilidad |
||
| Alteraciones del sistema musculoesquelético, tejido conjuntivo y huesos |
Alteraciones musculares (por ejemplo mialgia, miositis, calambres musculares y debilidad) |
|||
| Alteraciones del sistema reproductor y de las glándulas mamarias |
Disfunción sexual |
|||
| Anomalías detectadas en análisis de laboratorio |
Aumento del nivel de homocisteína en sangre*** |
Aumento del nivel de creatinina en sangre |
Aumento del nivel de urea en sangre |
* En el estudio FIELD, un estudio aleatorizado controlado con placebo que incluyó a 9795 pacientes con diabetes mellitus tipo 2, se observó un aumento estadísticamente significativo en la frecuencia de pancreatitis en pacientes que recibieron fenofibrato en comparación con el grupo que recibió placebo (0,8 % frente a 0,5 %, respectivamente; p = 0,031).
** Se observó un aumento estadísticamente significativo en la frecuencia de embolia pulmonar (0,7 % en el grupo placebo frente a 1,1 % en el grupo fenofibrato; p = 0,022) y un aumento estadísticamente no significativo en la frecuencia de trombosis venosa profunda (1,0 % en el grupo placebo [48/4900 pacientes] frente a 1,4 % en el grupo fenofibrato [67/4895 pacientes]; p = 0,074).
*** El aumento medio del nivel de homocisteína en sangre en pacientes que recibieron fenofibrato fue de 6,5 µmol/l y fue reversible tras la interrupción del tratamiento con fenofibrato. El riesgo aumentado de eventos trombóticos venosos podría estar relacionado con niveles elevados de homocisteína. La relevancia clínica de este hallazgo no ha sido determinada.
Además de los eventos observados en estudios clínicos, durante el período poscomercialización del medicamento Tricor® 145 mg se han recibido notificaciones espontáneas de reacciones adversas que se indican a continuación; la frecuencia exacta no puede determinarse con los datos disponibles, por lo que se ha clasificado como «desconocida».
Alteraciones del sistema respiratorio, del tórax y del mediastino: enfermedad pulmonar intersticial.
Alteraciones del sistema musculoesquelético y del tejido conectivo: rabdomiólisis.
Alteraciones del sistema hepatobiliar: ictericia, complicaciones de la enfermedad biliar litiasica (por ejemplo, colecistitis, colangitis, cólico biliar).
Alteraciones de la piel y del tejido subcutáneo: reacciones cutáneas graves (por ejemplo, eritema multiforme, síndrome de Stevens-Johnson, necrólisis epidérmica tóxica).
\u>Alteraciones del sistema nervioso: fatiga.
Si se presentan cualquier reacción adversa durante el uso del medicamento o reclamaciones sobre la calidad del producto, rogamos notificarlo a la empresa Abbott Ukraine LLC al teléfono: +38 044-498-60-80 o por correo electrónico: [email protected].
Período de validez. 3 años.
Condiciones de almacenamiento. Conservar en el envase original a una temperatura no superior a 25 °C, en un lugar inaccesible para los niños.
Envase. 10 comprimidos en blíster, 2 o 3 blísteres en caja de cartón.
Categoría de dispensación. Bajo receta médica.
Fabricantes. Irish Fournier Laboratories Limited, Irlanda / Fournier Laboratories Ireland Limited, Ireland.
Astrea Fontaine, Francia / Astrea Fontaine, France.
Domicilio de los fabricantes y direcciones de los lugares de ejercicio de su actividad. Anngrove, Carrigtwohill, Co. Cork, Irlanda / Anngrove, Carrigtwohill, Co. Cork, Ireland.
Rue Des Pres Potets, Fontaine Les Dijon, 21121, Francia / Rue Des Pres Potets, Fontaine Les Dijon, 21121, France.