TriCor® 145 mg
Ucraina
Indice
ISTRUZIONI PER L'USO MEDICINALE DEL MEDICINALE TRICOR® 145 MG (TRICOR® 145 MG)
Composizione:
Principio attivo: fenofibrato;
1 compressa contiene 145 mg di fenofibrato;
Eccipienti: idrossipropilmetilcellulosa, docusato di sodio, saccarosio, laurilsolfato di sodio, monoidrato di lattosio, cellulosa microcristallina silicatizzata, crospovidone, stearato di magnesio, Opatry® OY-B-28920 (alcool polivinilico, diossido di titanio (E 171), talco, lecitina di soia, gomma xantana).
Forma farmaceutica. Compresse rivestite con film.
Principali caratteristiche fisico-chimiche: compresse bianche, ovali, rivestite con film, con incisione «145» su un lato e con il logo dell'azienda «Fournier» sull'altro.
Gruppo farmacoterapeutico. Agenti ipolipemizzanti. Preparati che riducono il livello di colesterolo e trigliceridi nel siero del sangue. Fibrati. Codice ATC C10AB05.
Proprietà farmacologiche.
Farmacodinamica.
Dislipidemia
Il fenofibrato è un derivato dell'acido fibrico, i cui effetti lipidomodificanti nell'uomo sono mediati dall'attivazione del recettore per la proliferazione dei perossisomi di tipo alfa (PPARα).
L'attivazione del PPARα induce il fenofibrato a potenziare la lipolisi e l'eliminazione dalle particelle aterogene plasmatiche ricche di trigliceridi, attraverso l'attivazione della lipoproteina lipasi e la riduzione della sintesi dell'apoproteina CIII. L'attivazione del PPARα induce inoltre un aumento della sintesi delle apoproteine AI e AII.
Gli effetti sopra descritti del fenofibrato sui lipoproteine portano a una riduzione delle frazioni di lipoproteine a bassa e molto bassa densità (LDL e VLDL) contenenti apoproteina B e a un aumento della frazione di lipoproteine ad alta densità (HDL) contenenti apoproteine AI e AII.
Inoltre, modulando la sintesi e il catabolismo delle frazioni VLDL, il fenofibrato aumenta l'eliminazione delle LDL e riduce il numero di particelle di LDL dense e piccole, il cui livello è elevato in soggetti con fenotipo lipoproteico aterogenico, spesso presente in pazienti a rischio di malattia coronarica ischemica.
Negli studi clinici, l'uso del fenofibrato ha determinato una riduzione del colesterolo totale del 20-25%, dei trigliceridi del 40-55% e un aumento del colesterolo HDL del 10-30%.
Nei pazienti con ipercolesterolemia, nei quali il colesterolo LDL è ridotto del 20-35%, l'effetto complessivo sul colesterolo totale determina una riduzione dei rapporti colesterolo totale/colesterolo HDL, colesterolo LDL/colesterolo HDL o apoproteina B/apoproteina AI, che sono marcatori di rischio aterogenico.
Esistono evidenze che il trattamento con fibrati possa ridurre la frequenza degli eventi nella malattia coronarica ischemica, ma i fibrati non hanno dimostrato una riduzione della mortalità totale nella prevenzione primaria o secondaria delle malattie cardiovascolari.
Lo studio ACCORD lipid è stato uno studio randomizzato controllato con placebo, che ha coinvolto 5518 pazienti con diabete mellito di tipo 2 trattati con fenofibrato in aggiunta a simvastatina. La terapia con fenofibrato più simvastatina, rispetto alla monoterapia con simvastatina, non ha mostrato differenze significative sull'endpoint primario combinato (infarto miocardico non fatale, ictus non fatale e morte cardiovascolare) (rapporto di rischio [RR] 0,92, IC 95% 0,79-1,08, p = 0,32; riduzione del rischio assoluto: 0,74%). In una sottopopolazione predefinita di pazienti con dislipidemia, con i livelli più bassi di colesterolo HDL (≤ 34 mg/dl o 0,88 mmol/l) e i livelli più alti di trigliceridi (≥ 204 mg/dl o 2,3 mmol/l) all'inizio del trattamento, la terapia combinata con fenofibrato e simvastatina ha mostrato una riduzione relativa del 31% del rischio dell'endpoint primario combinato rispetto alla monoterapia con simvastatina (rapporto di rischio [RR] 0,69, IC 95% 0,49-0,97, p = 0,03; riduzione del rischio assoluto: 4,95%). Un'analisi di un'altra sottopopolazione predefinita ha evidenziato un'interazione statisticamente significativa tra trattamento e sesso (p = 0,01), suggerendo un possibile beneficio della terapia combinata negli uomini (p = 0,037), ma un potenziale rischio più elevato dell'endpoint primario nelle donne trattate con terapia combinata rispetto alla monoterapia con simvastatina (p = 0,069). Questo fenomeno non è stato osservato nella sottopopolazione sopra menzionata di pazienti con dislipidemia, ma non sono stati neppure ottenuti chiari dati a sostegno di un beneficio nelle donne con dislipidemia trattate con fenofibrato più simvastatina, e un possibile effetto dannoso in questa sottopopolazione non può essere escluso.
I depositi extravascolari di colesterolo (xantomi tendinei e tuberosi) possono ridursi significativamente o scomparire completamente durante il trattamento con fenofibrato.
Nei pazienti con livelli elevati di fibrinogeno, trattati con fenofibrato, si è osservata una significativa riduzione di questo parametro, così come nei pazienti con livelli elevati di lipoproteina (a). Il fenofibrato riduce i livelli di altri marcatori infiammatori, come la proteina C-reattiva.
L'effetto uricosurico del fenofibrato, che determina una riduzione del livello di acido urico di circa il 25%, deve essere considerato un effetto favorevole aggiuntivo del farmaco nei pazienti con dislipidemia e iperuricemia.
Un effetto antiaggregante del fenofibrato sui piastrini è stato osservato in studi sugli animali e in uno studio clinico, che hanno dimostrato una riduzione dell'aggregazione piastrinica indotta da ADP, acido arachidonico ed adrenalina.
Retinopatia diabetica
Diversi meccanismi sono stati proposti per spiegare gli effetti del fenofibrato nei pazienti con retinopatia diabetica proliferativa (PDR) e edema maculare diabetico (DME) in vitro e nei modelli animali. È stato dimostrato che il fenofibrato riduce l'espressione retinica del fattore di crescita dell'endotelio vascolare (VEGF), principale fattore angiogenico nella PDR, riduce la permeabilità vascolare e l'apoptosi dell'epitelio pigmentato retinico, entrambi coinvolti nello sviluppo del DME.
Lo studio FIELD è stato uno studio multicentrico randomizzato che ha coinvolto 9795 pazienti con diabete mellito di tipo 2. I pazienti arruolati sono stati randomizzati a ricevere fenofibrato 200 mg al giorno (n = 4895) o placebo (n = 4900). In uno studio sottostudio oftalmologico con 1012 pazienti sono state effettuate fotografie retiniche standardizzate, valutate secondo i criteri ETDRS (Early Treatment Diabetic Retinopathy Study) per determinare la frequenza cumulativa di sviluppo della retinopatia diabetica e delle sue singole manifestazioni. Le analisi sono state condotte su tutti i pazienti che hanno iniziato il trattamento. Nello studio sottostudio oftalmologico, l'endpoint primario, rappresentato da un aggravamento di due stadi della retinopatia, non differiva significativamente tra i due gruppi nel complesso (46 [9,6%] pazienti nel gruppo fenofibrato contro 57 [12,3%] nel gruppo placebo, p = 0,19) né nella sottopopolazione di pazienti senza retinopatia preesistente (43 [11,4%] contro 43 [11,7%], p = 0,87). Al contrario, nel gruppo di pazienti con retinopatia già esistente, un aggravamento di due stadi è stato osservato in un numero significativamente minore di pazienti nel gruppo fenofibrato rispetto al gruppo placebo (3 [3,1%] contro 14 [14,6%], p = 0,004).
I dati relativi al trattamento laser della retinopatia diabetica, predefinito come endpoint terziario nello studio principale, sono stati raccolti a ogni visita del paziente. La necessità di un primo trattamento laser per qualsiasi forma di retinopatia è risultata significativamente inferiore nel gruppo trattato con fenofibrato rispetto al placebo (164 [3,4%] pazienti nel gruppo fenofibrato contro 238 [4,9%] nel gruppo placebo; rapporto di rischio [RR] 0,69, IC 95% 0,56-0,84, p = 0,0002; riduzione del rischio assoluto: 1,5% [0,7-2,3]). Tale necessità non dipendeva dalla concentrazione plasmatica dei lipidi.
In una sottopopolazione di 2856 partecipanti allo studio ACCORD (ACCORD Eye) è stato valutato l'effetto di tre strategie terapeutiche sul progresso della retinopatia diabetica: trattamento intensivo o standard della glicemia (obiettivo di HbA1c <6,0% o 7,0-7,9% rispettivamente), trattamento della dislipidemia (terapia combinata con fenofibrato 160 mg/giorno più simvastatina o placebo più simvastatina) o controllo della pressione arteriosa sistolica (obiettivo <120 o <140 mmHg). Il progresso della retinopatia diabetica è stato definito dopo 4 anni come un aumento di almeno 3 punti sulla scala ETDRS (valutato mediante fotografie stereoscopiche a sette campi del fondo oculare) o lo sviluppo di retinopatia diabetica che richiedeva fotocoagulazione laser o vitrectomia.
La frequenza di progressione della retinopatia diabetica è stata del 6,5% nel gruppo con trattamento intensivo della dislipidemia con fenofibrato rispetto al 10,2% nel gruppo placebo (odds ratio aggiustato 0,60; IC 95% 0,42-0,87, p = 0,006). Si è concluso che la terapia combinata intensiva della dislipidemia riduce la frequenza di progressione della retinopatia diabetica.
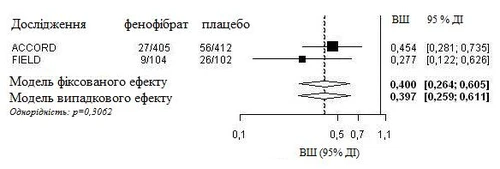
È stato effettuato un'analisi integrata dei dati individuali dei pazienti dello studio FIELD e delle informazioni pubblicate dello studio ACCORD Eye. L'endpoint primario combinato dello studio ACCORD Eye è stato applicato allo studio FIELD, cioè un aggravamento di 3 punti sulla scala ETDRS, fotocoagulazione o vitrectomia per il trattamento della retinopatia diabetica proliferativa. Entrambi gli studi sono risultati omogenei (modello a effetti fissi) e hanno mostrato una riduzione complessiva del 60% della progressione della retinopatia diabetica (odds ratio: 0,40, IC 95% 0,26-0,61).
Progressione della retinopatia diabetica (DR) in pazienti con DR all'inizio dello studio: analisi combinata degli studi FIELD PSP-DR e ACCORD Eye, utilizzando l'endpoint primario dello studio ACCORD Eye.
Farmacocinetica.
Assorbimento.
La concentrazione massima del farmaco nel plasma (Cmax) viene raggiunta entro 2-4 ore dopo somministrazione orale. La concentrazione plasmatica rimane stabile durante il trattamento cronico in tutti i pazienti.
A differenza dei precedenti farmaci a base di fenofibrato, con l'assunzione di TriCor® 145 mg in forma di nanoparticelle, la concentrazione massima nel plasma e l'esposizione totale non dipendono dall'assunzione di cibo; pertanto, TriCor® 145 mg può essere assunto indipendentemente dai pasti.
In uno studio sull'effetto del cibo sull'assorbimento del farmaco, dopo somministrazione della nuova formulazione di compresse da 145 mg di fenofibrato a soggetti sani a digiuno e con un pasto ricco di grassi, si è dimostrato che il cibo non influenza l'esposizione (AUC e Cmax) all'acido fenofibrico.
Distribuzione.
L'acido fenofibrico si lega in misura elevata all'albumina plasmatica (oltre il 99%).
Metabolismo ed eliminazione.
Dopo somministrazione orale, il fenofibrato viene rapidamente idrolizzato dalle esterasi all'acido fenofibrico, il metabolita attivo. Il fenofibrato inalterato non è rilevabile nel plasma. Il fenofibrato non è un substrato del CYP3A4 e non partecipa al metabolismo microsomiale epatico.
Il farmaco viene eliminato principalmente attraverso le urine. L'eliminazione è praticamente completa entro 6 giorni. Il fenofibrato viene eliminato principalmente sotto forma di acido fenofibrico e del suo coniugato glucuronidato. Nei pazienti anziani, la clearance plasmatica totale dell'acido fenofibrico non mostra variazioni significative.
Gli studi di cinetica dopo dose singola e trattamento cronico hanno dimostrato che il farmaco non si accumula nell'organismo. L'acido fenofibrico non viene eliminato mediante emodialisi.
Il tempo di dimezzamento dell'acido fenofibrico è di circa 20 ore.
Caratteristiche cliniche.
Indicazioni.
TriCor® 145 mg è indicato come complemento alla dieta e ad altre misure non farmacologiche (ad esempio esercizio fisico, riduzione del peso corporeo) nelle seguenti condizioni:
- grave ipertrigliceridemia con o senza bassi livelli di colesterolo delle lipoproteine ad alta densità;
- iperlipidemia mista nei casi in cui l'uso di statine è controindicato o vi è intolleranza alle statine;
- iperlipidemia mista in pazienti con elevato rischio cardiovascolare, in aggiunta alla terapia con statine, quando i livelli di trigliceridi e di colesterolo delle lipoproteine ad alta densità non sono adeguatamente controllati.
Retinopatia diabetica: TriCor® 145 mg è indicato per ridurre la progressione della retinopatia diabetica in pazienti con diabete mellito di tipo 2 e retinopatia diabetica preesistente.
Controindicazioni.
Insufficienza epatica (inclusa cirrosi biliare e alterazioni persistenti della funzionalità epatica di causa non nota).
Malattie conclamate della colecisti.
Gravi malattie renali croniche.
Pancreatite cronica o acuta, ad eccezione dei casi di pancreatite acuta indotta da grave ipertrigliceridemia.
Fotallergia o reazioni fototossiche accertate durante il trattamento con fibrati o ketoprofene.
Ipersensibilità al principio attivo o a uno qualsiasi degli eccipienti elencati nella sezione «Composizione».
Inoltre, TriCor® 145 mg non deve essere somministrato a pazienti con allergia all'arachide, all'olio di arachide, al lecitina di soia o prodotti simili, a causa del rischio di reazioni di ipersensibilità.
Interazioni con altri medicinali e altre forme di interazione.
Anticoagulanti orali.
Il fenofibrato potenzia l'effetto degli anticoagulanti orali e può aumentare il rischio di emorragia. Si raccomanda di ridurre la dose di anticoagulante di circa un terzo all'inizio del trattamento e di regolarla successivamente in modo graduale in base al rapporto normalizzato internazionale (INR).
Ciclosporina.
Sono stati riportati alcuni casi gravi, ma reversibili, di alterazione della funzionalità renale durante l'uso concomitante di fenofibrato e ciclosporina. Pertanto, la funzionalità renale deve essere attentamente monitorata nei pazienti che assumono questa combinazione e il trattamento con fenofibrato deve essere interrotto in caso di gravi alterazioni dei parametri di laboratorio.
Inibitori della HMG-CoA reduttasi e altri fibrati.
Il rischio di tossicità muscolare grave aumenta con l'uso concomitante di fibrati e inibitori della HMG-CoA reduttasi o di altri fibrati. Tale terapia combinata deve essere utilizzata con cautela e i pazienti devono essere attentamente monitorati per segni di tossicità muscolare (vedere sezione «Avvertenze speciali e precauzioni di impiego»).
Glitazoni.
Sono stati osservati casi di riduzione paradossale e inversa del colesterolo HDL durante l'uso concomitante di fenofibrato e glitazoni. Pertanto, si raccomanda di monitorare i livelli di colesterolo HDL durante l'uso combinato di questi farmaci e di interromperne l'assunzione qualora i livelli di colesterolo HDL diventino troppo bassi.
Enzimi del citocromo P450.
Studi in vitro condotti con microsomi epatici umani hanno dimostrato che il fenofibrato e l'acido fenofibrico non sono inibitori delle isoenzimi del citocromo P450 CYP3A4, CYP2D6, CYP2E1 o CYP1A2. Essi sono deboli inibitori di CYP2C19 e CYP2A6 e deboli o moderati inibitori di CYP2C9 alle concentrazioni terapeutiche.
Nei pazienti che assumono contemporaneamente fenofibrato e farmaci metabolizzati da CYP2C19, CYP2A6 e in particolare da CYP2C9, con un indice terapeutico stretto, è necessario un attento monitoraggio e, se necessario, un aggiustamento della dose di tali farmaci.
Caratteristiche d'uso.
Ipelipidemia secondaria.
Prima di iniziare la terapia con fenofibrato, è necessario trattare adeguatamente le condizioni sottostanti che causano l'ipercolesterolemia secondaria, come diabete mellito di tipo 2 non controllato, ipotiroidismo, sindrome nefrosica, disproteinemia, malattia epatica ostruttiva o alcolismo. L'iperlipidemia secondaria può verificarsi anche in pazienti in trattamento con diuretici, beta-bloccanti, estrogeni, progestinici, contraccettivi orali combinati, immunosoppressori e inibitori della proteasi. In tali casi, è necessario stabilire se l'iperlipidemia sia primaria o secondaria (possibile aumento dei livelli lipidici indotto dai suddetti farmaci).
Funzionalità epatica.
Come con altri farmaci ipolipemizzanti, in alcuni pazienti sono stati osservati aumenti dei livelli delle transaminasi. Nella maggior parte dei casi, tale aumento è stato temporaneo, lieve e asintomatico. Si raccomanda di controllare i livelli delle transaminasi ogni 3 mesi durante il primo anno di terapia e periodicamente successivamente. È necessario prestare attenzione ai pazienti con aumenti dei livelli di transaminasi e interrompere il trattamento se i livelli di AST e ALT superano di oltre 3 volte il limite superiore della norma. L'uso di fenofibrato deve essere interrotto in caso di comparsa di sintomi di epatite (ad esempio ittero, prurito) e conferma della diagnosi tramite esami di laboratorio.
Pancreas.
Sono stati riportati casi di pancreatite in pazienti trattati con fenofibrato (vedi sezioni «Controindicazioni» e «Effetti indesiderati»). Tale evento può essere conseguenza di un'insufficiente efficacia terapeutica nei pazienti con grave ipertrigliceridemia, di un effetto diretto del farmaco o di un fenomeno secondario mediato dalla formazione di calcoli biliari o di fango biliare con ostruzione del coledoco.
Muscoli.
Durante l'uso di fibrati e di altri farmaci ipolipemizzanti, sono stati riportati effetti tossici sul muscolo, inclusi rari casi di rabdomiolisi con o senza insufficienza renale. La frequenza di tale evento aumenta in presenza di ipoalbuminemia o di anamnesi di insufficienza renale. Il rischio di sviluppare rabdomiolisi può essere aumentato in pazienti con fattori predisponenti a miopatia e/o rabdomiolisi, tra cui età ≥ 70 anni, anamnesi personale o familiare di patologie muscolari ereditarie, compromissione della funzionalità renale, ipotiroidismo e abuso di alcol. In tali pazienti, è necessario valutare attentamente il rapporto beneficio/rischio prima di iniziare la terapia con fenofibrato.
Un effetto tossico muscolare deve essere sospettato in caso di mialgia diffusa, miosite, crampi muscolari e debolezza e/o di un marcato aumento dei livelli di CPK (oltre 5 volte il limite superiore della norma). In tali situazioni, il trattamento con fenofibrato deve essere interrotto.
Il rischio di effetti tossici muscolari può aumentare se il farmaco viene somministrato in associazione con un altro fibrato o con un inibitore dell'HMG-CoA reduttasi, specialmente in caso di preesistente patologia muscolare. Pertanto, la somministrazione concomitante di fenofibrato con un inibitore dell'HMG-CoA reduttasi o con un altro fibrato deve essere riservata a pazienti con grave dislipidemia combinata e alto rischio cardiovascolare, in assenza di anamnesi di patologie muscolari e con rigoroso monitoraggio degli effetti tossici muscolari.
Funzionalità renale.
Se il livello di creatinina aumenta di oltre il 50% rispetto al limite superiore della norma (LSN), il trattamento con fenofibrato deve essere interrotto. Si raccomanda di controllare i livelli di creatinina nei primi 3 mesi dall'inizio del trattamento e periodicamente successivamente (per quanto riguarda il dosaggio, vedere la sezione «Modalità di somministrazione e posologia»).
Sostanze eccipienti.
Il medicinale contiene lattosio; pertanto, non deve essere assunto da pazienti con rari disturbi ereditari come intolleranza al galattosio, deficienza di lattasi o malassorbimento di glucosio-galattosio.
Il medicinale contiene saccarosio; pertanto, non deve essere assunto da pazienti con rari disturbi ereditari come intolleranza al fruttosio, malassorbimento di glucosio-galattosio o deficienza di saccarasi-isomaltasi.
Uso durante la gravidanza o l'allattamento.
Gravidanza. Non vi sono dati sufficienti sull'uso di fenofibrato in donne in gravidanza. Negli studi sugli animali non è stato osservato alcun effetto teratogeno. Effetti embriotossici sono stati osservati con dosi tossiche per l'organismo materno. Il potenziale rischio nell'uomo è sconosciuto. Pertanto, TriCor® 145 mg deve essere usato durante la gravidanza solo dopo un'attenta valutazione del rapporto beneficio/rischio.
Allattamento. Non è noto se fenofibrato e/o i suoi metaboliti siano escreti nel latte materno umano. Non può essere escluso un rischio per i neonati allattati; pertanto, fenofibrato non deve essere usato durante l'allattamento.
Fertilità. Negli studi sugli animali sono stati osservati effetti reversibili sulla fertilità. Non vi sono dati clinici sull'effetto di TriCor® 145 mg sulla fertilità nell'uomo.
Effetti sulla capacità di guidare veicoli o di usare macchinari.
TriCor® 145 mg non ha alcun effetto oppure ha un effetto trascurabile sulla capacità di guidare veicoli o di usare macchinari.
Modalità e posologia.
TriCor® 145 mg può essere assunto in qualsiasi momento della giornata, indipendentemente dai pasti (vedere la sezione «Proprietà farmacologiche. Farmacocinetica»). Le compresse devono essere inghiottite intere, accompagnate da un bicchiere d’acqua.
La terapia dietetica iniziata prima della prescrizione del medicinale deve essere proseguita.
Durante il trattamento dell’iperlipidemia, l’efficacia terapeutica deve essere monitorata mediante la determinazione dei livelli lipidici nel siero sanguigno. Se dopo alcuni mesi (ad esempio dopo 3 mesi) non si ottiene una risposta adeguata al trattamento, devono essere considerate terapie aggiuntive o alternative.
Adulti
La dose raccomandata è di 1 compressa contenente 145 mg di fenofibrato, una volta al giorno. Ai pazienti che assumono 1 capsula contenente 200 mg di fenofibrato o 1 compressa contenente 160 mg di fenofibrato, si può sostituire con 1 compressa di TriCor® 145 mg senza ulteriore adeguamento della dose.
Se un paziente deve assumere fenofibrato per due indicazioni (iperlipidemia e retinopatia diabetica), deve assumere solo 1 compressa di TriCor® 145 mg al giorno.
Pazienti anziani
Ai pazienti anziani senza alterazioni della funzionalità renale si raccomanda la dose abituale per adulti.
Alterazioni della funzionalità renale
Nei pazienti con alterazioni della funzionalità renale è necessario ridurre la dose. L’uso di fenofibrato alla dose attuale di 145 mg non è raccomandato nei pazienti con insufficienza renale cronica di grado moderato (clearance della creatinina da 30 a 60 ml/min).
L’uso di fenofibrato è controindicato nei pazienti con grave insufficienza renale cronica (clearance della creatinina < 30 ml/min).
Alterazioni della funzionalità epatica
TriCor® 145 mg non è raccomandato nei pazienti con alterazioni della funzionalità epatica a causa della mancanza di dati.
Età pediatrica
La sicurezza e l’efficacia dell’uso di fenofibrato nei bambini e negli adolescenti (di età inferiore ai 18 anni) non sono state stabilite e i dati pertinenti sono mancanti. Pertanto, l’uso di fenofibrato non è raccomandato nei bambini e negli adolescenti (di età inferiore ai 18 anni).
Sovradosaggio.
Sono stati riportati solo singoli casi di sovradosaggio di fenofibrato. Nella maggior parte dei casi non sono stati osservati sintomi di sovradosaggio.
Non esiste un antidoto specifico. In caso di sospetto di sovradosaggio, si deve effettuare un trattamento sintomatico e adottare le opportune misure di supporto se necessario. Il fenofibrato non viene eliminato mediante emodialisi.
Effetti indesiderati.
Le reazioni avverse più frequentemente osservate durante la terapia con fenofibrato sono disturbi del sistema gastrointestinale, in particolare a carico dello stomaco o dell’intestino.
I seguenti effetti indesiderati sono stati osservati negli studi clinici controllati con placebo (n=2344) con la frequenza indicata:
| Classe di sistema e organo secondo MedDRA |
Molto frequente ≥ 1/10 |
Frequente ≥ 1/100, <1/10 |
Non comune ≥ 1/1000, <1/100 |
Raro ≥ 1/10000, <1/1000 |
| Disturbi del sistema emolinfopoietico |
Diminuzione dei livelli di emoglobina Diminuzione del numero di globuli bianchi nel sangue |
|||
| Disturbi del sistema immunitario |
Ipersensibilità (inclusa reazione anafilattica) |
|||
| Disturbi del sistema nervoso |
Cefalea |
|||
| Disturbi del sistema vascolare |
Tromboembolia (embolia polmonare, trombosi venosa profonda)** |
|||
| Disturbi del tratto gastrointestinale |
Segni e sintomi a carico degli organi digestivi (dolore addominale, nausea, vomito, diarrea, meteorismo) |
Pancreatite* |
||
| Disturbi del sistema epatobiliare |
Aumento dei livelli delle transaminasi (vedere il paragrafo «Informazioni importanti») |
Calcolosi biliare (vedere il paragrafo «Informazioni importanti») |
Epatite |
|
| Disturbi della cute e del tessuto sottocutaneo |
Reazioni cutanee di ipersensibilità (ad esempio eruzioni, prurito, orticaria) |
Alopecia Reazioni di fotosensibilità |
||
| Disturbi del sistema muscoloscheletrico e del tessuto connettivo |
Disturbi muscolari (ad esempio mialgia, miosite, crampi e debolezza muscolare) |
|||
| Disturbi del sistema riproduttivo e delle ghiandole mammarie |
Dismorfia sessuale |
|||
| Anomalie riscontrate negli esami di laboratorio |
Incremento dei livelli ematici di omocisteina*** |
Incremento dei livelli ematici di creatinina |
Incremento dei livelli ematici di urea |
* Nello studio FIELD – uno studio randomizzato controllato con placebo, a cui hanno partecipato 9795 pazienti con diabete mellito di tipo 2 – nei pazienti trattati con fenofibrato è stata osservata una maggiore incidenza statisticamente significativa di pancreatite rispetto ai pazienti del gruppo placebo (0,8% e 0,5% rispettivamente, p = 0,031).
** È stata osservata una maggiore incidenza statisticamente significativa di embolia polmonare (0,7% nel gruppo placebo e 1,1% nel gruppo fenofibrato; p = 0,022) e un aumento statisticamente non significativo dell'incidenza di trombosi venosa profonda (1,0% nel gruppo placebo [48/4900 pazienti] e 1,4% nel gruppo fenofibrato [67/4895 pazienti]; p = 0,074).
*** L'aumento medio dei livelli plasmatici di omocisteina nei pazienti trattati con fenofibrato è stato di 6,5 µmol/l ed è risultato reversibile dopo l'interruzione della terapia con fenofibrato. Un aumentato rischio di eventi trombotici venosi potrebbe essere correlato all'aumento dei livelli di omocisteina. L'importanza clinica di questo fenomeno non è stata chiarita.
Oltre agli effetti osservati negli studi clinici, durante il periodo post-marketing con l'uso del medicinale TriCor® 145 mg sono state segnalate spontaneamente le seguenti reazioni avverse riportate di seguito; la loro frequenza esatta non può essere determinata sulla base dei dati disponibili, pertanto è stata classificata come «non nota».
Patologie dell'apparato respiratorio, torace e mediastino: malattia polmonare interstiziale.
Patologie del sistema muscoloscheletrico e del tessuto connettivo: rabdomiolisi.
Patologie del sistema epatobiliare: ittero, complicanze della malattia litiasica biliare (ad esempio colecistite, colangite, colica biliare).
Patologie della cute e del tessuto sottocutaneo: gravi reazioni cutanee (ad esempio eritema multiforme, sindrome di Stevens-Johnson, necrolisi epidermica tossica).
Patologie del sistema nervoso: affaticamento.
In caso di insorgenza di qualsiasi reazione avversa durante l'uso del medicinale o di eventuali reclami sulla qualità del prodotto, si prega di informare la società Abbott Ukraine S.R.L. al numero telefonico: +38 044-498-60-80 oppure via e-mail all'indirizzo: [email protected].
Periodo di validità. 3 anni.
Condizioni di conservazione. Conservare nella confezione originale a una temperatura non superiore a 25 °C, in un luogo inaccessibile ai bambini.
Confezionamento. 10 compresse in un blister, 2 o 3 blister in una confezione di cartone.
Categoria di rilascio. Su prescrizione medica.
Produttori. Irish Laboratories Fournier Limited, Irlanda / Fournier Laboratories Ireland Limited, Ireland.
Astrea Fontaine, Francia / Astrea Fontaine, France.
Sede dei produttori e indirizzi dei luoghi di esercizio della loro attività. Anngrove, Carrigtwohill, Co. Cork, Irlanda / Anngrove, Carrigtwohill, Co. Cork, Ireland.
Rue Des Pres Potets, Fontaine Les Dijon, 21121, Francia / Rue Des Pres Potets, Fontaine Les Dijon, 21121, France.