Tezpair
UkrainaSpis treści
INSTRUKCJA dotyczaca stosowania leku Tezpair (TEZSPIRE®)
Skład:
substancja czynna: tezepelumab (tezepelumab);
szykan wstępnie napełniony
1 szykan wstępnie napełniony zawiera 210 mg tezepelumabu w 1,91 ml roztworu (110 mg/ml);
długopis strzykawkowy wstępnie napełniony
1 długopis strzykawkowy wstępnie napełniony zawiera 210 mg tezepelumabu w 1,91 ml roztworu (110 mg/ml);
substancje pomocnicze:
szykan wstępnie napełniony
L-prolin, kwas octowy lodowaty, polisorbat 80, wodorotlenek sodu, woda do wstrzykiwań;
długopis strzykawkowy wstępnie napełniony
L-prolin, kwas octowy lodowaty, polisorbat 80, wodorotlenek sodu, woda do wstrzykiwań.
Postać farmaceutyczna. Roztwór do wstrzykiwań.
Główne właściwości fizykochemiczne: roztwór od przezroczystego do oplalizującego, od bezbarwnego do jasnożółtego.
Grupa farmakoterapeutyczna. Leki stosowane w leczeniu chorób obturacyjnych dróg oddechowych. Inne leki stosowane systemowo w chorobach obturacyjnych dróg oddechowych. Kod ATC R03D X11.
Właściwości farmakologiczne.
Farmakodynamika.
Mechanizm działania
Tezepelumab jest ludzkim monoklonalnym przeciwciałem IgG2λ, wytwarzanym metodą rekombinacji DNA w komórkach jajnika chomika chińskiego (CHO). Działanie tezepelumabu skierowane jest przeciwko tymicznemu stromalnemu limfopoetyce (TSLP), zapobiegając jego wiązaniu się z heterodimerowym receptorem TSLP. W przypadku astmy oskrzelowej zarówno alergiczne, jak i niealergiczne czynniki wyzwalające indukują powstawanie TSLP. Blokowanie TSLP przez tezepelumab zmniejsza szeroki zakres biomarkerów i cytokin związanych z zapaleniem dróg oddechowych (np. eozynofili we krwi, eozynofili w błonie podśluzowej dróg oddechowych, IgE, FeNO, IL-5 oraz IL-13); jednakże mechanizm działania tezepelumabu w astmie oskrzelowej nie został ostatecznie ustalony.
Skutki farmakodynamiczne
Wpływ na eozynofile we krwi oraz biomarkery i cytokiny związane z zapaleniem
W badaniach klinicznych stosowanie tezepelumabu w dawce 210 mg podskórnie co 4 tygodnie prowadziło do obniżenia liczby eozynofili we krwi, poziomu FeNO, stężenia IL-5, stężenia IL-13 oraz stężenia IgE w surowicy krwi w porównaniu do placebo. Te markery osiągnęły bliskie maksymalnemu stężenie po 2 tygodniach leczenia, z wyjątkiem IgE, które obniżało się wolniej. Efekty te utrzymywały się przez cały okres leczenia.
Wpływ na eozynofile w błonie podśluzowej dróg oddechowych
W trakcie badania klinicznego stosowanie tezepelumabu w dawce 210 mg podskórnie co 4 tygodnie zmniejszyło liczbę eozynofili w warstwie podśluzowej o 89% w porównaniu do zmniejszenia o 25% przy stosowaniu placebo. Obniżenie to było stałe niezależnie od początkowego poziomu biomarkerów zapalenia.
Immunogenność
W badaniu NAVIGATOR przeciwciała przeciwko lekowi (APL) stwierdzono w dowolnym czasie u 26 (4,9%) z 527 pacjentów otrzymujących tezepelumab w zalecanym trybie dawkowania przez 52-tygodniowy okres badania. Spośród tych 26 pacjentów u 10 osób (1,9% spośród otrzymujących tezepelumab) rozwinęły się APL powstające w trakcie leczenia, a u 1 pacjenta (0,2% spośród otrzymujących tezepelumab) pojawiły się przeciwciała neutralizujące. Tytry APL były zazwyczaj niskie i często przejściowe. Nie zaobserwowano dowodów wpływu APL na farmakokinetykę, farmakodynamikę, skuteczność ani profil bezpieczeństwa.
Skuteczność kliniczna
Skuteczność tezepelumabu oceniano w dwóch randomizowanych, podwójnie ślepych, kontrolowanych placebo badaniach klinicznych z równoległymi grupami (PATHWAY i NAVIGATOR) trwających 52 tygodnie, w których wzięło udział 1609 pacjentów w wieku od 12 lat z ciężką astmą oskrzelową. W obu badaniach nie stawiano pacjentom wymogu co do minimalnego początkowego poziomu eozynofili we krwi ani innych biomarkerów zapalenia (np. FeNO lub IgE).
PATHWAY – to 52-tygodniowe badanie zaostrzeń, przeprowadzone wśród 550 pacjentów (w wieku od 18 lat) z niekontrolowaną ciężką astmą oskrzelową, którzy otrzymywali leczenie tezepelumabem w dawce 70 mg podskórnie co 4 tygodnie, w dawce 210 mg podskórnie co 4 tygodnie, w dawce 280 mg podskórnie co 2 tygodnie lub placebo. Pacjenci musieli mieć w wywiadzie 2 lub więcej przypadków zaostrzeń astmy oskrzelowej wymagających leczenia kortykosteroidami doustnymi lub systemowymi, albo 1 przypadek zaostrzenia astmy oskrzelowej, który doprowadził do hospitalizacji w ciągu ostatnich 12 miesięcy.
NAVIGATOR – to 52-tygodniowe badanie przeprowadzone wśród 1061 pacjentów (dorośli i dzieci w wieku od 12 lat) z niekontrolowaną ciężką astmą oskrzelową, którzy otrzymywali leczenie tezepelumabem w dawce 210 mg podskórnie co 4 tygodnie lub placebo. Pacjenci musieli mieć w wywiadzie 2 lub więcej przypadków zaostrzeń astmy oskrzelowej wymagających leczenia kortykosteroidami doustnymi lub systemowymi, albo które doprowadziły do hospitalizacji w ciągu ostatnich 12 miesięcy.
W badaniach PATHWAY i NAVIGATOR pacjenci musieli mieć wynik 1,5 punktu lub więcej w kwestionariuszu kontroli astmy oskrzelowej (ACQ-6) podczas skriningu oraz obniżoną funkcję płuc na poziomie wyjściowym (przedszybkość przepływu wydechowego FEV1 poniżej 80% wartości przewidywanej u dorosłych i poniżej 90% wartości przewidywanej u dzieci). Pacjenci musieli otrzymywać regularne leczenie średnimi lub wysokimi dawkami inhalacyjnych kortykosteroidów (ICS) oraz co najmniej jedną dodatkową terapię w celu kontroli astmy oskrzelowej, z zastosowaniem lub bez doustnych kortykosteroidów (OCS). Wysoką dawkę ICS zdefiniowano jako > 500 µg propionianu fluktykazonu lub równowartości na dobę. Średnią dawkę ICS zdefiniowano jako od > 250 do 500 µg propionianu fluktykazonu lub równowartości na dobę w badaniu PATHWAY oraz jako 500 µg propionianu fluktykazonu lub równowartości na dobę w badaniu NAVIGATOR. Pacjenci kontynuowali leczenie tło astmy oskrzelowej przez cały okres badań.
Charakterystyki demograficzne i wyjściowe obu badań przedstawiono w tabeli 1.
Tabela 1
Charakterystyki demograficzne i wyjściowe badań astmy oskrzelowej
| PATHWAY |
NAVIGATOR |
|
| Średnia wieku (w latach) (SD) |
52 (12) |
50 (16) |
| Kobiety (%) |
66 |
64 |
| Rasa kaukaska (%) |
92 |
62 |
| Rasa czarna lub Afroamerykanie (%) |
3 |
6 |
| Rasa mongolska (%) |
3 |
28 |
| Osoby pochodzenia hiszpańskiego lub latynoamerykańskiego (%) |
1 |
15 |
| Średni czas trwania astmy oskrzelowej (lat) (SD) |
17 (12) |
22 (16) |
| Nigdy nie palący (%) |
81 |
80 |
| Stosowanie ICS w wysokiej dawce (%) |
49 |
75 |
| Stosowanie SCS (%) |
9 |
9 |
| Średnia liczba zaostrzeń w poprzednim roku (SD) |
2,4 (1,2) |
2,8 (1,4) |
| Średnia wartość wyjściowego FEV1 w odniesieniu do wartości przewidywanej (%) (SD) |
60 (13) |
63 (18) |
| Średnia wartość FEV1 przed zastosowaniem leku rozszerzającego oskrzela (l) (SD) |
1,9 (0,6) |
1,8 (0,7) |
| Średnia wartość odwracalności FEV1 po zastosowaniu leku rozszerzającego oskrzela (%) (SD) |
23 (20) |
15 (15) |
| Średnia liczba eoz. we krwi na poziomie wyjściowym (komórek/μl) (SD) |
371 (353) |
340 (403) |
| Liczba eoz. we krwi ≥ 150 komórek/μl (%) |
76 |
74 |
| Pozytywny status alergiczny (%)a |
46 |
64 |
| Średnia wartość FeNO (ppb) (SD) |
35 (39) |
44 (41) |
| FeNO ≥ 25 ppb (%) |
44 |
59 |
| Średnia wartość ACQ-6 (SD) |
2,7 (0,8) |
2,8 (0,8) |
| Liczba eoz. we krwi ≥ 150 komórek/μl i FeNO ≥ 25 ppb (%) |
38 |
47 |
a Pozytywny status alergiczy, określony pozytywnym wynikiem surowicy IgE, specyficznym dla każdego z wieloletnich aler genów powietrznych w panelu FIFА.
ACQ-6 — Kwestionariusz Kontroli Astmy Osobistej 6; eoz. — eozynofile; FIFА — fluorescencyjny test immunoenzymatyczny; FeNO — stężenie tlenku azotu w wydychanym powietrzu; ОFV1 — objętość wymuszonego wydechu w ciągu jednej sekundy; IKS — inhalacyjny kortykosteroid; IgE — immunoglobulina E; PKS — doustny kortykosteroid; млрд–1 — cząstek na miliard; SW — odchylenie standardowe.
Poniższe uogólnione wyniki dotyczą zalecanego schematu stosowania tezepelumabu w dawce 210 mg podskórnie co 4 tygodnie.
Zaostrzenia
Pierwotnym punktem końcowym w badaniach PATHWAY i NAVIGATOR była częstość zaostrzeń astmy oskrzelowej ciężkiego stopnia, mierzona przez 52 tygodnie. Zaostrzenie astmy oskrzelowej ciężkiego stopnia definiowano jako pogorszenie przebiegu astmy oskrzelowej, wymagające zastosowania lub zwiększenia dawki doustnych lub systemowych kortykosteroidów przez co najmniej 3 dni lub jednorazowego zastrzyku depot kortykosteroidów, oraz/lub wizytę w izbie przyjęć wymagającą zastosowania doustnych lub systemowych kortykosteroidów, oraz/lub hospitalizację.
W badaniach PATHWAY i NAVIGATOR pacjenci otrzymujący tezepelumab wykazali istotne zmniejszenie rocznej częstości zaostrzeń astmy oskrzelowej ciężkiego stopnia w porównaniu z odpowiednimi wartościami uzyskanymi przy stosowaniu placebo (tabela 2 i tabela 3). U pacjentów otrzymujących tezepelumab odnotowano również mniejszą liczbę zaostrzeń wymagających wizyty w izbie przyjęć i/lub hospitalizacji w porównaniu z pacjentami otrzymującymi placebo. W badaniach PATHWAY i NAVIGATOR częstość zaostrzeń astmy oskrzelowej ciężkiego stopnia, które wymagały wizyty w izbie przyjęć i/lub hospitalizacji, zmniejszyła się odpowiednio o 85% i 79% przy stosowaniu tezepelumabu w dawce 210 mg podskórnie co 4 tygodnie.
Tabela 2
Częstość zaostrzeń ciężkiego stopnia w 52. tygodniu badania NAVIGATORa
| Tezpairumab (N = 528) |
Placebo (N = 531) |
|
| Roczna częstość zaostrzeń astmy ciężkiej |
||
| Częstość |
0,93 |
2,10 |
| Stosunek częstości (95 % CI) |
0,44 (0,37; 0,53) |
|
| Wartość p |
< 0,001 |
|
a Czas ryzyka określa się jako całkowity czas, w którym może wystąpić nowe nasilenie (tj. całkowity czas obserwacji pomniejszony o czas trwania nasilenia i 7 dni po nim).
CI — przedział ufności.
Tabela 3
Częstość nasileń ciężkiego stopnia w 52. tygodniu w badaniu PATHWAYa
| Tezepelumab (N = 137) |
Placebo (N = 138) |
|
| Roczna częstość zaostrzeń ciężkiej astmy oskrzelowej |
||
| Częstość |
0,20 |
0,72 |
| Stosunek częstości (95 % CI) |
0,29 (0,16; 0,51) |
|
| Wartość p |
< 0,001 |
|
a Czas ryzyka definiuje się jako całkowity czas obserwacji.
CI – przedział ufności.
Analiza w podgrupach
W badaniu NAVIGATOR tezepelumab wykazał zmniejszenie częstości zaostrzeń ciężkiej astmy niezależnie od wyjściowego poziomu eozynofili we krwi, FeNO oraz statusu atopowego (określonego na podstawie poziomu specyficznego IgE dla alergenów sezonowych). Podobne wyniki zaobserwowano w badaniu PATHWAY, patrz rysunek 1.
W badaniu NAVIGATOR zmniejszenie częstości zaostrzeń ciężkiej astmy było większe wraz ze wzrostem wyjściowej liczby eozynofili we krwi i wartości FeNO (stosunek częstości = 0,79 [95 % CI: 0,48; 1,28] u pacjentów z wyjściową liczbą eozynofili we krwi < 150 komórek/μl i wyjściowym poziomem FeNO < 25 ppb; stosunek częstości = 0,30 [95 % CI: 0,23; 0,40] u pacjentów z wyjściową liczbą eozynofili we krwi ≥ 150 komórek/μl i wyjściowym poziomem FeNO ≥ 25 ppb).
Rysunek 1
Stosunek rocznej częstości zaostrzeń ciężkiej astmy w okresie 52 tygodni w zależności od różnych wyjściowych biomarkerów dla analizy pełnej populacji (połączone dane badań NAVIGATOR i PATHWAY)a
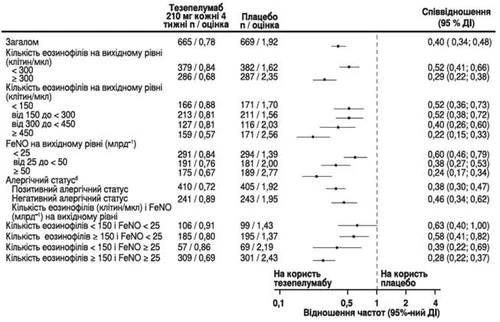
a Czas ryzyka definiuje się jako całkowity czas trwania, w którym może wystąpić nowe zaostrzenie (tzn. całkowity czas obserwacji pomniejszony o czas trwania zaostrzenia oraz 7 dni po nim).
b Status alergiczny, określony na podstawie poziomu surowicy IgE specyficznego dla dowolnego alergenu sezonowego w teście FIFAX.
Funkcja płuc
W badaniu NAVIGATOR oceniano zmianę FEV1 od wartości wyjściowej jako wtórny punkt końcowy. W porównaniu z placebo tezepelumab zapewniał klinicznie istotne poprawy średniej zmiany FEV1 od wartości wyjściowej (tabela 4).
Wyniki zgłaszane przez pacjentów
W badaniu NAVIGATOR oceniano zmiany od wartości wyjściowej w punktach ACQ-6, w Standaryzowanym Kwestionariuszu Jakości Życia u pacjentów z astmą oskrzelową w wieku od 12 lat [AQLQ(S) 12+] oraz w średnim tygodniowym wyniku w Dzienniku Symptomów Astmy (ASD) jako punkty końcowe wtórne. Nasilenie świstów, duszności, kaszlu i uczucia ucisku w klatce piersiowej oceniano dwa razy dziennie (rano i wieczorem). Przebudzenia w nocy oraz aktywność oceniano codziennie. Ogólny wynik ASD obliczano jako średnią z 10 punktów (tabela 4).
Poprawa w ACQ-6 i AQLQ(S) 12+ obserwowano odpowiednio już po 2 i 4 tygodniach od zastosowania tezepelumabu i utrzymywała się do 52. tygodnia w obu badaniach.
Tabela 4
Wyniki dla kluczowych wtórnych punktów końcowych w 52. tygodniu badania NAVIGATORa
| Tezpair |
Placebo |
|
| FEV1 przed zastosowaniem leku rozszerzającego oskrzela |
||
| N |
527 |
531 |
| Średnia zmiana od wartości wyjściowej, obliczona metodą najmniejszych kwadratów (l) |
0,23 |
0,10 |
| Średnia różnica w porównaniu z placebo, obliczona metodą najmniejszych kwadratów (l) (95 % CI) |
0,13 (0,08; 0,18) |
|
| Wartość p |
< 0,001 |
|
| Łączna punktacja AQLQ(S) 12+ |
||
| N |
525 |
526 |
| Średnia zmiana od wartości wyjściowej, obliczona metodą najmniejszych kwadratów |
1,48 |
1,14 |
| Różnica w porównaniu z placebo (95 % CI) |
0,33 (0,20; 0,47) |
|
| Wartość p |
< 0,001 |
|
| Punktacja ACQ-6 |
||
| N |
527 |
531 |
| Średnia zmiana od wartości wyjściowej, obliczona metodą najmniejszych kwadratów |
-1,53 |
-1,20 |
| Różnica w porównaniu z placebo (95 % CI) |
-0,33 (-0,46; -0,20) |
|
| Wartość p |
< 0,001 |
|
| ASD |
||
| N |
525 |
531 |
| Średnia zmiana od wartości wyjściowej, obliczona metodą najmniejszych kwadratów |
-0,70 |
-0,59 |
| Różnica w porównaniu z placebo (95 % CI) |
-0,11 (-0,19; -0,04) |
|
| Wartość p |
0,004 |
|
a Oceny uzyskano na podstawie mieszанego modelu dla pomiarów powtarzalnych (Mixed Model for Repeated Measures (MMRM)) z wykorzystaniem wszystkich dostępnych danych pacjentów z co najmniej jedną zmianą od wartości wyjściowej, w tym danych po przerwaniu leczenia.
ACQ-6 — Opytujnik kontroli astmy oskrzelowej 6; AQLQ(S) 12+ — Standardowy opytujnik jakości życia chorych na astmę oskrzelową od 12 roku życia; ASD — Dziennik objawów astmy oskrzelowej; CI — przedział ufności; OBJ — objętość wymuszona wydechu w ciągu jednej sekundy; MNK — metoda najmniejszych kwadratów; N — liczba pacjentów, których dane zostały uwzględnione w analizie (FA), z co najmniej jedną zmianą od wartości wyjściowej
Pacjenci w wieku podeszłym (≥ 65 lat)
Spośród 665 pacjentów z astmą oskrzelową, którzy otrzymywali tezepelumab w dawce 210 mg podskórnie co 4 tygodnie w badaniach PATHWAY i NAVIGATOR, łącznie 119 pacjentów miało co najmniej 65 lat, w tym 32 pacjentów miało co najmniej 75 lat. Profil bezpieczeństwa w tych grupach wiekowych był podobny do profilu w ogólnej populacji badawczej. Skuteczność w tych grupach wiekowych była podobna do skuteczności w ogólnej populacji badawczej w badaniu NAVIGATOR. Badanie PATHWAY nie obejmowało wystarczającej liczby pacjentów w wieku co najmniej 65 lat, aby można było określić skuteczność w tej grupie wiekowej.
Populacja pediatryczna
Łącznie 82 dzieci w wieku od 12 do 17 lat z ciężką, niekontrolowaną astmą oskrzelową zostało włączonech do badania NAVIGATOR i otrzymywało tezepelumab (n = 41) lub placebo (n = 41). Spośród 41 dzieci, 15 dzieci z grupy otrzymującej tezepelumab przyjmowało wysokie dawki IKS na poziomie wyjściowym. Roczna częstość zaostrzeń astmy oskrzelowej obserwowana u dzieci otrzymujących tezepelumab wynosiła 0,68 w porównaniu do 0,97 u tych, którzy przyjmowali placebo (stosunek częstości 0,70; 95 % CI 0,34; 1,46). Średnia obliczona metodą MNK zmiana od wartości wyjściowej OBJ obserwowana u dzieci otrzymujących tezepelumab wynosiła 0,44 l w porównaniu do 0,27 l u tych, którzy otrzymywali placebo (średnia różnica obliczona metodą MNK, 0,17 l; 95 % CI 0,01; 0,35). Odpowiedzi farmakodynamiczne u dzieci były ogólnie podobne do odpowiedzi w ogólnej populacji badawczej.
Europejska Agencja Leków odroczyła obowiązek przedstawienia wyników badań leku Tezpair w jednej lub kilku podgrupach populacji pediatrycznej w przypadku astmy oskrzelowej (informacje dotyczące stosowania leku u dzieci znajdują się w sekcji „Dzieci”).
Farmakokinetyka.
Farmakokinetyka tezepelumabu była proporcjonalna do dawki po podaniu podskórnym w zakresie dawek od 2,1 mg do 420 mg.
Wchłanianie
Po jednorazowym podaniu podskórnym maksymalne stężenie w surowicy osiągane było w przybliżeniu po 3–10 dniach. Na podstawie populacyjnej analizy farmakokinetycznej obliczona absolutna biodostępność wynosiła około 77%. Nie stwierdzono klinicznie istotnej różnicy w biodostępności przy wstrzykiwaniu w różne miejsca (brzuch, uda lub ramię).
Rozkład
Na podstawie populacyjnej analizy farmakokinetycznej, objętość rozkładu centralna i obwodowa tezepelumabu wynosiła odpowiednio 3,9 l i 2,2 l dla osoby o masie ciała 70 kg.
Metabolizm
Tezepelumab to ludzkie monoklonalne przeciwciało (IgG2λ), które ulega proteolitycznemu rozszczepieniu przez enzymy szeroko rozpowszechnione w organizmie i nie jest metabolizowane przez enzymy wątrobowe.
Eliminacja
Tezepelumab, będąc ludzkim monoklonalnym przeciwciałem, jest wydzielany z organizmu drogą wewnątrzkomórkowego katabolizmu, a nie ma dowodów na eliminację zależną od celu działania. Na podstawie populacyjnej analizy farmakokinetycznej, obliczony klirens tezepelumabu wynosił 0,17 l/dobę dla osoby o masie ciała 70 kg. Okres półtrwania wynosił około 26 dni.
Specjalne kategorie pacjentów
Wiek, płeć, rasa
Na podstawie populacyjnej analizy farmakokinetycznej, wiek, płeć i rasa pacjenta nie miały klinicznie istotnego wpływu na farmakokinetykę tezepelumabu.
Masa ciała
Na podstawie populacyjnej analizy farmakokinetycznej, większa masa ciała pacjenta była związana z niższą ekspozycją na lek. Jednak wpływ masy ciała na ekspozycję nie ma istotnego znaczenia dla skuteczności ani profilu bezpieczeństwa i nie wymaga korekty dawki.
Dzieci
Na podstawie populacyjnej analizy farmakokinetycznej, nie stwierdzono klinicznie istotnej różnicy związanej z wiekiem pacjenta w farmakokinetyce tezepelumabu między dorosłymi a dziećmi w wieku od 12 do 17 lat. Farmakokinetyka tezepelumabu nie była badana u dzieci poniżej 12 roku życia (patrz sekcja „Sposób stosowania i dawki”).
Pacjenci w wieku podeszłym (≥ 65 lat)
Na podstawie populacyjnej analizy farmakokinetycznej, nie stwierdzono klinicznie istotnej różnicy w farmakokinetyce tezepelumabu między pacjentami w wieku co najmniej 65 lat a młodszych pacjentów.
Naruszenie funkcji nerek
Nie przeprowadzono oficjalnych badań klinicznych w celu zbadania wpływu tezepelumabu na naruszenie funkcji nerek. Na podstawie populacyjnej analizy farmakokinetycznej, klirens tezepelumabu był podobny u pacjentów z łagodnym zaburzeniem funkcji nerek (klirens kreatyniny od 60 do < 90 ml/min), umiarkowanym zaburzeniem funkcji nerek (klirens kreatyniny od 30 do < 60 ml/min) oraz u pacjentów z normalną funkcją nerek (klirens kreatyniny ≥ 90 ml/min). Tezepelumab nie był badany u pacjentów z ciężkim zaburzeniem funkcji nerek (klirens kreatyniny < 30 ml/min); jednak tezepelumab nie jest wydzielany przez nerki.
Naruszenie funkcji wątroby
Nie przeprowadzono oficjalnych badań klinicznych w celu zbadania wpływu tezepelumabu na zaburzenia funkcji wątroby. Monoklonalne przeciwciała IgG są głównie wydzielane niezależnie od wątroby; oczekuje się, że zmiana funkcji wątroby nie wpłynie na klirens tezepelumabu. Na podstawie populacyjnej analizy farmakokinetycznej, wyjściowe biomarkery funkcji wątroby (ALT, AST i bilirubina) nie wpływały na klirens tezepelumabu.
Charakterystyka kliniczna.
Wskazania.
Tezpair wskazany jest jako dodatkowa terapia wspomagająca u dorosłych i dzieci w wieku od 12 lat z ciężką astmą oskrzelową, która nie jest odpowiednio kontrolowana mimo stosowania wysokich dawek kortykosteroidów wziewnych w połączeniu z innym lekiem stosowanym w terapii utrzymującej.
Przeciwwskazania.
Nadwrażliwość na substancje czynne lub na którąkolwiek z substancji pomocniczych wymienionych w sekcji „Skład”.
Interakcje z innymi lekami i inne rodzaje interakcji.
Badania kliniczne dotyczące interakcji z innymi lekami nie były prowadzone.
Pacjentom otrzymującym tezepelumab należy unikać stosowania żywych osłabionych szczepionek.
W randomizowanym, podwójnie ślepych badaniach z równoległymi grupami, w których wzięło udział 70 pacjentów w wieku od 12 do 21 lat z umiarkowaną i ciężką astmą oskrzelową, leczenie tezepelumabem nie wpływało na indukowaną sezonową czterowalentną szczepionką przeciw grypie produkcję przeciwciał humoralnych.
Nie oczekuje się klinicznie istotnego wpływu tezepelumabu na farmakokinetykę leków stosowanych jednocześnie w leczeniu astmy oskrzelowej. Wyniki analizy farmakokinetyki populacyjnej wskazują, że leki stosowane zazwyczaj równolegle w leczeniu astmy oskrzelowej (w szczególności antagoniści receptorów leukotrienowych, teofilina/aminofilina oraz kortykosteroidy doustne) nie wpływały na klirens tezepelumabu.
Szczególne ostrzeżenia i środki ostrożności podczas stosowania.
Śledzenie
W celu ułatwienia śledzenia leków biologicznych należy dokładnie odnotować nazwę handlową i numer serii zastosowanego leku w dokumentacji medycznej pacjenta.
Zaostrzenie astmy oskrzelowej
Leku Tezpair nie należy stosować w leczeniu zaostrzeń astmy oskrzelowej.
Podczas leczenia mogą wystąpić objawy związane z astmą oskrzelową lub zaostrzenie astmy oskrzelowej. Pacjentów należy poinstruować, aby w przypadku braku kontroli nad astmą oskrzelową lub jej nasilenia po włączeniu leczenia, skontaktowali się z lekarzem.
Kortykosteroidy
Nie zaleca się nagłego odstawienia kortykosteroidów po rozpoczęciu terapii. Redukcja dawki kortykosteroidów, jeśli jest to wskazane, powinna być stopniowa i odbywać się pod kontrolą lekarza.
Reakcje nadwrażliwości
Po zastosowaniu tezepelumabu mogą wystąpić reakcje nadwrażliwości (w tym anafilaksja, wysypka) (zob. rozdział „Działania niepożądane”). Mogą one pojawić się w ciągu kilku godzin po podaniu leku, ale w niektórych przypadkach ich początek może być opóźniony (np. po kilku dniach).
Anafilaksja w wywiadzie, niezwiązana z tezepelumabem, może stanowić czynnik ryzyka wystąpienia anafilaksji po podaniu leku Tezpair. Zgodnie z praktyką kliniczną, pacjenci powinni pozostawać pod obserwacją przez odpowiedni czas po podaniu leku Tezpair.
W przypadku wystąpienia ciężkiej reakcji nadwrażliwości (np. anafilaksji) należy natychmiast przerwać stosowanie tezepelumabu i rozpocząć leczenie odpowiednie do sytuacji klinicznej.
Ciężkie infekcje
Blokada tymicznego stromalnego limfopoetyny (TSLP) teoretycznie może zwiększać ryzyko ciężkich infekcji. W badaniach kontrolowanych placebo nie zaobserwowano zwiększenia częstości ciężkich infekcji podczas stosowania tezepelumabu.
Pacjenci z istniejącymi ciężkimi infekcjami powinni ukończyć leczenie przed rozpoczęciem terapii tezepelumabem. Jeśli podczas leczenia tezepelumabem u pacjenta rozwinie się ciężka infekcja, terapię tezepelumabem należy przerwać do czasu wygojenia się ciężkiej infekcji.
Ciężkie zdarzenia sercowe
W długoterminowym badaniu klinicznym zaobserwowano nierównowagę ciężkich działań niepożądanych ze strony serca u pacjentów otrzymujących tezepelumab w porównaniu z pacjentami otrzymującymi placebo. Związek przyczynowo-skutkowy między tezepelumabem a takimi zdarzeniami nie został ustalony, a także nie określono populacji pacjentów z ryzykiem wystąpienia takich zdarzeń.
Pacjentów należy poinformować o objawach wskazujących na zdarzenie kardiologiczne (np. ból w klatce piersiowej, duszność, osłabienie, uczucie zawrotów głowy lub stanu przedobrzuczkowego) oraz o konieczności natychmiastowego skontaktowania się z lekarzem w przypadku ich wystąpienia. Jeśli podczas leczenia tezepelumabem u pacjenta rozwinie się ciężkie zdarzenie kardiologiczne, terapię tezepelumabem należy przerwać do czasu ustabilizowania się stanu ostrego.
Obecnie brak danych dotyczących ponownego leczenia pacjentów, u których wystąpiło ciężkie zdarzenie kardiologiczne lub ciężka infekcja.
Infekcja pasożytnicza (helmintoza)
TSLP może uczestniczyć w odpowiedzi immunologicznej na niektóre infekcje helminthowe. Pacjenci z rozpoznanymi infekcjami helminthowymi byli wykluczani z udziału w badaniach klinicznych. Nie wiadomo, czy tezepelumab może wpływać na odpowiedź pacjenta na leczenie infekcji helminthowych.
Pacjenci z istniejącymi infekcjami helminthowymi powinni ukończyć leczenie przed rozpoczęciem terapii tezepelumabem. Jeśli pacjenci zostaną zainfekowani podczas leczenia i nie odpowiadają na leczenie przeciwpasożytnicze, terapię tezepelumabem należy przerwać do czasu wygojenia się infekcji.
Zawartość sodu
Ten lek zawiera mniej niż 1 mmol sodu (23 mg) na dawkę 210 mg, co oznacza, że praktycznie nie zawiera sodu.
Stosowanie w okresie ciąży lub karmienia piersią.
Ciąża
Brak danych lub dostępne dane są ograniczone (mniej niż 300 wyników dotyczących ciąży) dotyczące stosowania tezepelumabu u kobiet w ciąży. Badania na zwierzętach nie wskazują na bezpośredni lub pośredni szkodliwy wpływ w odniesieniu do toksyczności rozrodczej.
Przeciwciała IgG człowieka, takie jak tezepelumab, przenikają przez barierę łożyskową; dlatego lek Tezpair może być przenoszony od ciężarnej do rozwijającego się płodu.
Z powodów ostrożności zaleca się unikanie stosowania leku Tezpair w czasie ciąży, z wyjątkiem przypadków, gdy oczekiwana korzyść dla kobiety ciężarnej przewyższa potencjalne ryzyko dla płodu.
Karmienie piersią
Nie wiadomo, czy tezepelumab wydzielany jest z mlekiem matki. Wiadomo, że ludzkie IgG wydzielają się z mlekiem matki w pierwszych dniach po porodzie, a ich stężenia szybko spadają do niskiego poziomu. W związku z tym w tym krótkim okresie nie można wykluczyć ryzyka dla dziecka karmionego piersią.
Decyzję o przerwaniu lub kontynuowaniu terapii tezepelumabem należy podjąć, biorąc pod uwagę korzyści płynące z karmienia piersią dla dziecka oraz korzyści z terapii dla kobiety.
W późniejszym okresie tezepelumab można stosować podczas karmienia piersią, jeśli istnieje konieczność kliniczna.
Plodność
Brak danych dotyczących wpływu na płodność u ludzi. Badania na zwierzętach nie wykazały negatywnego wpływu leczenia tezepelumabem na płodność.
Wpływ na zdolność do kierowania pojazdami i obsługiwanie maszyn.
Lek Tezpair nie wpływa lub ma nieznaczny wpływ na zdolność kierowania pojazdami i obsługiwanie maszyn.
Sposób stosowania i dawki
Leczenie powinien rozpoczynać lekarz posiadający doświadczenie w rozpoznawaniu i leczeniu ciężkiej astmy oskrzelowej.
Dawkowanie
Dorośli i dzieci (od 12. roku życia)
Zalecana dawka to 210 mg tezepelumabu podawanych co 4 tygodnie w formie iniekcji podskórnej.
Lek Tezpair przeznaczony jest do długotrwałego leczenia. Decyzję o kontynuowaniu terapii należy podejmować co najmniej raz w roku, biorąc pod uwagę stopień kontroli astmy oskrzelowej u pacjenta.
Pominięcie dawki
W przypadku pominięcia dawki należy ją podać jak najszybciej. Następnie pacjent może powrócić do zaplanowanego harmonogramu podawania leku. Jeśli nadszedł czas na kolejną dawkę, należy podać lek zgodnie z planem. Nie wolno podawać podwójnej dawki.
Pacjenci z grup szczególnych
Pacjenci w wieku powyżej 65. roku życia
U pacjentów w wieku powyżej 65. roku życia nie jest wymagana korekta dawki (patrz sekcja „Farmakokinetyka”).
Zaburzenia funkcji nerek i wątroby
U pacjentów z zaburzeniami funkcji nerek lub wątroby nie jest wymagana korekta dawki (patrz sekcja „Farmakokinetyka”).
Sposób stosowania
Lek Tezpair podaje się w formie iniekcji podskórnej.
Pacjent może samodzielnie wykonać sobie iniekcję lub lek może podać opiekun pacjenta po przejściu szkolenia z techniki wykonywania iniekcji podskórnej. Pacjentom i/lub opiekunom należy przejść odpowiednie szkolenie dotyczące przygotowania i podania leku Tezpair przed jego zastosowaniem zgodnie z „Instrukcją stosowania”.
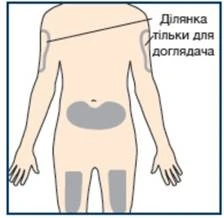
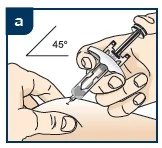
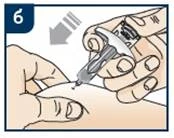
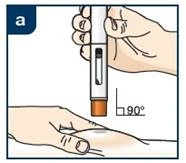
Lek Tezpair należy podawać podskórnie w przednią część uda lub brzuch, z wyjątkiem obszaru 5 cm wokół pępka. Jeśli iniekcję wykonuje pracownik służby zdrowia lub opiekun, lek może być również podawany w okolice ramienia. Pacjent nie powinien samodzielnie wykonywać iniekcji w ramię. Leku nie należy podawać w obszary z siniakami, wrażliwe, z rumieniem lub z twardą skórą. Zaleca się zmienianie miejsca iniekcji przy każdym podaniu.
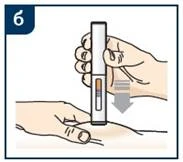
Szczegółowe instrukcje dotyczące podania leku za pomocą wypełnionego wcześniej strzykawki lub wypełnionego wcześniej pióra strzykawkowego zawarte są w „Instrukcji stosowania”.
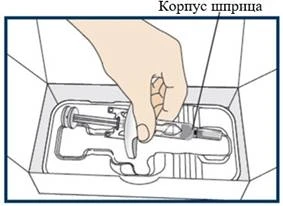
Instrukcja stosowania (wypełniona wcześniej strzykawka)
Niniejsza „Instrukcja stosowania” zawiera informacje dotyczące sposobu podawania leku Tezpair.
Przed zastosowaniem leku Tezpair za pomocą wypełnionej wcześniej strzykawki pracownik służby zdrowia musi pokazać pacjentowi lub jego opiekunowi, jak prawidłowo z niej korzystać.
Przed rozpoczęciem stosowania leku Tezpair za pomocą wypełnionej wcześniej strzykawki oraz za każdym razem przed wykonaniem kolejnej iniekcji należy przeczytać niniejszą „Instrukcję stosowania”. Może ona zawierać nowe informacje. Niniejsza informacja nie zastępuje konsultacji z pracownikiem służby zdrowia w sprawie stanu zdrowia lub leczenia.
W przypadku pojawienia się jakichkolwiek pytań u pacjenta lub opiekuna należy skontaktować się z pracownikiem służby zdrowia.
Ważne informacje, które należy znać przed podaniem leku Tezpair.
Lek Tezpair należy przechowywać w lodówce w temperaturze od 2 do 8 °C w opakowaniu kartonowym zewnętrznym do momentu przygotowania do zastosowania. Lek Tezpair można przechowywać w temperaturze pokojowej w zakresie od 20 do 25 °C w opakowaniu kartonowym zewnętrznym przez maksymalnie 30 dni.
Gdy lek Tezpair osiągnie temperaturę pokojową, nie należy ponownie umieszczać go w lodówce.
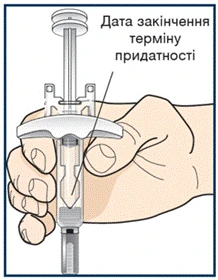
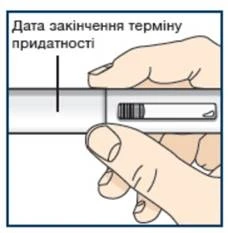
Lek Tezpair, który był przechowywany w temperaturze pokojowej ponad 30 dni, należy wyrzucić (utylizować) (patrz krok 10).
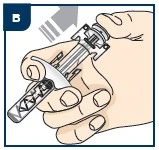
| Nie stosuj leku Tezpair za pomocą wstępnie wypełnionego strzykawki, jeśli: |
|
|
Nie wstrząsaj wstępnie wypełnioną strzykawką. |
|
Nie przekazuj wstępnie wypełnionej strzykawki innym osobom do użycia. |
|
Nie używaj wstępnie wypełnionej strzykawki więcej niż 1 raz. |
|
Nie narażaj leku Tezpair w wstępnie wypełnionej strzykawce na działanie ciepła. |
Jeśli wystąpi coś z wymienionego powyżej, wyrzuć strzykawkę do odpornego na przebicie pojemnika na ostre przedmioty i użyj nowej wstępnie napełnionej strzykawki z lekiem Tezpair.
Każda wstępnie napełniona strzykawka z lekiem Tezpair zawiera 1 dawkę leku Tezpair, którą można zastosować tylko 1 raz.
Przechowuj lek Tezpair we wstępnie napełnionej strzykawce oraz wszystkie leki w miejscu niedostępnym dla dzieci i poza ich zasięgiem wzroku.
Lek Tezpair podaje się wyłącznie przez wstrzyknięcie pod skórę (podskórnie).
Wstępnie napełniona strzykawka z lekiem Tezpair
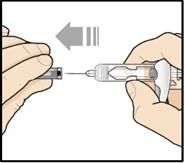
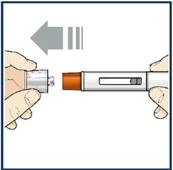
Nie zdejmuj osłonki igły przed krokiem 7 niniejszej instrukcji, gdy będziesz gotowy podać lek Tezpair.
Nie dotykaj zatrzasków aktywujących mechanizm ochrony igły. Pozwoli to uniknąć przedwczesnego aktywowania urządzenia zabezpieczającego (mechanizmu ochrony igły).
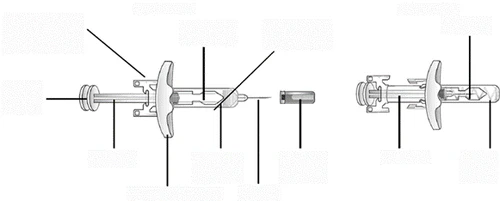
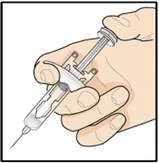
Przygotowanie do podania leku Tezpair.
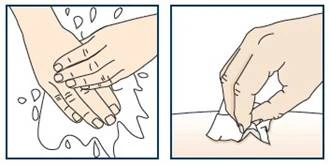
Krok 1. Zbierz niezbędne materiały
- 1 wstępnie napełniona strzykawka z lekiem Tezpair, wyjęta z lodówki;
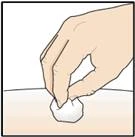
- 1 chusteczka alkoholowa;
- 1 waty lub gaza;
- 1 plaster (opcjonalnie);
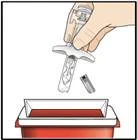
- 1 odporny na przebicie pojemnik na utylizację ostrych przedmiotów. Instrukcje dotyczące bezpiecznego wyrzucenia (utylizacji) użytej wstępnie napełnionej strzykawki z lekiem Tezpair znajdują się w kroku 10.
|
|
|
|
|
|
| Wstępnie wypełniona strzykawka |
Wacik alkoholowy |
Wacik watowy lub gaz |
Plaster |
Kontener |



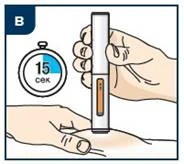
| Krok 2. Przygotowanie do stosowania wypełnionego wcześniej strzykawki z lekiem Tezpair Przed wstrzyknięciem pozwól lekowi Tezpair ogrzać się do temperatury pokojowej w zakresie od 20 do 25 °C przez około 60 minut lub dłużej (nie dłużej niż 30 dni). Przechowuj wypełnioną wcześniej strzykawkę w zewnętrznej tekturowej osłonie, aby chronić ją przed światłem. Nie podgrzewaj wypełnionej wcześniej strzykawki w żaden inny sposób. Na przykład nie podgrzewaj jej w kuchence mikrofalowej ani w gorącej wodzie, nie stawiaj w pobliżu innych źródeł ciepła. |
Nie wkładaj wypełnionego wcześniej strzykawki z lekiem Tezpair z powrotem do lodówki po osiągnięciu przez nią temperatury pokojowej. Wyrzuć (zutylizuj) wypełnioną wcześniej strzykawkę z lekiem Tezpair, która była przechowywana w temperaturze pokojowej dłużej niż 30 dni. Nie zdejmuj nakrywki igły przed krokiem 7.
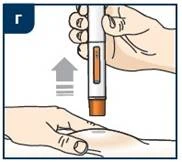
Unieważnianie leku Tezpair
Instrukcje dotyczące utylizacji Utylizuj pojemnik zgodnie z wskazaniami pracownika medycznego lub farmaceuty. Nie wyrzucaj używanego pojemnika na przedmioty ostrych do odpadów komunalnych, chyba że zasady lokalne pozwalają na takie postępowanie. Nie poddawaj recyklingowi używanego pojemnika na przedmioty ostre. Instrukcja stosowania (szczepionka wstępnie napełniona w piórze strzykawkowym) Niniejsza „Instrukcja stosowania” zawiera informacje dotyczące podawania leku Tezpair. Zanim zaczniesz stosować lek Tezpair za pomocą wstępnie napełnionego pióra strzykawkowego, pracownik medyczny powinien pokazać tobie lub twojemu opiekunowi, jak poprawnie z niego korzystać. Przed pierwszym zastosowaniem leku Tezpair za pomocą wstępnie napełnionego pióra strzykawkowego oraz za każdym razem przed wykonaniem kolejnej iniekcji przeczytaj uważnie niniejszą „Instrukcję stosowania”. Może ona zawierać nowe informacje. Niniejsze informacje nie zastępują konsultacji z pracownikiem medycznym dotyczącej stanu zdrowia lub leczenia. W przypadku jakichkolwiek pytań ty lub twój opiekun powinni skontaktować się z pracownikiem medycznym. Ważne informacje, które należy znać przed podaniem leku Tezpair Przechowuj lek Tezpair w lodówce w temperaturze od 2 do 8 °C w opakowaniu kartonowym aż do momentu zastosowania. Lek Tezpair można przechowywać w temperaturze pokojowej w zakresie od 20 do 25 °C w opakowaniu kartonowym przez maksymalnie 30 dni. Gdy lek Tezpair osiągnie temperaturę pokojową, nie wkładaj go ponownie do lodówki. Wyrzuć (utylizuj) lek Tezpair, który był przechowywany w temperaturze pokojowej ponad 30 dni (patrz krok 10).
Jeśli wystąpi coś z wymienionego powyżej, usuń wypełnioną wcześniej strzykawkę-ручkę do odpornego na przebitcie pojemnika na ostre przedmioty i użyj nowej wypełnionej wcześniej strzykawki-ручki z lekiem Tezpair. Każda wstępnie wypełniona strzykawka-ручка zawiera 1 dawkę leku Tezpair, którą można zastosować tylko raz. Przechowuj lek Tezpair w wstępnie wypełnionej strzykawce-ручce oraz wszystkie leki w miejscu niedostępnym dla dzieci i poza ich zasięgiem wzroku. Lek Tezpair podaje się wyłącznie przez iniekcję podskórną. Wstępnie wypełniona strzykawka-ручka z lekiem Tezpair Nie zdejmuj nakładki do kroku 6 tej instrukcji, gdy będziesz gotowy podać lek Tezpair. 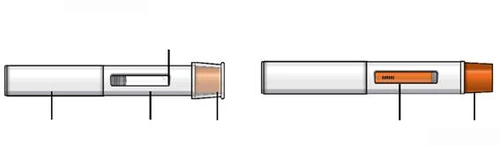
Przygotowanie do podania leku Tezpair Krok 1. Zbierz niezbędne materiały
Krok 2. Przygotowanie do zastosowania wstępnie napełnionej strzykawki-pióra z lekiem Tezpair
Nie wkładaj leku Tezpair z powrotem do lodówki po osiągnięciu temperatury pokojowej. Usuń (utylizuj) lek Tezpair, który był przechowywany w temperaturze pokojowej dłużej niż 30 dni. Nie zdejmuj nakrętki do kroku 6.
Instrukcje dotyczące utylizacji Utylizuj pełny pojemnik zgodnie z instrukcjami pracownika medycznego lub farmaceuty. Nie wyrzucaj używanego pojemnika na przedmioty ostrych do odpadów domowych, chyba że zasady wspólnotowe zezwalają na takie działanie. Nie przetwarzaj ponownie używanego pojemnika na przedmioty ostre. Dzieci. Bezpieczeństwo i skuteczność stosowania leku Tezpair u dzieci poniżej 12. roku życia nie zostały ustalone. Brak danych. Przedawkowanie. W badaniach klinicznych pacjenci z astmą oskrzelową otrzymywali lek w dawkach do 280 mg podskórnie co 2 tygodnie oraz w dawkach do 700 mg dożylnie co 4 tygodnie, bez obserwacji objawów toksyczności zależnych od dawki. Nie ma specyficznego leczenia w przypadku przedawkowania tezepelumabem. W przypadku przedawkowania należy podjąć leczenie wspierające z odpowiednim nadzorem, jeśli to konieczne. Niepożądane działania.▼ Ten lek podlega dodatkowemu nadzorowi. Umożliwia to szybkie uzyskiwanie nowych informacji dotyczących bezpieczeństwa. Osoby sprawujące opiekę zdrowotną prosi się o zgłaszanie wszelkich podejrzewanych niepożądanych działań. Streszczenie profilu bezpieczeństwa Najczęściej obserwowanymi niepożądanymi działaniami podczas leczenia były artrealgia (3,8%) i zapalenie gardła (4,1%). Wykaz niepożądanych działań przedstawiony w tabeli W badaniach klinicznych z udziałem pacjentów z ciężkim astmą oskrzelowym, łącznie 665 pacjentów otrzymało co najmniej jedną dawkę leku Tezpair w badaniach trwających 52 tygodnie. Częstość występowania niepożądanych działań określa się następująco: bardzo często (≥ 1/10); często (od ≥ 1/100 do < 1/10); rzadko (od ≥ 1/1000 do < 1/100); niezwykle rzadko (od ≥ 1/10 000 do < 1/1 000); bardzo rzadko (< 1/10 000); częstość nieznana (nie można oszacować na podstawie dostępnych danych). W ramach każdej kategorii częstości niepożądane działania są wymienione w kolejności zmniejszającego się nasilenia. Tabela 5 Wykaz niepożądanych działań
a Zapalenie gardła było definiowane następującymi pogrupowanymi wyrazami preferowanymi: zapalenie gardła, bakteryjne zapalenie gardła, zapalenie gardła streptokokowe i wirusowe zapalenie gardła. b Wysypka była definiowana następującymi pogrupowanymi wyrazami preferowanymi: wysypka, wysypka z świądem, wysypka rumieniowa, wysypka makularna, wysypka makulopapularna, wysypka plamista. c Zobacz sekcję „Opis poszczególnych działań niepożądanych”. Opis poszczególnych działań niepożądanych Reakcje w miejscu wstrzyknięcia Zgodnie z połączonymi danymi dotyczącymi profilu bezpieczeństwa uzyskanymi na podstawie badań PATHWAY i NAVIGATOR, reakcje w miejscu wstrzyknięcia (np. zaczerwienienie w miejscu wstrzyknięcia, obrzęk w miejscu wstrzyknięcia, ból w miejscu wstrzyknięcia) występowały z częstością 3,8% u pacjentów otrzymujących tezepelumab w dawce 210 mg podskórnie co 4 tygodnie. Dzieci Ogółem 82 dzieci w wieku od 12 do 17 lat z ciężką, niekontrolowaną astmą zostało włączonych do 52-tygodniowego badania fazy 3 NAVIGATOR (zobacz sekcję „Właściwości farmakologiczne”). Profil bezpieczeństwa u dzieci był ogólnie podobny do profilu w ogólnej populacji badawczej. Zgłaszanie działań niepożądanych Zgłaszanie działań niepożądanych po rejestracji produktu leczniczego ma istotne znaczenie. Umożliwia to monitorowanie stosunku korzyści do ryzyka w przypadku stosowania tego produktu leczniczego. Pracownicy medyczni i farmaceutyczni, a także pacjenci lub ich ustawowi przedstawiciele, powinni zgłaszać wszystkie przypadki podejrzewanych działań niepożądanych oraz braku skuteczności leku za pośrednictwem Zautomatyzowanego Systemu Informacyjnego nadzoru farmakologicznego pod adresem: https://aisf.dec.gov.ua. Okres ważności. 36 miesięcy. Produkt leczniczy Tezpair może być przechowywany w temperaturze pokojowej do 25 °C przez maksymalnie 30 dni. Po wyjęciu z lodówki produkt leczniczy Tezpair należy użyć w ciągu 30 dni lub poddać utylizacji. Warunki przechowywania. Przechowywać w temperaturze od 2 do 8 °C (zalecane 5 °C). Przechowywać wstępnie napełniony strzykacz lub wstępnie napełnioną strzykawkę-pensyl w tekturowym pudełku w celu ochrony przed działaniem światła. Nie zamrażać. Nie wstrząsać. Nie narażać na działanie ciepła. Przechowywać w miejscu niedostępnym dla dzieci. Opakowanie. Wstępnie napełniony strzykacz Roztwór do wstrzykiwań w jednorazowym wstępnie napełnionym strzykaczu z niezdejmowaną igłą, osłonką ochronną i ogranicznikiem ruchu tłoka. Po 1 jednorazowym wstępnie napełnionym strzykaczu w blisterze termoformowanym; po 1 blisterze w tekturowym pudełku z oznakowaniem w języku ukraińskim. Wstępnie napełniona strzykawka-pensyl Roztwór do wstrzykiwań w jednorazowej wstępnie napełnionej strzykawce-pensylu do automatycznego wstrzyknięcia z niezdejmowaną igłą, osłonką ochronną i ogranicznikiem ruchu tłoka. Po 1 jednorazowej wstępnie napełnionej strzykawce-pensylu do automatycznego wstrzyknięcia w tekturowym pudełku z oznakowaniem w języku ukraińskim. Kategoria wydania. Na receptę. Producent. AstraZeneca AB / AstraZeneca AB. Miejsce położenia producenta oraz adres miejsca prowadzenia działalności. Gertuneweggen, Sodertalje, 152 57, Szwecja / Gartunavagen, Sodertalje, 152 57, Sweden. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||