Sibrava
Ukraina
Spis treści
INSTRUKCJA dotycząca stosowania leku SIBRAVA (SYBRAVA)
Skład:
substancja czynna: inclisiran (inclisiran);
1 przedwypełniony strzykawka zawiera sodową sól inclisiranu w ilości odpowiadającej 284 mg inclisiranu, w 1,5 ml roztworu;
1 ml roztworu zawiera sodową sól inclisiranu w ilości odpowiadającej 189 mg inclisiranu;
substancje pomocnicze: woda do wstrzykiwań, wodorotlenek sodu, kwas fosforowy stężony.
Postać leku. Roztwór do wstrzykiwań w przedwypełnionej strzykawce.
Główne właściwości fizykochemiczne: przezroczysty roztwór, bezbarwny lub bladożółty, praktycznie bez cząstek.
Grupa farmakoterapeutyczna.
Leki wpływające na układ sercowo-naczyniowy. Środki obniżające stężenie lipidów we krwi. Leki hipolipidemiczne jednoskładnikowe. Inne leki hipolipidemiczne.
Kod ATC C10A X16.
Właściwości farmakologiczne.
Mechanizm działania
Inclisiran to dwuniciowa mała interferująca kwas rybonukleinowy (siRNA), który obniża poziom cholesterolu, skonjugowany z trzycukrowym N-acetylogalaktozaminem (GalNAc) na łańcuchu kodującym w celu ułatwienia wchłaniania przez hepatocyty. W hepatocytach inclisiran wykorzystuje mechanizm interferencji RNA i kieruje katalityczny rozpad mRNA do proproteinowej konwertazy subtylizyny/peptydazy typu 9. Powoduje to zwiększenie recyrkulacji i ekspresji receptorów LDL-C (cholesterol lipoprotein o niskiej gęstości) na powierzchni komórek hepatocytów, co zwiększa wychwyt LDL-C i obniża jego poziom w krążeniu.
Farmakodynamika
Po pojedynczym podaniu podskórnej dawki 284 mg inclisiranu obniżenie poziomu LDL-C było widoczne już po 14 dniach. Średnie obniżenie poziomu LDL-C o 48–51 % obserwowano w okresie 30–60 dni po podaniu dawki. Po 180 dniach poziom LDL-C nadal był obniżony o około 53 %.
Skuteczność kliniczna i bezpieczeństwo
W badaniach klinicznych oraz niektórych publikacjach dawka 284 mg inclisiranu jest równoważna i oznaczana jako 300 mg soli sodowej inclisiranu.
Skuteczność inclisiranu oceniano w trzech badaniach fazy III u pacjentów z chorobami sercowo-naczyniowymi miażdżycowymi (ASCVD) (choroba wieńcowa, choroba naczyń mózgowych lub choroba tętnic obwodowych), równoważnikami ryzyka ASCVD (cukrzyca typu 2, hipercholesterolemia rodzinna lub 10-letnie ryzyko zdarzeń sercowo-naczyniowych ≥20 %, oszacowane według skali ryzyka Framinghama lub równoważnej skali) oraz/lub hipercholesterolemią rodzinną (FH). Pacjenci otrzymywali maksymalną tolerowaną dawkę statyny, z dodatkową terapią modyfikującą lipidy lub bez niej, i wymagali dalszego obniżenia poziomu LDL-C (pacjenci, u których nie osiągnięto celów terapeutycznych). Około 17 % pacjentów miało nietolerancję statyn. Pacjentom podawano podskórne wstrzyknięcia 284 mg inclisiranu lub placebo w dniu 1., 90., 270. i 450. Obserwację prowadzono do dnia 540.
Wpływ inclisiranu na chorobę sercowo-naczyniową i śmiertelność nie został określony.
W analizie podsumowującej badania fazy III, inclisiran podawany podskórnie obniżał poziom LDL-C o 50–55 % już od dnia 90. (rys. 1), a ten efekt utrzymywał się w trakcie długotrwałej terapii. Maksymalne obniżenie poziomu LDL-C osiągnięto w dniu 150. po drugiej dawce. Niewielkie, ale istotne statystycznie zwiększenia LDL-C do 65 % były związane z niższymi wyjściowymi poziomami LDL-C (około < 2 mmol/l [77 mg/dl]), wyższymi wyjściowymi poziomami PCSK9, wyższymi dawkami statyn i ich intensywnością.
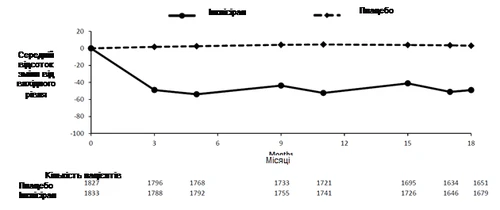
Rys. 1. Średni procentowy poziom zmiany LDL-C od wartości wyjściowej u pacjentów z pierwotną hipercholesterolemią i dyslipidemią mieszaną, otrzymujących inclisiran, w porównaniu z pacjentami otrzymującymi placebo (analiza podsumowująca).
ASCVD i równoważniki ryzyka ASCVD
Przeprowadzono dwa badania z udziałem pacjentów z ASCVD i równoważnikami ryzyka ASCVD (ORION-10 i ORION-11). Pacjenci otrzymywali maksymalną tolerowaną dawkę statyny, z dodatkową terapią modyfikującą lipidy (np. ezetymib) lub bez niej, i wymagali dalszego obniżenia poziomu LDL-C. Ponieważ obniżenie poziomu LDL-C ma poprawiać wyniki sercowo-naczyniowe, wtórnymi punktami końcowymi w każdym badaniu był procentowy poziom zmiany LDL-C od wartości wyjściowej do dnia 510. w porównaniu z placebo oraz skorygowany względem czasu procentowy poziom zmiany LDL-C od wartości wyjściowej po dniu 90. do dnia 540. w celu oceny sumarycznego wpływu na LDL-C w czasie.
ORION-10 to wieloośrodkowe, podwójnie ślepe, randomizowane, kontrolowane placebo badanie trwające 18 miesięcy, w którym wzięło udział 1561 pacjentów z ASCVD.
Średni wiek pacjentów na początku badania wynosił 66 lat (zakres od 35 do 90 lat), 60 % pacjentów miało co najmniej 65 lat, 31 % stanowiły kobiety, 86 % to biała rasa, 13 % – czarna rasa, 1 % – Azjaci, 14 % – pochodzenie hiszpańskie lub latynoskie. Średni wyjściowy poziom LDL-C wynosił 2,7 mmol/l (105 mg/dl). 69 % pacjentów otrzymywało wysokie dawki statyn, 19 % – średnie dawki, 1 % – niskie dawki, a 11 % nie przyjmowało statyn. Najczęściej stosowanymi statynami były atorwastatyna i rosuvastatyna.
Inclisiran istotnie zmniejszył średni procentowy poziom zmiany LDL-C od wartości wyjściowej do dnia 510. w porównaniu z placebo o 52 % (95 % CI: -56 %, -49 %; p < 0,0001) (tabela 2).
Inclisiran istotnie zmniejszył również skorygowany względem czasu procentowy poziom zmiany LDL-C od wartości wyjściowej po dniu 90. do dnia 540. o 54 % w porównaniu z placebo (95 % CI: -56 %, -51 %; p < 0,0001). Dodatkowe wyniki przedstawiono w tabeli 1.
Tabela 1
Średni procentowy poziom zmiany od wartości wyjściowej i różnica w porównaniu z placebo dla parametrów lipidowych w dniu 510. w badaniu ORION-10
| Grupa leczenia |
Ch-LPNHG |
Cholesterol całkowity |
Nie Ch-LPWHG |
Apo-B |
Lp(a)* |
| Średnia wartość wyjściowa, mg/dl** |
105 |
181 |
134 |
94 |
122 |
| Dzień 510 (średni procentowy spadek od poziomu wyjściowego) |
|||||
| Placebo (n = 780) |
1 |
0 |
0 |
-2 |
4 |
| Inklisiran (n = 781) |
-51 |
-34 |
-47 |
-45 |
-22 |
| Różnica względem placebo (średni LS) (95 % CI) |
-52 (-56, -49) |
-33 (-35, -31) |
-47 (-50, -44) |
-43 (-46, -41) |
-26 (-29, -22) |
| * Dzień 540; mediana procentowej zmiany wartości Lp(a). ** Średnia wartość wyjściowa Lp(a), nmol/l. |
|||||
W dniu 510 celowy poziom cholesterolu LPNiskiego (CH-LPN) < 1,8 mmol/l (70 mg/dl) osiągnięto u 84 % pacjentów z chorobą tętnic wieńcowych (CTW) w grupie inklicyranu w porównaniu do 18 % pacjentów w grupie placebo.
Obserwowano ciągłe i statystycznie istotne (p < 0,0001) zmniejszenie procentowej zmiany CH-LPN od wartości wyjściowej do dnia 510 oraz czasowo skorygowanej procentowej zmiany CH-LPN od wartości wyjściowej po dniu 90. do dnia 540 we wszystkich podgrupach niezależnie od wyjściowych danych demograficznych, wyjściowych cech choroby (w tym płci, wieku, wskaźnika masy ciała, rasy pacjenta oraz stosowania statyn), chorób współistniejących i regionu geograficznego.
ORION-11 to międzynarodowe, wieloośrodkowe, podwójnie ślepe, randomizowane, kontrolowane placebo badanie trwające 18 miesięcy, w którym wzięło udział 1617 pacjentów z CTW lub równoważnymi czynnikami ryzyka CTW. Ponad 75 % pacjentów otrzymywało leczenie wspomagające wysokimi dawkami statyn, 87 % pacjentów miało CTW, a 13 % – równoważne czynniki ryzyka CTW.
Średni wiek pacjentów na początku badania wynosił 65 lat (zakres od 20 do 88 lat), 55 % pacjentów miało co najmniej 65 lat, 28 % stanowiły kobiety, 98 % – białych, 1 % – czarnoskórych, 1 % – Azjatów oraz 1 % – pochodzenia hiszpańskiego i latynoamerykańskiego. Średni wyjściowy poziom CH-LPN wynosił 2,7 mmol/l (105 mg/dl). 78 % pacjentów otrzymywało wysokie dawki statyn, 16 % – średnie dawki statyn, 0,4 % – niskie dawki, a 5 % nie stosowało statyn. Najczęściej stosowanymi statynami były atorwastatyna i rozuwastatyna.
Inklicyran istotnie zmniejszał średni procentowy spadek CH-LPN od wartości wyjściowej do dnia 510 w porównaniu do placebo – o 50 % (95% CI -53 %, -47 %; p < 0,0001) (tabela 3).
Inklicyran istotnie zmniejszał również czasowo skorygowany procentowy spadek CH-LPN od wartości wyjściowej po dniu 90. do dnia 540 – o 49 % w porównaniu do placebo (95 % CI -52 %, -48 %; p < 0,0001). Dodatkowe wyniki przedstawiono w tabeli 2.
Tabela 2
Średni procentowy spadek od wartości wyjściowej oraz różnica w porównaniu do placebo dla parametrów lipidowych w dniu 510 w badaniu ORION-11
| Grupa leczenia |
Ch-LPNH |
Cholesterol całkowity |
Nie-Ch-LPHW |
Apo-B |
Lp(a)* |
| Średnia wartość wyjściowa, mg/dl** |
105 |
185 |
136 |
96 |
107 |
| Dzień 510 (średni procentowy spadek od poziomu wyjściowego) |
|||||
| Placebo (n = 807) |
4 |
2 |
2 |
1 |
0 |
| Inklisiran (n = 810) |
-46 |
-28 |
-41 |
-38 |
-19 |
| Różnica w porównaniu z placebo (średnia LS) (95 % CI) |
-50 (-53, -47) |
-30 (-32, -28) |
-43 (-46, -41) |
-39 (-41, -37) |
-19 (-21, -16) |
| * Dzień 540; mediana procentowej zmiany wartości Lp(a). ** Średnia wartość wyjściowa Lp(a), nmol/l. |
|||||
Na 510. dzień celowy poziom cholesterolu LPNL < 1,8 mmol/l (70 mg/dl) osiągnięto u 82% pacjentów z ASCVD w grupie inklisiranu w porównaniu do 16% pacjentów w grupie placebo. U pacjentów z równoważnikiem ryzyka ASCVD celowy poziom cholesterolu LPNL < 2,6 mmol/l (100 mg/dl) osiągnięto u 78% pacjentów w grupie inklisiranu w porównaniu do 31% pacjentów w grupie placebo.
Obserwowano spójne i statystycznie istotne (p < 0,05) zmniejszenie procentowej zmiany cholesterolu LPNL od wartości wyjściowej do 510. dnia oraz skorygowanej względem czasu procentowej zmiany cholesterolu LPNL od wartości wyjściowej po 90. dniu i do 540. dnia u wszystkich podgrup niezależnie od wyjściowych danych demograficznych, wyjściowych cech choroby (w tym płci, wieku, wskaźnika masy ciała, pochodzenia rasowego pacjenta i stosowania statyn), chorób współistniejących oraz regionu geograficznego.
Heterozygotyczna hipercholesterolemia rodzinna
ORION-9 to międzynarodowe, wieloośrodkowe, podwójnie ślepe, randomizowane, kontrolowane placebo 18-miesięczne badanie z udziałem 482 pacjentów z heterozygotyczną hipercholesterolemią rodzinną (HeFH). Wszyscy pacjenci otrzymywali maksymalne dawki statyn tolerowane indywidualnie, z dodatkową terapią modyfikującą lipidy (np. ezetymib) lub bez niej, i wymagali dodatkowego obniżenia cholesterolu LPNL. Rozpoznanie HeFH stawiano na podstawie genotypowania lub kryteriów klinicznych („potwierdzona FH”, stosując kryteria Simon Broome lub WHO/Sieć Dutch Lipid Network).
Pierwotnymi punktami końcowymi była procentowa zmiana cholesterolu LPNL od wartości wyjściowej do 510. dnia w porównaniu do placebo oraz skorygowana względem czasu procentowa zmiana cholesterolu LPNL od wartości wyjściowej po 90. dniu i do 540. dnia w celu oceny całkowitego wpływu na cholesterol LPNL w czasie. Kluczowymi wtórnymi punktami końcowymi były zmiana absolutna cholesterolu LPNL od wartości wyjściowej do 510. dnia, skorygowana względem czasu zmiana absolutna cholesterolu LPNL od wartości wyjściowej po 90. dniu i do 540. dnia oraz procentowa zmiana PCSK9, cholesterolu ogólnego, Apo-B i nie-HDL-C od wartości wyjściowej do 510. dnia. Dodatkowe wtórne punkty końcowe obejmowały indywidualną odpowiedź na inklisiran oraz odsetek pacjentów, którzy osiągnęli ogólne cele lipidowe z uwzględnieniem ich poziomu ryzyka ASCVD.
Średni wiek pacjentów na początku badania wynosił 55 lat (zakres od 21 do 80 lat), 22% pacjentów miało co najmniej 65 lat, 53% stanowiły kobiety, 94% – biała rasa, 3% – czarna rasa, 3% – Azjaci oraz 3% – pochodzenie hiszpańskie i latynoamerykańskie. Średnie wyjściowe stężenie cholesterolu LPNL wynosiło 4,0 mmol/l (153 mg/dl). 74% pacjentów otrzymywało wysokie dawki statyn, 15% – średnie dawki statyn, a 10% – nie stosowało statyn. 52% pacjentów otrzymywało ezetymib. Najczęściej stosowanymi statynami były atorwastatyna i rosuvastatyna.
Inklisiran istotnie zmniejszał średni procentowy spadek cholesterolu LPNL od wartości wyjściowej do 510. dnia w porównaniu do placebo o 48% (95% CI: -54%, -42%; p < 0,0001) (tabela 4).
Inklisiran istotnie zmniejszał również skorygowaną względem czasu procentową zmianę cholesterolu LPNL od wartości wyjściowej po 90. dniu i do 540. dnia o 44% w porównaniu do placebo (95% CI: -48%, -40%; p < 0,0001). Dodatkowe wyniki przedstawiono w tabeli 3.
Tabela 3
Średnia procentowa zmiana od wartości wyjściowej oraz różnica względem placebo dla parametrów lipidowych na 510. dzień w badaniu ORION-9
| Grupa leczenia |
Ch-LPNH |
Cholesterol całkowity |
Nie-Ch-LPHW |
Apo-B |
Lp(a)* |
|
| Średnia wartość wyjściowa, mg/dl** |
153 |
231 |
180 |
124 |
121 |
|
| Dzień 510 (średni procentowy udział zmiany od poziomu wyjściowego) |
||||||
| Placebo (n = 240) |
8 |
7 |
7 |
3 |
4 |
|
| Inklisiran (n = 242) |
-40 |
-25 |
-35 |
-33 |
-13 |
|
| Różnica w porównaniu z placebo (średnie LS) (95% CI) |
-48 (-54, -42) |
-32 (-36, -28) |
-42 (-47, -37) |
-36 (-40, -32) |
-17 (-22, -12) |
|
| * Na 540. dzień; mediana procentowej zmiany wartości Lp(a). ** Średnia wyjściowa wartość Lp(a), nmol/l. |
||||||
Na 510. dzień celowy poziom cholesterolu LPNiskiego (CLDL) < 1,8 mmol/l (70 mg/dl) osiągnięto u 52,5% pacjentów z chorobą sercowo-naczyniową (ASCVD) w grupie leczonej inclisiranem w porównaniu do 1,4% pacjentów z ASCVD w grupie placebo, natomiast w populacji z równoważnym ryzykiem ASCVD celowy poziom CLDL < 2,6 mmol/l (100 mg/dl) osiągnięto u 66,9% pacjentów w grupie leczonej inclisiranem w porównaniu do 8,9% pacjentów w grupie placebo.
Obserwowano spójne i statystycznie istotne (p < 0,05) zmniejszenie procentowej zmiany CLDL od wartości wyjściowej do 510. dnia oraz czasowo skorygowanej procentowej zmiany CLDL od wartości wyjściowej po 90. i do 540. dnia u wszystkich podgrup niezależnie od wyjściowych danych demograficznych, wyjściowych cech choroby (w tym płci, wieku, wskaźnika masy ciała, pochodzenia rasowego pacjenta oraz stosowania statyn), chorób współistniejących i regionu geograficznego.
Dzieci
Europejska Agencja Leków zwolniła producenta z obowiązku przedstawienia wyników badań inclisiranu w jednej lub kilku podgrupach dzieci w leczeniu podwyższonych poziomów cholesterolu (informacje dotyczące stosowania u dzieci znajdują się w sekcji „Sposób stosowania i dawka”).
Farmakokinetyka
Wchłanianie
Po pojedynczym podaniu podskórnie wpływ systemowy inclisiranu zwiększał się w przybliżeniu proporcjonalnie do dawki w zakresie od 24 mg do 756 mg. Przy zalecanym schemacie dawkowania 284 mg stężenie w osoczu osiągało szczyt około 4 godziny po podaniu, ze średnią wartością Cmax wynoszącą 509 ng/ml. Stężenia osiągały poziom poniżej granicy wykrywalności w ciągu 48 godzin po podaniu. Średnia powierzchnia pod krzywą „stężenie w osoczu/czas”, ekstrapolowana do nieskończoności, wynosiła 7980 ng*godz/ml. Wyniki farmakokinetyczne po wielokrotnym podawaniu podskórnym inclisiranu były podobne do tych po dawce pojedynczej.
Rozkład
Inclisiran in vitro wiąże się z białkami osocza w 87% przy odpowiednich stężeniach klinicznych w osoczu. Po pojedynczym podaniu podskórnym 284 mg inclisiranu zdrowym dorosłym średnie pozorne objętości rozkładu wynosiły około 500 l. Na podstawie danych nieklinicznych stwierdzono, że inclisiran jest dobrze wchłaniany i charakteryzuje się wysoką selektywnością wobec wątroby – organu docelowego w obniżaniu poziomu cholesterolu.
Biotransformacja
Inclisiran jest głównie metabolizowany przez nukleazy do krótszych, nieaktywnych nukleotydów różnej długości. Inclisiran nie jest substratem typowych transporterów leków i, mimo że badania in vitro nie były prowadzone, nie przewiduje się, że jest substratem cytochromu P450.
Wydalanie
Okres półtrwania elimi nacji inclisiranu wynosi około 9 godzin; przy wielokrotnym dawkowaniu nie obserwuje się jego akumulacji. 16% dawki inclisiranu wydala się z moczem.
Liniowość/nieliniowość
W badaniu klinicznym fazy I obserwowano przybliżoną proporcjonalność wpływu inclisiranu do dawki po podaniu podskórnym w zakresie od 24 mg do 756 mg. Po wielokrotnym podawaniu podskórnym inclisiranu nie obserwowano akumulacji ani zmian zależnych od czasu.
Związek farmakokinetyka/farmakodynamika
W badaniu klinicznym fazy I nie stwierdzono związku między parametrami farmakokinetycznymi inclisiranu a farmakodynamicznym wpływem na CLDL. Selektywne dostarczanie inclisiranu do hepatocytów, gdzie wchodzi on w skład indukowanego przez RNA kompleksu ciszącego (RISC), powoduje długotrwały efekt, który przekracza oczekiwany na podstawie okresu półtrwania w osoczu wynoszącego 9 godzin. Maksymalny wpływ na obniżenie CLDL obserwowano przy dawce 284 mg, przy czym wyższe dawki nie powodowały większego efektu.
Grupy specjalne pacjentów
Niewydolność nerek
W analizie farmakokinetycznej danych z badania specjalnego u pacjentów z niewydolnością nerek stwierdzono około 2,3-, 2,0- i 3,3-krotne zwiększenie Cmax oraz około 1,6-, 1,8- i 2,3-krotne zwiększenie AUC inclisiranu u pacjentów z niewydolnością nerek lekką (klirens kreatyniny [CrCL] od 60 ml/min do 89 ml/min), umiarkowaną (CrCL od 30 ml/min do 59 ml/min) i ciężką (CrCL od 15 ml/min do 29 ml/min) odpowiednio, w porównaniu z pacjentami z prawidłową funkcją nerek. Mimo wyższego przemijającego narażenia w osoczu przez 48 godzin, obniżenie CLDL było podobne we wszystkich grupach pod względem funkcji nerek. Na podstawie modelowania populacyjnego farmakodynamicznego nie zaleca się korekty dawki u pacjentów z niewydolnością nerek w stadium końcowym. Ze względu na właściwości farmakokinetyczne i farmakodynamiczne oraz ocenę bezpieczeństwa, korekta dawki nie jest wymagana u pacjentów z niewydolnością nerek lekką, umiarkowaną i ciężką. Wpływ hemodializy na farmakokinetykę inclisiranu nie był badany. Ponieważ inclisiran wydala się z moczem, hemodializy nie należy przeprowadzać co najmniej przez 72 godziny po podaniu inclisiranu.
Niewydolność wątroby
W analizie farmakokinetycznej danych z badania specjalnego u pacjentów z niewydolnością wątroby stwierdzono około 1,1- i 2,1-krotne zwiększenie Cmax oraz około 1,3- i 2,0-krotne zwiększenie AUC inclisiranu odpowiednio u pacjentów z niewydolnością wątroby lekką (klasa A wg klasyfikacji Childa-Pagha) i umiarkowaną (klasa B wg klasyfikacji Childa-Pagha) w porównaniu z pacjentami z prawidłową funkcją wątroby. Mimo wyższego przemijającego narażenia w osoczu przez 48 godzin, obniżenie CLDL było podobne w grupach pacjentów z prawidłową funkcją wątroby i lekką niewydolnością wątroby leczonych inclisiranem. U pacjentów z umiarkowaną niewydolnością wątroby wyjściowe poziomy PCSK9 były istotnie niższe, a obniżenie CLDL mniejsze niż u pacjentów z prawidłową funkcją wątroby. Korekta dawki nie jest wymagana u pacjentów z niewydolnością wątroby lekką i umiarkowaną (klasa A i B wg klasyfikacji Childa-Pagha). Nie badano stosowania inclisiranu u pacjentów z ciężką niewydolnością wątroby (klasa C wg klasyfikacji Childa-Pagha).
Inne kategorie specjalne
Analiza populacyjna farmakodynamiczna została przeprowadzona na podstawie danych 4328 pacjentów. Stwierdzono, że wiek, masa ciała, płeć, pochodzenie rasowe pacjenta oraz klirens kreatyniny nie mają istotnego wpływu na farmakodynamikę inclisiranu. Korekta dawki u pacjentów z uwzględnieniem tych danych demograficznych nie jest zalecana.
Charakterystyka kliniczna.
Wskazania.
Lek Sibrava wskazany jest w leczeniu dorosłych pacjentów z pierwotną hipercholesterolemią (heterozygotyczną rodzinną i nieryzyczną) lub z mieszaną dyslipidemią jako uzupełnienie diety:
- w połączeniu ze statyną lub statyną i innymi lekami obniżającymi stężenie lipidów, gdy nie można osiągnąć docelowych poziomów cholesterolu w LPNŻ przy maksymalnej dawce statyny, którą pacjent toleruje, lub
- samodzielnie lub w połączeniu z innymi lekami obniżającymi stężenie lipidów u pacjentów z nietolerancją lub przeciwwskazaniami do stosowania statyn.
Przeciwwskazania.
Podwyższona wrażliwość na substancję czynną lub którykolwiek z substancji pomocniczych.
Interakcje z innymi lekami i inne rodzaje interakcji.
Inklicyran nie jest substancją oddziałującą na typowe przenośniki leków i, choć badania in vitro nie były prowadzone, nie przewiduje się, że jest substancją oddziałującą na cytochrom P450. Inklicyran nie jest inhibitorem ani induktorem enzymów cytochromu P450 ani typowych przenośników leków. Dlatego nie przewiduje się klinicznie istotnych interakcji inklicyranu z innymi lekami. Ze względu na ograniczone dane, nie przewiduje się klinicznie istotnych interakcji z atorwastatyną, rowazastatyną ani innymi statynami.
Особливости stosowania.
Hemodializa
Wpływ hemodializy na farmakokinetykę inklisirany nie był badany. Ze względu na to, że inklisiran jest wydalany z moczem, hemodializy nie należy przeprowadzać przez co najmniej 72 godziny po podaniu inklisirany.
Zawartość sodu
Ten lek zawiera mniej niż 1 mmol sodu (23 mg) na dawkę, co oznacza, że jest uważany za pozbawiony sodu.
Stosowanie w czasie ciąży lub karmienia piersią.
Ciąża
Brak danych lub są one ograniczone co do stosowania inklisirany u ciężarnych. Badania na zwierzętach nie wskazują na szkodliwe działanie bezpośrednie lub pośrednie dotyczące toksyczności reprodukcyjnej. Jako środek ostrożności zaleca się unikanie stosowania inklisirany w czasie ciąży.
Karmienie piersią
Nie wiadomo, czy inklisiran przenika do mleka matki. Dane farmakodynamiczne/toksykologiczne uzyskane u zwierząt wskazują na wydzielanie inklisirany do mleka. Nie można wykluczyć ryzyka dla noworodków/niemowląt.
Decyzję o zaprzestaniu karmienia piersią lub o zaprzestaniu/zawstrzymaniu terapii inklisiranem należy podjąć, biorąc pod uwagę korzyści z karmienia piersią dla dziecka oraz korzyści z leczenia dla kobiety.
Funkcja rozrodcza
Brak danych dotyczących wpływu inklisirany na funkcję rozrodczą człowieka. Badania na zwierzętach nie wykazały wpływu na płodność.
Wpływ na zdolność prowadzenia pojazdów i obsługiwanie maszyn.
Inklisiran nie wpływa lub wpływa nieznacznie na szybkość reakcji podczas prowadzenia pojazdów lub pracy z innymi maszynami.
Sposób stosowania i dawki
Dawkowanie
Zalecana dawka wynosi 284 mg inklisiranu, podawanego w formie pojedynczej iniekcji podskórnej na początku leczenia, następnie po 3 miesiącach, a potem co 6 miesięcy.
Pominięcie dawki
Jeśli podanie dawki zostało pominięte i od zaplanowanego terminu minęło mniej niż 3 miesiące, inklisirana należy podać i kontynuować leczenie zgodnie z pierwotnym harmonogramem pacjenta.
Jeśli od zaplanowanego terminu podania leku minęło więcej niż 3 miesiące, należy rozpocząć nowy harmonogram leczenia: inklisirana należy podać na początku, następnie po 3 miesiącach, a potem co 6 miesięcy.
Przejście z leczenia inhibitorami monoklonalnych przeciwciał przeciwko PCSK9
Inklisirana można podawać bezpośrednio po ostatniej dawce inhibitora monoklonalnych przeciwciał przeciwko PCSK9. W celu utrzymania obniżonego poziomu cholesterolu LPNL zaleca się podawanie inklisiranu w ciągu 2 tygodni od ostatniej dawki inhibitora monoklonalnych przeciwciał przeciwko PCSK9.
Osoby z grup szczególnych
Pacjenci w wieku powyżej 65 lat
U pacjentów w wieku powyżej 65 lat nie jest wymagana korekta dawki.
Niewydolność wątroby
U pacjentów z łagodną (klasa A wg klasyfikacji Childa-Pugh) lub umiarkowaną (klasa B wg klasyfikacji Childa-Pugh) niewydolnością wątroby nie jest wymagana korekta dawki. Brak danych dotyczących pacjentów z ciężką niewydolnością wątroby (klasa C wg klasyfikacji Childa-Pugh). Inklisirana należy stosować z ostrożnością u pacjentów z ciężką niewydolnością wątroby.
Niewydolność nerek
U pacjentów z łagodną, umiarkowaną lub ciężką niewydolnością nerek oraz z nerek w stadium końcowym nie jest wymagana korekta dawki. Doświadczenie w stosowaniu inklisiranu u pacjentów z ciężką niewydolnością nerek jest ograniczone. U tych pacjentów inklisirana należy stosować z ostrożnością. Informacje dotyczące środków ostrożności podczas hemodializy znajdują się w sekcji „Szczególne wskazania dotyczące stosowania”.
Sposób podania
Lek przeznaczony jest do podania podskórnego.
Inklisirana należy podawać podskórnie w okolicy brzucha; alternatywnymi miejscami iniekcji są ramię lub uda. Nie należy wykonywać zastrzyków w obszary skóry dotknięte chorobą lub urazem, takie jak oparzenia słoneczne, wysypki, stan zapalny lub infekcje skóry.
Każdą dawkę 284 mg podaje się za pomocą jednej jednorazowej strzykawki wstępnie napełnionej. Każda wstępnie napełniona strzykawka przeznaczona jest wyłącznie do jednorazowego użytku.
Inklisirana powinien podawać personel medyczny.
Dzieci
Bezpieczeństwo i skuteczność stosowania inklisiranu u dzieci (do 18 roku życia) nie zostały ustalone.
Brak danych.
Przedawkowanie
Nie zaobserwowano żadnych klinicznie istotnych działań niepożądanych u zdrowych ochotników, którzy otrzymywali inklisirana w dawkach przekraczających dawkę terapeutyczną. Nie ma specjalnego przeciwdziałwa na przedawkowanie inklisiranem. W przypadku przedawkowania stosuje się leczenie objawowe oraz, w razie potrzeby, środki wspomagające.
Niepożądane działania.
Streszczenie profilu bezpieczeństwa
Jedynymi niepożądanymi działaniami związanymi z zastosowaniem inklisiranu były niepożądane reakcje w miejscu wstrzyknięcia (8,2%).
Wykaz niepożądanych działań
Niepożądane działania wymieniono według głównych klas narządów i układów (tabela 4). Częstość określono następująco: bardzo często (≥ 1/10); często (≥ 1/100 – < 1/10); rzadko (≥ 1/1000 – < 1/100); bardzo rzadko (≥ 1/10000 – < 1/1000); nieznane (nie można oszacować na podstawie dostępnych danych).
Tabela 4
Niepożądane działania występujące u pacjentów otrzymujących inklisiran
| Klasa układów narządów |
Reakcja niepożądana |
Kategoria częstości |
|
| Zaburzenia ogólne i reakcje w miejscu wstrzyknięcia |
Reakcje niepożądane w miejscu wstrzyknięcia1 |
Często |
|
| 1 Zob. sekcję „Opis niektórych reakcji niepożądanych” |
|||
Opis niektórych działań niepożądanych
Działania niepożądane w miejscu wstrzyknięcia
W trakcie badań podstawowych działania niepożądane w miejscu wstrzyknięcia występowały u 8,2 % i 1,8 % pacjentów odpowiednio w grupie leczonej inklisiranem i w grupie placebo. Odsetek pacjentów w każdej grupie, którzy przerwali leczenie z powodu działań niepożądanych w miejscu wstrzyknięcia, wynosił odpowiednio 0,2 % i 0,0 %. Wszystkie te działania niepożądane miały łagodny lub umiarkowany stopień ciężkości, były przejściowe i ustępowały bez konsekwencji. Najczęstsze działania niepożądane w miejscu wstrzyknięcia u pacjentów otrzymujących inklisiran to były reakcje w miejscu wstrzyknięcia (3,1 %), ból w miejscu wstrzyknięcia (2,2 %), zaczerwienienie w miejscu wstrzyknięcia (1,6 %) oraz wysypka w miejscu wstrzyknięcia (0,7 %).
Specjalne kategorie pacjentów
Pacjenci w podeszłym wieku
Spośród 1833 pacjentów, którzy otrzymywali inklisiran w trakcie badań podstawowych, 981 (54 %) miało co najmniej 65 lat, a 239 (13 %) – co najmniej 75 lat. Nie zaobserwowano ogólnych różnic w zakresie bezpieczeństwa między tymi pacjentami a pacjentami młodszej grupy wiekowej.
Immunogenność
W trakcie badań testy na obecność przeciwciał do leku przeprowadzono u 1830 pacjentów. Potwierdzona pozytywność stwierdzona była u 1,8 % (33/1830) pacjentów przed podaniem dawki oraz u 4,9 % (90/1830) pacjentów w ciągu 18 miesięcy leczenia inklisiranem. Nie zaobserwowano klinicznie istotnych różnic w profilach skuteczności klinicznej, bezpieczeństwa i farmakodynamiki inklisiranu u pacjentów z pozytywnym wynikiem testu na obecność przeciwciał przeciwko inklisiranowi.
Wskazania laboratoryjne
W trakcie badań klinicznych fazy III częściej obserwowano podwyższenia stężenia transaminaz w surowicy od > 1 × górna granica normy (GGN) do ≤ 3 × GGN u pacjentów otrzymujących inklisiran (ALT 19,7 %; AST 17,2 %) w porównaniu z pacjentami otrzymującymi placebo (ALT 13,6 %; AST 11,1 %). Te podwyższenia nie przekraczały klinicznie istotnego progu 3 × GGN, przebiegały bezobjawowo i nie były związane z działaniami niepożądanymi ani innymi objawami zaburzeń funkcji wątroby.
Zgłaszanie podejrzeń działań niepożądanych
Zgłaszanie działań niepożądanych po rejestracji produktu leczniczego ma istotne znaczenie. Umożliwia monitorowanie stosunku korzyści do ryzyka w przypadku stosowania tego produktu leczniczego. Pracownicy medyczni i farmaceutyczni, a także pacjenci lub ich ustawowo upoważnieni przedstawiciele, powinni zgłaszać wszystkie przypadki podejrzewanych działań niepożądanych oraz braku skuteczności leku za pośrednictwem zautomatyzowanego systemu informacyjnego nadzoru farmakologicznego pod adresem: https://aisf.dec.gov.ua/
Okres ważności.
3 lata.
Warunki przechowywania.
Produkt leczniczy nie wymaga szczególnych warunków przechowywania. Nie mrozić. Przechowywać w miejscu niedostępnym dla dzieci.
Opakowanie.
Po 1,5 ml roztworu w strzykawce wstępnie napełnionej; po 1 strzykawce wstępnie napełnionej w blisterze; po 1 blisterze w pudełku kartonowym.
Kategoria wydania. Na receptę.
Producent.
Novartis Pharmaceuticals Manufacturing GmbH, Austria.
Adres producenta i miejsce prowadzenia działalności.
Biochemiestrasse 10, Langkampfen, 6336, Austria.