Sibavra
Ucraina
Indice
ISTRUZIONE per l'uso medicinale del medicinale SIBRAVA (SYBRAVA)
Composizione:
Principio attivo: inclisiran;
1 siringa preriempita contiene sodio inclisiran corrispondente a 284 mg di inclisiran, in 1,5 ml di soluzione;
1 ml di soluzione contiene sodio inclisiran corrispondente a 189 mg di inclisiran;
Eccipienti: acqua per preparazioni iniettabili, idrossido di sodio, acido fosforico concentrato.
Forma farmaceutica. Soluzione iniettabile in siringa preriempita.
Principali caratteristiche fisico-chimiche: soluzione limpida, incolore o leggermente giallastra, praticamente priva di particelle.
Gruppo farmacoterapeutico.
Farmaci che agiscono sul sistema cardiovascolare. Farmaci ipolipemizzanti. Farmaci ipolipemizzanti monocomponente. Altri farmaci ipolipemizzanti.
Codice ATC C10A X16.
Proprietà farmacologiche
Mecanismo d'azione
Inclisiran è un acido ribonucleico interferente a doppio filamento (siRNA) coniugato, sulla catena codificante, con un trisaccaride di N-acetilgalattosamina (GalNAc) per facilitare l'assorbimento dagli epatociti. Negli epatociti, inclisiran sfrutta il meccanismo di interferenza dell'RNA (RNAi) e indirizza la degradazione catalitica dell'mRNA della proproteina convertasi subtilisina/chymotripsina tipo 9 (PCSK9). Ciò aumenta il ricircolo e l'espressione dei recettori per il colesterolo LDL (lipoproteine a bassa densità) sulla superficie delle cellule epatiche, incrementando l'assorbimento delle LDL-C e riducendone il livello circolante.
Farmacodinamica
Dopo una singola somministrazione sottocutanea di 284 mg di inclisiran, la riduzione delle LDL-C è risultata evidente entro 14 giorni. Una riduzione media delle LDL-C del 48–51% è stata osservata tra i 30 e i 60 giorni successivi alla somministrazione. Al giorno 180, il livello delle LDL-C risultava ancora ridotto di circa il 53%.
Efficacia clinica e sicurezza
Nei trial clinici e in alcune pubblicazioni, la dose di 284 mg di inclisiran è considerata equivalente e indicata come 300 mg di sale sodico di inclisiran.
L'efficacia di inclisiran è stata valutata in tre studi di fase III su pazienti con malattie cardiovascolari aterosclerotiche (ASCVD) (malattia coronarica, malattia cerebrovascolare o malattia arteriosa periferica), equivalenti di rischio ASCVD (diabete mellito di tipo 2, ipercolesterolemia familiare o rischio a 10 anni di eventi cardiovascolari ≥20%, calcolato con il punteggio di rischio Framingham o scale equivalenti) e/o ipercolesterolemia familiare (FH). I pazienti assumevano la dose massima tollerata di statina, con o senza ulteriore terapia modificatrice dei lipidi, e necessitavano di un’ulteriore riduzione delle LDL-C (pazienti nei quali non erano stati raggiunti gli obiettivi terapeutici). Circa il 17% dei pazienti presentava intolleranza alle statine. Ai pazienti è stata somministrata una iniezione sottocutanea di 284 mg di inclisiran o placebo ai giorni 1, 90, 270 e 450. Il follow-up è stato effettuato fino al giorno 540.
L’impatto di inclisiran sulla morbilità e mortalità cardiovascolare non è stato determinato.
In un’analisi pooled di fase III, inclisiran somministrato per via sottocutanea ha ridotto le LDL-C del 50–55% già al giorno 90 (fig. 1), mantenendo questo effetto durante la terapia prolungata. La massima riduzione delle LDL-C è stata raggiunta al giorno 150, dopo la seconda somministrazione. Aumenti modesti ma statisticamente significativi delle LDL-C fino al 65% sono stati associati a livelli basali più bassi di LDL-C (circa < 2 mmol/l [77 mg/dl]), livelli basali più elevati di PCSK9, dosi più alte di statina e maggiore intensità del trattamento con statine.
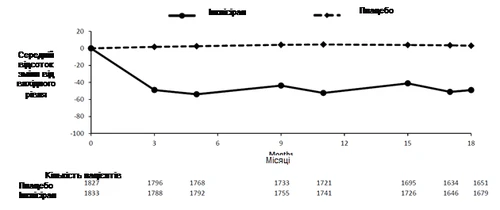
Fig. 1. Percentuale media di variazione rispetto ai livelli basali di LDL-C in pazienti con ipercolesterolemia primaria e dislipidemia mista trattati con inclisiran rispetto a quelli trattati con placebo (analisi pooled).
ASCVD ed equivalenti di rischio ASCVD
Sono stati condotti due studi su pazienti con ASCVD ed equivalenti di rischio ASCVD (ORION-10 e ORION-11). I pazienti assumevano la dose massima tollerata di statina, con o senza ulteriore terapia modificatrice dei lipidi (ad esempio ezetimiba), e necessitavano di un’ulteriore riduzione delle LDL-C. Poiché la riduzione delle LDL-C dovrebbe migliorare gli esiti cardiovascolari, gli endpoint primari co-primari in ciascuno studio erano: la percentuale di variazione delle LDL-C rispetto ai livelli basali al giorno 510 rispetto al placebo e la percentuale di variazione delle LDL-C aggiustata per il tempo dal basale dopo il giorno 90 fino al giorno 540, per valutare l’effetto cumulativo nel tempo sulle LDL-C.
ORION-10 è uno studio multicentrico, randomizzato, in doppio cieco, controllato con placebo, della durata di 18 mesi, che ha coinvolto 1561 pazienti con ASCVD.
L’età media dei pazienti al basale era di 66 anni (range da 35 a 90 anni), il 60% dei pazienti aveva un’età ≥65 anni, il 31% erano donne, l’86% caucasici, il 13% afrodiscendenti, l’1% asiatici e il 14% di origine ispanica o latinoamericana. Il livello medio basale di LDL-C era di 2,7 mmol/l (105 mg/dl). Il 69% dei pazienti assumeva statine ad alta intensità, il 19% a intensità media, l’1% a bassa intensità e l’11% non assumeva statine. Le statine più comunemente utilizzate erano atorvastatina e rosuvastatina.
Inclisiran ha ridotto in modo significativo la percentuale media di variazione delle LDL-C rispetto al basale al giorno 510 rispetto al placebo, con una riduzione del 52% (IC 95%: -56%, -49%; p < 0,0001) (tabella 2).
Inclisiran ha ridotto in modo significativo anche la percentuale di variazione delle LDL-C aggiustata per il tempo dal basale dopo il giorno 90 fino al giorno 540, con una riduzione del 54% rispetto al placebo (IC 95%: -56%, -51%; p < 0,0001). Per ulteriori risultati, vedere tabella 1.
Tabella 1
Percentuale media di variazione rispetto ai livelli basali e differenza rispetto al placebo nei parametri lipidici al giorno 510 nello studio ORION-10
| Gruppo terapeutico |
XC-LPDG |
Colesterolo totale |
Non XC-LPVG |
Apo-B |
Lp(a)* |
| Valore medio basale, mg/dl** |
105 |
181 |
134 |
94 |
122 |
| Giorno 510 (percentuale media di cambiamento rispetto al valore basale) |
|||||
| Placebo (n = 780) |
1 |
0 |
0 |
-2 |
4 |
| Inclisiran (n = 781) |
-51 |
-34 |
-47 |
-45 |
-22 |
| Differenza rispetto al placebo (media LS) (IC 95%) |
-52 (-56, -49) |
-33 (-35, -31) |
-47 (-50, -44) |
-43 (-46, -41) |
-26 (-29, -22) |
| * Al giorno 540; mediana della percentuale di cambiamento dei valori di Lp(a). ** Valore medio basale di Lp(a), nmol/l. |
|||||
Alla giornata 510, l'obiettivo terapeutico di C-LDL < 1,8 mmol/l (70 mg/dl) è stato raggiunto nell'84 % dei pazienti con ASCVD nel gruppo inclisiran rispetto all'18 % dei pazienti nel gruppo placebo.
Una riduzione progressiva e statisticamente significativa (p < 0,0001) della percentuale di variazione del C-LDL rispetto al basale fino al giorno 510 e della percentuale di variazione del C-LDL corretta per il tempo rispetto al basale dopo il giorno 90 fino al giorno 540 è stata osservata in tutti i sottogruppi, indipendentemente dalle caratteristiche demografiche basali, dalle caratteristiche della malattia al basale (inclusi sesso, età, indice di massa corporea, razza del paziente e uso di statine), dalle comorbidità e dalla regione geografica.
ORION-11 è uno studio internazionale, multicentrico, in doppio cieco, randomizzato, controllato con placebo, della durata di 18 mesi, condotto su 1617 pazienti con ASCVD o equivalenti di rischio ASCVD. Oltre il 75 % dei pazienti assumeva un trattamento di base con statine ad alto dosaggio, l'87 % aveva ASCVD e il 13 % presentava equivalenti di rischio ASCVD.
L'età media dei pazienti al basale era di 65 anni (range da 20 a 88 anni), il 55 % dei pazienti aveva un'età ≥ 65 anni, il 28 % erano donne, il 98 % caucasici, l'1 % afrodiscendenti, l'1 % asiatici e l'1 % di origine ispanica e latinoamericana. Il livello medio basale di C-LDL era di 2,7 mmol/l (105 mg/dl). Il 78 % dei pazienti assumeva statine ad alto dosaggio, il 16 % dosaggi medi, lo 0,4 % dosaggi bassi e il 5 % non assumeva statine. Le statine più comunemente utilizzate erano atorvastatina e rosuvastatina.
Inclisiran ha ridotto in modo significativo la percentuale media di variazione del C-LDL rispetto al basale fino al giorno 510 rispetto al placebo, del 50 % (IC 95 % -53 %, -47 %; p < 0,0001) (tabella 3).
Inclisiran ha ridotto in modo significativo anche la percentuale di variazione del C-LDL corretta per il tempo rispetto al basale dopo il giorno 90 fino al giorno 540, del 49 % rispetto al placebo (IC 95 % -52 %, -48 %; p < 0,0001). Per ulteriori risultati, vedere tabella 2.
Tabella 2
Percentuale media di variazione rispetto al basale e differenza rispetto al placebo nei parametri lipidici al giorno 510 nello studio ORION-11
| Gruppo di trattamento |
Col-CVLP |
Colesterolo totale |
Non Col-CVLP |
Apo-B |
Lp(a)* |
| Valore medio basale, mg/dL** |
105 |
185 |
136 |
96 |
107 |
| Giorno 510 (percentuale media di variazione rispetto al valore basale) |
|||||
| Placebo (n = 807) |
4 |
2 |
2 |
1 |
0 |
| Inclisiran (n = 810) |
-46 |
-28 |
-41 |
-38 |
-19 |
| Differenza rispetto al placebo (media LS) (IC 95%) |
-50 (-53, -47) |
-30 (-32, -28) |
-43 (-46, -41) |
-39 (-41, -37) |
-19 (-21, -16) |
| * Al giorno 540; mediana della percentuale di variazione dei valori di Lp(a). ** Valore medio basale di Lp(a), nmol/L. |
|||||
Al giorno 510, l'obiettivo di colesterolo LDL < 1,8 mmol/l (70 mg/dl) è stato raggiunto nell'82% dei pazienti con malattia cardiovascolare (MCV) nel gruppo trattato con inclisiran, rispetto al 16% dei pazienti nel gruppo placebo. Nei pazienti con rischio cardiovascolare equivalente, l'obiettivo di colesterolo LDL < 2,6 mmol/l (100 mg/dl) è stato raggiunto nel 78% dei pazienti nel gruppo inclisiran rispetto al 31% nel gruppo placebo.
Una riduzione progressiva e statisticamente significativa (p < 0,05) della percentuale di variazione del colesterolo LDL rispetto al basale fino al giorno 510 e della percentuale di variazione corretta per il tempo rispetto al basale tra il giorno 90 e il giorno 540 è stata osservata in tutte le sottopopolazioni, indipendentemente dalle caratteristiche demografiche basali, dalle caratteristiche della malattia basali (inclusi sesso, età, indice di massa corporea, razza del paziente e uso di statine), dalle comorbidità e dalla regione geografica.
Ipercolesterolemia familiare eterozigote
ORION-9 è uno studio internazionale, multicentrico, in doppio cieco, randomizzato, controllato con placebo, della durata di 18 mesi, che ha coinvolto 482 pazienti con ipercolesterolemia familiare eterozigote (IFH). Tutti i pazienti assumevano la dose massima tollerata di statina, con o senza ulteriore terapia lipidomodulante (ad esempio ezetimibe), e necessitavano di un ulteriore abbassamento del colesterolo LDL. La diagnosi di IFH è stata stabilita sulla base del genotipizzazione o di criteri clinici ("FH confermata", utilizzando i criteri di Simon Broome o dell'OMS/rete Dutch Lipid Network).
Gli endpoint primari secondari erano la percentuale di riduzione del colesterolo LDL rispetto al basale al giorno 510 rispetto al placebo e la percentuale di riduzione corretta per il tempo del colesterolo LDL rispetto al basale dal giorno 90 al giorno 540, per valutare l'effetto cumulativo nel tempo sul colesterolo LDL. Gli endpoint secondari principali includevano la variazione assoluta del colesterolo LDL dal basale al giorno 510, la variazione assoluta corretta per il tempo del colesterolo LDL dal basale dal giorno 90 al giorno 540 e la percentuale di variazione di PCSK9, colesterolo totale, Apo-B e colesterolo non-HDL rispetto al basale al giorno 510. Gli endpoint secondari aggiuntivi includevano la risposta individuale all'inclisiran e la quota di pazienti che hanno raggiunto i livelli lipidici obiettivo raccomandati in base al loro livello di rischio cardiovascolare.
L'età media dei pazienti al basale era di 55 anni (intervallo da 21 a 80 anni), il 22% dei pazienti aveva ≥ 65 anni, il 53% erano donne, il 94% erano di razza bianca, il 3% afrodiscendenti, il 3% asiatici e il 3% di origine ispanica e latinoamericana. Il valore medio basale di colesterolo LDL era di 4,0 mmol/l (153 mg/dl). Il 74% dei pazienti assumeva dosi elevate di statine, il 15% dosi medie di statine e il 10% non assumeva statine. Il 52% dei pazienti assumeva ezetimibe. Le statine più comunemente utilizzate erano atorvastatina e rosuvastatina.
L'inclisiran ha ridotto in modo significativo la percentuale media di variazione del colesterolo LDL rispetto al basale al giorno 510 rispetto al placebo del 48% (IC 95%: -54%, -42%; p < 0,0001) (tabella 4).
L'inclisiran ha ridotto in modo significativo anche la percentuale di variazione corretta per il tempo del colesterolo LDL rispetto al basale dal giorno 90 al giorno 540 del 44% rispetto al placebo (IC 95%: -48%, -40%; p < 0,0001). Per ulteriori risultati, vedere tabella 3.
Tabella 3
Percentuale media di variazione rispetto al basale e differenza rispetto al placebo dei parametri lipidici al giorno 510 nello studio ORION-9
| Gruppo terapeutico |
LDL-C |
Colesterolo totale |
non-HDL-C |
Apo-B |
Lp(a)* |
|
| Valore medio basale, mg/dl** |
153 |
231 |
180 |
124 |
121 |
|
| Giorno 510 (percentuale media di variazione rispetto al valore basale) |
||||||
| Placebo (n = 240) |
8 |
7 |
7 |
3 |
4 |
|
| Inclisiran (n = 242) |
-40 |
-25 |
-35 |
-33 |
-13 |
|
| Differenza rispetto al placebo (media LS) (IC 95%) |
-48 (-54, -42) |
-32 (-36, -28) |
-42 (-47, -37) |
-36 (-40, -32) |
-17 (-22, -12) |
|
| * Al giorno 540; mediana della percentuale di variazione dei valori di Lp(a). ** Valore medio basale di Lp(a), nmol/l. |
||||||
Al giorno 510, l'obiettivo di colesterolo LDL < 1,8 mmol/l (70 mg/dl) è stato raggiunto nel 52,5% dei pazienti con ASCVD nel gruppo trattato con inclisiran rispetto all'1,4% dei pazienti con ASCVD nel gruppo placebo, mentre nella popolazione con rischio equivalente di ASCVD l'obiettivo di colesterolo LDL < 2,6 mmol/l (100 mg/dl) è stato raggiunto nel 66,9% dei pazienti nel gruppo inclisiran rispetto all'8,9% dei pazienti nel gruppo placebo.
Una riduzione progressiva e statisticamente significativa (p < 0,05) della percentuale di variazione del colesterolo LDL dal valore basale fino al giorno 510 e della percentuale di variazione corretta per il tempo dal valore basale dopo il giorno 90 fino al giorno 540 è stata osservata in tutti i sottogruppi, indipendentemente dalle caratteristiche demografiche basali, dalle caratteristiche della malattia basali (inclusi sesso, età, indice di massa corporea, razza del paziente e uso di statine), dalle comorbidità e dalla regione geografica.
Popolazione pediatrica
L’Agenzia europea per i medicinali ha concesso una deroga dall'obbligo di presentare i risultati degli studi con inclisiran in una o più sottopopolazioni pediatriche per il trattamento dell'aumento dei livelli di colesterolo (per informazioni sull'uso in età pediatrica, vedere la sezione «Modalità di somministrazione e posologia»).
Farmacocinetica
Assorbimento
Dopo somministrazione sottocutanea singola, l'esposizione sistemica ad inclisiran aumentava in modo approssimativamente proporzionale alla dose nell'intervallo da 24 mg a 756 mg. Con il regime posologico raccomandato di 284 mg, la concentrazione plasmatica raggiungeva un picco circa 4 ore dopo la somministrazione, con un valore medio di Cmax di 509 ng/ml. Le concentrazioni scendevano al di sotto della soglia di rilevabilità entro 48 ore dalla somministrazione. L'area media sotto la curva concentrazione plasmatica-tempo, estrapolata all'infinito, era di 7980 ng*h/ml. I risultati farmacocinetici dopo somministrazione sottocutanea ripetuta di inclisiran erano simili a quelli osservati dopo dose singola.
Distribuzione
In vitro, inclisiran è legato alle proteine plasmatiche per l'87% alle concentrazioni cliniche appropriate. Dopo somministrazione sottocutanea singola di 284 mg di inclisiran in adulti sani, il volume apparente di distribuzione è di circa 500 l. Sulla base di dati preclinici, inclisiran viene ben assorbito e presenta un'elevata selettività per il fegato, l'organo bersaglio per la riduzione del colesterolo.
Biotrasformazione
Inclisiran è principalmente metabolizzato da nucleasi in nucleotidi inattivi più corti di varia lunghezza. Inclisiran non è substrato dei comuni trasportatori di farmaci e, sebbene non siano stati condotti studi in vitro, non si prevede che sia un substrato del citocromo P450.
Eliminazione
Il tempo di emivita terminale di inclisiran è di circa 9 ore; non si verifica accumulo con somministrazione ripetuta. Il 16% della dose di inclisiran viene eliminato nelle urine.
Linearità/non linearità
In uno studio clinico di Fase I, è stato osservato un aumento approssimativamente proporzionale alla dose dell'esposizione ad inclisiran dopo somministrazione sottocutanea di dosi nell'intervallo da 24 mg a 756 mg. Dopo somministrazione sottocutanea ripetuta di inclisiran, non è stato osservato accumulo né variazioni dipendenti dal tempo.
Rapporto farmacocinetica/farmacodinamica
In uno studio clinico di Fase I, non è stata osservata correlazione tra i parametri farmacocinetici di inclisiran e l'effetto farmacodinamico sul colesterolo LDL. La somministrazione selettiva di inclisiran agli epatociti, dove entra nel complesso di silenziamento mediato da RNA (RISC), determina un effetto prolungato che supera quello atteso in base al tempo di emivita plasmatico di 9 ore. L'effetto massimo sulla riduzione del colesterolo LDL è stato osservato con la dose di 284 mg, mentre dosi superiori non hanno determinato un effetto maggiore.
Gruppi particolari di pazienti
Insufficienza renale
Nell’analisi farmacocinetica di uno studio specifico in pazienti con insufficienza renale, è stato riportato un aumento della Cmax di inclisiran di circa 2,3, 2,0 e 3,3 volte e un aumento dell'AUC di circa 1,6, 1,8 e 2,3 volte rispettivamente in pazienti con insufficienza renale lieve (clearance della creatinina [CrCL] da 60 ml/min a 89 ml/min), moderata (CrCL da 30 ml/min a 59 ml/min) e grave (CrCL da 15 ml/min a 29 ml/min), rispetto ai pazienti con funzionalità renale normale. Nonostante una maggiore esposizione plasmatica transitoria entro 48 ore, la riduzione del colesterolo LDL è stata simile in tutti i gruppi per funzionalità renale. Sulla base di una modellizzazione farmacodinamica di popolazione, non è raccomandata alcuna modifica posologica per pazienti con insufficienza renale allo stadio terminale. Sulla base delle proprietà farmacocinetiche e farmacodinamiche e della valutazione della sicurezza, non è necessaria alcuna modifica posologica per pazienti con insufficienza renale lieve, moderata o grave. L’impatto dell’emodialisi sulla farmacocinetica di inclisiran non è stato studiato. Poiché inclisiran viene eliminato dai reni, l’emodialisi non deve essere effettuata almeno entro 72 ore dalla somministrazione di inclisiran.
Insufficienza epatica
Nell’analisi farmacocinetica di uno studio specifico in pazienti con insufficienza epatica, è stato riportato un aumento della Cmax di inclisiran di circa 1,1 e 2,1 volte e un aumento dell’AUC di circa 1,3 e 2,0 volte rispettivamente in pazienti con insufficienza epatica lieve (classe A secondo la classificazione di Child-Pugh) e moderata (classe B secondo la classificazione di Child-Pugh), rispetto ai pazienti con funzionalità epatica normale. Nonostante una maggiore esposizione plasmatica transitoria entro 48 ore, la riduzione del colesterolo LDL è stata simile nei gruppi di pazienti con funzionalità epatica normale e con insufficienza epatica lieve trattati con inclisiran. Nei pazienti con insufficienza epatica moderata, i livelli basali di PCSK9 erano notevolmente più bassi e la riduzione del colesterolo LDL minore rispetto ai pazienti con funzionalità epatica normale. Non è necessaria alcuna modifica posologica per pazienti con insufficienza epatica lieve o moderata (classe A e B secondo la classificazione di Child-Pugh). L’uso di inclisiran in pazienti con insufficienza epatica grave (classe C secondo la classificazione di Child-Pugh) non è stato studiato.
Altre categorie particolari
È stato effettuato un’analisi farmacodinamica di popolazione basata sui dati di 4328 pazienti. È stato stabilito che età, peso corporeo, sesso, razza del paziente e clearance della creatinina non influenzano in modo significativo la farmacodinamica di inclisiran. Non è raccomandata alcuna modifica posologica in base a questi parametri demografici.
Caratteristiche cliniche.
Indicazioni.
Il medicinale Sibavra è indicato per il trattamento di adulti con ipercolesterolemia primaria (ipercolesterolemia familiare eterozigote e non familiare) o dislipidemia mista, come integrazione alla dieta:
- in associazione con una statina o una statina e altri agenti ipolipemizzanti quando il livello target di colesterolo-LDL non può essere raggiunto con la dose massima tollerata di statina, oppure
- da solo o in associazione con altri agenti ipolipemizzanti nel caso di intolleranza o controindicazioni alle statine.
Controindicazioni.
Ipersensibilità al principio attivo o a uno qualsiasi degli eccipienti.
Interazioni con altri medicinali e altre forme di interazione.
Inclisiran non è un substrato dei comuni trasportatori di farmaci e, sebbene non siano stati effettuati studi in vitro, non si prevede che sia un substrato del citocromo P450. Inclisiran non è un inibitore né un induttore degli enzimi del citocromo P450 né dei comuni trasportatori di farmaci. Pertanto, non sono previste interazioni clinicamente rilevanti tra inclisiran e altri medicinali. Alla luce dei dati limitati, non sono previste interazioni clinicamente rilevanti con atorvastatina, rosuvastatina o altre statine.
Caratteristiche di impiego.
Emodialisi
L'effetto dell'emodialisi sulla farmacocinetica di inclisiran non è stato studiato. Poiché inclisiran viene escreto dai reni, l'emodialisi non deve essere effettuata per almeno 72 ore dopo la somministrazione di inclisiran.
Contenuto di sodio
Questo medicinale contiene meno di 1 mmol di sodio (23 mg) per dose, ovvero è considerato praticamente privo di sodio.
Uso durante la gravidanza o l’allattamento.
Gravidanza
I dati sull'uso di inclisiran durante la gravidanza sono assenti o limitati. Gli studi sugli animali non indicano effetti tossici riproduttivi diretti o indiretti. Come misura precauzionale, si raccomanda di evitare l'uso di inclisiran durante la gravidanza.
Allattamento
Non è noto se inclisiran passi nel latte materno. I dati farmacodinamici/toxicologici disponibili sugli animali hanno mostrato l'escrezione di inclisiran nel latte. Il rischio per neonati/lattanti non può essere escluso.
La decisione di interrompere l’allattamento al seno o di interrompere/astenersi dal trattamento con inclisiran deve essere presa tenendo in considerazione il beneficio dell’allattamento per il neonato e il beneficio della terapia per la donna.
Funzione riproduttiva
Non sono disponibili dati sull'effetto di inclisiran sulla funzione riproduttiva nell'uomo. Gli studi sugli animali non hanno evidenziato alcun effetto sulla fertilità.
Capacità di influire sulla velocità di reazione nel guidare o nell’uso di macchinari.
Inclisiran non ha alcun effetto oppure ha un effetto trascurabile sulla velocità di reazione nel guidare veicoli o nell’uso di macchinari.
Modalità e posologia di somministrazione.
Posologia
La dose raccomandata è di 284 mg di inclisiran, somministrata come iniezione sottocutanea singola all'inizio del trattamento, poi dopo 3 mesi e successivamente ogni 6 mesi.
Salto della dose
Se la somministrazione della dose viene saltata e dal momento previsto sono trascorsi meno di 3 mesi, inclisiran deve essere somministrato e il trattamento deve proseguire secondo il programma iniziale del paziente.
Se dal momento previsto per la somministrazione del medicinale sono trascorsi più di 3 mesi, è necessario avviare un nuovo schema terapeutico: inclisiran deve essere somministrato all'inizio, poi dopo 3 mesi e successivamente ogni 6 mesi.
Passaggio dal trattamento con inibitori monoclonali anticorpi anti-PCSK9
Inclisiran può essere somministrato immediatamente dopo l'ultima dose di inibitore monoclonale anticorpi anti-PCSK9. Per mantenere la riduzione dei livelli di colesterolo LDL, si raccomanda di somministrare inclisiran entro 2 settimane dall'ultima dose di inibitore monoclonale anticorpi anti-PCSK9.
Categorie speciali di pazienti
Pazienti anziani (≥ 65 anni)
Non è necessaria alcuna modifica della dose nei pazienti anziani.
Insufficienza epatica
Non è necessaria alcuna modifica della dose nei pazienti con insufficienza epatica lieve (classe A secondo la classificazione di Child-Pugh) o moderata (classe B secondo la classificazione di Child-Pugh). Non sono disponibili dati nei pazienti con insufficienza epatica grave (classe C secondo la classificazione di Child-Pugh). Inclisiran deve essere utilizzato con cautela nei pazienti con insufficienza epatica grave.
Insufficienza renale
Non è necessaria alcuna modifica della dose nei pazienti con insufficienza renale lieve, moderata o grave e in quelli con insufficienza renale allo stadio terminale. L'esperienza nell'uso di inclisiran nei pazienti con insufficienza renale grave è limitata. Inclisiran deve essere utilizzato con cautela in tali pazienti. Per informazioni sulle precauzioni in caso di emodialisi, vedere la sezione «Informazioni importanti sulla somministrazione».
Modalità di somministrazione
Il medicinale è destinato alla somministrazione sottocutanea.
Inclisiran è destinato alla somministrazione sottocutanea nell'area addominale; alternative sono il braccio o la coscia. Non somministrare iniezioni in aree di patologie cutanee o lesioni, come scottature solari, eruzioni cutanee, infiammazioni o infezioni cutanee.
Ogni dose da 284 mg deve essere somministrata tramite un'unica siringa preriempita. Ogni siringa preriempita è destinata all'uso monouso.
Inclisiran deve essere somministrato da un operatore sanitario.
Uso pediatrico.
La sicurezza e l'efficacia di inclisiran nei bambini (età inferiore a 18 anni) non sono state stabilite.
Dati non disponibili.
Sovradosaggio.
Nessuna reazione avversa clinicamente significativa è stata osservata in volontari sani che avevano ricevuto inclisiran a dosi superiori a quelle terapeutiche. Non esiste un antidoto specifico in caso di sovradosaggio con inclisiran. In caso di sovradosaggio, si deve effettuare un trattamento sintomatico e, se necessario, misure di supporto.
Effetti indesiderati.
Riassunto del profilo di sicurezza
Gli unici effetti indesiderati associati all'uso di inclisiran sono stati reazioni nel sito di iniezione (8,2%).
Elenco degli effetti indesiderati
Gli effetti indesiderati sono elencati in base alle classi di sistemi organici (tabella 4). La frequenza è definita come segue: molto comune (≥ 1/10); comune (≥ 1/100 – < 1/10); non comune (≥ 1/1000 – < 1/100); raro (≥ 1/10000 – < 1/1000); molto raro (< 1/10000); non noto (non può essere stimato sulla base dei dati disponibili).
Tabella 4
Effetti indesiderati osservati nei pazienti trattati con inclisiran
| Classe di organi e sistemi |
Reazione avversa |
Categoria di frequenza |
|
| Disturbi generali e reazioni nel sito di somministrazione |
Reazioni avverse nel sito di somministrazione1 |
Comune |
|
| 1 Vedere paragrafo «Descrizione di alcune reazioni avverse» |
|||
Descrizione di alcune reazioni avverse
Reazioni avverse nel sito di somministrazione
Negli studi di riferimento, le reazioni avverse nel sito di somministrazione si sono verificate nell’8,2% e nell’1,8% dei pazienti rispettivamente nel gruppo trattato con inclisiran e nel gruppo placebo. La percentuale di pazienti in ciascun gruppo che hanno interrotto il trattamento a causa di reazioni avverse nel sito di somministrazione è stata rispettivamente dello 0,2% e dello 0,0%. Tutte queste reazioni avverse erano di grado lieve o moderato, temporanee e si sono risolte senza conseguenze. Le reazioni avverse più comuni nel sito di somministrazione nei pazienti trattati con inclisiran sono state reazioni nel sito di iniezione (3,1%), dolore nel sito di iniezione (2,2%), eritema nel sito di iniezione (1,6%) e eruzione cutanea nel sito di iniezione (0,7%).
Categorie speciali di pazienti
Pazienti anziani
Dei 1833 pazienti che hanno ricevuto inclisiran negli studi di riferimento, 981 (54%) avevano un’età pari o superiore a 65 anni e 239 (13%) avevano un’età pari o superiore a 75 anni. Non sono state osservate differenze generali di sicurezza tra questi pazienti e quelli di età inferiore.
Immunogenicità
Negli studi, 1830 pazienti sono stati sottoposti a test per la ricerca di anticorpi contro il farmaco. Una positività confermata è stata osservata nell’1,8% (33/1830) dei pazienti prima della somministrazione della dose e nel 4,9% (90/1830) dei pazienti entro 18 mesi di trattamento con inclisiran. Non sono state osservate differenze clinicamente rilevanti nei profili di efficacia clinica, sicurezza e farmacodinamica di inclisiran nei pazienti con risultato positivo per anticorpi anti-inclisiran.
Parametri di laboratorio
Negli studi clinici di Fase III, si sono osservati aumenti più frequenti dei livelli sierici delle transaminasi epatiche da > 1 × limite superiore della norma (LSN) a ≤ 3 × LSN nei pazienti trattati con inclisiran (ALT 19,7%; AST 17,2%) rispetto a quelli trattati con placebo (ALT 13,6%; AST 11,1%). Tali aumenti non hanno superato la soglia clinicamente rilevante di 3 × LSN, si sono manifestati in modo asintomatico e non sono stati associati a reazioni avverse né ad altri segni di disfunzione epatica.
Segnalazione delle sospette reazioni avverse
La segnalazione delle reazioni avverse dopo l’immissione in commercio del medicinale è importante. Permette di monitorare il rapporto beneficio/rischio del medicinale. I professionisti sanitari e farmaceutici, nonché i pazienti o i loro rappresentanti legali, devono segnalare tutti i casi sospetti di reazioni avverse e di mancata efficacia del medicinale attraverso il sistema informatizzato automatizzato di farmacovigilanza al seguente indirizzo: https://aisf.dec.gov.ua/
Periodo di validità.
3 anni.
Condizioni di conservazione.
Il medicinale non richiede particolari condizioni di conservazione. Non congelare. Conservare in un luogo inaccessibile ai bambini.
Confezionamento.
1,5 ml di soluzione in una siringa preriempita; 1 siringa preriempita in un blister; 1 blister in una scatola di cartone.
Categoria di rilascio. Su prescrizione medica.
Produttore.
Novartis Pharmaceutical Manufacturing GmbH, Austria.
Sede del produttore e suo indirizzo operativo.
Biochemiestrasse 10, Langkampfen, 6336, Austria.