Pergoveris®
Ukraina
Spis treści
INSTRUKCJA DOTYCZĄCA STOSOWANIA LEKU DO ZASTOSOWANIA W MEDYCYNE Pergoveris® (PERGOVERIS®)
Skład:
Substancje czynne: foltropina alfa, lutropina alfa;
1 fiolka z proszkiem zawiera 150 JM (równowartość 11 µg) foltropiny alfa (rekombinowanego ludzkiego hormonu folikulotropowego, rhFSH) oraz 75 JM (równowartość 3 µg) lutropiny alfa (rekombinowanego ludzkiego hormonu luteinizującego, rhLH);
Substancje pomocnicze: sacharoza, fosforan sodu dwuwodny, fosforan sodu dwuwodny monohydrat, metionina, polisorbat 20, kwas fosforowy stężony, wodorotlenek sodu;
Roztwórnik: 1 ml wody do wstrzykiwań.
Postać leku. Proszek i roztwórnik do sporządzenia roztworu do wstrzykiwań.
Główne właściwości fizykochemiczne: lek to liofilizat w postaci tabletki o barwie białej lub prawie białej; rozcieńczalnik to klarowna, bezbarwna ciecz.
Grupa farmakoterapeutyczna. Hormony płciowe i modyfikatory układu rozrodczego. Gonadotropiny i inne stymulatory owulacji. Gonadotropiny. Kombinacje.
Kod ATC G03G A30.
Właściwości farmakologiczne.
Farmakodynamika.
Pergoveris® to lek zawierający rekombinacyjny hormon folikulotropowy (follitropina alfa, r-hFSH) oraz hormon luteinizujący (lutropina alfa, r-hLH), wytworzone w komórkach jajnika chomika chińskiego metodą rekombinowanego DNA.
Mechanizm działania
Hormon luteinizujący (LH) i hormon folikulotropowy (FSH) są wydzielane przez przednią płaszczyzna przysadki pod wpływem hormonu uwalniającego gonadotropiny (GnRH) i odgrywają pomocniczą rolę w rozwoju pęcherzyków i owulacji. W komórkach tkanki theca LH stymuluje wydzielanie androgenów, które przenikają do komórek ziarnistych, gdzie są przekształcane w estradiol (E2) za pomocą aromatazy. W komórkach ziarnistych FSH stymuluje rozwój pęcherzyków jajnikowych, natomiast LH uczestniczy w rozwoju pęcherzyków, sterydogenezie i dojrzewaniu.
Skutki farmakodynamiczne
Po podaniu r-hFSH obserwuje się wzrost stężenia inhibiny i estradiolu, co prowadzi do indukcji rozwoju pęcherzyków. Stężenia inhibiny w surowicy szybko rosną i mogą być wykrywalne już trzeciego dnia po podaniu r-hFSH, natomiast wzrost stężenia estradiolu wymaga dłuższego czasu i stwierdza się go dopiero od czwartego dnia leczenia. Całkowita objętość pęcherzyków zaczyna wzrastać około 4–5 dnia codziennego podawania r-hFSH, a maksymalny efekt osiągany jest u większości pacjentek około 10 dnia od rozpoczęcia podawania gonadotropiny. Pierwotnym skutkiem podania r-hLH jest zależny od dawki wzrost wydzielania E2 oraz wzmocnienie działania r-hFSH na wzrost pęcherzyków.
Skuteczność kliniczna
W badaniach klinicznych u pacjentek z ciężkim niedoborem FSH i LH, potwierdzonym stężeniem LH w surowicy < 1,2 MIU/ml, oznaczonym w centralnym laboratorium, częstość owulacji na cykl wynosiła 70–75%. Należy jednak pamiętać, że wyniki pomiarów stężenia LH uzyskane w różnych laboratoriach mogą się różnić.
Przeprowadzono badanie kliniczne doboru dawki r-hLH u kobiet z hipogonadotropowym hipogonadyzmem i endogennym stężeniem LH w surowicy poniżej 1,2 MIU/ml. Podawanie codziennie dawki 75 MIU r-hLH (w połączeniu z 150 MIU r-hFSH) prowadziło do odpowiedniego rozwoju pęcherzyków i wydzielania estrogenów. Natomiast podawanie dawki dobowej 25 MIU r-hLH (w połączeniu z 150 MIU r-hFSH) prowadziło do niewystarczającego rozwoju pęcherzyków. W związku z tym codzienne podawanie zawartości mniejszej niż jedno opakowanie może nie zapewnić odpowiedniego rozwoju pęcherzyków.
Chociaż w większości przypadków u pacjentek poddawanych protokołom technik rozrodu wspomaganego (ART) skuteczna jest monoterapia z zastosowaniem rekombinowanego FSH, opublikowane dane wskazują na korzyści z łączonego stosowania r-hFSH i r-hLH u pacjentek, które nie odpowiadają odpowiednio na monoterapię r-hFSH (grupa pacjentek z suboptymalną odpowiedzią na r-hFSH). Dodanie r-hLH do terapii ma na celu zwiększenie wrażliwości jajników na działanie r-hFSH, wspomaganie wydzielania estradiolu przez pęcherzyki przedowulacyjne i tym samym wzrostu endometrium, a także wspieranie późnej fazy luteinizacji pęcherzyków, co sprzyja osiągnięciu normalnego poziomu progesteronu w fazie ciałka żółtego.
Farmakokinetyka.
Podczas jednoczesnego podawania follitropiny alfa i lutropiny alfa nie występuje interakcja farmakokinetyczna.
Follitropina alfa
Rozkład
Po wstrzyknięciu dożylnym follitropina alfa rozkłada się w płynie międzykomórkowym z początkowym okresem półtrwania wynoszącym około 2 godzin, a wydalana jest z okresem półtrwania końcowego od 14 do 17 godzin. Objętość rozkładu w stanie stacjonarnym mieści się w zakresie od 9 do 11 l.
Po podaniu podskórnej absolutna biodostępność wynosi 66%, a pozorny okres półtrwania końcowego mieści się w zakresie od 24 do 59 godzin. Po podaniu podskórnej stwierdzono zależność proporcjonalną od dawki dla dawek do 900 MIU. Powtarzane podawanie follitropiny alfa prowadzi do trzykrotnego wzrostu jej akumulacji, osiągając stan równowagi w ciągu 3–4 dni.
Wydalanie
Całkowity klirens wynosi 0,6 l/h, a około 12% dawki follitropiny alfa wydala się z moczem.
Lutropina alfa
Rozkład
Po wstrzyknięciu dożylnym lutropina alfa szybko się rozkłada z początkowym okresem półtrwania wynoszącym około 1 godziny i wydalana jest z okresem półtrwania końcowego wynoszącym około 9–11 godzin. Objętość rozkładu w stanie stacjonarnym mieści się w zakresie od 5 do 14 l. Lutropina alfa charakteryzuje się kinetyką liniową, tzn. wartość AUC jest wprost proporcjonalna do dawki.
Po podaniu podskórnej absolutna biodostępność wynosi 56%, a pozorny okres półtrwania końcowego mieści się w zakresie od 8 do 21 godzin. Po podaniu podskórnej stwierdzono zależność proporcjonalną od dawki dla dawek do 450 MIU. Farmakokinetyka lutropiny alfa jest porównywalna po pojedynczym i wielokrotnym podaniu, a współczynnik akumulacji lutropiny alfa jest minimalny.
Wydalanie
Całkowity klirens mieści się w zakresie od 1,7 do 1,8 l/godz., przy czym mniej niż 5% podanej dawki wydala się z moczem.
Charakterystyka kliniczna.
Wskazania.
- Stymulacja rozwoju pęcherzyków Graafa u kobiet z ciężkim niedoborem hormonu люteinizującego (LH) i hormonu folikulotropowego (FSH).
- Kontrolowana stymulacja jajników u pacjentek z podoptymalną odpowiedzią na leczenie w ramach stosowania technik rozrodu wspomaganego (ART), takich jak zapłodnienie in vitro (IVF), wewnątrzokrężne wprowadzenie plemnika do cytoplazmy oocytu (ICSI), przeszczepienie gamet do jajowodu (GIFT) i przeszczepienie zygoty do jajowodu (ZIFT). W badaniach klinicznych podoptymalną odpowiedź na leczenie określano według następujących parametrów:
- mniej niż 7 pęcherzyków przedowulacyjnych lub oocytów oraz/lub
- stosowanie wysokich dawek FSH (≥ 3000 MI na cykl) oraz/lub
- późny wiek reprodukcyjny matki (od 35 lat).
Przeciwwskazania.
- Nadwrażliwość na folitropinę alfa i lutropinę alfa lub na którąkolwiek substancję pomocniczą leku;
- guzy podwzgórza i przysadki mózgu;
- powiększenie jajników lub torbielowatość niezwiązane z zespołem policystycznych jajników;
- krwawienia ginekologiczne nieznanej etiologii;
- raka jajników, macicy lub gruczołów piersiowych.
Nie należy stosować leku Pergoveris® również w przypadkach, w których nie można uzyskać skutecznej odpowiedzi na leczenie, np. w przypadku:
- pierwotnego niewydolności jajników;
- wrodzonych wad narządów płciowych niezgodnych z ciążą;
- mięśniaków macicy niezgodnych z ciążą.
Interakcje z innymi lekami i inne rodzaje interakcji.
Nie należy wstrzykiwać leku Pergoveris® w połączeniu z innymi lekami w formie jednej iniekcji, z wyjątkiem folitropiny alfa, dla której badania wykazały, że jednoczesne podawanie nie wpływa istotnie na aktywność, stabilność, właściwości farmakokinetyczne lub farmakodynamiczne substancji czynnych.
Szczególne wytyczne dotyczące stosowania.
Śledzenie
W celu poprawy śledzenia leków biologicznych należy dokładnie odnotowywać nazwę i numer serii stosowanego leku.
Ogólne zalecenia
Preparat Pergoveris® zawiera substancje o znacznej aktywności gonadotropowej, które mogą powodować działania niepożądane od łagodnego do ciężkiego stopnia nasilenia, dlatego może być przepisywany wyłącznie przez lekarzy dobrze obeznanych z problemami niepłodności oraz metodami ich leczenia.
Przed rozpoczęciem leczenia bezpłodna para powinna przejść badania mające na celu wykrycie istniejących lub możliwych przeciwwskazań do zajścia w ciążę. W szczególności pacjentów należy przebadać pod kątem obecności hipotyreozę, niewydolności nadnerczy, hiperprolaktynemii i odpowiednio przepisać specyficzne leczenie.
Terapia gonadotropinami wymaga określonego nakładu czasu od lekarzy i innych personelu medycznego, a także odpowiedniego sprzętu do monitorowania leczenia. Bezpieczne i skuteczne stosowanie preparatu Pergoveris® u kobiet wymaga regularnego monitorowania reakcji jajników za pomocą ultrasonografii, najlepiej równocześnie z oznaczaniem stężenia estradiolu we krwi. Reakcja pacjentek na podanie FSH/LH ma charakter indywidualny, przy czym niektóre pacjentki słabo reagują na FSH/LH. Kobiety powinny stosować najniższą skuteczną dawkę preparatu, odpowiednią do celu leczenia.
Porfiria
Pacjentki z porfirią lub z przypadkami porfirii w rodzinie powinny być poddawane starannemu nadzorowi medycznemu podczas leczenia preparatem Pergoveris®, ponieważ leczenie to może zwiększać ryzyko napadów ostrych. W przypadku pierwszych objawów rozwoju tego stanu lub jego nasilenia może zaistnieć potrzeba przerwania leczenia.
Zespół nadreaktywności jajników (OHSS)
Oczekiwanym skutkiem kontrolowanej stymulacji jajników jest pewne zwiększenie ich rozmiarów. Zjawisko to, które występuje najczęściej u kobiet z zespołem policystycznych jajników, zazwyczaj ustępuje bez specjalnego leczenia.
W przeciwieństwie do niepowikłanego zwiększenia rozmiarów jajników, OHSS to zespół, który rozwija się stopniowo, o różnym stopniu nasilenia. Objawia się wyraźnym zwiększeniem rozmiarów jajników, wysokimi stężeniami sterydów płciowych we krwi oraz wzrostem przepuszczalności naczyń krwionośnych, co może prowadzić do gromadzenia się płynu w jamie brzusznej, opłucnej i rzadziej – w osierdziu.
W ciężkich przypadkach OHSS mogą występować następujące objawy: ból i uczucie napięcia w jamie brzusznej, znaczne zwiększenie rozmiarów jajników, przyrost masy ciała, duszność, oliguria oraz objawy ze strony przewodu pokarmowego, w tym nudności, wymioty i biegunka.
Podczas badania klinicznego mogą być wykryte hipowolemia, zagęszczenie krwi, zaburzenia elektrolitowe, wodobrzusze, hemoperitoneum, wylewy do opłucnej, wodopłucne lub ostry zespół ostrej niewydolności oddechowej, a także powikłania zakrzepowo-zatorowe.
W bardzo rzadkich przypadkach ciężki OHSS może być powikłany skręceniem jajnika oraz powikłaniami zakrzepowo-zatorowymi, takimi jak zator tętnicy płucnej, udar niedokrwienny czy zawał mięśnia sercowego.
Nieuzależnione czynniki ryzyka rozwoju OHSS obejmują młody wiek, szczupłą budowę ciała, zespół policystycznych jajników, wysokie dawki egzogennych gonadotropin, wysokie bezwzględne lub szybko rosnące stężenia estradiolu we krwi (> 900 pg/ml lub > 3300 pmol/l przy anowulacji), wcześniejsze epizody OHSS, dużą liczbę rosnących folikuli jajnikowych (> 3 folikule o średnicy ≥ 14 mm przy anowulacji) lub dużą liczbę uzyskanych oocytów w cyklach Eko.
Dokładne przestrzeganie zalecanych dawek preparatów Pergoveris® i FSH oraz zalecanego schematu podawania może zminimalizować ryzyko rozwoju nadreaktywności jajników. W celu wczesnego wykrycia odpowiednich czynników ryzyka zaleca się monitorowanie cykli stymulacji za pomocą ultrasonografii oraz oznaczania poziomu estradiolu.
Istnieją podstawy do przypuszczeń, że hCG odgrywa kluczową rolę w inicjowaniu OHSS i że zespół ten może być cięższy i dłuższy, jeśli dojdzie do zajścia w ciążę. Dlatego przy obecności objawów OHSS, takich jak stężenie estradiolu we krwi > 5500 pg/ml lub > 20200 pmol/l oraz/lub rozwój ≥ 40 folikuli łącznie, zaleca się odwołanie podania hCG i doradzenie pacjentce powstrzymania się od stosunków płciowych lub stosowania barierowych metod antykoncepcji przez co najmniej 4 dni. OHSS może szybko postępować (w ciągu 24 godzin) i w ciągu kilku dni stać się poważnym powikłaniem medycznym. Zazwyczaj występuje po zakończeniu leczenia hormonalnego i osiąga maksymalną częstość około 7–10 dnia po zakończeniu leczenia. Zwykle OHSS ustępuje spontanicznie po nadejściu menstruacji. Dlatego po podaniu hCG pacjentki powinny być poddawane nadzorowi medycznemu przez co najmniej 2 tygodnie.
W przypadku wystąpienia ciężkiej postaci OHSS należy przerwać leczenie gonadotropinami, jeśli nadal trwa. Pacjentkę należy hospitalizować i rozpocząć specyficzną terapię OHSS.
W przypadku podejrzenia ryzyka rozwoju OHSS należy rozważyć możliwość przerwania leczenia.
Skręcenie jajnika
Zgłaszano przypadki skręcenia jajnika po leczeniu innymi preparatami gonadotropin. Mogą one być związane z innymi czynnikami ryzyka, takimi jak OHSS, ciąża, wcześniejsze operacje brzuszne, skręcenie jajnika w wywiadzie, wcześniejsze lub istniejące torbie jajnika lub zespół policystycznych jajników. Uszkodzenie jajnika z powodu niedokrwienia można zminimalizować dzięki wczesnej diagnostyce i natychmiastowemu rozkręceniu.
Ciąża mnoga
U pacjentek poddawanych indukcji owulacji częstość ciąży mnogich i urodzeń jest zwiększona w porównaniu do naturalnego zapłodnienia. Większość ciąży mnogich to bliźniaki. Ciąża mnoga, szczególnie wyższego rzędu, wiąże się ze zwiększonym ryzykiem niekorzystnych wyników porodu i okresu okołoporodowego.
W celu zminimalizowania ryzyka ciąży mnogiej zaleca się staranne monitorowanie reakcji jajników.
Pacjentki powinny zostać poinformowane o potencjalnym ryzyku urodzenia dzieci mnogich przed rozpoczęciem leczenia. W przypadku podejrzenia ryzyka ciąży mnogiej należy rozważyć możliwość przerwania leczenia.
Przerwanie ciąży
Częstość przypadków poronień lub samoistnych aborcji jest wyższa u pacjentek poddawanych stymulacji wzrostu folikuli w celu indukcji owulacji niż w ogólnej populacji.
Ciąża ektopowa
Kobiety z chorobami jajowodów w wywiadzie są narażone na ryzyko ciąży ektopowego niezależnie od tego, czy nastąpi on w wyniku naturalnego zapłodnienia czy leczenia niepłodności. Zgłaszano, że częstość ciąży ektopowych po przeprowadzeniu Eko jest wyższa niż w ogólnej populacji.
Nowotwory układu rozrodczego
Istnieją doniesienia o przypadkach zarówno nowotworów łagodnych, jak i złośliwych jajników oraz innych narządów układu rozrodczego u kobiet, które w leczeniu niepłodności stosowały kilka leków. Nie ustalono jeszcze, czy leczenie gonadotropinami zwiększa podstawowe ryzyko rozwoju takich nowotworów u kobiet niepłodnych.
Wady wrodzone
Częstość wad wrodzonych po przeprowadzeniu Eko może być nieco wyższa niż po naturalnym zapłodnieniu. Uważa się, że jest to skutkiem różnic w charakterystyce rodziców (np. wiek matki, jakość nasienia) oraz ciąży mnogich.
Zjawiska zakrzepowo-zatorowe
U kobiet z niedawnymi lub istniejącymi chorobami zakrzepowo-zatorowymi oraz u kobiet, u których ogólnie występują czynniki ryzyka rozwoju zjawisk zakrzepowo-zatorowych, takimi jak przypadki w rodzinie lub osobiste, trombofilia lub ciężka postać otyłości (wskaźnik masy ciała > 30 kg/m²), leczenie gonadotropinami może prowadzić do dalszego wzrostu tego ryzyka. U takich kobiet należy ocenić korzyści z zastosowania gonadotropin w stosunku do ryzyka rozwoju takich zjawisk. Należy jednak zaznaczyć, że sama ciąża oraz OHSS zwiększają ryzyko rozwoju powikłań zakrzepowo-zatorowych.
Preparat Pergoveris® zawiera mniej niż 1 mmol sodu (23 mg) w jednej dawce, co oznacza, że jest praktycznie pozbawiony sodu.
Stosowanie w czasie ciąży lub karmienia piersią.
Ciąża
W czasie ciąży nie ma wskazań do stosowania preparatu Pergoveris®. Dane uzyskane dla niewielkiej liczby przypadków stosowania preparatu w czasie ciąży wskazują na brak negatywnego wpływu foltropiny alfa i lutropiny alfa na ciążę, rozwój embrionu lub płodu, poród i okres poporodowy po przeprowadzonej kontrolowanej stymulacji jajników. W przypadku stosowania w czasie ciąży dane kliniczne nie pozwalają jednoznacznie wykluczyć efektu teratogennego preparatu Pergoveris®.
Karmienie piersią
Preparat Pergoveris® nie jest wskazany do stosowania w czasie karmienia piersią.
Wpływ na zdolność prowadzenia pojazdów lub obsługiwania maszyn.
Pergoveris® nie wpływa lub prawie nie wpływa na zdolność pacjentek do prowadzenia samochodu lub pracy z maszynami.
Sposób stosowania i dawki
Leczenie z zastosowaniem leku Pergoveris® należy rozpoczynać pod nadzorem lekarza dobrze obeznanego w leczeniu zaburzeń płodności.
Kobiety z ciężkim niedoborem sekrecji LH i FSH
U kobiet z niedoborem sekrecji LH i FSH celem terapii z zastosowaniem leku Pergoveris® jest wspomaganie rozwoju pęcherzyków z późniejszym dojrzewaniem po podaniu ludzkiego gonadotropiny chorionicznej (hCG). Jeśli takie pacjentki cierpią na amenoreę i mają niski poziom endogennych estrogenów, leczenie można rozpocząć w dowolnym czasie.
Pergoveris® podaje się w postaci cyklu codziennych wstrzyknięć. Leczenie prowadzi się z uwzględnieniem indywidualnej reakcji pacjentki, ocenianej na podstawie wyników ultrasonografii wielkości pęcherzyków oraz oznaczenia poziomu estrogenów. Zalecany tryb leczenia polega na rozpoczęciu codziennego podawania zawartości jednego fiolki leku Pergoveris® (150 j.m. rekombinowanego FSH i 75 j.m. rekombinowanego LH). W przypadku podania mniejszej dawki dziennego leku reakcja pęcherzykowa może być niewystarczająca z powodu niedostatecznej ilości lutropiny alfa.
Jeśli uznaje się za konieczne zwiększenie dawki FSH, najlepiej zmieniać ją w odstępach 7–14-dniowych o 37,5–75 j.m., stosując zarejestrowany lek zawierający folitropinę alfa. Dopuszczalne jest przedłużenie czasu stymulacji w ramach jednego cyklu leczenia do 5 tygodni.
Po osiągnięciu optymalnej reakcji, w ciągu 24–48 godzin po ostatniej iniekcji leku Pergoveris®, należy podać pojedynczą dawkę 250 µg rekombinowanego hCG (r-hCG) lub od 5000 do 10 000 j.m. hCG. Pacjentce zaleca się współżycie w dniu podania hCG oraz w dniu następnym. Alternatywnie można przeprowadzić inseminację wewnątrzmaciczną lub inną procedurę wspomaganych technik rozrodczych zgodnie z decyzją lekarza podejmowaną indywidualnie w każdym przypadku.
Należy rozważyć możliwość wspomagania fazy lutealnej, ponieważ niedobór substancji o działaniu luteotropowym (LH/hCG) po owulacji może prowadzić do przedwczesnej niedostateczności ciałka żółtego.
W przypadku nadmiernej reakcji leczenie należy przerwać i nie podawać hCG. W kolejnym cyklu leczenia należy rozpocząć od niższej dawki FSH niż w poprzednim cyklu (patrz sekcja „Szczególne ostrzeżenia i środki ostrożności”).
Pacjentki z suboptymalną odpowiedzią na leczenie w ramach procedur ART
Zaleca się rozpoczęcie leczenia od podania 300 j.m. rekombinowanego FSH codziennie przez pierwsze 5–7 dni cyklu leczenia. Od 6–8 dnia kontrolowanej stymulacji jajników iniekcje FSH należy zastąpić codziennym podawaniem zawartości dwóch fiol leku Pergoveris® (300 j.m. rekombinowanego FSH i 150 j.m. rekombinowanego LH).
Alternatywny schemat leczenia polega na rozpoczęciu codziennego podawania zawartości dwóch fiol leku Pergoveris® (300 j.m. rekombinowanego FSH i 150 j.m. rekombinowanego LH) od pierwszego dnia kontrolowanej stymulacji jajników, przeprowadzanej po desensytyzacji przysadki.
Leczenie kontynuuje się aż do osiągnięcia odpowiedniego rozwoju pęcherzyków (ocenianego na podstawie stężenia estrogenów w surowicy i/lub danych z badania ultrasonograficznego), a dawkę FSH dostosowuje się do odpowiedzi pacjentki. Zazwyczaj całkowita dzienna dawka rekombinowanego FSH nie powinna przekraczać 450 j.m.
Po osiągnięciu odpowiedniego rozwoju pęcherzyków w celu indukcji końcowego dojrzewania pęcherzyków niezbędnego do pobrania oocytów należy podać hCG. Aby zmniejszyć ryzyko wystąpienia zespołu hiperstimulacji jajników (OHSS), podanie hCG należy odłożyć, jeśli w ostatnim dniu terapii stwierdza się patologiczne powiększenie jajników.
W przypadku nadmiernej reakcji leczenie należy przerwać i nie podawać hCG. W kolejnym cyklu leczenia należy rozpocząć od niższej dawki FSH niż w poprzednim cyklu.
Osobne grupy pacjentów
Pacjentki w wieku podeszłym
Nie ma odpowiednich wskazań do stosowania leku Pergoveris® u pacjentek w wieku podeszłym. Bezpieczeństwo i skuteczność stosowania leku u tych pacjentek nie zostały ustalone.
Pacjentki z zaburzeniami funkcji nerek lub wątroby
Bezpieczeństwo, skuteczność oraz parametry farmakokinetyczne leku Pergoveris® u pacjentek z zaburzeniami funkcji nerek lub wątroby nie zostały ustalone.
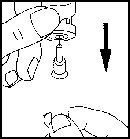
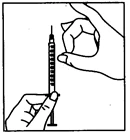
W przypadku samodzielnego podawania leku Pergoveris® należy przeczytać i wykonać poniższe instrukcje.
Lek Pergoveris® przeznaczony jest do podania podskórnego. Bezpośrednio przed zastosowaniem proszek należy rozpuścić w rozpuszczalniku dołączonym do opakowania z lekiem. Każda fiolka przeznaczona jest do jednorazowego użytku. Rozcieńczony roztwór leku powinien być klarowny i nie zawierać cząstek.
Samodzielne podawanie leku Pergoveris® mogą przeprowadzać wyłącznie dobrze poinstruowane i odpowiednio wyszkolone pacjentki, które w razie potrzeby mają możliwość skonsultowania się z fachowcem. Pierwszą iniekcję leku Pergoveris® należy wykonać pod bezpośrednim nadzorem personelu medycznego.
- Umij ręce. Ważne jest, aby ręce i wszystko, co będzie używane, były jak najczystsze.
- Przygotuj wszystkie niezbędne materiały. Na czystej powierzchni połóż jedną fiolkę z lekiem, jedną fiolkę z rozpuszczalnikiem, dwa waciki nasączone alkoholem, jeden strzykawkę, jedną igłę do przygotowania roztworu i jedną cienką igłę do wstrzyknięć podskórnych oraz pojemnik na zużyte szkło i igły.
- Zdejmij ochronny korek z fiolki z rozpuszczalnikiem. Następnie dołącz do strzykawki igłę do przygotowania roztworu i naciśnij tłokiem powietrze, wciskając tłok do około oznaczenia 1 ml. Następnie włóż igłę do fiolki, wciskając tłokiem powietrze, odwróć fiolkę do góry dnem i ostrożnie wciągnij cały rozpuszczalnik do strzykawki. Ostrożnie, nie dotykając igły, połóż strzykawkę na powierzchnię roboczą.
|
|
|
|
|
|
|
|
|
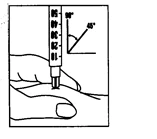
- Unieś wszystkie używane przedmioty: natychmiast po zakończeniu iniekcji umieść wszystkie igły i puste fiolki szklane w pojemniku na ostre przedmioty. Należy również unieść wszelkie niewykorzystane roztwory.
Dzieci.
Nie ma odpowiednich wskazań do stosowania leku Pergoveris® u pacjentek z grupy pediatrycznej.
Przedawkowanie.
Efekty przedawkowania leku Pergoveris® są nieznane, jednak istnieje możliwość rozwoju zespołu nadreakcji jajników (patrz sekcja „Szczególne ostrzeżenia i środki ostrożności przed zastosowaniem”).
Leczenie objawowe.
Działania niepożądane.
Ogólny opis profilu bezpieczeństwa
Najczęściej zgłaszane działania niepożądane podczas stosowania leku to ból głowy, torbiele jajników oraz reakcje miejscowe w miejscu wstrzyknięcia (np. ból, zaczerwienienie, krwiak, obrzęk i/lub podrażnienie w miejscu wstrzyknięcia).
Często występuje zespół hiperstymulacji jajników (ZHGJ) o łagodnym lub umiarkowanym nasileniu, który należy traktować jako nieodłączne ryzyko procedury stymulacji. Ciężkie postacie ZHGJ są rzadkie.
Bardzo rzadko mogą występować przypadki zjawisk tromboembolicznych, które zazwyczaj są związane z ciężkimi postaciami ZHGJ.
Lista działań niepożądanych
Działania niepożądane obserwowane podczas stosowania leku wymieniono poniżej i sklasyfikowano według układów narządów oraz częstości występowania, która jest określana następująco: bardzo często (≥ 1/10), często (od ≥ 1/100 do < 1/10), rzadko (od ≥ 1/1000 do < 1/100), bardzo rzadko (od ≥ 1/10000 do < 1/1000), niezbyt rzadko (< 1/10000), częstość nieznana (dokładna częstość nie może być ustalona na podstawie dostępnych danych).
Zaburzenia układu odpornościowego
Bardzo rzadko: reakcje nadwrażliwości od łagodnego do ciężkiego nasilenia, w tym reakcje anafilaktyczne i wstrząs.
Zaburzenia układu nerwowego
Bardzo często: ból głowy.
Zaburzenia naczyniowe
Bardzo rzadko: zjawiska tromboemboliczne, zazwyczaj związane z ciężkim ZHGJ.
Zaburzenia układu oddechowego
Bardzo rzadko: nasilenie się lub pogorszenie astmy.
Zaburzenia układu pokarmowego
Często: ból i uczucie napięcia w jamie brzusznej, dyskomfort brzuszny, nudności, wymioty, biegunka.
Zaburzenia układu rozrodczego i gruczołów mlekowych
Bardzo często: torbiele jajników.
Często: ból piersi, ból w okolicy miednicy, ZHGJ o łagodnym do umiarkowanego nasilenia (wraz z objawami towarzyszącymi).
Rzadko: ciężki ZHGJ (wraz z objawami towarzyszącymi) (patrz sekcja „Szczególne środki ostrożności”).
Bardzo rzadko: powikłania ciężkiego ZHGJ.
Zaburzenia ogólne oraz reakcje w miejscu podania
Bardzo często: reakcje w miejscu wstrzyknięcia o łagodnym do ciężkiego nasilenia (np. ból, zaczerwienienie, krwiak, obrzęk i/lub podrażnienie w miejscu wstrzyknięcia).
Zgłaszanie działań niepożądanych po rejestracji leku ma istotne znaczenie. Umożliwia to monitorowanie stosunku korzyści do ryzyka związanego ze stosowaniem tego leku. Osoby medyczne i farmaceutyczne, a także pacjenci lub ich prawni przedstawiciele powinni zgłaszać wszystkie przypadki podejrzewanych działań niepożądanych oraz braku skuteczności leku poprzez Automatyczny System Informacyjny Nadzoru Farmakologicznego pod adresem: https://aisf.dec.gov.ua.
Okres ważności. 3 lata.
Lek przeznaczony jest do natychmiastowego i jednorazowego użycia bezpośrednio po pierwszym otwarciu fiolki i rozcieńczeniu.
Nie stosować po upływie terminu ważności podanego na opakowaniu.
Warunki przechowywania.
Przechowywać w temperaturze nie wyższej niż 25 °C.
Przechowywać we wkładce oryginalnego opakowania w celu ochrony przed światłem.
Przechowywać w miejscu niedostępnym dla dzieci.
Niezgodność.
Lek Pergoveris® można mieszać z folitropiną alfa i podawać oba leki w jednym wstrzyknięciu.
Opakowanie.
- proszek do sporządzenia roztworu do wstrzykiwań w fiolkach nr 1 lub nr 3 w zestawie z rozcieńczalnikiem (1 ml wody do wstrzykiwań) w fiolkach nr 1 lub nr 3 w opakowaniu konturowym bąbelkowym; po 1 opakowaniu konturowym bąbelkowym w pudełku kartonowym.
- proszek do sporządzenia roztworu do wstrzykiwań w fiolkach nr 5 w zestawie z rozcieńczalnikiem (1 ml wody do wstrzykiwań) w fiolkach nr 5 w opakowaniu konturowym bąbelkowym; po 2 opakowania konturowe bąbelkowe w pudełku kartonowym.
Kategoria receptury. Na receptę.
Producent.
Merck Serono S.A., oddział w Aubonne / Merck Serono S.A., Succursale d’Aubonne.
Miejsce produkcji oraz adres siedziby producenta.
Zone Industrielle de l’Ouriettaz, 1170 Aubonne, Szwajcaria / Zone Industrielle de l’Ouriettaz, 1170 Aubonne, Switzerland.