Pergoveris®
Ucraina
Indice
ISTRUZIONE PER L'USO MEDICO DEL MEDICINALE Pergoveris® (PERGOVERIS®)
Composizione:
Principi attivi: follitropina alfa, lutropina alfa;
1 flaconcino con polvere contiene 150 UI (equivalenti a 11 µg) di follitropina alfa (ormone follicolostimolante ricombinante umano r-FSH) e 75 UI (equivalenti a 3 µg) di lutropina alfa (ormone luteinizzante ricombinante umano r-LH);
Eccipienti: saccarosio, fosfato disodico diidrato, fosfato monosodico monoidrato, metionina, polisorbato 20, acido fosforico concentrato, idrossido di sodio;
Solvente: 1 ml di acqua per preparazioni iniettabili.
Forma farmaceutica. Polvere e solvente per soluzione iniettabile.
Principali caratteristiche fisico-chimiche: il medicinale è un liofilizzato di colore bianco o quasi bianco sotto forma di pellet; solvente: liquido trasparente incolore.
Gruppo farmacoterapeutico. Ormoni sessuali e modulatori del sistema riproduttivo. Gonadotropine e altri stimolanti dell'ovulazione. Gonadotropine. Combinazioni.
Codice ATC G03G A30.
Proprietà farmacologiche.
Farmacodinamica.
Pergoveris® è un medicinale contenente ormone follicolo-stimolante ricombinante (follitropina alfa, r-hFSH) e ormone luteinizzante ricombinante (lutropina alfa, r-hLH), prodotti mediante tecnologia del DNA ricombinante in cellule ovariche di criceto cinese.
Meccanismo d'azione
L'ormone luteinizzante (LH) e l'ormone follicolo-stimolante (FSH) sono secreti dall'ipofisi anteriore in risposta all'ormone rilasciante della gonadotropina (GnRH) e svolgono un ruolo chiave nello sviluppo follicolare e nell'ovulazione. Nelle cellule della teca, l'LH stimola la secrezione di androgeni, che vengono trasportati alle cellule della granulosa per essere convertiti in estradiolo (E2) tramite l'aromatasi. Nelle cellule della granulosa, l'FSH stimola lo sviluppo dei follicoli ovarici, mentre l'LH è coinvolto nello sviluppo follicolare, nella steroidogenesi e nella maturazione.
Effetti farmacodinamici
Dopo somministrazione di r-hFSH si osserva un aumento dei livelli di inibina ed estradiolo, con conseguente induzione dello sviluppo follicolare. I livelli sierici di inibina aumentano rapidamente e possono essere rilevati già dal terzo giorno dopo la somministrazione di r-hFSH, mentre l'aumento dei livelli di estradiolo richiede più tempo e si manifesta solo a partire dal quarto giorno di trattamento. Il volume follicolare totale inizia ad aumentare circa dopo 4-5 giorni di somministrazione giornaliera di r-hFSH e, in base alla risposta della paziente, l'effetto massimo si raggiunge circa dopo 10 giorni dall'inizio della somministrazione della gonadotropina. L'effetto primario della somministrazione di r-hLH è un aumento dose-dipendente della secrezione di E2 e un potenziamento dell'azione di r-hFSH sulla crescita follicolare.
Efficacia clinica
Negli studi clinici condotti su pazienti con grave carenza di FSH e LH, definita da un livello sierico endogeno di LH < 1,2 UI/l misurato in un laboratorio centrale, la frequenza di ovulazione per ciclo è risultata compresa tra il 70 e il 75%. Tuttavia, va considerato che i risultati della misurazione dei livelli di LH possono variare tra laboratori diversi.
È stato condotto uno studio clinico di determinazione della dose ottimale di r-hLH in donne con ipogonadismo ipogonadotropo e livelli sierici endogeni di LH inferiori a 1,2 UI/l. La somministrazione giornaliera di 75 UI di r-hLH (in associazione con 150 UI di r-hFSH) ha determinato uno sviluppo follicolare e una secrezione estrogenica adeguati. Al contrario, la somministrazione giornaliera di 25 UI di r-hLH (in associazione con 150 UI di r-hFSH) ha determinato uno sviluppo follicolare insufficiente. Pertanto, la somministrazione giornaliera di una quantità inferiore al contenuto di un flaconcino potrebbe non garantire uno sviluppo follicolare adeguato.
Sebbene per la maggior parte delle pazienti sottoposte a protocolli di tecnologie di riproduzione assistita (ART) sia accettabile una terapia monodose con FSH ricombinante, dati pubblicati indicano i vantaggi dell'uso combinato di r-hFSH e r-hLH in pazienti che rispondono in modo inadeguato alla monoterapia con r-hFSH soltanto (gruppo di pazienti con risposta subottimale a r-hFSH). L'aggiunta di r-hLH alla terapia ha lo scopo di aumentare la sensibilità ovarica all'azione di r-hFSH, favorire la secrezione di estradiolo da parte dei follicoli preovulatori e quindi la crescita dell'endometrio, nonché sostenere la tardiva luteinizzazione dei follicoli, favorendo il raggiungimento di livelli normali di progesterone nella fase luteinica.
Farmacocinetica.
Non si verifica alcuna interazione farmacocinetica quando follitropina alfa e lutropina alfa vengono somministrate contemporaneamente.
Follitropina alfa
Distribuzione
Dopo somministrazione endovenosa, la follitropina alfa si distribuisce nel liquido interstiziale con una fase iniziale di emivita di circa 2 ore e viene eliminata con un'emivita terminale compresa tra 14 e 17 ore. Il volume di distribuzione allo stato stazionario varia tra 9 e 11 litri.
Dopo somministrazione sottocutanea, la biodisponibilità assoluta è del 66% e l'emivita terminale apparente varia tra 24 e 59 ore. Dopo somministrazione sottocutanea è stata dimostrata una relazione proporzionale alla dose per dosi fino a 900 UI. La somministrazione ripetuta di follitropina alfa determina un accumulo triplo, raggiungendo uno stato stazionario in 3-4 giorni.
Eliminazione
La clearance totale è di 0,6 l/ora e circa il 12% della dose di follitropina alfa viene escreto nelle urine.
Lutropina alfa
Distribuzione
Dopo somministrazione endovenosa, la lutropina alfa si distribuisce rapidamente con un'emivita iniziale di circa 1 ora e viene eliminata con un'emivita terminale di circa 9-11 ore. Il volume di distribuzione allo stato stazionario varia tra 5 e 14 litri. La lutropina alfa presenta una farmacocinetica lineare, ovvero l'AUC è direttamente proporzionale alla dose somministrata.
Dopo somministrazione sottocutanea, la biodisponibilità assoluta è del 56% e l'emivita terminale apparente varia tra 8 e 21 ore. Dopo somministrazione sottocutanea è stata dimostrata una relazione proporzionale alla dose per dosi fino a 450 UI. La farmacocinetica della lutropina alfa è comparabile dopo somministrazione singola e ripetuta, e il coefficiente di accumulo della lutropina alfa è minimo.
Eliminazione
La clearance totale varia tra 1,7 e 1,8 l/ora, con meno del 5% della dose somministrata escreta nelle urine.
Caratteristiche cliniche.
Indicazioni.
- Stimolazione dello sviluppo follicolare nelle donne con grave insufficienza dell'ormone luteinizzante (LH) e dell'ormone follicolo-stimolante (FSH).
- Stimolazione ovarica controllata in pazienti con risposta subottimale al trattamento durante l'impiego di tecniche di riproduzione assistita (ART), come la fecondazione in vitro (IVF), l'iniezione intracitoplasmatica dello spermatozoo (ICSI), il trasferimento gametico in tuba (GIFT) e il trasferimento della zigote in tuba (ZIFT). Nei trial clinici, la risposta subottimale al trattamento è stata definita in base ai seguenti parametri:
- meno di 7 follicoli preovulatori o ovociti e/o
- utilizzo di alte dosi di FSH (≥ 3000 UI per ciclo) e/o
- età riproduttiva avanzata della madre (a partire dai 35 anni).
Controindicazioni.
- Ipersensibilità al follitropina alfa e al lutropina alfa o a qualsiasi eccipiente del medicinale;
- tumori dell'ipotalamo o dell'ipofisi;
- ingrandimento delle ovaie o cisti non correlate al sindrome dell'ovaio policistico;
- emorragie ginecologiche di origine sconosciuta;
- carcinoma delle ovaie, dell'utero o delle ghiandole mammarie.
Il medicinale Pergoveris® non deve inoltre essere utilizzato nei casi in cui non è possibile ottenere una risposta efficace al trattamento, ad esempio in presenza di:
- insufficienza ovarica primaria;
- malformazioni congenite degli organi genitali incompatibili con la gravidanza;
- fibromi uterini incompatibili con la gravidanza.
Interazioni con altri medicinali ed altre forme di interazione.
Pergoveris® non deve essere iniettato in associazione con altri farmaci nella stessa iniezione, eccetto che con follitropina alfa, per la quale studi hanno dimostrato che la somministrazione concomitante non altera in modo significativo l'attività, la stabilità, le proprietà farmacocinetiche o farmacodinamiche delle sostanze attive.
Caratteristiche particolari di impiego.
Tracciabilità
Al fine di migliorare la tracciabilità dei medicinali biologici, è necessario registrare chiaramente il nome e il numero di lotto del medicinale utilizzato.
Raccomandazioni generali
Il medicinale Pergoveris® contiene sostanze con elevata attività gonadotropa, in grado di provocare reazioni avverse da lievi a gravi; pertanto, può essere prescritto soltanto da medici esperti nei problemi di infertilità e nei metodi per il loro trattamento.
Prima dell'inizio del trattamento, la coppia infertile deve sottoporsi a un'accurata valutazione al fine di individuare eventuali controindicazioni esistenti o potenziali alla gravidanza. In particolare, i pazienti devono essere sottoposti a esami per verificare la presenza di ipotiroidismo, insufficienza surrenalica, iperprolattinemia e devono ricevere un trattamento specifico appropriato.
La terapia con gonadotropine richiede un impegno temporale da parte dei medici e di altri operatori sanitari, nonché un'adeguata strumentazione per il monitoraggio del trattamento. L'uso sicuro ed efficace di Pergoveris® nelle donne richiede un monitoraggio regolare della risposta ovarica mediante ecografia, preferibilmente associata alla determinazione dei livelli sierici di estradiolo. La risposta delle pazienti all'FSH/LH è di carattere individuale e alcune pazienti rispondono all'FSH/LH in modo molto scarso. Le donne devono ricevere la dose più bassa efficace in relazione all'obiettivo terapeutico.
Porfiria
Le pazienti con porfiria o con anamnesi familiare di porfiria devono essere sottoposte a un rigoroso monitoraggio medico durante il trattamento con Pergoveris®, poiché tale trattamento può aumentare il rischio di crisi acute. In caso di comparsa dei primi segni di tale condizione o di peggioramento dello stato, potrebbe rendersi necessario interrompere il trattamento.
Sindrome da iperstimolazione ovarica (OHSS)
Un aumento delle dimensioni ovariche rappresenta un effetto atteso della stimolazione ovarica controllata. Questo fenomeno, più comune nelle donne affette da sindrome dell'ovaio policistico, di solito si risolve spontaneamente senza trattamento specifico.
A differenza dell'aumento ovarico non complicato, l'OHSS è una sindrome che si manifesta con un'escalation di gravità. Essa comprende un evidente aumento delle dimensioni delle ovaie, elevati livelli sierici di steroidi sessuali e un aumento della permeabilità vascolare, che può causare l'accumulo di liquido nella cavità addominale, pleurica e, raramente, pericardica.
Nei casi gravi, l'OHSS può manifestarsi con sintomi quali dolore e sensazione di distensione addominale, marcato aumento delle dimensioni delle ovaie, aumento di peso, dispnea, oliguria e sintomi gastrointestinali, inclusi nausea, vomito e diarrea.
All'esame clinico possono essere riscontrati ipovolemia, emocoagulazione, squilibrio elettrolitico, ascite, emoperitoneo, versamenti pleurici, idrotorace o sindrome da distress respiratorio acuto, nonché complicanze tromboemboliche.
In casi molto rari, un'OHSS grave può complicarsi con torsione ovarica e complicanze tromboemboliche, come embolia polmonare, ictus ischemico e infarto miocardico.
I fattori di rischio indipendenti per lo sviluppo dell'OHSS includono giovane età, costituzione esile, sindrome dell'ovaio policistico, alte dosi di gonadotropine esogene, elevati livelli sierici assoluti o in rapida crescita di estradiolo (> 900 pg/ml o > 3300 pmol/l in caso di anovulazione), episodi precedenti di OHSS, elevato numero di follicoli ovarici in crescita (> 3 follicoli con diametro ≥ 14 mm in caso di anovulazione) o elevato numero di ovociti recuperati nei cicli di FIV.
L'aderenza alle dosi e alle modalità di somministrazione raccomandate per Pergoveris® e FSH può minimizzare il rischio di iperstimolazione ovarica. Per individuare precocemente i fattori di rischio associati, si raccomanda il monitoraggio dei cicli di stimolazione mediante ecografia e determinazione del livello di estradiolo.
Esistono motivi per ritenere che l'LH ricombinante svolga un ruolo chiave nell'insorgenza dell'OHSS e che tale sindrome possa aggravarsi e protrarsi ulteriormente in caso di gravidanza. Pertanto, in presenza di segni di OHSS, come livelli sierici di estradiolo > 5500 pg/ml o > 20200 pmol/l e/o sviluppo di ≥ 40 follicoli in totale, si raccomanda di sospendere l'assunzione di LH ricombinante e di consigliare alla paziente di astenersi dai rapporti sessuali o di utilizzare metodi contraccettivi barriera per almeno 4 giorni. L'OHSS può progredire rapidamente (entro 24 ore) e diventare una seria complicanza medica entro pochi giorni. Si verifica più frequentemente dopo l'interruzione del trattamento ormonale e raggiunge la massima incidenza circa 7-10 giorni dopo la fine del trattamento. Di solito, l'OHSS si risolve spontaneamente con l'arrivo delle mestruazioni. Pertanto, dopo l'iniezione di LH ricombinante, le pazienti devono essere sottoposte a monitoraggio medico per almeno 2 settimane.
In caso di OHSS grave, il trattamento con gonadotropine deve essere interrotto se ancora in corso. La paziente deve essere ricoverata e deve essere avviata una terapia specifica per l'OHSS.
In caso di sospetto rischio di sviluppo di OHSS, si deve considerare la possibilità di interrompere il trattamento.
Torsione ovarica
Sono stati riportati casi di torsione ovarica dopo trattamento con altri medicinali a base di gonadotropine. Tali eventi possono essere associati ad altri fattori di rischio, come OHSS, gravidanza, precedenti interventi chirurgici addominali, torsione ovarica in anamnesi, cisti ovariche preesistenti o sindrome dell'ovaio policistico. I danni all'ovaio dovuti a insufficiente apporto ematico possono essere minimizzati mediante diagnosi precoce e immediata detorsione.
Gravidanza multipla
Nei pazienti sottoposti a induzione dell'ovulazione, la frequenza di gravidanze multiple e di parti multipli è aumentata rispetto al concepimento spontaneo. La maggior parte delle gravidanze multiple riguarda gemelli. La gravidanza multipla, in particolare di ordine superiore, comporta un rischio aumentato di esiti sfavorevoli del parto e del periodo perinatale.
Per minimizzare il rischio di gravidanza multipla, si raccomanda un attento monitoraggio della risposta ovarica.
Le pazienti devono essere informate del potenziale rischio di parti multipli prima dell'inizio del trattamento. In caso di sospetto rischio di gravidanza multipla, si deve considerare la possibilità di interrompere il trattamento.
Interruzione della gravidanza
La frequenza di aborti spontanei o di perdita della gravidanza è maggiore nelle pazienti sottoposte a stimolazione follicolare per l'induzione dell'ovulazione rispetto alla popolazione generale.
Gravidanza extrauterina
Le donne con anamnesi di patologie tubariche hanno un rischio di gravidanza extrauterina indipendentemente dal fatto che la gravidanza sia avvenuta spontaneamente o in seguito a trattamento per l'infertilità. È stato riportato che l'incidenza di gravidanza extrauterina dopo trattamento di riproduzione assistita (ART) è maggiore rispetto alla popolazione generale.
Neoplasie del sistema riproduttivo
Sono stati riportati casi sia di neoplasie benigne che maligne delle ovaie e di altri organi del sistema riproduttivo in donne trattate con diversi farmaci per l'infertilità. Non è ancora chiaro se il trattamento con gonadotropine aumenti il rischio basale di sviluppare tali tumori nelle donne infertili.
Malformazioni congenite
L'incidenza di malformazioni congenite dopo trattamento di riproduzione assistita (ART) può essere leggermente superiore rispetto al concepimento spontaneo. Si ritiene che ciò sia dovuto a differenze nelle caratteristiche dei genitori (ad esempio, età materna, qualità dello sperma) e alla gravidanza multipla.
Eventi tromboembolici
Nelle donne con tromboembolie recenti o preesistenti e in quelle con fattori di rischio generali per eventi tromboembolici, come storia personale o familiare, trombofilia o grave obesità (indice di massa corporea > 30 kg/m²), il trattamento con gonadotropine può ulteriormente aumentare tale rischio. In queste pazienti, deve essere valutato il beneficio dell'uso di gonadotropine rispetto al rischio di sviluppare tali eventi. Tuttavia, si deve notare che la gravidanza stessa e l'OHSS aumentano il rischio di complicanze tromboemboliche.
Pergoveris® contiene meno di 1 mmol di sodio (23 mg) per dose, ovvero è praticamente privo di sodio.
Uso durante la gravidanza o l'allattamento.
Gravidanza
Non vi sono indicazioni per l'uso di Pergoveris® durante la gravidanza. I dati disponibili, relativi a un numero limitato di casi di uso del medicinale durante la gravidanza, indicano l'assenza di effetti negativi di follitropina alfa e lutropina alfa sulla gravidanza, sullo sviluppo dell'embrione o del feto, sul parto e sullo sviluppo postnatale dopo stimolazione ovarica controllata. Tuttavia, in caso di uso durante la gravidanza, i dati clinici non sono sufficienti per escludere un effetto teratogeno di Pergoveris®.
Allattamento
Pergoveris® non è indicato per l'uso durante l'allattamento.
Capacità di guidare veicoli o di usare macchinari.
Pergoveris® non ha alcun effetto o un effetto trascurabile sulla capacità delle pazienti di guidare veicoli o di usare macchinari.
Modalità di somministrazione e dosaggio.
Il trattamento con Pergoveris® deve essere iniziato sotto la supervisione di un medico esperto nella gestione dei disturbi della fertilità.
Donne con grave carenza di secrezione di LH e FSH
Nelle donne con carenza di secrezione di LH e FSH, l’obiettivo del trattamento con Pergoveris® è promuovere lo sviluppo follicolare, seguito dalla maturazione finale dopo somministrazione di gonadotropina corionica umana (hCG). Se queste pazienti soffrono di amenorrea e presentano bassi livelli di estrogeni endogeni, il trattamento può essere iniziato in qualsiasi momento.
Pergoveris® viene somministrato sotto forma di iniezioni giornaliere. Il trattamento deve essere personalizzato in base alla risposta individuale della paziente, valutata mediante ecografia per determinare la dimensione dei follicoli e misurazione dei livelli ematici di estrogeni. Il regime raccomandato prevede l’iniezione giornaliera del contenuto di un flacone di Pergoveris® (150 UI di FSH ricombinante e 75 UI di LH ricombinante). Se viene somministrata una dose giornaliera inferiore, la risposta follicolare potrebbe risultare inadeguata a causa della quantità insufficiente di lutropina alfa.
Se si ritiene necessario aumentare la dose di FSH, è preferibile farlo a intervalli di 7-14 giorni, aumentando la dose di 37,5-75 UI, utilizzando un prodotto registrato di follitropina alfa. È accettabile prolungare la durata della stimolazione fino a un massimo di 5 settimane all’interno di un singolo ciclo di trattamento.
Una volta raggiunta la risposta ottimale, entro 24-48 ore dall’ultima iniezione di Pergoveris®, deve essere somministrata una dose singola di 250 mcg di hCG ricombinante oppure da 5000 a 10.000 UI di hCG. Si raccomanda alla paziente di avere rapporti sessuali nel giorno dell’iniezione di hCG e nel giorno successivo. In alternativa, può essere effettuata un’inseminazione intrauterina o un’altra procedura di medicina riproduttiva assistita, secondo decisione medica individuale in ogni singolo caso.
Si dovrebbe considerare la possibilità di un supporto alla fase luteinica, poiché la carenza di sostanze con attività luteotropa (LH/hCG) dopo l’ovulazione potrebbe causare una precoce insufficienza del corpo luteo.
Se si verifica una risposta eccessiva, il trattamento deve essere interrotto e l’iniezione di hCG deve essere annullata. Nel ciclo successivo, il trattamento deve essere iniziato con una dose di FSH inferiore rispetto a quella utilizzata nel ciclo precedente (vedere il paragrafo «Avvertenze e precauzioni speciali»).
Pazienti con risposta subottimale al trattamento nell’ambito di procedure di riproduzione assistita
Il trattamento dovrebbe iniziare con 300 UI di FSH ricombinante somministrati giornalmente per i primi 5-7 giorni del ciclo di trattamento. A partire dal 6°-8° giorno di stimolazione ovarica controllata, le iniezioni di FSH devono essere sostituite con la somministrazione giornaliera del contenuto di due flaconi di Pergoveris® (300 UI di FSH ricombinante e 150 UI di LH ricombinante).
Un regime alternativo prevede la somministrazione giornaliera del contenuto di due flaconi di Pergoveris® (300 UI di FSH ricombinante e 150 UI di LH ricombinante) già dal primo giorno della stimolazione ovarica controllata, effettuata dopo la desensibilizzazione ipofisaria.
Il trattamento prosegue fino al raggiungimento di un adeguato sviluppo follicolare (valutato mediante livelli sierici di estrogeni e/o ecografia) e la dose di FSH viene regolata in base alla risposta della paziente. Generalmente, la dose giornaliera totale di FSH ricombinante non dovrebbe superare le 450 UI.
Una volta raggiunto un adeguato sviluppo follicolare, per indurre la maturazione finale dei follicoli necessaria al recupero degli ovociti, deve essere somministrata hCG. Per ridurre il rischio di sviluppare la sindrome da iperstimolazione ovarica (OHSS), l’iniezione di hCG deve essere annullata in caso di aumento anomalo delle dimensioni delle ovaie nell’ultimo giorno di terapia.
Se si verifica una risposta eccessiva, il trattamento deve essere interrotto e l’iniezione di hCG deve essere annullata. Nel ciclo successivo, il trattamento deve essere iniziato con una dose di FSH inferiore rispetto a quella utilizzata nel ciclo precedente.
Gruppi di pazienti particolari
Pazienti anziane
Non vi sono indicazioni specifiche per l’uso di Pergoveris® in pazienti anziane. La sicurezza e l’efficacia del farmaco in queste pazienti non sono state stabilite.
Pazienti con compromissione della funzionalità renale o epatica
La sicurezza, l’efficacia e i parametri farmacocinetici di Pergoveris® in pazienti con compromissione della funzionalità renale o epatica non sono stati stabiliti.
Prima della somministrazione autonoma di Pergoveris®, leggere e seguire attentamente le istruzioni riportate di seguito.
Pergoveris® è destinato alla somministrazione sottocutanea. Immediatamente prima dell’uso, la polvere deve essere ricostituita con il solvente fornito nell’imballaggio. Ogni flacone è destinato a un solo uso. La soluzione ricostituita deve essere limpida e priva di particelle.
La somministrazione autonoma di Pergoveris® può essere effettuata solo da pazienti ben informate e adeguatamente formate, che abbiano la possibilità di consultare un professionista qualora necessario. La prima iniezione di Pergoveris® deve essere eseguita sotto diretta supervisione di un operatore sanitario.
- Lavarsi le mani. È importante che mani e materiali siano il più possibile puliti.
- Preparare tutti i materiali necessari. Su una superficie pulita, posizionare un flacone del medicinale, un flacone di solvente, due tamponi imbevuti di alcol, una siringa, un’ago per la ricostituzione della soluzione e un ago sottile per iniezioni sottocutanee, nonché un contenitore per lo smaltimento di vetro e aghi usati.
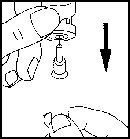
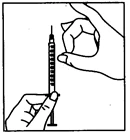
- Rimuovere il tappo protettivo dal flacone del solvente. Collegare quindi all’ago della siringa l’ago per la ricostituzione, aspirare una certa quantità di aria nella siringa arretrando lo stantuffo fino al segno di circa 1 ml. Inserire poi l’ago nel flacone, espellere l’aria spingendo lo stantuffo, capovolgere il flacone e aspirare con attenzione tutto il solvente nella siringa. Posizionare con cautela la siringa, senza toccare l’ago, sulla superficie di lavoro.
|
|
|
|
|
|
|
|
|
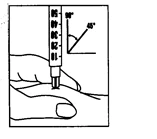
- Smaltimento degli oggetti utilizzati: immediatamente dopo l'iniezione, smaltire tutti gli aghi e le fiale di vetro vuote in un contenitore per oggetti taglienti. Smaltire anche qualsiasi soluzione non utilizzata.
Età pediatrica.
Non esistono indicazioni appropriate per l'uso di Pergoveris® in pazienti pediatriche.
Sovradosaggio.
Gli effetti di un sovradosaggio con Pergoveris® non sono noti, ma è possibile il rischio di sviluppare un sindrome da iperstimolazione ovarica (vedere il paragrafo «Informazioni importanti sull'uso del medicinale»).
Il trattamento è sintomatico.
Effetti indesiderati.
Descrizione generale del profilo di sicurezza
Durante l'uso del medicinale, gli effetti indesiderati più frequentemente riportati sono stati cefalea, cisti ovariche e reazioni locali nel sito di iniezione (ad esempio dolore, eritema, ematoma, gonfiore e/o irritazione nel sito di iniezione).
È stata spesso riportata la sindrome da iperstimolazione ovarica (OHSS) di grado lieve o moderato, che deve essere considerata un rischio intrinseco della procedura di stimolazione. Le forme gravi di OHSS sono rare.
Molto raramente, possono verificarsi casi di tromboembolia, generalmente associati a forme gravi di OHSS.
Elenco degli effetti indesiderati
Gli effetti indesiderati osservati durante l'uso del medicinale sono riportati di seguito e classificati per classi di organi e sistemi e per frequenza, definita come segue: molto comune (≥ 1/10), comune (da ≥ 1/100 a < 1/10), non comune (da ≥ 1/1000 a < 1/100), raro (da ≥ 1/10000 a < 1/1000), molto raro (< 1/10000), frequenza non nota (la frequenza esatta non può essere determinata sulla base dei dati disponibili).
Dal sistema immunitario
Molto raro: reazioni di ipersensibilità da lievi a gravi, comprese reazioni anafilattiche e shock anafilattico.
Dal sistema nervoso
Molto comune: cefalea.
Disturbi vascolari
Molto raro: tromboembolia, generalmente associata a OHSS grave.
Dal sistema respiratorio
Molto raro: peggioramento o esacerbazione dell'asma.
Dal sistema gastrointestinale
Comune: dolore e sensazione di distensione addominale, disagio addominale, nausea, vomito, diarrea.
Dal sistema riproduttivo e dalle ghiandole mammarie
Molto comune: cisti ovariche.
Comune: dolore al seno, dolore pelvico, OHSS da lieve a moderato (inclusi i sintomi associati).
Non comune: OHSS grave (inclusi i sintomi associati) (vedere la sezione «Precauzioni per l’uso»).
Raro: complicanze da OHSS grave.
Disturbi generali e reazioni nel sito di somministrazione
Molto comune: reazioni nel sito di iniezione da lievi a gravi (ad esempio dolore, eritema, ematoma, gonfiore e/o irritazione nel sito di iniezione).
La segnalazione degli effetti indesiderati dopo l’autorizzazione del medicinale è di fondamentale importanza. Permette un monitoraggio continuo del rapporto beneficio/rischio del medicinale. I professionisti sanitari e farmaceutici, così come i pazienti o i loro rappresentanti legali, devono segnalare qualsiasi sospetto effetto indesiderato o mancanza di efficacia del medicinale attraverso il Sistema Informatizzato Automatico di Farmacovigilanza al seguente indirizzo: https://aisf.dec.gov.ua.
Durata della validità. 3 anni.
Il medicinale è destinato all’uso immediato e monouso dopo la prima apertura del flacone e la ricostituzione.
Non utilizzare oltre la data di scadenza indicata sull’imballaggio.
Condizioni di conservazione.
Conservare a una temperatura non superiore a 25 °C.
Conservare nell’imballaggio originale al riparo dalla luce.
Conservare fuori dalla portata dei bambini.
Incompatibilità.
Il medicinale Pergoveris® può essere miscelato con folitropina alfa e entrambi i medicinali possono essere somministrati in un’unica iniezione.
Confezione.
- Polvere per soluzione iniettabile in flaconcini n. 1 o n. 3, in combinazione con solvente (1 ml di acqua per preparazioni iniettabili) in flaconcini n. 1 o n. 3, in confezione blister; 1 confezione blister per scatola di cartone.
- Polvere per soluzione iniettabile in flaconcini n. 5, in combinazione con solvente (1 ml di acqua per preparazioni iniettabili) in flaconcini n. 5, in confezione blister; 2 confezioni blister per scatola di cartone.
Categoria di fornitura. Sotto prescrizione medica.
Produttore.
Merck Serono S.A., succursale di Aubonne / Merck Serono S.A., Succursale d’Aubonne.
Sede del produttore e indirizzo del luogo di attività.
Zone Industrielle de l’Ouriettaz, 1170 Aubonne, Svizzera / Zone Industrielle de l’Ouriettaz, 1170 Aubonne, Switzerland.