Mavenklad®
Ukraina
Spis treści
INSTRUKCJA dotyczÄ cÄ a stosowania leku Mavenklad® (Mavenklad®)
SkÅ ad:
substancja czynna: kladyryna;
1 tabletka zawiera 10 mg kladyryny;
substancje pomocnicze: hydroksypropylobetaksytryna, sorbitol (E 420), stearynian magnezu.
Postać leku. Tabletki.
GÅ owne wÅ aÅ ciwoÅ ci fizykochemiczne: biaÅ e, okrÄ gÅ e, podwójnie wypukÅ e tabletki o Å rodku 8,5 mm z oznaczeniem „C” z jednej strony i „10” z drugiej.
Grupa farmakoterapeutyczna. Selektywne leki immunosupresyjne. Kladyryna.
Kod ATC L04A A40.
Właściwości farmakologiczne.
Farmakodynamika.
Mechanizm działania
Kladrybina jest analogiem nukleozydowym dezoksyadenozytu. Podstawienie atomu chloru w pierścieniu purynowym chroni kladrybinę przed rozkładem przez adenozynę deaminazę, zwiększając czas przebywania proleków kladrybiny wewnątrzkomórkowo. Dalsze fosforylowanie kladrybiny prowadzące do powstania jej aktywnej formy – trifosforanu 2-chlorodezoksyadenozytu (Cd-ATP) – osiągane jest szczególnie skutecznie w limfocytach, ponieważ w tych komórkach występują stałe wysokie poziomy deoksycytydynokinazy (DCK) oraz względnie niskie poziomy 5'-nukleotydazy (5'-NTazy). Wysoka wartość stosunku DCK do 5'-NTazy sprzyja kumulacji Cd-ATP, co czyni limfocyty szczególnie wrażliwymi na śmierć komórkową. Ze względu na niższy stosunek DCK/5'-NTaza inne komórki pochodzące z szpiku kostnego są uszkadzane w mniejszym stopniu niż limfocyty. DCK jest enzymem ograniczającym szybkość przemiany kladrybiny do jej aktywnej formy trifosforanowej, co prowadzi do selektywnego wyczerpywania się limfocytów T i B, zarówno dzielących się, jak i niepodzielonych.
Głównym mechanizmem działania Cd-ATP, indukującym apoptozę, jest bezpośredni i pośredni wpływ na syntezę DNA oraz funkcję mitochondriów. W komórkach dzielących się Cd-ATP wpływa na syntezę DNA poprzez hamowanie rybonukleotydoreduktazy oraz konkuruje z dezoksyadenozynotrifosforanem o wbudowanie do DNA za pośrednictwem polimeraz DNA. W komórkach pozostających w stanie spoczynku kladrybina powoduje jednoniciowe pęknięcia DNA, szybkie wyczerpanie nikotynoamidoadeninodinukleotydu, rozszczepienie ATP oraz śmierć komórkową. Istnieją dowody, że kladrybina może również powodować bezpośredni, zależny i niezależny od kaspaz, proces apoptotyczny poprzez uwalnianie cytochromu C oraz czynnika indukującego apoptozę do cytozolu w komórkach niepodzielonych.
Patogeneza stwardnienia rozsianego (SR) obejmuje złożony kaskadę zjawisk, w których ważną rolę odgrywają różne typy komórek immunologicznych, w tym autoreaktywne limfocyty T i B. Mechanizm działania terapeutycznego kladrybiny w SR nie został jeszcze w pełni poznany, ale uważa się, że jej główny wpływ na limfocyty B i T przerywa kaskadę zjawisk immunologicznych mających decydujące znaczenie dla SR.
Różne poziomy ekspresji DCK i 5'-NTazy w podtypach komórek immunologicznych mogą wyjaśniać różnicę w wrażliwości komórek immunologicznych na kladrybinę. Dzięki tym poziomom ekspresji komórki układu odporności wrodzonej są mniej uszkadzane niż komórki układu odporności nabytej.
Efekty farmakodynamiczne
Wykazano, że kladrybina wykazuje długotrwały efekt, wpływając głównie na limfocyty oraz procesy autoimmunologiczne zaangażowane w patofizjologię SR.
W badaniach największa część pacjentów z limfopenią stopnia 3 lub 4 (od < 500 do 200 komórek/mm³ lub < 200 komórek/mm³) występowała 2 miesiące po podaniu pierwszej dawki kladrybiny w każdym roku, co wskazuje na istnienie okresu czasowego pomiędzy obecnością kladrybiny w osoczu a maksymalnym efektem hematologicznym.
Dane uzyskane w badaniach klinicznych przy zaproponowanej dawce kumulacyjnej 3,5 mg/kg masy ciała wskazują na stopniowe przywracanie mediany liczby limfocytów do zakresu normy w 84. tygodniu po podaniu pierwszej dawki kladrybiny (około 30 tygodni po podaniu ostatniej dawki kladrybiny). Liczba limfocytów u ponad 75% pacjentów powracała do zakresu normy do 144. tygodnia po podaniu pierwszej dawki kladrybiny (około 90 tygodni po podaniu ostatniej dawki kladrybiny).
Leczenie doustną formą kladrybiny prowadzi do szybkiego zmniejszenia się liczby komórek CD4+ i CD8+ w krwi. W przypadku komórek CD8+ obserwuje się mniej wyraźne zmniejszenie i szybszą regenerację niż w przypadku komórek CD4+, co prowadzi do tymczasowego zmniejszenia się stosunku CD4 do CD8. Kladrybina zmniejsza liczbę komórek B CD19+ oraz komórek naturalnych zabójców CD16+/CD56+, których liczba również przywracana jest szybciej niż liczba limfocytów CD4+.
Skuteczność i bezpieczeństwo kliniczne
SR z przebiegiem rzutowo-remisyjnym
Skuteczność i bezpieczeństwo doustnego stosowania kladrybiny oceniano w randomizowanym, podwójnie ślepych, kontrolowanym placebo badaniu klinicznym (CLARITY) z udziałem 1326 pacjentów z przebiegiem rzutowo-remisyjnym SR. Celem badania była ocena skuteczności kladrybiny w porównaniu z placebo w zakresie zmniejszenia rocznej częstości rzutów (ARR) (główny punkt końcowy), spowolnienia postępującego niepełnosprawstwa oraz zmniejszenia aktywnych ognisk w badaniu MRI.
Pacjenci przyjmowali albo placebo (n = 437), albo dawkę kumulacyjną kladrybiny 3,5 mg/kg (n = 433) lub 5,25 mg/kg masy ciała (n = 456) przez okres 96-tygodniowy (2-letni), składający się z dwóch cykli leczenia. Pacjentów losowano do przyjmowania dawki kumulacyjnej 3,5 mg/kg w pierwszym cyklu leczenia w tygodniach 1 i 5 pierwszego roku oraz w drugim cyklu leczenia w tygodniach 1 i 5 drugiego roku. Pacjentów losowano do przyjmowania dawki kumulacyjnej 5,25 mg/kg w dodatkowym okresie leczenia w tygodniach 9 i 13 pierwszego roku. Większość pacjentów w grupie placebo (87,0%) oraz w grupach leczonych kladrybiną w dawkach 3,5 mg/kg (91,9%) i 5,25 mg/kg (89,0%) ukończyła pełny 96-tygodniowy okres badania.
Pacjenci musieli mieć co najmniej jeden rzut w ciągu ostatnich 12 miesięcy. W całej populacji badawczej mediana wieku pacjentów wynosiła 39 lat (zakres od 18 do 65 lat), a stosunek kobiet do mężczyzn wynosił około 2 : 1. Średnia długość trwania choroby SR przed włączeniem do badania wynosiła 8,7 roku, a mediana wyjściowego wskaźnika niepełnosprawności neurologicznej we wszystkich grupach leczenia według rozszerzonej skali stanu niepełnosprawstwa Kurtzkego (EDSS) wynosiła 3,0 punktu (zakres od 0 do 6,0 punktu). Ponad dwie trzecie pacjentów przed badaniem nie przyjmowało leków modyfikujących przebieg SR. Pozostali pacjenci wcześniej leczono interferonem beta-1a, interferonem beta-1b, acetatem glatyrameru lub natalizumabem.
U pacjentów z przebiegiem rzutowo-remisyjnym SR przyjmujących kladrybinę w dawce 3,5 mg/kg obserwowano istotne statystycznie poprawy w porównaniu z pacjentami przyjmującymi placebo w zakresie rocznej częstości rzutów, odsetka pacjentów bez rzutów w ciągu 96 tygodni, odsetka pacjentów bez trwałego niepełnosprawstwa w ciągu 96 tygodni oraz czasu do 3-miesięcznego postępu EDSS (patrz tabela 1).
| Tabela 1 Wyniki kliniczne badania CLARITY (96 tygodni) |
|||
| Parametr |
Placebo |
Kumulacyjna dawka kladyrybiny |
|
| 3,5 mg/kg |
5,25 mg/kg |
||
| Uśredniona roczna częstość nawrotów (95 % CI) |
0,33 (0,29; 0,38) |
0,14* (0,12; 0,17) |
0,15* (0,12; 0,17) |
|
57,6 % |
54,5 % |
|
| Część pacjentów bez nawrotów przez 96 tygodni |
60,9 % |
79,7 % |
78,9 % |
| Czas do 3-miesięcznego postępu osłabienia EDSS, 10. percentyl (miesiące) |
10,8 |
13,6 |
13,6 |
|
0,67 (0,48; 0,93) p = 0,018 |
0,69 (0,49; 0,96) p = 0,026 |
|
| CI – przedział ufności * p < 0,001 w porównaniu z placebo. |
|||
Wyniki leczenia kladrybinem w dawce 3,5 mg/kg były istotnie statystycznie lepsze niż placebo pod względem liczby i względnego zmniejszenia ognisk T1 z kontrastem Gd+, ognisk T2 oraz kombinowanych unikalnych ognisk w obrazowaniu rezonansu magnetycznego (MRI) mózgu przez pełne 96 tygodni badania. Pacjenci przyjmujący kladrybinę wykazali względne zmniejszenie średniej liczby ognisk T1 z kontrastem Gd+ o 86 % (skorygowana średnia liczba dla grupy kladrybiny 3,5 mg/kg i grupy placebo wynosiła odpowiednio 0,12 i 0,91), względne zmniejszenie średniej liczby ognisk T2 o 73 % (skorygowana średnia liczba dla grupy kladrybiny 3,5 mg/kg i grupy placebo wynosiła odpowiednio 0,38 i 1,43) oraz względne zmniejszenie średniej liczby kombinowanych unikalnych ognisk o 74 % na skan pacjenta (skorygowana średnia liczba dla grupy kladrybiny 3,5 mg/kg i grupy placebo wynosiła odpowiednio 0,43 i 1,72) (p < 0,001 dla wszystkich trzech wyników MRI).
Retrospektywna analiza czasu do potwierdzonego postępującego przez 6 miesięcy pogorszenia według skali EDSS wykazała zmniejszenie ryzyka niepełnosprawności o 47 % w grupie przyjmującej kladrybinę w dawce 3,5 mg/kg w porównaniu z grupą placebo (stosunek ryzyka 0,53; 95 % przedział ufności [0,36; 0,79], p < 0,05). W grupie placebo 10. percentyl postępującej niepełnosprawności osiągnięto po 245 dniach, podczas gdy w grupie kladrybiny 3,5 mg/kg nie osiągnięto go w całym okresie badania.
Jak pokazano w tabeli 1, wyższe dawki kumulacyjne kladrybiny nie dawały żadnych klinicznie istotnych korzyści, ale były związane z większą częstością limfopenii stopnia ≥ 3 (44,9 % w grupie 5,25 mg/kg w porównaniu z 25,6 % w grupie 3,5 mg/kg).
Pacjenci, którzy zakończyli udział w badaniu CLARITY, mogli wziąć udział w badaniu CLARITY Extension, którego głównym celem była ocena bezpieczeństwa. W tym rozszerzonym badaniu 806 pacjentów przyjmowało albo placebo, albo dawkę kumulacyjną kladrybiny 3,5 mg/kg (w trybie podobnym do stosowanego w CLARITY) przez okres 96 tygodni.
Wielkość efektu w zakresie zmniejszenia częstości nawrotów i spowolnienia postępującej niepełnosprawności u pacjentów przyjmujących dawkę 3,5 mg/kg przez 2 lata była utrzymywana w 3. i 4. roku.
Skuteczność leczenia pacjentów z wysoką aktywnością choroby
Retrospektywnie przeprowadzono analizę skuteczności w podgrupach pacjentów z wysoką aktywnością choroby, którzy przyjmowali kladrybinę doustnie w zalecanej dawce kumulacyjnej 3,5 mg/kg. Do tych podgrup włączono:
- pacjentów z 1 nawrotem w poprzednim roku i co najmniej 1 ogniskiem T1 z kontrastem Gd+ lub 9 lub więcej ogniskami T2 podczas terapii innymi lekami modyfikującymi przebieg choroby,
- pacjentów z 2 lub więcej nawrotami w poprzednim roku, niezależnie od tego, czy byli leczeni lekami modyfikującymi przebieg choroby, czy nie.
Analiza danych uzyskanych w badaniu CLARITY wykazała podobny wpływ leczenia na częstość nawrotów, przy czym częstość nawrotów przeliczona na rok wynosiła od 0,16 do 0,18 w grupach kladrybiny i od 0,47 do 0,50 w grupie placebo (p < 0,0001). W porównaniu z populacją ogólną obserwowano większy wpływ leczenia na czas do trwałego pogorszenia niepełnosprawności przez 6 miesięcy, przy czym kladrybina zmniejszała ryzyko postępującej niepełnosprawności o 82 % (stosunek ryzyka 0,18; 95 % przedział ufności [0,07; 0,47]). W grupie placebo 10. percentyl postępującej niepełnosprawności osiągnięto między 16 a 23 tygodniem, podczas gdy w grupach kladrybiny nie osiągnięto go w całym okresie badania.
Wtórnego postępujący stwardnienie rozsiane z nawrotami
Dodatkowe badanie przyjmowania kladrybiny w połączeniu z interferonem beta w porównaniu z placebo i interferonem beta obejmowało również ograniczoną liczbę pacjentów ze wtórnym postępującym przebiegiem stwardnienia rozsianego (26 pacjentów). U tych pacjentów leczenie kladrybiną w dawce 3,5 mg/kg prowadziło do zmniejszenia częstości nawrotów przeliczonej na rok w porównaniu z placebo (0,03 w porównaniu z 0,30; stosunek ryzyka: 0,11; p < 0,05). Nie zaobserwowano różnic w częstości nawrotów przeliczonej na rok między pacjentami z nawrotowo-remisyjnym stwardnieniem rozsianym a pacjentami ze wtórnym postępującym przebiegiem stwardnienia rozsianego z nawrotami. W żadnej z tych podgrup nie wykazano wpływu leku na postęp niepełnosprawności.
Pacjenci ze wtórnym postępującym przebiegiem stwardnienia rozsianego zostali wykluczeni z badania CLARITY. Jednak retrospektywna analiza mieszanych grup pacjentów z badań CLARITY i ONWARD, których zakwalifikowano na podstawie wyniku wyjściowego w skali EDSS ≥ 3,5 jako wskaźnik stwardnienia rozsianego ze wtórnym postępującym przebiegiem, wykazała podobne zmniejszenie częstości nawrotów przeliczonej na rok w porównaniu z pacjentami z wynikiem EDSS poniżej 3.
Farmakokinetyka.
Kladrybina należy do leków prolek i musi być fosforylowana wewnątrz komórki, aby osiągnąć aktywność biologiczną. Farmakokinetykę kladrybiny badano po podaniu doustnym i dożylnym pacjentom ze stwardnieniem rozsianym oraz pacjentom z nowotworami złośliwymi, a także w systemach in vitro.
Wchłanianie
Po doustnym przyjęciu kladrybina jest szybko wchłaniana. Przyjęcie 10 mg kladrybiny prowadzi do średniego Cmax kladrybiny w zakresie od 22 do 29 ng/ml oraz odpowiadającego mu średniego AUC w zakresie od 80 do 101 ng·h/ml.
Po doustnym przyjęciu kladrybiny na czczo mediana Tmax wynosiła 0,5 godziny (zakres od 0,5 do 1,5 godziny). Po przyjęciu z tłustym posiłkiem wchłanianie kladrybiny było opóźnione (mediana Tmax 1,5 godziny, zakres od 1 do 3 godziny) i Cmax było zmniejszone o 29 %, podczas gdy wartość AUC pozostała niezmieniona. Biologiczna dostępność doustnej dawki 10 mg kladrybiny wynosiła około 40 %.
Rozkład
Kladrybina ma dużą objętość rozkładu, co wskazuje na jej rozległy rozkład do tkanek i wewnątrzkomórkowe wychwytowanie. W badaniach stwierdzono, że średnia objętość rozkładu kladrybiny wynosiła od 480 do 490 l. Wiązanie z białkami osocza wynosi 20 % i nie zależy od stężenia kladrybiny w osoczu.
Rozkład kladrybiny przez błony biologiczne odbywa się za pomocą różnych białek transportowych, w tym ENT1, CNT3 i BCRP.
W badaniach in vitro wykazano, że efliuks kladrybiny jest jedynie minimalnie związany z P-gp, dlatego nie przewiduje się klinicznie istotnych interakcji z inhibitorami P-gp.
Badania in vitro wykazały nieznaczne przenikanie kladrybiny do hepatocytów człowieka, pośredniczone przez transportery.
Kladrybina ma potencjał do przenikania przez barierę krew-mózg. Niewielkie badanie z udziałem pacjentów z nowotworem wykazało, że stosunek stężenia kladrybiny w płynie mózgowo-rdzeniowym do osocza wynosi około 0,25.
Kladrybina i/lub jej fosforylowane metabolity w znacznym stopniu gromadzą się i są utrzymywane w limfocytach człowieka. W warunkach in vitro stwierdzono, że stosunek wewnątrzkomórkowego do zewnątrzkomórkowego nagromadzenia wynosi około 30–40 już po 1 godzinie od początku ekspozycji.
Biotransformacja
Metabolizm kladrybiny badano u pacjentów ze stwardnieniem rozsianym po przyjęciu jednej tabletki 10 mg oraz jednorazowym dożylnej dawce 3 mg. Po podaniu doustnym i dożylnym głównym składnikiem wykrytym w osoczu i moczu była pierwotna związek kladrybina. Zawartość jej metabolitu 2-chloroadeniny w osoczu i moczu była nieistotna. W osoczu i moczu można było znaleźć jedynie śladowe ilości innych metabolitów kladrybiny.
W wątrobowych systemach in vitro obserwowano nieistotny metabolizm kladrybiny (co najmniej 90 % stanowiła niezmieniona kladrybina).
Kladrybina nie jest istotnym substratem enzymów cytochromu P450 i nie wykazuje istotnego potencjału jako inhibitor CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, CYP2E1 oraz CYP3A4. Nie przewiduje się, że inhibicja tych enzymów lub polimorfizm genetyczny (np. CYP2D6, CYP2C9 lub CYP2C19) będzie prowadzić do klinicznie istotnego wpływu na farmakokinetykę lub ekspozycję kladrybiny. Kladrybina nie wykazuje klinicznie istotnego wpływu indukcyjnego na enzymy CYP1A2, CYP2B6 oraz CYP3A4.
Po wejściu do komórek docelowych kladrybina jest fosforylowana przy udziale DCK (a także przy udziale deoksyguanozynokinazy w mitochondriach) do kladrybiny monofosforanu (Cd-AMP), a następnie do kladrybiny difosforanu (Cd-ADP) i kladrybiny trifosforanu (Cd-ATP). Defosforylacja i dezaktywacja Cd-AMP są katalizowane przez cytoplazmatyczną 5'-NTazę. W badaniu wewnątrzkomórkowej farmakokinetyki Cd-AMP i Cd-ATP z udziałem pacjentów z przewlekłą białaczką szpikową stężenia Cd-ATP wynosiły około połowy stężeń Cd-AMP.
Wewnątrzkomórkowy okres półtrwania wynosił 15 godzin dla Cd-AMP i 10 godzin dla Cd-ATP.
Wydalanie
Na podstawie połączonych populacyjnych danych farmakokinetycznych zebranych w różnych badaniach, medianowe wartości wydalania wynosiły 22,2 l/h dla klirensu nerkowego i 23,4 l/h dla klirensu pozanerkowego. Klirens nerkowy przekraczał szybkość filtracji kłębuszkowej, co wskazuje na aktywną wydzielanie kanalikowe kladrybiny.
Pozanerkowa część wydalania kladrybiny (około 50 %) obejmuje nieistotny metabolizm wątrobowy i rozległy wewnątrzkomórkowy rozkład oraz wychwyt aktywnego pochodnego kladrybiny (Cd-ATP) przez docelowy wewnątrzkomórkowy kompartment (tj. limfocyty), z późniejszym wydalaniem wewnątrzkomórkowego Cd-ATP zgodnie z cyklem życia i drogami wydalania z tych komórek.
Szacowany końcowy okres półtrwania u typowego pacjenta z populacji, dla której oceniano parametry farmakokinetyczne, wynosi około 1 dobę. Jednak nie prowadzi to do żadnej kumulacji leku po codziennym przyjmowaniu dawki, ponieważ ten okres półtrwania dotyczy jedynie niewielkiej części AUC.
Zależność od dawki i czasu
Po doustnym przyjęciu kladrybiny w dawkach od 3 do 20 mg Cmax i AUC wzrastały w zależności od dawki, co pozwala założyć, że dla dawek doustnych do 20 mg wchłanianie nie jest ograniczane przez procesy ograniczające szybkość lub pojemność.
Po wielokrotnym przyjmowaniu dawek nie obserwowano znaczącej kumulacji kladrybiny w osoczu. Nie ma wskazówek, że farmakokinetyka kladrybiny po wielokrotnym przyjmowaniu może zmieniać się w czasie.
Osobne grupy pacjentów
Ocena farmakokinetyki kladrybiny u pacjentów ze stwardnieniem rozsianym w wieku dziecięcym lub starszym nie była przeprowadzana, tak samo jak u pacjentów z zaburzeniami funkcji nerek lub wątroby.
Analiza populacyjna kinetyki nie wykazała wpływu wieku (zakres od 18 do 65 lat) lub płci pacjenta na farmakokinetykę kladrybiny.
Zaburzenia funkcji nerek
Wykazano, że klirens nerkowy kladrybiny zależy od klirensu kreatyniny. Na podstawie analizy populacyjnej farmakokinetyki, która uwzględniała dane pacjentów z normalną funkcją nerek i łagodnym uszkodzeniem nerek, oczekuje się, że całkowity klirens kladrybiny u pacjentów z łagodnym uszkodzeniem nerek (CLCR = 60 ml/min) będzie umiarkowanie zmniejszony, co prowadzi do wzrostu ekspozycji o 25 %.
Zaburzenia funkcji wątroby
Rola funkcji wątroby w wydalaniu kladrybiny uznawana jest za nieistotną.
Interakcje farmakokinetyczne
Badania interakcji lekowych u pacjentów ze stwardnieniem rozsianym wykazały, że biologiczna dostępność 10 mg kladrybiny po doustnym przyjęciu nie zmienia się przy jednoczesnym stosowaniu z pantoprazolem.
Charakterystyka kliniczna.
Wskazania.
Mavenklad® jest wskazany w leczeniu dorosłych pacjentów z nawrotową postacią stwardnienia rozsianego (SR) o wysokiej aktywności choroby, ustalonej na podstawie badań klinicznych lub wizualizacyjnych.
Przeciwwskazania.
- Nadwrażliwość na substancję czynną lub którykolwiek z substancji pomocniczych leku;
- zakażenie wirusem niedoboru odporności człowieka (HIV);
- aktywne przewlekłe infekcje (gruźlica lub zapalenie wątroby);
- rozpoczęcie leczenia kladyrybina u pacjentów z osłabionym układem odpornościowym, w tym u pacjentów przyjmujących terapię immunosupresyjną lub mielosupresyjną;
- aktywne nowotwory złośliwe;
- umiarkowane lub ciężkie uszkodzenie nerek (klirens kreatyniny < 60 ml/min);
- okres ciąży i karmienia piersią.
Interakcje z innymi lekami i inne rodzaje interakcji.
Mavenklad® zawiera hydroksypropylobetadekstrynę, która może tworzyć kompleksy z innymi lekami, co potencjalnie prowadzi do wzrostu biodostępności takich leków (szczególnie leków o niskiej rozpuszczalności). Dlatego w ciągu ograniczonej liczby dni przyjmowania kladyrybiny zaleca się przyjmowanie wszelkich leków doustnych najmniej 3-godzinny odstęp przed lub po przyjęciu leku Mavenklad®.
Leki immunosupresyjne
Pacjentom z osłabionym układem odpornościowym, w tym pacjentom poddawanym terapii immunosupresyjnej lub mielosupresyjnej, np. metotreksatem, cyklofosfamidem, cyklosporyną lub azatiopryną, lub tym, którzy stale stosują kortykosteroidy, przeciwwskazane jest rozpoczynanie leczenia kladyrybinem ze względu na ryzyko dodatkowego wpływu na układ odpornościowy.
Podczas leczenia kladyrybinem można stosować intensywną krótkoterminową terapię kortykosteroidami systemowymi.
Inne leki modyfikujące przebieg choroby
Stosowanie kladyrybiny w połączeniu z interferonem beta wiąże się ze zwiększonym ryzykiem limfopenii. Bezpieczeństwo i skuteczność stosowania kladyrybiny w połączeniu z innymi lekami modyfikującymi przebieg SR nie zostały ustalone, dlatego nie zaleca się takiego jednoczesnego leczenia.
Leki hematotoksyczne
Ponieważ kladyrybina powoduje zmniejszenie liczby limfocytów, przyjmowanie kladyrybiny przed lub jednocześnie z innymi substancjami wpływającymi na profil hematologiczny (takimi jak karbamazepina) może prowadzić do rozwoju dodatkowych odczynów hematologicznych. W takich przypadkach zaleca się staranne monitorowanie parametrów hematologicznych.
Szczepionki żywe lub żywe osłabione
Nie należy rozpoczynać leczenia lekiem Mavenklad® w ciągu 4–6 tygodni po szczepieniu szczepionkami żywymi lub żywymi osłabionymi ze względu na ryzyko ostrych infekcji szczepionkowych. W trakcie i po leczeniu kladyrybinem należy unikać szczepień szczepionkami żywymi lub żywymi osłabionymi aż do momentu powrotu liczby limfocytów pacjenta do normy.
Silne inhibitory transporterów ENT1, CNT3 i BCRP
Jedynym prawdopodobnym źródłem interakcji mających znaczenie kliniczne na poziomie absorpcji kladyrybiny jest białko odporności nowotworu piersi (BCRP lub ABCG2). Inhibicja BCRP w przewodzie pokarmowym może zwiększać doustną biodostępność i działanie ogólnoustrojowe kladyrybiny. Znane inhibitory BCRP, które in vivo mogą zmieniać farmakokinetykę substratów BCRP o 20%, obejmują eltrombopag.
Badania in vitro wskazują, że kladyrybina jest substancją podlegającą działaniu transporterów nukleozydowych: równoważnego (ENT1) i koncentrującego (CNT3). W związku z tym teoretycznie biodostępność, dystrybucja wewnątrzkomórkowa i wydalanie nerkowe kladyrybiny mogą być wpływowane przez silne inhibitory transporterów ENT1, CNT3 i BCRP, takie jak dilazep, nifedypina, nimodypina, cylostażol, sulindak i rezerpina. Jednak końcowy wpływ, wyrażony w zmianie potencjalnego działania kladyrybiny, trudno przewidzieć.
Chociaż znaczenie kliniczne takich interakcji jest nieznane, w trakcie 4- lub 5-dniowego leczenia kladyrybiną zaleca się unikanie jednoczesnego przyjmowania silnych inhibitorów ENT1, CNT3 i BCRP. Jeśli jest to niemożliwe, należy rozważyć dobór alternatywnych leków do jednoczesnego stosowania, które nie mają lub mają minimalne właściwości inhibitorów wobec transporterów ENT1, CNT3 i BCRP. Jeśli to niemożliwe, zaleca się zmniejszenie dawki leków zawierających takie substancje do minimalnej dawki koniecznej, rozdzielenie czasu przyjmowania leków oraz staranne monitorowanie stanu pacjenta.
Silne induktory transporterów BCRP i P-gp
Wpływ silnych induktorów transporterów eflukswych BCRP i białka glikoproteiny P (P-gp) na biodostępność i rozkład kladyrybiny nie był badany w specjalnych badaniach. Przy jednoczesnym przyjmowaniu silnych induktorów transporterów BCRP (np. kortykosteroidów) lub P-gp (np. ryfampicyny, zieleńsia pospolitego) należy uwzględnić możliwość osłabienia działania kladyrybiny.
Środki antykoncepcyjne hormonalne
Przy jednoczesnym przyjmowaniu kladyrybiny z doustnymi środkami antykoncepcyjnymi (etyniloestradiol i lewonorgestrel) nie stwierdzono klinicznie istotnej interakcji farmakokinetycznej z kladyrybiną. Dlatego nie należy oczekiwać, że jednoczesne stosowanie z kladyrybiną spowoduje zmniejszenie skuteczności środków antykoncepcyjnych hormonalnych (patrz rozdział „Stosowanie w okresie ciąży lub karmienia piersią”).
Szczególne ostrzeżenia i środki ostrożności.
Monitorowanie hematologiczne
Mechanizm działania kladyrydyny jest ściśle związany ze zmniejszeniem liczby limfocytów. Wpływ na liczbę limfocytów jest zależny od dawki. W badaniach klinicznych obserwowano również zmniejszenie liczby neutrofili, liczby erytrocytów, hematokrytu, hemoglobiny oraz liczby płytek krwi w porównaniu z wartościami wyjściowymi, choć te parametry zazwyczaj pozostawały w granicach normy.
Podczas stosowania kladyrydyny razem z innymi substancjami wpływającymi na profil hematologiczny, należy spodziewać się dodatkowych działań niepożądanych.
Liczba limfocytów powinna być oznaczana:
- przed rozpoczęciem leczenia w roku 1,
- przed rozpoczęciem leczenia w roku 2,
- po 2 i 6 miesiącach od rozpoczęcia leczenia w każdym roku leczenia; jeśli liczba limfocytów jest niższa niż 500 komórek/mm³, ten parametr należy aktywnie monitorować, aż do ponownego wzrostu wartości.
Informacje dotyczące decyzji terapeutycznych opartych na liczbie limfocytów znajdują się w sekcji „Sposób podania i dawki” oraz w podsekcji „Infekcje” poniżej.
Infekcje
Kladyrydyna może osłabiać odporność organizmu, zwiększając tym samym ryzyko wystąpienia infekcji. Podczas leczenia lekiem Mavenklad® obserwowano poważne, ciężkie i oportunistyczne infekcje, w tym przypadki zakończone śmiercią. Zakażenie HIV, aktywny gruźlica oraz aktywne zapalenie wątroby należy wykluczyć przed rozpoczęciem leczenia kladyrydyną (patrz sekcja „Przeciwwskazania”).
Podczas leczenia mogą aktywizować się infekcje utajone, w tym gruźlica lub zapalenie wątroby. Dlatego przed rozpoczęciem terapii w roku 1 i roku 2 należy przeprowadzić badania w kierunku obecności infekcji utajonych, w szczególności gruźlicy oraz zapalenia wątroby typu B i C. Rozpoczęcie leczenia lekiem Mavenklad® należy odłożyć do skutecznego wyleczenia infekcji.
U pacjentów z ostrymi infekcjami należy również rozważyć odroczenie rozpoczęcia leczenia kladyrydyną, aż do pełnego kontrolowania infekcji.
Szczególną uwagę należy zwrócić na pacjentów, którzy nie mieli kontaktu z wirusem ospy wietrznej w wywiadzie. Przed rozpoczęciem terapii kladyrydyną zaleca się szczepienie pacjentów bez przeciwciał przeciwko osie wietrznej. W takim przypadku rozpoczęcie leczenia lekiem Mavenklad® należy odłożyć o 4–6 tygodni, aby osiągnąć pełny efekt szczepienia.
U pacjentów przyjmujących kladyrydynę obserwowano zwiększoną częstość występowania opryszczki półpasiec. Jeśli liczba limfocytów spadnie poniżej 200 komórek/mm³, należy rozważyć profilaktykę przeciwwirusową przeciwko wirusowi opryszczki zgodnie z lokalnymi standardowymi protokołami podczas limfopenii stopnia 4.
U pacjentów z liczbą limfocytów poniżej 500 komórek/mm³ należy aktywnie monitorować objawy i oznaki sugerujące obecność infekcji, w szczególności opryszczki półpasiec. Jeśli takie objawy i oznaki występują, należy rozpocząć leczenie infekcji zgodnie z wskazaniami klinicznymi. Można rozważyć wstrzymanie lub odroczenie leczenia lekiem Mavenklad® do skutecznego wyleczenia infekcji.
Zgłaszano przypadki postępującej wielofocalnej leukoencefalopatii (PML) podczas stosowania leków kladyrydyny dożylnej u pacjentów leczonych z powodu białaczki włosieniastej według innych schematów leczenia.
Chociaż nie zgłaszano przypadków PML podczas stosowania tabletek kladyrydyny, przed rozpoczęciem przyjmowania tabletek Mavenklad® (zazwyczaj w ciągu 3 miesięcy) należy wykonać badanie MRI jako podstawowe.
Nowotwory złośliwe
W badaniach klinicznych przypadki nowotworów złośliwych obserwowano częściej u pacjentów przyjmujących kladyrydynę w porównaniu z pacjentami przyjmującymi placebo.
Mavenklad® jest przeciwwskazany u pacjentów z SM z aktywnymi nowotworami złośliwymi (patrz sekcja „Przeciwwskazania”). U pacjentów z wcześniejszymi nowotworami złośliwymi należy przeprowadzić indywidualną ocenę korzyści i ryzyka stosowania leku Mavenklad® przed rozpoczęciem leczenia. Pacjentom przyjmującym kladyrydynę należy zalecić przestrzeganie standardowych zaleceń dotyczących badań przesiewowych na nowotwory.
Funkcja wątroby
Rzadko zgłaszano przypadki uszkodzenia wątroby, w tym poważne przypadki, u pacjentów leczonych lekiem Mavenklad®.
Przed przepisaniem leku Mavenklad® należy wziąć pod uwagę pełen wywiad pacjenta, biorąc pod uwagę wcześniejsze epizody uszkodzenia wątroby innymi lekami oraz istniejące zaburzenia wątrobowe. Przed rozpoczęciem leczenia w roku 1 i roku 2 należy oznaczyć poziomy aminotransferaz, fosfatazy alkalicznej oraz bilirubiny ogólnej w surowicy krwi pacjenta. Podczas leczenia należy monitorować enzymy wątrobowe i bilirubinę na podstawie objawów i objawów klinicznych.
Jeśli u pacjenta wystąpią objawy kliniczne, wzrost poziomu enzymów wątrobowych o nieustalonej przyczynie lub objawy sugerujące dysfunkcję wątroby (np. nudności, wymioty, ból brzucha, osłabienie, anoreksja o nieustalonej przyczynie lub żółtaczka i/lub ciemne zabarwienie moczu), należy natychmiast oznaczyć poziomy transaminaz surowicy i bilirubiny ogólnej. W razie potrzeby leczenie lekiem Mavenklad® należy wstrzymać lub przerwać.
Antykoncepcja
Przed rozpoczęciem leczenia w roku 1 i roku 2 należy poinformować kobiety w wieku rozrodczym oraz mężczyzn, którzy potencjalnie mogą zostać ojcami, o potencjalnym poważnym ryzyku dla płodu oraz konieczności zastosowania skutecznych środków antykoncepcyjnych.
Kobiety w wieku rozrodczym powinny zapobiegać zajściu w ciążę, stosując skuteczne środki antykoncepcyjne podczas leczenia kladyrydyną i co najmniej przez 6 miesięcy po przyjęciu ostatniej dawki.
Pacjenci-mężczyźni powinni podejmować środki zapobiegające zajściu w ciążę u swojej partnerki podczas leczenia kladyrydyną i co najmniej przez 6 miesięcy po przyjęciu ostatniej dawki.
Przetaczanie krwi
U pacjentów wymagających przetaczania krwi zaleca się napromieniowanie komórkowych składników krwi przed podaniem w celu zapobiegania chorobie przeszczep przeciwko gospodarzowi. Zaleca się konsultację z lekarzem hematologiem.
Przejście na leczenie kladyrydyną i przejście z niego
Przed rozpoczęciem leczenia pacjentów, którzy wcześniej przyjmowali leki immunomodulujące lub immunosupresyjne, należy wziąć pod uwagę mechanizm działania oraz długość działania tych leków. Potencjalny dodatkowy wpływ na układ odpornościowy należy również uwzględnić, jeśli takie leki są stosowane po leczeniu lekiem Mavenklad®.
Przy przejściu z innego leku stosowanego w leczeniu SM zaleca się wykonanie podstawowego badania MRI (patrz podsekcja „Infekcje” powyżej).
Zaburzenia funkcji wątroby
Stosowanie kladyrydyny nie jest zalecane u pacjentów z umiarkowanym lub ciężkim zaburzeniem funkcji wątroby (klasa Childa-Puga > 6).
Sorbit
Mavenklad® zawiera sorbit. W przypadku stwierdzonej nietolerancji niektórych cukrów należy skonsultować się z lekarzem przed rozpoczęciem przyjmowania tego leku.
Należy wziąć pod uwagę dodatkowy wpływ leków stosowanych równocześnie, które zawierają sorbit (lub fruktozę), oraz spożycie sorbitu (lub fruktozy) z pożywieniem.
Obecność sorbitu w lekach doustnych może wpływać na biodostępność innych leków doustnych stosowanych równocześnie.
Stosowanie w czasie ciąży lub karmienia piersią.
Antykoncepcja u mężczyzn i kobiet
Przed rozpoczęciem leczenia w roku 1 i roku 2 należy poinformować kobiety w wieku rozrodczym oraz mężczyzn, którzy potencjalnie mogą zostać ojcami, o potencjalnym poważnym ryzyku dla płodu oraz konieczności zastosowania skutecznych środków antykoncepcyjnych.
U kobiet w wieku rozrodczym przed rozpoczęciem leczenia lekiem Mavenklad® w latach 1 i 2 należy wykluczyć ciążę, a zajściu w ciążę należy zapobiegać, stosując skuteczne środki antykoncepcyjne podczas leczenia kladyrydyną i co najmniej przez 6 miesięcy po przyjęciu ostatniej dawki. Kobiety, które zajęły w ciążę podczas leczenia lekiem Mavenklad®, powinny przerwać terapię.
Ponieważ kladyrydyna wpływa na syntezę DNA, należy spodziewać się negatywnego wpływu na gametogenezę u ludzi. Dlatego pacjenci-mężczyźni powinni podejmować środki zapobiegające zajściu w ciążę u swojej partnerki podczas leczenia kladyrydyną i co najmniej przez 6 miesięcy po przyjęciu ostatniej dawki.
Ciąża
Ze względu na doświadczenie z zastosowania innych substancji hamujących syntezę DNA u ludzi, kladyrydyna może powodować wady wrodzone u płodu w przypadku przyjmowania w czasie ciąży. W badaniach na zwierzętach wykazano toksyczność reprodukcyjną kladyrydyny.
Mavenklad® jest przeciwwskazany u kobiet w ciąży (patrz sekcja „Przeciwwskazania”).
Karmienie piersią
Ograniczone dane uzyskane z doniesień spontanicznych wskazują, że kladyrydyna wydzielana jest w mleku matki, choć nie ustalono jeszcze, w jakiej ilości. Ze względu na potencjalne poważne działania niepożądane u niemowląt karmionych piersią, karmienie piersią jest przeciwwskazane podczas leczenia lekiem Mavenklad® i przez 1 tydzień po przyjęciu ostatniej dawki (patrz sekcja „Przeciwwskazania”).
Niepłodność
U myszy nie obserwowano wpływu kladyrydyny na płodność ani funkcję rozrodczą potomstwa, jednak u myszy i małp obserwowano wpływ na funkcję jąder.
Ponieważ kladyrydyna wpływa na syntezę DNA, należy spodziewać się negatywnego wpływu na gametogenezę u ludzi. Dlatego pacjenci-mężczyźni powinni podejmować środki zapobiegające zajściu w ciążę u swojej partnerki podczas leczenia kladyrydyną i co najmniej przez 6 miesięcy po przyjęciu ostatniej dawki.
Wpływ na zdolność prowadzenia pojazdów lub obsługiwanie maszyn.
Mavenklad® nie wpływa lub prawie nie wpływa na zdolność pacjentów do prowadzenia samochodów i obsługiwanie innych mechanizmów.
Sposób stosowania i dawki
Leczenie powinno być rozpoczynane i prowadzone pod nadzorem lekarza posiadającego doświadczenie w leczeniu SM.
Dawki
Zalecana dawka kumulacyjna leku wynosi 3,5 mg/kg masy ciała w ciągu 2 lat i jest stosowana w postaci jednego cyklu leczenia o dawce 1,75 mg/kg rocznie. Każdy cykl leczenia składa się z 2 tygodni leczenia: jeden na początku pierwszego miesiąca, a drugi na początku drugiego miesiąca odpowiedniego roku leczenia. W przypadku wskazań medycznych (np. w celu przywrócenia liczby limfocytów) cykl leczenia w roku 2 może zostać odłożony do 6 miesięcy. Każdy tydzień leczenia składa się z 4 lub 5 dni, w których pacjent przyjmuje 10 mg lub 20 mg (jedną lub dwie tabletki) w postaci pojedynczej dawki dobowej, w zależności od masy ciała. Dalsze szczegółowe informacje podano w tabelach 2 i 3.
Po zakończeniu 2 cykli leczenia dalsze leczenie kladrabiny w latach 3 i 4 nie jest wymagane. Ponowne rozpoczęcie leczenia po 4 latach nie było badane.
Kryteria rozpoczęcia i kontynuacji terapii
Liczba limfocytów musi być:
- w normie przed rozpoczęciem leczenia w roku 1,
- co najmniej 800 komórek/mm³ przed rozpoczęciem leczenia w roku 2.
W razie potrzeby, w celu przywrócenia liczby limfocytów, cykl leczenia w roku 2 może być odłożony do 6 miesięcy. Jeśli przywrócenie to wymaga więcej niż 6 miesięcy, pacjent nie może więcej przyjmować kladrabiny.
Podział dawki
Podział całkowitej dawki w ciągu 2 lat leczenia przedstawiono w tabeli 2. U niektórych pacjentów, ze względu na masę ciała, liczba tabletek może różnić się w pierwszym i drugim tygodniu leczenia. Stosowanie doustnych form kladrabiny u pacjentów o masie ciała mniejszej niż 40 kg nie było badane.
Tabela 2 Dawka kladrabiny w zależności od masy ciała pacjenta w tygodniu leczenia każdego roku leczenia
| Zakres masy ciała, kg |
Dawka w mg (liczba tabletek 10 mg) w tydzień leczenia |
|
| Tydzień leczenia 1 |
Tydzień leczenia 2 |
|
| od 40 do < 50 |
40 mg (4 tabletki) |
40 mg (4 tabletki) |
| od 50 do < 60 |
50 mg (5 tabletek) |
50 mg (5 tabletek) |
| od 60 do < 70 |
60 mg (6 tabletek) |
60 mg (6 tabletek) |
| od 70 do < 80 |
70 mg (7 tabletek) |
70 mg (7 tabletek) |
| od 80 do < 90 |
80 mg (8 tabletek) |
70 mg (7 tabletek) |
| od 90 do < 100 |
90 mg (9 tabletek) |
80 mg (8 tabletek) |
| od 100 do < 110 |
100 mg (10 tabletek) |
90 mg (9 tabletek) |
| od 110 i więcej |
100 mg (10 tabletek) |
100 mg (10 tabletek) |
W tabeli 3 pokazano, jak rozłożyć ogólną liczbę tabletek na tydzień leczenia na poszczególne dni. Zaleca się przyjmowanie każdej dobowej dawki kladyrybiny w każdym tygodniu leczenia w odstępach 24 godzin, mniej więcej o tej samej porze dnia. Jeśli dawka dobową stanowią dwie tabletki, obie tabletki przyjmuje się razem jako jedną dawkę.
| Tabela 3 Rozkład tabletek według dni tygodnia |
|||||
| Ogólna liczba tabletek w ciągu tygodnia |
Dzień 1 |
Dzień 2 |
Dzień 3 |
Dzień 4 |
Dzień 5 |
| 4 |
1 |
1 |
1 |
1 |
0 |
| 5 |
1 |
1 |
1 |
1 |
1 |
| 6 |
2 |
1 |
1 |
1 |
1 |
| 7 |
2 |
2 |
1 |
1 |
1 |
| 8 |
2 |
2 |
2 |
1 |
1 |
| 9 |
2 |
2 |
2 |
2 |
1 |
| 10 |
2 |
2 |
2 |
2 |
2 |
Pominiętą dawkę należy przyjąć jak najszybciej po stwierdzeniu pominięcia tego samego dnia zgodnie z harmonogramem leczenia.
Nie należy przyjmować pominiętej dawki jednocześnie z kolejną dawką według harmonogramu leczenia następnego dnia. W przypadku pominiętej dawki pacjent powinien przyjąć ją następnego dnia i wydłużyć liczbę dni leczenia w tym tygodniu leczenia. Jeśli pominięto dwie kolejne dawki, stosuje się ten sam sposób postępowania, a liczba dni w tygodniu leczenia wydłuża się o dwa dni.
Stosowanie równoczesne innych leków doustnych
W ciągu ograniczonej liczby dni przyjmowania kladyrybiny zaleca się oddzielenie przyjmowania innych leków doustnych od przyjmowania leku Mavenklad® o co najmniej 3 godziny.
Osoby szczególnej grupy pacjentów
Pacjenci z zaburzeniem funkcji nerek
Nie przeprowadzono specjalnych badań leczenia pacjentów z zaburzeniem funkcji nerek.
U pacjentów z łagodnym zaburzeniem funkcji nerek (klirens kreatyniny od 60 do 89 ml/min) nie jest konieczna korekta dawki.
Nie badano bezpieczeństwa i skuteczności stosowania kladyrybiny u pacjentów z umiarkowanym lub ciężkim zaburzeniem funkcji nerek. Dlatego kladyrybina jest przeciwwskazana tym pacjentom (patrz sekcja „Przeciwwskazania”).
Pacjenci z zaburzeniem funkcji wątroby
Nie przeprowadzono badań leczenia pacjentów z zaburzeniem funkcji wątroby.
Nie jest konieczna korekta dawki u pacjentów z łagodnym zaburzeniem funkcji wątroby, ponieważ znaczenie funkcji wątroby w wydalaniu kladyrybiny uważa się za nieistotne. Z powodu braku danych stosowanie kladyrybiny nie jest zalecane pacjentom z umiarkowanym lub ciężkim zaburzeniem funkcji wątroby (wskaźnik Childa-Puga > 6).
Pacjenci w wieku podeszłym
U pacjentów w wieku podeszłym kladyrybinę należy stosować z ostrożnością ze względu na potencjalnie większą częstość występowania zaburzeń funkcji wątroby lub nerek, choroby współistniejące oraz inne rodzaje terapii.
Sposób stosowania
Mavenklad® jest przeznaczony do stosowania doustnego. Tabletki należy przyjmować z wodą i połykać nie żując. Tabletki można przyjmować niezależnie od posiłku.
Ponieważ tabletki nie są powleczone powłoką, należy je połykać natychmiast po wyjęciu z blistrów, nie pozostawiać na powierzchni ani nie trzymać w rękach dłużej niż to konieczne do przyjęcia dawki. Jeśli tabletka została pozostawiona na powierzchni lub jeśli pękniętą lub zmiażdżoną tabletę wyjęto z blistra, należy dokładnie wypłukać wodą powierzchnię, z którą tabletka miała kontakt.
Pacjent powinien brać tabletki suchymi rękami, a następnie dokładnie umyć ręce.
Instrukcja samodzielnego przyjmowania tabletek Mavenklad® przez pacjenta
- Przygotuj szklankę wody i upewnij się, że Twoje ręce są suche i czyste.
- Weź pudełko z lekiem, obracając je stroną z drukowanymi instrukcjami w Twoją stronę.

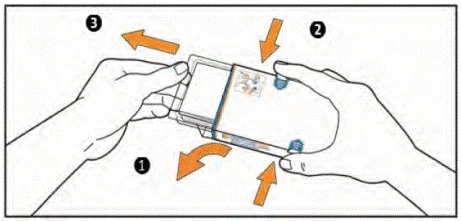
- (1) Otwórz klapkę z lewej strony pudełka.
(2) Jednocześnie naciśnij haczyki na pudełku z obu stron wskazującym i dużym palcem i trzymaj je w tej pozycji.
(3) Wyciągnij opakowanie foliowe, aż się przesunie. UWAGA: nie wyciągaj opakowania foliowego z pudełka.
- Wyjmij ulotkę dołączoną do leku. Upewnij się, że dokładnie zapoznałeś się z jej treścią, w tym z instrukcją samodzielnego przyjmowania tabletek.
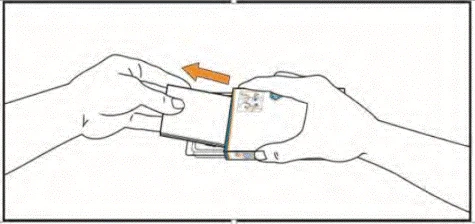
- Podnieś blister, naciskając palcami na otwór w opakowaniu foliowym. Umieść dłoń pod blisterem i wyciśnij do dłoni 1 lub 2 tabletki, zgodnie z zaleceniem lekarza.
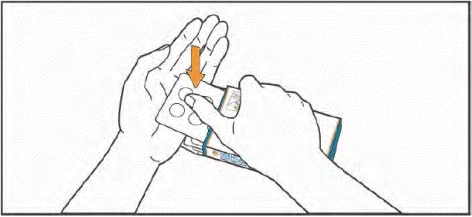
- Przyjmij tabletki, popijając je wodą. Tabletki należy połykać całe, nie żując i nie dopuszczając do ich rozpuszczenia w jamie ustnej. Kontakt tabletek z powierzchnią skóry należy ograniczyć. Podczas przyjmowania tabletek nie dotykaj rękami nosa, oczu ani innych części ciała.
- Po przyjęciu tabletek dokładnie umyj ręce wodą z mydłem.
- Włóż opakowanie foliowe z powrotem do pudełka. Przechowuj je w oryginalnym opakowaniu w celu ochrony przed wilgocią.
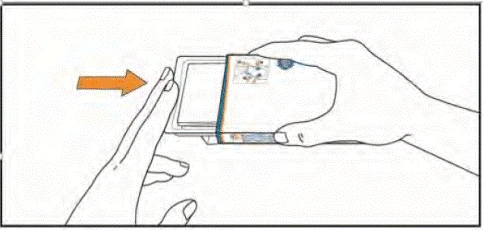
Do następnego przyjęcia dawki przechowuj tabletki w blisterze. Nie wyciskaj tabletek z blistera i nie przechowuj ich w innym pojemniku.
Dzieci.
U dzieci (do 18 roku życia) bezpieczeństwo i skuteczność stosowania leku Mavenklad® nie zostały ustalone. Brak odpowiednich danych.
Przedawkowanie.
Doświadczenie w zakresie przedawkowania kladyrybiny przy doustnym przyjmowaniu jest ograniczone. Wiadomo, że limfopenia, która się wówczas rozwija, jest zależna od dawki.
U pacjentów z przedawkowaniem kladyrybiny zaleca się szczególnie staranne monitorowanie parametrów hematologicznych.
Nie znano specyficznego antydota do kladyrybiny. Leczenie polega na starannym obserwowaniu stanu pacjenta i podejmowaniu odpowiednich działań wspomagających. Należy wziąć pod uwagę możliwość konieczności odstawienia leku Mavenklad®. Ze względu na szybki i rozległy wewnątrzkomórkowy oraz tkankowy rozdział mało prawdopodobne jest znaczne wydalenie kladyrybiny w trakcie dializy krwi.
Efekty uboczne
Streszczenie profilu bezpieczeństwa
Najbardziej klinicznie istotnymi efektami ubocznymi są limfopenia (25,6%) oraz opryszcz zoster (3,0%). Częstość występowania opryszczu zoster była wyższa w okresie, w którym obserwowano limfopenię stopnia 3 lub 4 (od < 500 do 200 komórek/mm³ lub < 200 komórek/mm³) w porównaniu z okresem, w którym pacjenci nie mieli limfopenii stopnia 3 lub 4.
Wykaz efektów ubocznych
Informacja o efektach ubocznych opisanych poniżej pochodzi z połączonych danych z badań klinicznych leczenia SM lekiem doustnym kladyrybinem stosowanym jako monoterapia w dawce skumulowanej 3,5 mg/kg. Baza danych bezpieczeństwa z tych badań obejmuje dane 923 pacjentów. Efekty uboczne ustalone w okresie pozarejestracyjnym oznaczono znakiem przypisu (*).
Do określenia częstości występowania efektów ubocznych stosowana jest następująca klasyfikacja: bardzo często (≥ 1/10), często (od ≥ 1/100 do < 1/10), rzadko (od ≥ 1/1000 do < 1/100), pojedyncze przypadki (od ≥ 1/10000 do < 1/1000), bardzo rzadko (< 1/10000), częstość nieznana (nie można ustalić na podstawie dostępnych danych).
Infekcje i inwazje
Często: opryszcz ustny, opryszcz zoster skóry.
Bardzo rzadko: gruźlica.
Zaburzenia układu krwi i chłonnego
Bardzo często: limfopenia.
Często: zmniejszenie liczby neutrofili.
Zaburzenia układu odpornościowego
Często: reakcje nadwrażliwości*, w tym świąd, pokrzywka, wysypka oraz pojedyncze przypadki obrzęku naczynioruchowego.
Zaburzenia wątrobowo-żółciowe
Rzadko: uszkodzenie wątroby*.
Zaburzenia skóry i tkanki podskórnej
Często: wysypka, wypadanie włosów.
Opis wybranych efektów ubocznych
Limfopenia
W badaniach klinicznych tymczasowa limfopenia stopnia 3 lub 4 rozwinęła się u 20–25% pacjentów przyjmujących skumulowaną dawkę kladyrybiny 3,5 mg/kg przez 2 lata jako monoterapię. Limfopenia stopnia 4 występowała u mniej niż 1% pacjentów. Największy odsetek pacjentów z limfopenią stopnia 3 lub 4 stwierdzono po 2 miesiącach od przyjęcia pierwszej dawki kladyrybiny w każdym roku (4,0% i 11,3% pacjentów z limfopenią stopnia 3 w roku 1 i roku 2, 0% i 0,4% pacjentów z limfopenią stopnia 4 w roku 1 i roku 2). U większości pacjentów oczekuje się przywrócenia liczby limfocytów do normy lub do limfopenii stopnia 1 w ciągu 9 miesięcy.
W celu zmniejszenia ryzyka rozwoju ciężkiej limfopenii należy określać liczbę limfocytów przed, w trakcie i po leczeniu kladyrybiną oraz należy przestrzegać rygorystycznych kryteriów rozpoczęcia i kontynuacji leczenia kladyrybiną.
Nowotwory złośliwe
W badaniach klinicznych oraz w długim okresie dalszej obserwacji pacjentów przyjmujących skumulowaną dawkę kladyrybiny 3,5 mg/kg doustnie, przypadki nowotworów złośliwych występowały częściej u pacjentów leczonych kladyrybiną (10 przypadków na 3414 pacjentolat [0,29 przypadku na 100 pacjentolat]) niż u pacjentów przyjmujących placebo (3 przypadki na 2022 pacjentolat [0,15 przypadku na 100 pacjentolat]).
Nadwrażliwość
W badaniach klinicznych przypadki nadwrażliwości występowały częściej u pacjentów przyjmujących skumulowaną dawkę 3,5 mg/kg kladyrybiny doustnie (11,8%) niż u pacjentów przyjmujących placebo (8,4%). Ciężkie przypadki nadwrażliwości występowały u 0,3% pacjentów przyjmujących kladyrybinę i nie występowały u pacjentów przyjmujących placebo. Efekty uboczne w postaci nadwrażliwości prowadziły do przerwania leczenia u 0,4% pacjentów przyjmujących kladyrybinę oraz u 0,3% pacjentów przyjmujących placebo.
Uszkodzenie wątroby
W okresie pozarejestracyjnym zgłaszano rzadkie przypadki uszkodzenia wątroby, w tym przypadki ciężkie oraz prowadzące do przerwania leczenia, które miały związek czasowy z zastosowaniem leku Mavenklad®. Tymczasowy wzrost stężenia aminotransferaz surowicy zwykle przekraczał górny limit normy (GLN) ponad 5-krotnie. Obserwowano pojedyncze przypadki tymczasowego wzrostu stężenia aminotransferaz surowicy do 40-krotnie wyższego niż GLN i/lub zapalenia wątroby z objawami oraz tymczasowym wzrostem stężenia bilirubiny i żółtaczką. Zdarzenia te występowały w różnym czasie od rozpoczęcia leczenia, jednak większość przypadków obserwowano w ciągu 8 tygodni od pierwszego cyklu leczenia (patrz sekcja „Szczególne ostrzeżenia i środki ostrożności stosowania”).
Zgłaszanie podejrzewanych efektów ubocznych po rejestracji leku ma istotne znaczenie. Umożliwia to monitorowanie stosunku korzyści do ryzyka związanego z zastosowaniem tego leku. Osoby medyczne i farmaceutyczne, a także pacjenci lub ich prawni przedstawiciele powinni zgłaszać wszystkie przypadki podejrzewanych efektów ubocznych oraz braku skuteczności leku poprzez Automatyczny System Informacyjny nadzoru farmakologicznego pod adresem: https://aisf.dec.gov.ua.
Okres ważności. 4 lata.
Nie stosować po upływie terminu ważności podanego na opakowaniu.
Warunki przechowywania.
Przechowywać w oryginalnym opakowaniu w celu ochrony przed wilgocią.
Przechowywać w miejscu niedostępnym dla dzieci.
Opakowanie. Po 1, 4 lub 6 tabletek w blistrze aluminiowym, zapieczętowanym w tekturowej kopercie, umieszczonych w opakowaniu konturowym i włożonych do tekturowego pudełka zabezpieczonego przed dostępem dzieci.
Kategoria wydania. Na receptę.
Producent. NerPharMa S.R.L. / NerPharMa S.R.L.
Miejsce produkcji i adres siedziby.
Viale Pasteur 10 (loc. Nerviano), 20014 Milan (MI), Włochy /
Viale Pasteur 10 (loc. Nerviano), 20014 Milan (MI), Italy.