Mavenclad®
Ucrania
Contenido
INSTRUCCIONES PARA USO MÉDICO DEL MEDICAMENTO MAVENKLAD® (MAVENKLAD®)
Composición:
Principio activo: cladribina;
1 tableta contiene 10 mg de cladribina;
Sustancias auxiliares: hidroxipropilbetadex, sorbitol (E 420), estearato de magnesio.
Forma farmacéutica. Tabletas.
Propiedades físico-químicas principales: tabletas blancas, redondas, biconvexas, de 8,5 mm de diámetro, con la impresión «C» en un lado y «10» en el otro.
Grupo farmacoterapéutico. Inmunosupresores selectivos. Cladribina.
Código ATC L04A A40.
Propiedades farmacodinámicas.
Farmacodinámica.
Mecanismo de acción
Cladribina es un análogo nucleósido de la desoxiadenosina. La sustitución por un átomo de cloro en el anillo de purina protege a la cladribina de la degradación por la adenosina desaminasa, aumentando el tiempo intracelular de permanencia de los profármacos de cladribina. La posterior fosforilación de cladribina, que conduce a la formación de su forma activa, el trifosfato 2-cloro-desoxiadenosina trifosfato (Cd-ATP), se logra especialmente de manera eficaz en los linfocitos debido a que estos poseen niveles constitutivamente altos de desoxicitidina quinasa (DCK) y niveles relativamente bajos de 5'-nucleotidasa (5'-NTasa). La elevada relación DCK/5'-NTasa favorece la acumulación de Cd-ATP, lo que hace que los linfocitos sean particularmente sensibles a la muerte celular. Debido a la menor relación DCK/5'-NTasa, otras células derivadas de la médula ósea se ven afectadas en menor grado que los linfocitos. La DCK es la enzima limitante en la conversión de cladribina a su forma trifosfato activa, lo que conduce a un agotamiento selectivo de linfocitos T y B, tanto en división como en reposo.
El mecanismo principal de acción de Cd-ATP que induce apoptosis ejerce un efecto directo e indirecto sobre la síntesis de ADN y la función mitocondrial. En células en división, Cd-ATP interfiere con la síntesis de ADN mediante la inhibición de la ribonucleótido reductasa y compite con el desoxiadenosina trifosfato por su incorporación en el ADN mediante las ADN polimerasas. En células en reposo, la cladribina provoca rupturas de cadena simple en el ADN, un rápido agotamiento de nicotinamida-adenina dinucleótido, escisión de ATP y muerte celular. Existen evidencias de que la cladribina también puede inducir apoptosis directa, dependiente e independiente de caspasa, mediante la liberación de citocromo C y del factor inductor de apoptosis al citosol de células no proliferantes.
La patogénesis de la esclerosis múltiple (EM) implica una cascada compleja de fenómenos en los que intervienen diversos tipos de células inmunitarias, incluyendo linfocitos T y B autorreactivos. El mecanismo terapéutico de acción de la cladribina en la EM aún no se ha comprendido completamente, pero se considera que su efecto principal sobre los linfocitos T y B interrumpe la cascada de fenómenos inmunitarios clave en la EM.
Diferentes niveles de expresión de DCK y 5'-NTasa en subtipos de células inmunitarias podrían explicar la diferencia en la sensibilidad de estas células a la cladribina. Debido a estos niveles de expresión, las células del sistema inmunitario innato se ven menos afectadas que las del sistema inmunitario adaptativo.
Efectos farmacodinámicos
Se ha demostrado que la cladribina ejerce un efecto prolongado, afectando principalmente a los linfocitos y a los procesos autoinmunitarios implicados en la fisiopatología de la EM.
En los estudios clínicos, la mayor proporción de pacientes con linfopenia grado 3 o 4 (entre < 500 y 200 células/mm³ o < 200 células/mm³) se observó a los 2 meses después de la primera dosis de cladribina cada año, lo que indica un intervalo de tiempo entre la presencia de cladribina en plasma y el efecto hematológico máximo.
Los datos obtenidos en estudios clínicos con la dosis acumulativa propuesta de 3,5 mg/kg de peso corporal indican una recuperación gradual de la mediana del recuento de linfocitos hasta el rango normal a la semana 84 tras la primera dosis de cladribina (aproximadamente 30 semanas tras la última dosis de cladribina). El recuento de linfocitos volvió al rango normal en más del 75 % de los pacientes para la semana 144 tras la primera dosis de cladribina (aproximadamente 90 semanas tras la última dosis de cladribina).
El tratamiento con el fármaco oral de cladribina conduce a una rápida reducción en sangre de los linfocitos T CD4+ y CD8+. La reducción es menos pronunciada y la recuperación más rápida en los linfocitos CD8+ que en los CD4+, lo que provoca una disminución temporal de la relación CD4/CD8. La cladribina reduce también el número de linfocitos B CD19+ y de células asesinas naturales CD16+/CD56+, cuyos recuentos también se recuperan más rápidamente que los de los linfocitos T CD4+.
Eficacia y seguridad clínicas
EM recurrente-remitente
La eficacia y seguridad del uso oral de cladribina se evaluaron en un estudio clínico aleatorizado, doble ciego, controlado con placebo (CLARITY) con 1326 pacientes con EM recurrente-remitente. El objetivo del estudio fue evaluar la eficacia de la cladribina frente a placebo en la reducción de la tasa anualizada de recaídas (ARR) (punto final primario), la desaceleración de la progresión de la discapacidad y la reducción de focos activos en resonancia magnética (RM).
Los pacientes recibieron o bien placebo (n = 437), o bien una dosis acumulativa de cladribina de 3,5 mg/kg (n = 433) o 5,25 mg/kg de peso corporal (n = 456) durante un período de estudio de 96 semanas (2 años), compuesto por dos ciclos de tratamiento. Los pacientes fueron aleatorizados para recibir una dosis acumulativa de 3,5 mg/kg durante el primer ciclo de tratamiento en las semanas 1 y 5 del primer año y durante el segundo ciclo de tratamiento en las semanas 1 y 5 del segundo año. Otros pacientes fueron aleatorizados para recibir una dosis acumulativa de 5,25 mg/kg durante un período adicional de tratamiento en las semanas 9 y 13 del primer año. La mayoría de los pacientes en el grupo placebo (87,0 %) y en los grupos tratados con cladribina a dosis de 3,5 mg/kg (91,9 %) y 5,25 mg/kg (89,0 %) completaron el período completo de 96 semanas del estudio.
Los pacientes debían haber presentado al menos una recaída en los 12 meses previos. En la población total estudiada, la mediana de edad de los pacientes fue de 39 años (rango de 18 a 65 años), y la proporción mujeres/hombres fue aproximadamente 2:1. La duración media de la enfermedad de EM antes de la inclusión en el estudio fue de 8,7 años, y la puntuación mediana basal de discapacidad neurológica, medida mediante la Escala Ampliada de Estado de Discapacidad (EDSS) en todos los grupos de tratamiento, fue de 3,0 puntos (rango de 0 a 6,0 puntos). Más de dos tercios de los pacientes no habían recibido previamente tratamientos modificadores del curso de la EM antes del estudio. El resto de los pacientes había sido tratado previamente con interferón beta-1a, interferón beta-1b, acetato de glatiramero o natalizumab.
En pacientes con EM recurrente-remitente que recibieron cladribina a la dosis de 3,5 mg/kg, se observó una mejora estadísticamente significativa en comparación con los pacientes que recibieron placebo, respecto a la tasa anualizada de recaídas, la proporción de pacientes sin recaídas durante 96 semanas, la proporción de pacientes sin discapacidad sostenida durante 96 semanas y el tiempo hasta la progresión de la EDSS durante 3 meses (ver tabla 1).
| Tabla 1 Resultados clínicos del estudio CLARITY (96 semanas) |
|||
| Parámetro |
Placebo |
Dosis acumulada de cladribina |
|
| 3,5 mg/kg |
5,25 mg/kg |
||
| Frecuencia anualizada de recaídas (IC 95 %) |
0,33 (0,29; 0,38) |
0,14* (0,12; 0,17) |
0,15* (0,12; 0,17) |
|
57,6 % |
54,5 % |
|
| Proporción de pacientes sin recaídas durante 96 semanas |
60,9 % |
79,7 % |
78,9 % |
| Tiempo hasta la progresión confirmada del EDSS durante 3 meses, percentil 10 (meses) |
10,8 |
13,6 |
13,6 |
|
0,67 (0,48; 0,93) p = 0,018 |
0,69 (0,49; 0,96) p = 0,026 |
|
| IC – intervalo de confianza * p < 0,001 frente al placebo. |
|||
Los resultados del tratamiento con cladribina a una dosis de 3,5 mg/kg fueron estadísticamente superiores al placebo respecto al número y reducción relativa de lesiones activas en T1 con realce de gadolinio (T1 Gd+), lesiones activas en T2 y lesiones únicas combinadas en la resonancia magnética cerebral durante los 96 semanas completas del estudio. Los pacientes que recibieron cladribina mostraron, en comparación con el grupo placebo, una reducción relativa del 86 % en el número medio de lesiones T1 Gd+ (la media ajustada para el grupo de cladribina 3,5 mg/kg y el grupo placebo fue de 0,12 y 0,91, respectivamente), una reducción relativa del 73 % en el número medio de lesiones activas en T2 (la media ajustada para el grupo de cladribina 3,5 mg/kg y el grupo placebo fue de 0,38 y 1,43, respectivamente) y una reducción relativa del 74 % en el número medio de lesiones únicas combinadas por escáner cerebral del paciente (la media ajustada para el grupo de cladribina 3,5 mg/kg y el grupo placebo fue de 0,43 y 1,72, respectivamente) (p < 0,001 en los tres resultados de resonancia magnética).
Un análisis retrospectivo del tiempo hasta la progresión confirmada a los 6 meses según la puntuación EDSS reveló una reducción del 47 % en el riesgo de progresión de la discapacidad en el grupo que recibió cladribina a una dosis de 3,5 mg/kg en comparación con el grupo placebo (riesgo relativo 0,53; IC del 95 % [0,36; 0,79], p < 0,05). En el grupo placebo, el percentil 10 para la progresión de la discapacidad se alcanzó a los 245 días, mientras que en el grupo de cladribina 3,5 mg/kg no se alcanzó durante todo el período del estudio.
Como se muestra en la tabla 1, dosis acumuladas más altas de cladribina no proporcionaron ventajas clínicamente significativas, pero se asociaron con una mayor frecuencia de linfopenia de grado ≥ 3 (44,9 % en el grupo de 5,25 mg/kg frente al 25,6 % en el grupo de 3,5 mg/kg).
Los pacientes que finalizaron su participación en el estudio CLARITY pudieron ser incluidos en el estudio CLARITY Extension, cuyo objetivo primario fue evaluar la seguridad. En este estudio ampliado, 806 pacientes recibieron placebo o una dosis acumulada de cladribina de 3,5 mg/kg (según un régimen similar al utilizado en CLARITY) durante un período de 96 semanas.
El efecto sobre la reducción de la frecuencia de recaídas y la desaceleración de la progresión de la discapacidad en pacientes que recibieron una dosis de 3,5 mg/kg durante 2 años se mantuvo durante los años 3 y 4.
Eficacia del tratamiento en pacientes con alta actividad de la enfermedad
Se realizó retrospectivamente un análisis de eficacia en subgrupos de pacientes con alta actividad de la enfermedad que recibieron cladribina por vía oral en la dosis acumulada recomendada de 3,5 mg/kg. Estos subgrupos incluyeron:
- pacientes con 1 recaída durante el año anterior y al menos 1 lesión T1 Gd+ o 9 o más lesiones T2 durante el tratamiento con otros fármacos modificadores del curso de la enfermedad,
- pacientes con 2 o más recaídas durante el año anterior, independientemente de si estaban o no en tratamiento con fármacos modificadores del curso de la enfermedad.
El análisis de los datos obtenidos en el estudio CLARITY mostró un impacto similar del tratamiento sobre la frecuencia de recaídas, siendo la tasa anualizada de recaídas de entre 0,16 y 0,18 en los grupos de cladribina y de entre 0,47 y 0,50 en el grupo placebo (p < 0,0001). En comparación con la población general, se observó un mayor impacto del tratamiento sobre el tiempo hasta la discapacidad establecida a los 6 meses, donde la cladribina redujo el riesgo de progresión de la discapacidad en un 82 % (riesgo relativo 0,18; IC del 95 % [0,07; 0,47]). En el grupo placebo, el percentil 10 para la progresión de la discapacidad se alcanzó entre las semanas 16 y 23, mientras que en los grupos de cladribina no se alcanzó durante todo el período del estudio.
Esclerosis múltiple secundariamente progresiva con recaídas
Un estudio adicional sobre la administración de cladribina junto con interferón beta frente a placebo más interferón beta también incluyó un número limitado de pacientes con esclerosis múltiple secundariamente progresiva (26 pacientes). En estos pacientes, el tratamiento con cladribina a una dosis de 3,5 mg/kg redujo la tasa anualizada de recaídas en comparación con el placebo (0,03 frente a 0,30; razón de riesgos: 0,11; p < 0,05). No se observaron diferencias en la tasa anualizada de recaídas entre pacientes con esclerosis múltiple recurrente-remitente y pacientes con esclerosis múltiple secundariamente progresiva con recaídas. No se demostró el impacto del fármaco sobre la progresión de la discapacidad en ninguno de estos subgrupos.
Los pacientes con esclerosis múltiple secundariamente progresiva fueron excluidos del estudio CLARITY. Sin embargo, un análisis retrospectivo de grupos mixtos de pacientes de los estudios CLARITY y ONWARD, definidos por una puntuación basal en la escala EDSS ≥ 3,5 como indicador de esclerosis múltiple secundariamente progresiva, mostró una reducción similar en la tasa anualizada de recaídas en comparación con pacientes con puntuación EDSS menor de 3.
Farmacocinética.
La cladribina pertenece a los pro-fármacos y debe fosforilarse dentro de la célula para alcanzar su actividad biológica. La farmacocinética de la cladribina se ha estudiado tras administración oral e intravenosa en pacientes con esclerosis múltiple y en pacientes con neoplasias malignas, así como en sistemas in vitro.
Absorción
Tras la administración oral, la cladribina se absorbe rápidamente. La ingesta de 10 mg de cladribina produce una Cmax media de cladribina en plasma en el rango de 22 a 29 ng/ml y un valor medio de AUC correspondiente entre 80 y 101 ng·h/ml.
Tras la administración oral en ayunas, el valor mediano de Tmax fue de 0,5 horas (rango de 0,5 a 1,5 horas). Tras la ingesta con alimentos grasos, la absorción de cladribina se retrasó (valor mediano de Tmax de 1,5 horas, rango de 1 a 3 horas) y la Cmax se redujo en un 29 %, mientras que el valor de AUC permaneció sin cambios. La biodisponibilidad de una dosis oral de 10 mg de cladribina fue aproximadamente del 40 %.
Distribución
La cladribina tiene un volumen de distribución elevado, lo que indica una distribución extensa hacia los tejidos y una captación intracelular. En estudios realizados, se ha observado que el volumen medio de distribución de la cladribina se encuentra entre 480 y 490 l. La unión a proteínas plasmáticas es del 20 % y no depende de las concentraciones de cladribina en plasma.
La distribución de la cladribina a través de membranas biológicas se realiza mediante distintas proteínas de transporte, incluyendo ENT1, CNT3 y BCRP.
En estudios in vitro se ha demostrado que el eflujo de cladribina está mínimamente relacionado con P-gp, por lo que no se esperan interacciones clínicamente relevantes con inhibidores de P-gp.
Estudios in vitro mostraron una entrada insignificante de cladribina mediada por transportadores en hepatocitos humanos.
La cladribina tiene potencial para atravesar la barrera hematoencefálica. Un pequeño estudio en pacientes con cáncer mostró que la relación de concentraciones en líquido cefalorraquídeo/plasma es aproximadamente de 0,25.
La cladribina y/o sus metabolitos fosforilados se acumulan y retienen significativamente en los linfocitos humanos. En condiciones in vitro, se ha observado que la relación de acumulación intracelular/extracelular es aproximadamente de 30 a 40 ya a la hora tras el inicio de la exposición.
Biotransformación
El metabolismo de la cladribina se ha estudiado en pacientes con esclerosis múltiple tras la ingesta de una tableta de 10 mg y una dosis única intravenosa de 3 mg. Tras la administración oral e intravenosa, el componente principal detectado en plasma y orina fue el compuesto original de cladribina. El contenido de su metabolito 2-cloroadenina en plasma y orina fue insignificante. En plasma y orina solo se detectaron trazas de otros metabolitos de cladribina.
En sistemas hepáticos in vitro se observó un metabolismo insignificante de cladribina (al menos el 90 % fue cladribina sin modificar).
La cladribina no es un sustrato significativo de las enzimas del citocromo P450 y no muestra potencial significativo como inhibidor de CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, CYP2E1 y CYP3A4. No se espera que la inhibición de estas enzimas o el polimorfismo genético (por ejemplo, CYP2D6, CYP2C9 o CYP2C19) produzca un impacto clínicamente significativo sobre la farmacocinética o exposición a cladribina. La cladribina no tiene un efecto inductivo clínicamente significativo sobre las enzimas CYP1A2, CYP2B6 y CYP3A4.
Tras su entrada en las células diana, la cladribina se fosforila mediante DCK (y también mediante desoxiguanosina quinasa en mitocondrias) a monofosfato de cladribina (Cd-AMP), y posteriormente a difosfato de cladribina (Cd-ADP) y trifosfato de cladribina (Cd-ATP). La defosforilación y desactivación de Cd-AMP es catalizada por la 5'-NTasa citoplasmática. En un estudio de farmacocinética intracelular de Cd-AMP y Cd-ATP en pacientes con leucemia mieloide crónica, los niveles de Cd-ATP fueron aproximadamente la mitad de los niveles de Cd-AMP.
El periodo de semivida intracelular fue de 15 horas para Cd-AMP y de 10 horas para Cd-ATP.
Eliminación
Basándose en datos farmacocinéticos poblacionales combinados recopilados en diversos estudios, los valores medianos de eliminación fueron de 22,2 l/h para el aclaramiento renal y 23,4 l/h para el aclaramiento no renal. El aclaramiento renal superó la velocidad de filtración glomerular, lo que indica una secreción tubular renal activa de cladribina.
La fracción no renal de la eliminación de cladribina (aproximadamente el 50 %) incluye un metabolismo hepático insignificante, una distribución intracelular extensa y la captación del derivado activo de cladribina (Cd-ATP) en el compartimento intracelular diana (es decir, linfocitos), con posterior eliminación del Cd-ATP intracelular según el ciclo vital y las vías de eliminación de estas células.
Según estimaciones, el periodo de semivida final en un paciente típico de la población evaluada para los parámetros farmacocinéticos es de aproximadamente 1 día. Sin embargo, esto no conduce a ninguna acumulación del fármaco tras la dosificación diaria, ya que este periodo de semivida se refiere únicamente a una pequeña fracción del AUC.
Dependencia de la dosis y del tiempo
Tras la administración oral de cladribina en dosis de 3 a 20 mg, la Cmax y el AUC aumentaron de forma dependiente de la dosis, lo que sugiere que para dosis orales hasta 20 mg no existen procesos que limiten la velocidad o capacidad de absorción.
Tras la administración de dosis repetidas, no se observó acumulación significativa de cladribina en plasma. No hay indicios de que la farmacocinética de cladribina tras la administración repetida cambie con el tiempo.
Grupos de pacientes especiales
No se ha evaluado la farmacocinética de cladribina en pacientes con esclerosis múltiple de edad pediátrica o avanzada, ni en pacientes con afectación de la función renal o hepática.
El análisis poblacional de la farmacocinética no mostró ningún efecto de la edad (rango de 18 a 65 años) o del sexo del paciente sobre la farmacocinética de cladribina.
Alteración de la función renal
Se ha demostrado que el aclaramiento renal de cladribina depende del aclaramiento de creatinina. Según el análisis de farmacocinética poblacional que incluyó datos de pacientes con función renal normal y afectación renal leve, se espera que el aclaramiento total de cladribina en pacientes con afectación renal leve (CLCR = 60 ml/min) se reduzca moderadamente, lo que provocará un aumento del 25 % en la exposición.
Alteración de la función hepática
Se considera que el papel de la función hepática en la eliminación de cladribina es insignificante.
Interacciones farmacocinéticas
Los estudios de interacciones medicamentosas en pacientes con esclerosis múltiple mostraron que la biodisponibilidad de 10 mg de cladribina por vía oral no se modifica al administrarse simultáneamente con pantoprazol.
Características clínicas.
Indicaciones.
Mavenclad® está indicado para el tratamiento de pacientes adultos con formas recidivantes de esclerosis múltiple (EM) con alta actividad de la enfermedad, confirmada mediante evaluaciones clínicas o de imagen.
Contraindicaciones.
- Hipersensibilidad al principio activo o a cualquiera de los excipientes del medicamento;
- Infección por el virus de la inmunodeficiencia humana (VIH);
- Infecciones crónicas activas (tuberculosis o hepatitis);
- Inicio del tratamiento con cladribina en pacientes con el sistema inmunitario debilitado, incluyendo aquellos que reciben terapia inmunosupresora o mielosupresora;
- Neoplasias malignas activas;
- Afectación renal moderada o grave (depuración de creatinina < 60 ml/min);
- Periodo de embarazo y lactancia.
Interacción con otros medicamentos y otras formas de interacción.
Mavenclad® contiene hidroxipropilbetadex, que puede formar complejos con otros medicamentos, lo que potencialmente podría aumentar la biodisponibilidad de estos últimos (especialmente medicamentos con baja solubilidad). Por lo tanto, durante los pocos días de administración de cladribina, se recomienda tomar cualquier medicamento por vía oral con un intervalo mínimo de 3 horas antes o después de la administración de Mavenclad®.
Medicamentos inmunosupresores
Está contraindicado iniciar el tratamiento con cladribina en pacientes con el sistema inmunitario debilitado, incluyendo aquellos que reciben terapia inmunosupresora o mielosupresora (por ejemplo, metotrexato, ciclofosfamida, ciclosporina o azatioprina), o aquellos que usan corticosteroides de forma continuada, debido al riesgo de efectos adicionales sobre el sistema inmunitario.
Durante el tratamiento con cladribina, puede administrarse terapia intensiva de corticosteroides sistémicos a corto plazo.
Otros medicamentos modificadores del curso de la enfermedad
La administración conjunta de cladribina e interferón beta aumenta el riesgo de linfopenia. No se ha establecido la seguridad ni la eficacia del uso de cladribina en combinación con otros medicamentos modificadores del curso de la EM, por lo que no se recomienda este tratamiento simultáneo.
Medicamentos hematotóxicos
Dado que la cladribina induce una disminución del número de linfocitos, puede esperarse el desarrollo de reacciones adversas hematológicas adicionales si se administra cladribina antes o simultáneamente con otras sustancias que afectan al perfil hematológico (como la carbamazepina). En tales casos, se recomienda un control riguroso de los parámetros hematológicos.
Vacunas vivas o vivas atenuadas
No debe iniciarse el tratamiento con Mavenclad® dentro de las 4-6 semanas posteriores a la vacunación con vacunas vivas o vivas atenuadas, debido al riesgo de infección aguda por la vacuna. Durante y después del tratamiento con cladribina, debe evitarse la vacunación con vacunas vivas o vivas atenuadas hasta que el recuento de linfocitos del paciente haya regresado a valores normales.
Inhibidores potentes de los transportadores ENT1, CNT3 y BCRP
En cuanto a la absorción de cladribina, la única fuente probable de interacciones con relevancia clínica es la proteína de resistencia al cáncer de mama (BCRP o ABCG2). La inhibición de BCRP en el tracto gastrointestinal puede aumentar la biodisponibilidad oral y la acción sistémica de cladribina. Entre los inhibidores conocidos de BCRP que in vivo pueden modificar la farmacocinética de los sustratos de BCRP en un 20 %, se incluye el eltrombopag.
Estudios in vitro indican que la cladribina es sustrato del transportador equilibrador de nucleósidos (ENT1) y del transportador concentrador de nucleósidos (CNT3). Por lo tanto, la biodisponibilidad, la distribución intracelular y la excreción renal de cladribina podrían teóricamente verse afectadas por inhibidores potentes de los transportadores ENT1, CNT3 y BCRP, como dilazep, nifedipino, nimodipino, cilostazol, sulindac y reserpina. Sin embargo, es difícil predecir el efecto neto sobre la actividad potencial de cladribina.
Aunque la relevancia clínica de estas interacciones no se conoce, durante el tratamiento de 4 o 5 días con cladribina se recomienda evitar la administración concomitante de inhibidores potentes de ENT1, CNT3 y BCRP. Si esto no es posible, se debe considerar la selección de medicamentos alternativos para uso concomitante que carezcan o tengan propiedades inhibitorias mínimas frente a los transportadores ENT1, CNT3 y BCRP. Si tampoco es posible, se recomienda reducir la dosis de los medicamentos que contengan estos compuestos a la dosis mínima necesaria, separar la administración de los medicamentos en el tiempo y controlar cuidadosamente al paciente.
Inductores potentes de los transportadores BCRP y P-gp
El efecto de los inductores potentes de los transportadores de eflujo BCRP y glicoproteína P (P-gp) sobre la biodisponibilidad y distribución de cladribina no se ha estudiado en ensayos especiales. Al administrar conjuntamente inductores potentes de BCRP (por ejemplo, corticosteroides) o de P-gp (por ejemplo, rifampicina, hipérico o Hypericum perforatum) debe tenerse en cuenta la posibilidad de un efecto atenuado de cladribina.
Anticonceptivos hormonales
No se ha observado interacción farmacocinética clínicamente relevante entre cladribina y anticonceptivos orales (etinilestradiol y levonorgestrel). Por lo tanto, no se espera que la administración concomitante con cladribina reduzca la eficacia de los anticonceptivos hormonales (véase la sección «Uso durante el embarazo o la lactancia»).
Características de uso.
Monitorización hematológica
El mecanismo de acción de la cladribina está estrechamente relacionado con la reducción del número de linfocitos. El efecto sobre el número de linfocitos es dependiente de la dosis. En estudios clínicos también se observó una disminución del número de neutrófilos, número de eritrocitos, hematocrito, hemoglobina y número de plaquetas en comparación con los valores basales, aunque estos parámetros generalmente permanecieron dentro del rango normal.
Cuando se administra cladribina antes o simultáneamente con otras sustancias que afectan el perfil hematológico, puede esperarse el desarrollo de reacciones adversas adicionales.
El número de linfocitos debe determinarse:
- antes del inicio del tratamiento en el año 1,
- antes del inicio del tratamiento en el año 2,
- a los 2 y 6 meses después del inicio del tratamiento en cada año de tratamiento; si el número de linfocitos es inferior a 500 células/mm³, este parámetro debe monitorizarse activamente hasta que los valores vuelvan a aumentar.
Para obtener información sobre la toma de decisiones respecto al tratamiento basada en el número de linfocitos, véase la sección «Posología y forma de administración» y el apartado «Infecciones» más adelante.
Infecciones
La cladribina puede debilitar la defensa inmunitaria del organismo, aumentando así la probabilidad de infecciones. Durante el tratamiento con MAVENCLAD® se han observado infecciones graves, severas y oportunistas, incluyendo casos con desenlace letal. La infección por VIH, la tuberculosis activa y la hepatitis activa deben descartarse antes de iniciar el tratamiento con cladribina (véase la sección «Contraindicaciones»).
Durante el tratamiento pueden reactivarse infecciones latentes, incluyendo tuberculosis o hepatitis. Por lo tanto, antes del inicio del tratamiento en el año 1 y año 2 debe realizarse una evaluación para detectar infecciones latentes, especialmente tuberculosis y hepatitis B y C. El inicio del tratamiento con MAVENCLAD® debe retrasarse hasta que las infecciones hayan sido adecuadamente tratadas.
En pacientes con infecciones agudas también debe considerarse la posibilidad de posponer el inicio del tratamiento con cladribina hasta que las infecciones estén completamente controladas.
Se recomienda especial atención en pacientes sin antecedentes de exposición al virus de la varicela. Antes de iniciar la terapia con cladribina se recomienda vacunar a los pacientes que carezcan de anticuerpos frente a la varicela. En tal caso, el inicio del tratamiento con MAVENCLAD® debe retrasarse entre 4 y 6 semanas para lograr el efecto completo de la vacunación.
En pacientes que han recibido cladribina se ha observado un aumento en la frecuencia de casos de herpes zóster. Si el número de linfocitos desciende por debajo de 200 células/mm³, durante la linfopenia de grado 4 debe considerarse la profilaxis antiviral contra el herpes según los protocolos estándar locales.
En pacientes con número de linfocitos inferior a 500 células/mm³ debe realizarse un seguimiento activo de signos y síntomas que sugieran infección, especialmente herpes zóster. Si se observan tales signos y síntomas, debe iniciarse el tratamiento de la infección según indicaciones clínicas. Puede considerarse la conveniencia de interrumpir o posponer el tratamiento con MAVENCLAD® hasta que la infección haya sido adecuadamente tratada.
Se han notificado casos de leucoencefalopatía multifocal progresiva (LMP) con el uso de preparaciones parenterales de cladribina en pacientes tratados por leucemia de células peludas con otro régimen terapéutico.
Aunque no se han notificado casos de LMP con el uso de comprimidos de cladribina, antes de iniciar el tratamiento con MAVENCLAD® (generalmente dentro de los 3 meses previos) debe realizarse una resonancia magnética (RM) basal.
Neoplasias malignas
En estudios clínicos, los casos de neoplasias malignas se observaron con mayor frecuencia en pacientes que recibieron cladribina en comparación con pacientes que recibieron placebo.
MAVENCLAD® está contraindicado en pacientes con EM que presenten neoplasias malignas activas (véase la sección «Contraindicaciones»). En pacientes con antecedentes de neoplasias malignas debe realizarse una evaluación individual de los beneficios y riesgos del uso de MAVENCLAD® antes de iniciar el tratamiento. A los pacientes que toman cladribina se les debe recomendar seguir las recomendaciones estándar de cribado para cáncer.
Función hepática
Se han notificado raramente casos de afectación hepática, incluyendo casos graves, en pacientes tratados con MAVENCLAD®.
Antes de prescribir MAVENCLAD®, debe considerarse el historial completo del paciente, especialmente episodios previos de afectación hepática por otros medicamentos y trastornos hepáticos existentes. Antes del inicio del tratamiento en el año 1 y año 2 deben determinarse los niveles séricos de aminotransferasas, fosfatasa alcalina y bilirrubina total. Durante el tratamiento debe realizarse monitorización de las enzimas hepáticas y la bilirrubina basándose en signos y síntomas clínicos.
Si el paciente desarrolla signos clínicos, aumento de enzimas hepáticas de origen desconocido o síntomas que sugieran disfunción hepática (por ejemplo, náuseas, vómitos, dolor abdominal, debilidad, anorexia de origen desconocido o ictericia y/o oscurecimiento de la orina), deben determinarse inmediatamente los niveles séricos de transaminasas y bilirrubina total. Si fuera necesario, el tratamiento con MAVENCLAD® debe interrumpirse o suspenderse.
Anticoncepción
Antes del inicio del tratamiento en el año 1 y año 2, tanto las mujeres en edad fértil como los hombres con potencial reproductivo deben informarse sobre el riesgo potencialmente grave para el feto y la necesidad de adoptar medidas anticonceptivas eficaces.
Las mujeres en edad fértil deben evitar el embarazo mediante el uso de métodos anticonceptivos eficaces durante el tratamiento con cladribina y al menos durante 6 meses después de la última dosis.
Los pacientes hombres deben adoptar medidas para evitar el embarazo de su pareja durante el tratamiento con cladribina y al menos durante 6 meses después de la última dosis.
Transfusión de sangre
En pacientes que requieran transfusión sanguínea, se recomienda irradiar los componentes celulares de la sangre antes de su administración para prevenir enfermedades asociadas a la transfusión del tipo «injerto contra huésped». Se recomienda consultar con un hematólogo.
Transición al tratamiento con cladribina y desde él
Antes de iniciar el tratamiento en pacientes que previamente hayan recibido medicamentos inmunomoduladores o inmunosupresores, debe considerarse el mecanismo de acción y la duración del efecto de estos medicamentos. También debe considerarse el posible efecto aditivo sobre el sistema inmunitario si tales medicamentos se administran tras el tratamiento con MAVENCLAD®.
Al cambiar desde otro medicamento para el tratamiento de la EM, se recomienda realizar una RM basal (véase el apartado «Infecciones» anterior).
Alteración de la función hepática
No se recomienda el uso de cladribina en pacientes con alteración hepática moderada o grave (puntuación de Child-Pugh > 6).
Sorbitol
MAVENCLAD® contiene sorbitol. En caso de intolerancia a ciertos azúcares, debe consultarse con el médico antes de iniciar el tratamiento con este medicamento.
Debe considerarse el efecto adicional de otros medicamentos que se administren simultáneamente y que contengan sorbitol (o fructosa), así como el consumo alimentario de sorbitol (o fructosa).
La presencia de sorbitol en medicamentos para uso oral puede afectar la biodisponibilidad de otros medicamentos orales administrados simultáneamente.
Uso durante el embarazo o la lactancia.
Anticoncepción en hombres y mujeres
Antes del inicio del tratamiento en el año 1 y año 2, tanto las mujeres en edad fértil como los hombres con potencial reproductivo deben informarse sobre el riesgo potencialmente grave para el feto y la necesidad de adoptar medidas anticonceptivas eficaces.
En mujeres en edad fértil, antes del inicio del tratamiento con MAVENCLAD® en los años 1 y 2 debe descartarse el embarazo, y debe evitarse el embarazo mediante el uso de métodos anticonceptivos eficaces durante el tratamiento con cladribina y al menos durante 6 meses después de la última dosis. Las mujeres que queden embarazadas durante el tratamiento con MAVENCLAD® deben interrumpir la terapia.
Dado que la cladribina afecta la síntesis del ADN, puede esperarse un efecto negativo sobre la gametogénesis humana. Por lo tanto, los pacientes hombres deben adoptar medidas para evitar el embarazo de su pareja durante el tratamiento con cladribina y al menos durante 6 meses después de la última dosis.
Embarazo
Debido a la experiencia con otras sustancias que inhiben la síntesis del ADN en humanos, la cladribina puede causar malformaciones congénitas si se administra durante el embarazo. En estudios en animales se ha demostrado toxicidad reproductiva de la cladribina.
MAVENCLAD® está contraindicado en mujeres embarazadas (véase la sección «Contraindicaciones»).
Lactancia
Datos limitados procedentes de notificaciones espontáneas indican que la cladribina se excreta en la leche materna humana, aunque aún no se ha determinado en qué cantidad. Debido a las reacciones adversas potencialmente graves en lactantes amamantados, la lactancia está contraindicada durante el tratamiento con MAVENCLAD® y durante 1 semana después de la última dosis (véase la sección «Contraindicaciones»).
Fertilidad
En ratones no se observó efecto de la cladribina sobre la fertilidad o la función reproductiva de la descendencia, aunque en ratones y monos se observó un efecto sobre la función testicular.
Dado que la cladribina afecta la síntesis del ADN, puede esperarse un efecto negativo sobre la gametogénesis humana. Por lo tanto, los pacientes hombres deben adoptar medidas para evitar el embarazo de su pareja durante el tratamiento con cladribina y al menos durante 6 meses después de la última dosis.
Efecto sobre la capacidad para conducir y utilizar máquinas.
MAVENCLAD® no afecta o afecta mínimamente la capacidad de los pacientes para conducir vehículos o manejar maquinaria.
Vía de administración y dosis.
El tratamiento debe iniciarse y realizarse bajo la supervisión de un médico con experiencia en el tratamiento de la EM.
Dosis
La dosis acumulada recomendada del medicamento es de 3,5 mg/kg de peso corporal durante 2 años, administrada como un curso de tratamiento anual de 1,75 mg/kg. Cada curso de tratamiento consta de 2 semanas de tratamiento, una al comienzo del primer mes y otra al comienzo del segundo mes del año correspondiente de tratamiento. Por razones médicas (por ejemplo, para permitir la recuperación del recuento de linfocitos), el curso de tratamiento en el año 2 puede posponerse hasta 6 meses. Cada semana de tratamiento consta de 4 o 5 días, durante los cuales el paciente toma 10 mg u 20 mg (una o dos tabletas) como dosis única diaria, según el peso corporal. Más información detallada se proporciona en las tablas 2 y 3.
Tras completar los 2 cursos de tratamiento, no se requiere tratamiento adicional con cladribina en los años 3 y 4. No se ha estudiado la reiniciación del tratamiento tras 4 años.
Criterios para iniciar y continuar la terapia
El recuento de linfocitos debe ser:
- normal antes del inicio del tratamiento en el año 1,
- al menos 800 células/mm³ antes del inicio del tratamiento en el año 2.
Si fuera necesario, por motivos de recuperación del recuento de linfocitos, el curso de tratamiento en el año 2 puede retrasarse hasta 6 meses. Si la recuperación del recuento requiere más de 6 meses, el paciente no podrá volver a tomar cladribina.
Reparto de la dosis
El reparto de la dosis total durante los 2 años de tratamiento se muestra en la tabla 2. Debido al peso corporal, algunos pacientes pueden requerir un número diferente de tabletas durante la primera y segunda semana de tratamiento. No se ha estudiado la administración de formas orales de cladribina en pacientes con un peso corporal inferior a 40 kg.
Tabla 2. Dosis de cladribina según el peso corporal del paciente durante la semana de tratamiento de cada año de tratamiento
| Rango de peso corporal, kg |
Dosis en mg (número de comprimidos de 10 mg) durante la semana de tratamiento |
|
| Semana de tratamiento 1 |
Semana de tratamiento 2 |
|
| de 40 a < 50 |
40 mg (4 comprimidos) |
40 mg (4 comprimidos) |
| de 50 a < 60 |
50 mg (5 comprimidos) |
50 mg (5 comprimidos) |
| de 60 a < 70 |
60 mg (6 comprimidos) |
60 mg (6 comprimidos) |
| de 70 a < 80 |
70 mg (7 comprimidos) |
70 mg (7 comprimidos) |
| de 80 a < 90 |
80 mg (8 comprimidos) |
70 mg (7 comprimidos) |
| de 90 a < 100 |
90 mg (9 comprimidos) |
80 mg (8 comprimidos) |
| de 100 a < 110 |
100 mg (10 comprimidos) |
90 mg (9 comprimidos) |
| de 110 o más |
100 mg (10 comprimidos) |
100 mg (10 comprimidos) |
En la tabla 3 se muestra cómo distribuir la cantidad total de comprimidos por semana de tratamiento en los días individuales. Se recomienda tomar la dosis diaria de cladribina cada semana de tratamiento con intervalos de 24 horas, aproximadamente a la misma hora del día. Si la dosis diaria es de dos comprimidos, ambos comprimidos se toman juntos como una sola dosis.
| Tabla 3 Distribución de las tabletas por días de la semana |
|||||
| Cantidad total de tabletas por semana |
Día 1 |
Día 2 |
Día 3 |
Día 4 |
Día 5 |
| 4 |
1 |
1 |
1 |
1 |
0 |
| 5 |
1 |
1 |
1 |
1 |
1 |
| 6 |
2 |
1 |
1 |
1 |
1 |
| 7 |
2 |
2 |
1 |
1 |
1 |
| 8 |
2 |
2 |
2 |
1 |
1 |
| 9 |
2 |
2 |
2 |
2 |
1 |
| 10 |
2 |
2 |
2 |
2 |
2 |
La dosis olvidada debe tomarse lo antes posible después de darse cuenta del olvido, el mismo día, de acuerdo con el régimen de tratamiento.
No se debe tomar la dosis olvidada junto con la dosis siguiente según el esquema de tratamiento del día siguiente. Si se omite una dosis, el paciente debe tomarla al día siguiente y aumentar el número de días de tratamiento en esa semana terapéutica. Si se omiten dos dosis consecutivas, se aplica el mismo enfoque y el número de días de tratamiento en la semana terapéutica se incrementa en dos días.
Uso concomitante con otros medicamentos orales
Durante el número limitado de días de administración de cladribina, se recomienda separar la administración de cualquier otro medicamento oral de la administración de Mavencle® por un mínimo de 3 horas.
Grupos de pacientes específicos
Pacientes con alteración de la función renal
No se han realizado estudios específicos sobre el tratamiento de pacientes con alteración de la función renal.
En pacientes con alteración renal leve (aclaramiento de creatinina entre 60 y 89 ml/min), no se considera necesaria la ajuste de la dosis.
No se han estudiado la seguridad y eficacia del uso de cladribina en pacientes con alteración renal moderada o grave. Por lo tanto, la cladribina está contraindicada en estos pacientes (ver sección «Contraindicaciones»).
Pacientes con alteración de la función hepática
No se han realizado estudios sobre el tratamiento de pacientes con alteración de la función hepática.
No se requiere ajuste de la dosis en pacientes con alteración hepática leve, ya que se considera que la función hepática tiene poca importancia en la eliminación de la cladribina. Debido a la falta de datos, no se recomienda el uso de cladribina en pacientes con alteración hepática moderada o grave (índice de Child-Pugh > 6).
Pacientes de edad avanzada
En pacientes de edad avanzada, se recomienda usar cladribina con precaución debido a la frecuencia potencialmente mayor de disminución de la función hepática o renal, enfermedades concomitantes y otros tratamientos que se estén recibiendo.
Vía de administración
Mavencle® está indicado para administración oral. Las tabletas deben tomarse con agua y tragarse enteras, sin masticar. Las tabletas pueden tomarse independientemente de las comidas.
Dado que las tabletas no tienen recubrimiento, deben tragarse inmediatamente después de extraerlas del blíster, sin dejarlas sobre ninguna superficie ni manipularlas más tiempo del necesario para su administración. Si una tableta ha estado expuesta sobre una superficie o si se ha extraído del blíster una tableta partida o desmenuzada, la superficie de contacto debe lavarse cuidadosamente con agua.
El paciente debe tomar las tabletas con las manos secas y, posteriormente, lavarse bien las manos.
Instrucciones para la autoadministración de las tabletas Mavencle® por el paciente
- Prepare un vaso con agua y asegúrese de tener las manos secas y limpias.
- Tome la caja del medicamento, orientándola hacia usted por el lado donde están impresas las instrucciones.

- (1) Abra la solapa del lado izquierdo de la caja.
(2) Simultáneamente, presione los ganchos de la caja con el dedo índice y el pulgar de ambos lados y manténgalos en esa posición.
(3) Tire del envase blíster hasta que se mueva. ADVERTENCIA: no extraiga el envase blíster de la caja.
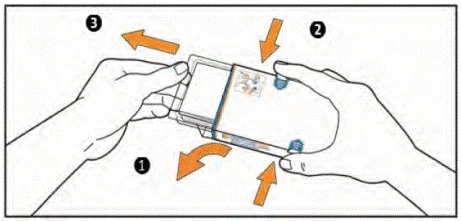
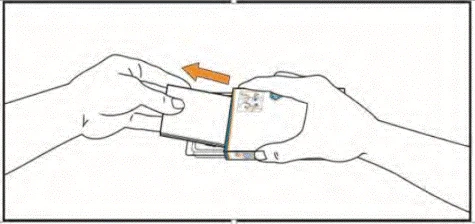
- Saque el prospecto del envase. Asegúrese de haber leído cuidadosamente su contenido, incluidas las instrucciones para la autoadministración de las tabletas.
- Levante el blíster presionando con los dedos a través del orificio del envase blíster. Coloque la mano debajo del blíster y empuje hacia fuera 1 o 2 tabletas, según la dosis indicada por su médico.
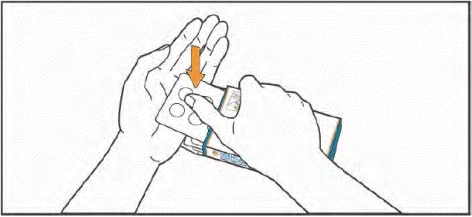
- Tome las tabletas con agua. Las tabletas deben tragarse enteras, sin masticar ni permitir que se disuelvan en la boca. El contacto de las tabletas con la piel debe limitarse. Durante la administración, evite tocar con las manos la nariz, los ojos y otras partes del cuerpo.
- Después de tomar las tabletas, lávese bien las manos con agua y jabón.
- Vuelva a colocar el envase blíster dentro de la caja. Guárdelo en su envase original para protegerlo de la humedad.
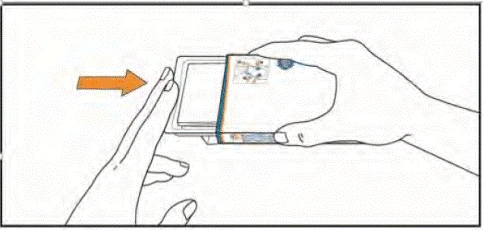
Guarde las tabletas en el blíster hasta el momento de la siguiente dosis. No destropee las tabletas del blíster ni las guarde en otro recipiente.
Niños
En niños (menores de 18 años), la seguridad y eficacia de Mavencle® no han sido establecidas. No existen datos adecuados.
Sobredosis
La experiencia con sobredosis de cladribina por vía oral es limitada. Se sabe que la linfopenia que se desarrolla como consecuencia es dependiente de la dosis.
En pacientes con sobredosis de cladribina, se recomienda un monitoreo hematológico especialmente riguroso.
No se conoce un antídoto específico para la cladribina. El tratamiento consiste en una observación cuidadosa del paciente y en la adopción de las medidas de apoyo adecuadas. Debe considerarse la posibilidad de suspender el tratamiento con Mavencle®. Debido a la rápida y extensa distribución intracelular y tisular, es poco probable que la hemodiálisis elimine significativamente la cladribina.
Reacciones adversas.
Perfil de seguridad resumido
Las reacciones adversas más clínicamente relevantes son la linfopenia (25,6 %) y el herpes zóster (3,0 %). La frecuencia de casos de herpes zóster fue mayor durante el período en que se observó linfopenia de grado 3 o 4 (desde < 500 hasta 200 células/mm³ o < 200 células/mm³) en comparación con el período en que los pacientes no presentaron linfopenia de grado 3 o 4.
Lista de reacciones adversas
La información sobre las reacciones adversas descritas a continuación se obtuvo a partir de datos combinados de estudios clínicos sobre el tratamiento de la EM con el fármaco oral cladribina, administrado como monoterapia en dosis acumulada de 3,5 mg/kg. La base de datos de seguridad de estos estudios incluye información de 923 pacientes. Las reacciones adversas identificadas durante el período poscomercialización se indican con un símbolo de nota al pie (*).
Para la clasificación de la frecuencia de las reacciones adversas se utiliza la siguiente clasificación: muy frecuentes (≥ 1/10), frecuentes (≥ 1/100 hasta < 1/10), poco frecuentes (≥ 1/1000 hasta < 1/100), raras (≥ 1/10000 hasta < 1/1000), muy raras (< 1/10000) y frecuencia no conocida (no puede determinarse con los datos disponibles).
Infecciones e infestaciones
Frecuentes: herpes oral, herpes zóster cutáneo.
Raras: tuberculosis.
Alteraciones de la sangre y del sistema linfático
Muy frecuentes: linfopenia.
Frecuentes: disminución del número de neutrófilos.
Alteraciones del sistema inmunitario
Frecuentes: reacciones de hipersensibilidad*, incluyendo prurito, urticaria, erupción cutánea y casos aislados de angioedema.
Alteraciones hepatobiliares
Poco frecuentes: afectación hepática*.
Alteraciones de la piel y del tejido subcutáneo
Frecuentes: erupción cutánea, alopecia.
Descripción de reacciones adversas individuales
Linfopenia
En estudios clínicos, linfopenia temporal de grado 3 o 4 se desarrolló en un 20–25 % de los pacientes que recibieron una dosis acumulada de cladribina de 3,5 mg/kg durante 2 años como monoterapia. La linfopenia de grado 4 se observó en menos del 1 % de los pacientes. La mayor proporción de pacientes con linfopenia de grado 3 o 4 se detectó a los 2 meses después de la primera dosis de cladribina cada año (4,0 % y 11,3 % de pacientes con linfopenia de grado 3 en el año 1 y año 2, respectivamente; 0 % y 0,4 % de pacientes con linfopenia de grado 4 en el año 1 y año 2, respectivamente). Se espera que en la mayoría de los pacientes el recuento de linfocitos se recupere a niveles normales o a linfopenia de grado 1 dentro de los 9 meses.
Para reducir el riesgo de linfopenia grave, se debe determinar el recuento de linfocitos antes, durante y después del tratamiento con cladribina, y se deben seguir estrictos criterios para iniciar y continuar el tratamiento con cladribina.
Neoplasias malignas
En estudios clínicos y durante el período prolongado de seguimiento posterior, en pacientes que recibieron una dosis acumulada oral de 3,5 mg/kg de cladribina, se observaron eventos de neoplasias malignas con mayor frecuencia en los pacientes tratados con cladribina (10 casos por 3414 paciente-años [0,29 casos por 100 paciente-años]) en comparación con los pacientes que recibieron placebo (3 casos por 2022 paciente-años [0,15 casos por 100 paciente-años]).
Hipersensibilidad
En estudios clínicos, los eventos de hipersensibilidad se observaron con mayor frecuencia en pacientes que recibieron una dosis acumulada oral de 3,5 mg/kg de cladribina (11,8 %) en comparación con los que recibieron placebo (8,4 %). Los eventos graves de hipersensibilidad se observaron en el 0,3 % de los pacientes que recibieron cladribina y no se observaron en los pacientes que recibieron placebo. Los eventos de hipersensibilidad llevaron a la interrupción del tratamiento en el 0,4 % de los pacientes que recibieron cladribina y en el 0,3 % de los que recibieron placebo.
Afectación hepática
Durante la vigilancia poscomercialización, se han notificado casos poco frecuentes de afectación hepática, incluyendo casos graves y casos que condujeron a la interrupción del tratamiento, temporalmente relacionados con la administración del medicamento Mavenclass®. Un aumento transitorio en los niveles séricos de transaminasas generalmente superó más de 5 veces el límite superior normal (LSN). Se han observado casos aislados de aumento transitorio de los niveles séricos de transaminasas hasta 40 veces por encima del LSN y/o hepatitis sintomática con aumento transitorio del nivel de bilirrubina y ictericia. Estos eventos ocurrieron en diferentes momentos tras el inicio del tratamiento, pero la mayoría de los casos se observaron dentro de las 8 semanas posteriores al primer ciclo de tratamiento (ver sección «Instrucciones de uso»).
La notificación de reacciones adversas tras la autorización del medicamento es de gran importancia, ya que permite continuar con el seguimiento de la relación beneficio/riesgo del medicamento. Los profesionales médicos y farmacéuticos, así como los pacientes o sus representantes legales, deben informar de todos los casos sospechosos de reacciones adversas y de falta de eficacia del medicamento a través del Sistema de Información Automatizado de Farmacovigilancia en el siguiente enlace: https://aisf.dec.gov.ua.
Período de validez. 4 años.
No utilizar después de la fecha de caducidad indicada en el envase.
Condiciones de conservación.
Conservar en el envase original para protegerlo de la humedad.
Mantener fuera del alcance y de la vista de los niños.
Envase. 1, 4 ó 6 comprimidos en blíster de aluminio, sellado en una funda de cartón, empaquetado en envase termoformado y colocado en una caja de cartón con protección contra el acceso de niños.
Categoría de dispensación. Bajo receta médica.
Fabricante. NerPharMa S.R.L. / NerPharMa S.R.L.
Dirección del fabricante y lugar de actividad.
Viale Pasteur 10 (localidad Nerviano), 20014 Milán (MI), Italia /
Viale Pasteur 10 (loc. Nerviano), 20014 Milan (MI), Italy.