Mavenclad®
Ukraine
Table of Contents
INSTRUCTIONS FOR MEDICAL USE OF THE MEDICINAL PRODUCT MAVENCLAD® (MAVENCLAD®)
Composition:
Active substance: cladribine;
1 tablet contains 10 mg of cladribine;
Excipients: hydroxypropylbetadex, sorbitol (E 420), magnesium stearate.
Medicinal form. Tablets.
Main physicochemical properties: white, round, biconvex tablets, 8.5 mm in diameter, with embossing "C" on one side and "10" on the other.
Pharmacotherapeutic group. Selective immunosuppressants. Cladribine.
ATC code L04A A40.
Pharmacological Properties
Pharmacodynamics
Mechanism of Action
Cladribine is a nucleoside analogue of deoxyadenosine. The substitution of a chlorine atom in the purine ring protects cladribine from degradation by adenosine deaminase, thereby increasing the intracellular residence time of cladribine prodrugs. Subsequent phosphorylation of cladribine leads to the formation of its active form, 2-chlorodeoxyadenosine triphosphate (Cd-ATP). This process occurs particularly efficiently in lymphocytes due to their constitutively high levels of deoxycytidine kinase (DCK) and relatively low levels of 5'-nucleotidase (5'-NTase). The high DCK to 5'-NTase ratio promotes the accumulation of Cd-AT0P, making lymphocytes especially susceptible to cell death. Other bone marrow-derived cells, which have a lower DCK/5'-NTase ratio, are less affected than lymphocytes. DCK is the rate-limiting enzyme in the conversion of cladribine into its active triphosphate form, resulting in selective depletion of both dividing and non-dividing T- and B-cells.
The primary mechanism of Cd-ATP-induced apoptosis involves direct and indirect effects on DNA synthesis and mitochondrial function. In dividing cells, Cd-ATP interferes with DNA synthesis by inhibiting ribonucleotide reductase and competes with deoxyadenosine triphosphate for incorporation into DNA by DNA polymerases. In resting cells, cladribine causes single-strand DNA breaks, rapid depletion of nicotinamide adenine dinucleotide (NAD), ATP breakdown, and cell death. Evidence suggests that cladribine may also induce direct, caspase-dependent and caspase-independent apoptosis by releasing cytochrome C and apoptosis-inducing factor into the cytosol of non-dividing cells.
The pathogenesis of multiple sclerosis (MS) involves a complex cascade of events in which various types of immune cells, including autoreactive T- and B-cells, play a crucial role. The therapeutic mechanism of action of cladribine in MS is not fully understood, but it is believed that its primary effect on T- and B-lymphocytes interrupts the immune cascade that is central to MS.
Differential expression levels of DCK and 5'-NTase among immune cell subtypes may explain the varying sensitivity of immune cells to cladribine. Due to these expression patterns, cells of the innate immune system are less affected than those of the adaptive immune system.
Pharmacodynamic Effects
Cladribine has been shown to exert a prolonged effect, primarily targeting lymphocytes and the autoimmune processes involved in the pathophysiology of MS.
In clinical studies, the highest proportion of patients with grade 3 or 4 lymphopenia (defined as < 500 to 200 cells/mm³ or < 200 cells/mm³) was observed 2 months after the first dose of cladribine each year, indicating a time lag between plasma presence of cladribine and maximal hematological effect.
Data from clinical trials using the proposed cumulative dose of 3.5 mg/kg body weight indicate a gradual recovery of median lymphocyte count to the normal range by week 84 after the first dose of cladribine (approximately 30 weeks after the last dose). Lymphocyte counts returned to the normal range in over 75% of patients by week 144 after the first dose (approximately 90 weeks after the last dose).
Oral cladribine treatment results in a rapid reduction in circulating CD4+ and CD8+ T-cells. CD8+ T-cells show a less pronounced reduction and faster recovery compared to CD4+ T-cells, leading to a temporary decrease in the CD4 to CD8 ratio. Cladribine reduces the number of CD19+ B-cells and CD16+/CD56+ natural killer (NK) cells, both of which recover more quickly than CD4+ T-cells.
Clinical Efficacy and Safety
Relapsing-Remitting Multiple Sclerosis
The efficacy and safety of oral cladribine were evaluated in a randomized, double-blind, placebo-controlled clinical trial (CLARITY) involving 1326 patients with relapsing-remitting multiple sclerosis. The primary objective of the study was to assess the efficacy of cladribine compared to placebo in reducing the annualized relapse rate (ARR) (primary endpoint), slowing disability progression, and reducing MRI-confirmed active lesions.
Patients received either placebo (n = 437), or cumulative doses of cladribine 3.5 mg/kg (n = 433) or 5.25 mg/kg body weight (n = 456) over a 96-week (2-year) study period consisting of two treatment courses. Patients were randomized to receive the cumulative dose of 3.5 mg/kg during the first treatment course at weeks 1 and 5 of the first year and the second treatment course at weeks 1 and 5 of the second year. Patients were also randomized to receive the cumulative dose of 5.25 mg/kg during an additional treatment period at weeks 9 and 13 of the first year. The majority of patients in the placebo group (87.0%) and in the cladribine treatment groups of 3.5 mg/kg (91.9%) and 5.25 mg/kg (89.0%) completed the full 96-week study period.
Patients were required to have experienced at least one relapse within the previous 12 months. In the overall study population, the median age was 39 years (range 18 to 65 years), and the female-to-male ratio was approximately 2:1. The mean duration of MS prior to study entry was 8.7 years, and the median baseline neurological disability score, assessed using the Expanded Disability Status Scale (EDSS) across all treatment groups, was 3.0 (range 0 to 6.0). More than two-thirds of patients had not received any disease-modifying therapy for MS prior to the study. The remaining patients had previously been treated with interferon beta-1a, interferon beta-1b, glatiramer acetate, or natalizumab.
In patients with relapsing-remitting MS receiving cladribine at a dose of 3.5 mg/kg, statistically significant improvements were observed compared to placebo recipients in terms of annualized relapse rate, proportion of patients relapse-free over 96 weeks, proportion of patients without sustained disability progression over 96 weeks, and time to 3-month progression on the EDSS (see Table 1).
| Table 1 Clinical results of the CLARITY study (96 weeks) |
|||
| Parameter |
Placebo |
Cumulative cladribine dose |
|
| 3.5 mg/kg |
5.25 mg/kg |
||
| Annualized relapse rate (95% CI) |
0.33 (0.29; 0.38) |
0.14* (0.12; 0.17) |
0.15* (0.12; 0.17) |
|
57.6% |
54.5% |
|
| Proportion of patients without relapses over 96 weeks |
60.9% |
79.7% |
78.9% |
| Time to 3-month EDSS progression, 10th percentile (months) |
10.8 |
13.6 |
13.6 |
|
0.67 (0.48; 0.93) p = 0.018 |
0.69 (0.49; 0.96) p = 0.026 |
|
| CI – confidence interval * p < 0.001 vs placebo. |
|||
Treatment outcomes with cladribine at a dose of 3.5 mg/kg were statistically significantly superior to placebo regarding the number and relative reduction of T1 Gd+ lesions, active T2 lesions, and combined unique lesions on brain MRI over the full 96-week study period. Patients receiving cladribine, compared to the placebo group, had a relative reduction of 86% in the mean number of T1 Gd+ lesions (adjusted mean number for the cladribine 3.5 mg/kg and placebo groups was 0.12 and 0.91, respectively), a relative reduction of 73% in the mean number of active T2 lesions (adjusted mean number for the cladribine 3.5 mg/kg and placebo groups was 0.38 and 1.43, respectively), and a relative reduction of 74% in the mean number of combined unique lesions per patient MRI scan (adjusted mean number for the cladribine 3.5 mg/kg and placebo groups was 0.43 and 1.72, respectively) (p < 0.001 for all three MRI outcomes).
A retrospective analysis of time to 6-month confirmed disability progression measured by EDSS revealed a 47% reduction in the risk of disability progression in the group receiving cladribine at 3.5 mg/kg compared to the placebo group (hazard ratio 0.53; 95% CI [0.36; 0.79], p < 0.05). In the placebo group, the 10th percentile for disability progression was reached by 245 days, whereas in the cladribine 3.5 mg/kg group it was not reached during the entire study period.
As shown in Table 1, higher cumulative doses of cladribine provided no clinically meaningful benefits but were associated with a higher frequency of grade ≥3 lymphopenia (44.9% in the 5.25 mg/kg group compared to 25.6% in the 3.5 mg/kg group).
Patients who completed participation in the CLARITY study could be enrolled in the CLARITY Extension study, whose primary objective was to assess safety. In this extension study, 806 patients received either placebo or a cumulative dose of cladribine 3.5 mg/kg (using a regimen similar to that used in CLARITY) over a 96-week period.
The treatment effect on reducing relapse rate and slowing disability progression in patients who received the 3.5 mg/kg dose over 2 years was maintained in years 3 and 4.
Efficacy in patients with highly active disease
A retrospective analysis of efficacy was conducted in subgroups of patients with highly active disease who received oral cladribine at the recommended cumulative dose of 3.5 mg/kg. These subgroups included:
- patients with 1 relapse in the prior year and at least 1 T1 Gd+ lesion or 9 or more T2 lesions while on other disease-modifying therapies,
- patients with 2 or more relapses in the prior year, regardless of whether they were on disease-modifying therapies or not.
Analysis of data from the CLARITY study showed a similar treatment effect on relapse rate, with annualized relapse rates ranging from 0.16 to 0.18 in the cladribine groups and from 0.47 to 0.50 in the placebo group (p < 0.0001). Compared to the overall population, a greater treatment effect was observed on the time to 6-month confirmed disability progression, with cladribine reducing the risk of disability progression by 82% (hazard ratio 0.18; 95% CI [0.07; 0.47]). In the placebo group, the 10th percentile for disability progression was reached between weeks 16 and 23, whereas in the cladribine groups it was not reached during the entire study period.
Secondary progressive MS with relapses
An additional study evaluating the addition of cladribine to interferon beta compared to placebo plus interferon beta also included a limited number of patients with secondary progressive MS (26 patients). In these patients, treatment with cladribine at a dose of 3.5 mg/kg resulted in a reduction in the annualized relapse rate compared to placebo (0.03 vs. 0.30; rate ratio: 0.11; p < 0.05). No difference in annualized relapse rate was observed between patients with relapsing-remitting MS and those with secondary progressive MS with relapses. The drug had no demonstrated effect on disability progression in either of these subgroups.
Patients with secondary progressive MS were excluded from the CLARITY study. However, a retrospective analysis of pooled patient groups from the CLARITY and ONWARD studies, identified by a baseline EDSS score ≥ 3.5 as an indicator of secondary progressive MS, showed a similar reduction in the annualized relapse rate compared to patients with an EDSS score below 3.
Pharmacokinetics
Cladribine is a prodrug that must be phosphorylated intracellularly to become biologically active. The pharmacokinetics of cladribine have been studied after oral and intravenous administration in patients with MS and in patients with malignancies, as well as in in vitro systems.
Absorption
After oral administration, cladribine is rapidly absorbed. A 10 mg dose of cladribine results in a mean Cmax of cladribine in the range of 22 to 29 ng/mL and a corresponding mean AUC in the range of 80 to 101 ng·h/mL.
Following oral administration on an empty stomach, the median Tmax was 0.5 hours (range 0.5 to 1.5 hours). When administered with a high-fat meal, absorption of cladribine was delayed (median Tmax 1.5 hours, range 1 to 3 hours), and Cmax was reduced by 29%, while AUC remained unchanged. The bioavailability of a 10 mg oral dose of cladribine was approximately 40%.
Distribution
Cladribine has a large volume of distribution, indicating extensive tissue distribution and intracellular uptake. Studies have shown that the mean volume of distribution of cladribine ranges from 480 to 490 L. Plasma protein binding is 20% and does not depend on cladribine plasma concentrations.
Distribution of cladribine across biological membranes is mediated by various transporter proteins, including ENT1, CNT3, and BCRP.
In vitro studies have shown that efflux of cladribine is only minimally related to P-gp; therefore, clinically significant interactions with P-gp inhibitors are not expected.
In vitro studies indicated negligible transporter-mediated uptake of cladribine into human hepatocytes.
Cladribine has the potential to cross the blood-brain barrier. A small study in cancer patients showed that the ratio of its concentration in cerebrospinal fluid to plasma is approximately 0.25.
Cladribine and/or its phosphorylated metabolites are substantially accumulated and retained in human lymphocytes. In vitro studies have shown that the ratio of intracellular to extracellular accumulation is approximately 30 to 40 within 1 hour of exposure.
Metabolism
The metabolism of cladribine was studied in patients with MS after a single 10 mg tablet and a single intravenous dose of 3 mg. After both oral and intravenous administration, the parent compound cladribine was the main component detected in plasma and urine. The levels of its metabolite 2-chloroadenine in both plasma and urine were negligible. Only trace amounts of other metabolites of cladribine were detectable in plasma and urine.
In in vitro hepatic systems, metabolism of cladribine was insignificant (at least 90% remained unchanged cladribine).
Cladribine is not a significant substrate for cytochrome P450 enzymes and does not show significant potential as an inhibitor of CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, CYP2E1, or CYP3A4. Inhibition of these enzymes or genetic polymorphisms (e.g., CYP2D6, CYP2C9, or CYP2C19) are not expected to result in clinically significant effects on the pharmacokinetics or exposure of cladribine. Cladribine has no clinically significant inductive effect on CYP1A2, CYP2B6, or CYP3A4 enzymes.
After entering target cells, cladribine is phosphorylated by DCK (and also by mitochondrial deoxyguanosine kinase) to cladribine monophosphate (Cd-AMP), then to cladribine diphosphate (Cd-ADP), and finally to cladribine triphosphate (Cd-ATP). Dephosphorylation and deactivation of Cd-AMP is catalyzed by cytoplasmic 5'-NTase. In an intracellular pharmacokinetic study of Cd-AMP and Cd-ATP in patients with chronic myelogenous leukemia, Cd-ATP levels were approximately half of Cd-AMP levels.
The intracellular half-life was 15 hours for Cd-AMP and 10 hours for Cd-ATP.
Elimination
Based on combined population pharmacokinetic data collected from various studies, median clearance values were 22.2 L/h for renal clearance and 23.4 L/h for non-renal clearance. Renal clearance exceeded the glomerular filtration rate, indicating active renal tubular secretion of cladribine.
The non-renal portion of cladribine elimination (approximately 50%) includes negligible hepatic metabolism and extensive intracellular distribution and uptake of the active cladribine derivative (Cd-ATP) into the target intracellular compartment (i.e., lymphocytes), followed by elimination of intracellular Cd-ATP according to the life cycle and elimination pathways of these cells.
The estimated terminal half-life in a typical patient from the population for which pharmacokinetic parameters were assessed is approximately 1 day. However, this does not result in any accumulation of the drug after daily dosing, as this half-life refers only to a small fraction of the AUC.
Dose- and time-dependence
After oral administration of cladribine in doses ranging from 3 to 20 mg, Cmax and AUC increased in a dose-dependent manner, suggesting that for oral doses up to 20 mg, absorption is not limited by processes affecting its rate or capacity.
After repeated dosing, no significant accumulation of cladribine in plasma was observed. There is no indication that the pharmacokinetics of cladribine after repeated administration change over time.
Special patient groups
Pharmacokinetic evaluation of cladribine in elderly or pediatric patients with MS, as well as in patients with impaired renal or hepatic function, has not been conducted.
Population pharmacokinetic analysis showed no effect of age (range 18 to 65 years) or patient sex on the pharmacokinetics of cladribine.
Renal impairment
Renal clearance of cladribine has been shown to depend on creatinine clearance. Based on population pharmacokinetic analysis, including data from patients with normal renal function and mild renal impairment, the total clearance of cladribine in patients with mild renal impairment (CLCR = 60 mL/min) is expected to be moderately reduced, resulting in a 25% increase in exposure.
Hepatic impairment
The role of hepatic function in the elimination of cladribine is considered negligible.
Pharmacokinetic interactions
Drug interaction studies in patients with MS showed that the bioavailability of 10 mg oral cladribine is not altered when co-administered with pantoprazole.
Clinical characteristics.
Indications.
Mavenclad® is indicated for the treatment of adult patients with relapsing forms of multiple sclerosis (MS) with high disease activity, established by clinical or imaging assessments.
Contraindications.
- Hypersensitivity to the active substance or to any of the excipients of the medicinal product;
- Human immunodeficiency virus (HIV) infection;
- Active chronic infections (tuberculosis or hepatitis);
- Initiating cladribine treatment in patients with impaired immunity, including patients receiving immunosuppressive or myelosuppressive therapy;
- Active malignancies;
- Moderate or severe renal impairment (creatinine clearance < 60 mL/min);
- Pregnancy and breastfeeding.
Interaction with other medicinal products and other forms of interaction.
Mavenclad® contains hydroxypropylbetadex, which may form complexes with other medicinal products, potentially leading to increased bioavailability of such agents (particularly medicinal products with low solubility). Therefore, during the limited number of days of cladribine administration, it is recommended to take any oral medicinal products at least 3 hours before or after taking Mavenclad®.
Immunosuppressive medicinal products
Initiating cladribine treatment is contraindicated in patients with impaired immunity, including patients receiving immunosuppressive or myelosuppressive therapy (e.g., methotrexate, cyclophosphamide, cyclosporine, or azathioprine), or those on long-term corticosteroid therapy, due to the risk of additive effects on the immune system.
Short-term intensive systemic corticosteroid therapy may be administered during cladribine treatment.
Other disease-modifying medicinal products
Concomitant use of cladribine with beta-interferon increases the risk of lymphopenia. The safety and efficacy of cladribine in combination with other disease-modifying treatments for MS have not been established; therefore, such concomitant therapy is not recommended.
Hematotoxic medicinal products
Since cladribine induces a reduction in lymphocyte count, additional hematological adverse reactions may be expected when cladribine is administered before or concurrently with other agents affecting the hematological profile (such as carbamazepine). In such cases, careful monitoring of hematological parameters is recommended.
Live or live attenuated vaccines
Treatment with Mavenclad® should not be initiated within 4–6 weeks after vaccination with live or live attenuated vaccines due to the risk of acute vaccine-related infection. Live or live attenuated vaccines should be avoided during and after cladribine treatment until the patient's lymphocyte count has returned to normal levels.
Strong inhibitors of ENT1, CNT3, and BCRP transporters
At the level of cladribine absorption, the only likely source of clinically relevant interactions is the breast cancer resistance protein (BCRP or ABCG2). Inhibition of BCRP in the gastrointestinal tract may increase the oral bioavailability and systemic exposure of cladribine. Known BCRP inhibitors that may alter the pharmacokinetics of BCRP substrates by 20% in vivo include eltrombopag.
In vitro studies indicate that cladribine is a substrate of the equilibrative nucleoside transporter (ENT1) and the concentrative nucleoside transporter (CNT3). Therefore, the bioavailability, intracellular distribution, and renal excretion of cladribine may theoretically be affected by strong inhibitors of ENT1, CNT3, and BCRP transporters, such as dipyridamole, nifedipine, nimodipine, cilostazol, sulindac, and reserpine. However, the net effect, expressed as a change in the potential activity of cladribine, is difficult to predict.
Although the clinical significance of such interactions is unknown, concomitant administration of strong inhibitors of ENT1, CNT3, and BCRP should be avoided during the 4- or 5-day cladribine treatment period. If avoidance is not possible, consideration should be given to selecting alternative concomitant medicinal products that lack or have minimal inhibitory properties toward ENT1, CNT3, and BCRP transporters. If this is not feasible, it is recommended to reduce the dose of medicinal products containing such compounds to the minimum required dose, separate the administration times of the drugs, and closely monitor the patient.
Strong inducers of BCRP and P-gp transporters
The effect of strong inducers of efflux transporters BCRP and P-glycoprotein (P-gp) on the bioavailability and distribution of cladribine has not been studied in dedicated trials. When concomitantly administered with strong inducers of BCRP transporters (e.g., corticosteroids) or P-gp (e.g., rifampicin, St. John’s wort), a potential reduction in cladribine efficacy should be considered.
Hormonal contraceptives
No clinically relevant pharmacokinetic interaction between cladribine and oral contraceptives (ethinylestradiol and levonorgestrel) has been observed. Therefore, concomitant use with cladribine is not expected to reduce the efficacy of hormonal contraceptives (see section "Use in pregnancy or breastfeeding").
Special precautions for use.
Hematological monitoring
The mechanism of action of cladribine is closely related to a reduction in lymphocyte count. The effect on lymphocyte count is dose-dependent. In clinical studies, reductions in neutrophil count, red blood cell count, hematocrit, hemoglobin, and platelet count were also observed compared to baseline values, although these parameters usually remained within normal limits.
When cladribine is administered concomitantly with other agents affecting the hematological profile, additional adverse reactions may be expected.
Lymphocyte counts should be determined:
- before starting treatment in year 1,
- before starting treatment in year 2,
- at 2 and 6 months after initiation of treatment in each treatment year; if the lymphocyte count is below 500 cells/mm³, this parameter should be actively monitored until values increase again.
For information on treatment decisions based on lymphocyte count, see section "Dosage and administration" and subsection "Infections" below.
Infections
Cladribine may weaken the body's immune defenses, thereby increasing the risk of infections. Serious, severe, and opportunistic infections, including fatal cases, have been observed during treatment with MAVENCLAD®. HIV infection, active tuberculosis, and active hepatitis must be excluded prior to initiating cladribine treatment (see section "Contraindications").
Latent infections, including tuberculosis or hepatitis, may be reactivated during treatment. Therefore, screening for latent infections, particularly tuberculosis and hepatitis B and C, should be performed before starting therapy in year 1 and year 2. Initiation of MAVENCLAD® treatment should be delayed until adequate treatment of infections has been completed.
In patients with acute infections, postponement of cladribine treatment initiation should also be considered until infections are fully controlled.
Particular attention is recommended for patients without a history of varicella-zoster virus exposure. Vaccination against varicella is recommended for patients lacking antibodies to varicella prior to starting cladribine therapy. In such cases, initiation of MAVENCLAD® treatment should be delayed by 4–6 weeks to allow full vaccine effect.
An increased incidence of herpes zoster has been observed in patients receiving cladribine. If lymphocyte count drops below 200 cells/mm³, antiviral prophylaxis against herpes should be considered during grade 4 lymphopenia according to local standard protocols.
Patients with lymphocyte counts below 500 cells/mm³ should be actively monitored for signs and symptoms suggestive of infection, including herpes zoster. If such signs or symptoms occur, appropriate treatment for infection should be initiated based on clinical judgment. Interruption or delay of MAVENCLAD® treatment may be considered until the infection is adequately treated.
Cases of progressive multifocal leukoencephalopathy (PML) have been reported with parenteral cladribine formulations used in patients with hairy cell leukemia treated under different treatment regimens.
Although PML has not been reported with oral cladribine tablets, a baseline MRI scan should be performed prior to initiating MAVENCLAD® tablets (usually within 3 months).
Malignant neoplasms
In clinical studies, cases of malignancies were observed more frequently in patients receiving cladribine compared to those receiving placebo.
MAVENCLAD® is contraindicated in patients with active malignancies (see section "Contraindications"). For patients with prior malignancies, individual benefit-risk assessment should be performed before initiating MAVENCLAD® treatment. Patients receiving cladribine should be advised to follow standard cancer screening recommendations.
Hepatic function
Hepatic events, including serious cases, have been infrequently reported in patients treated with MAVENCLAD®.
The patient's full medical history should be considered before prescribing MAVENCLAD®, particularly prior episodes of drug-induced liver injury and existing hepatic disorders. Serum levels of aminotransferases, alkaline phosphatase, and total bilirubin should be assessed before treatment initiation in year 1 and year 2. Hepatic enzymes and bilirubin should be monitored during treatment based on clinical signs and symptoms.
If clinical signs develop, unexplained increases in hepatic enzyme levels occur, or symptoms suggestive of hepatic dysfunction arise (e.g., nausea, vomiting, abdominal pain, fatigue, unexplained anorexia, or jaundice and/or dark urine), serum transaminases and total bilirubin levels should be promptly measured. If necessary, treatment with MAVENCLAD® should be interrupted or discontinued.
Contraception
Prior to starting treatment in year 1 and year 2, women of childbearing potential and men who may potentially father children should be informed about the potential serious risk to the fetus and the necessity of using effective contraceptive measures.
Women of childbearing potential must avoid pregnancy by using effective contraception during cladribine treatment and for at least 6 months after the last dose.
Male patients must take precautions to prevent pregnancy in their partners during cladribine treatment and for at least 6 months after the last dose.
Blood transfusion
For patients requiring blood transfusion, irradiation of cellular blood components prior to infusion is recommended to prevent transfusion-associated graft-versus-host disease. Consultation with a hematologist is recommended.
Transitioning to and from cladribine treatment
Before initiating treatment in patients previously treated with immunomodulatory or immunosuppressive medicinal products, the mechanism of action and duration of effect of these agents should be considered. The potential for additive effects on the immune system should also be considered when such medicinal products are used after MAVENCLAD® treatment.
When switching from another medicinal product for the treatment of MS, a baseline MRI scan is recommended (see subsection "Infections" above).
Hepatic impairment
The use of cladribine is not recommended in patients with moderate or severe hepatic impairment (Child-Pugh score > 6).
Sorbitol
MAVENCLAD® contains sorbitol. In case of known intolerance to certain sugars, consultation with a physician is recommended before initiating this medicinal product.
The additional impact of concomitantly administered medicinal products containing sorbitol (or fructose) and dietary intake of sorbitol (or fructose) should be considered.
The presence of sorbitol in oral medicinal products may affect the bioavailability of other concurrently administered oral medicinal products.
Use during pregnancy or breastfeeding.
Contraception in men and women
Prior to starting treatment in year 1 and year 2, women of childbearing potential and men who may potentially father children should be informed about the potential serious risk to the fetus and the necessity of using effective contraceptive measures.
In women of childbearing potential, pregnancy must be excluded before initiating MAVENCLAD® treatment in years 1 and 2, and effective contraception must be used during cladribine treatment and for at least 6 months after the last dose. Women who become pregnant during treatment with MAVENCLAD® must discontinue therapy.
Since cladribine affects DNA synthesis, a negative impact on human gametogenesis is expected. Therefore, male patients must take precautions to prevent pregnancy in their partners during cladribine treatment and for at least 6 months after the last dose.
Pregnancy
Based on experience with other agents that inhibit DNA synthesis in humans, cladribine may cause fetal developmental abnormalities if used during pregnancy. Reproductive toxicity of cladribine has been demonstrated in animal studies.
MAVENCLAD® is contraindicated in pregnant women (see section "Contraindications").
Breastfeeding
Limited data from spontaneous reports indicate that cladribine is excreted in human breast milk, although the quantity has not yet been established. Due to the potential for serious adverse reactions in breastfed infants, breastfeeding is contraindicated during treatment with MAVENCLAD® and for 1 week after the last dose (see section "Contraindications").
Fertility
No effects of cladribine on fertility or reproductive function of offspring were observed in mice; however, effects on testicular function were observed in mice and monkeys.
Since cladribine affects DNA synthesis, a negative impact on human gametogenesis is expected. Therefore, male patients must take precautions to prevent pregnancy in their partners during cladribine treatment and for at least 6 months after the last dose.
Ability to affect reaction speed when driving or operating machinery.
MAVENCLAD® has no or negligible influence on the ability of patients to drive or operate machinery.
Method of Administration and Dosage
Treatment should be initiated and supervised by a physician experienced in the management of multiple sclerosis (MS).
Dosage
The recommended cumulative dose of the medicinal product is 3.5 mg/kg body weight over 2 years, administered as one treatment course of 1.75 mg/kg body weight annually. Each treatment course consists of 2 treatment weeks—one at the beginning of the first month and another at the beginning of the second month of the respective treatment year. Based on medical necessity (e.g., to allow recovery of lymphocyte counts), the treatment course in year 2 may be delayed by up to 6 months. Each treatment week consists of 4 or 5 days during which the patient takes 10 mg or 20 mg (one or two tablets) as a single daily dose, depending on body weight. Further detailed information is provided in Tables 2 and 3.
After completion of 2 treatment courses, further treatment with cladribine in years 3 and 4 is not required. Re-initiation of treatment after 4 years has not been studied.
Criteria for Initiation and Continuation of Therapy
Lymphocyte counts must be:
- within normal range before initiation of treatment in year 1,
- at least 800 cells/mm³ before initiation of treatment in year 2.
If necessary, to allow recovery of lymphocyte counts, the treatment course in year 2 may be postponed for up to 6 months. If recovery takes longer than 6 months, the patient must not receive cladribine again.
Dosage Schedule
The distribution of the total dose over the 2 years of treatment is shown in Table 2. For some patients, depending on body weight, the number of tablets may differ between the first and second treatment weeks. The use of oral cladribine in patients with body weight below 40 kg has not been studied.
Table 2. Cladribine dosage according to patient body weight during each treatment week of the treatment years
| Body weight range, kg |
Dose in mg (number of 10 mg tablets) during treatment week |
|
| Treatment week 1 |
Treatment week 2 |
|
| from 40 to < 50 |
40 mg (4 tablets) |
40 mg (4 tablets) |
| from 50 to < 60 |
50 mg (5 tablets) |
50 mg (5 tablets) |
| from 60 to < 70 |
60 mg (6 tablets) |
60 mg (6 tablets) |
| from 70 to < 80 |
70 mg (7 tablets) |
70 mg (7 tablets) |
| from 80 to < 90 |
80 mg (8 tablets) |
70 mg (7 tablets) |
| from 90 to < 100 |
90 mg (9 tablets) |
80 mg (8 tablets) |
| from 100 to < 110 |
100 mg (10 tablets) |
90 mg (9 tablets) |
| from 110 and above |
100 mg (10 tablets) |
100 mg (10 tablets) |
Table 3 shows how to distribute the total number of tablets per treatment week across individual days. It is recommended that each daily dose of cladribine in each treatment week be administered at 24-hour intervals, approximately at the same time of day. If the daily dose consists of two tablets, both tablets should be taken together as a single dose.
| Table 3 Distribution of tablets by days of the week |
|||||
| Total number of tablets per week |
Day 1 |
Day 2 |
Day 3 |
Day 4 |
Day 5 |
| 4 |
1 |
1 |
1 |
1 |
0 |
| 5 |
1 |
1 |
1 |
1 |
1 |
| 6 |
2 |
1 |
1 |
1 |
1 |
| 7 |
2 |
2 |
1 |
1 |
1 |
| 8 |
2 |
2 |
2 |
1 |
1 |
| 9 |
2 |
2 |
2 |
2 |
1 |
| 10 |
2 |
2 |
2 |
2 |
2 |
A missed dose should be taken as soon as possible on the same day after the omission, according to the treatment schedule.
A missed dose must not be taken together with the next scheduled dose on the following day. In case of a missed dose, the patient should take it the next day and extend the number of treatment days by one day within that treatment week. If two consecutive doses are missed, the same approach should be applied, and the number of days in the treatment week should be extended by two days.
Concomitant use of other oral medicinal products
During the limited number of days of cladribine administration, it is recommended to separate the intake of any other oral medicinal products from the intake of Mavenclad® by at least 3 hours.
Specific patient groups
Patients with renal impairment
Specific studies on the treatment of patients with renal impairment have not been conducted.
For patients with mild renal impairment (creatinine clearance from 60 to 89 mL/min), dose adjustment is not considered necessary.
The safety and efficacy of cladribine in patients with moderate or severe renal impairment have not been studied. Therefore, cladribine is contraindicated in such patients (see section "Contraindications").
Patients with hepatic impairment
Studies on the treatment of patients with hepatic impairment have not been conducted.
Dose adjustment is not required in patients with mild hepatic impairment, as the role of liver function in the elimination of cladribine is considered to be negligible. Due to lack of data, the use of cladribine is not recommended in patients with moderate or severe hepatic impairment (Child–Pugh score > 6).
Elderly patients
Cladribine should be used with caution in elderly patients, taking into account the potential for a higher incidence of impaired hepatic or renal function, concomitant diseases, and concomitant therapies.
Method of administration
Mavenclad® is intended for oral use. The tablets should be taken with water and swallowed whole, without chewing. Tablets may be taken independently of food intake.
Since the tablets are not coated, they should be swallowed immediately after removal from the blister pack, without leaving them on a surface or holding them in the hands longer than necessary for administration. If a tablet has been left on a surface or if a broken or crushed tablet has been removed from the blister, the surface that came into contact with the tablet should be thoroughly washed with water.
The patient should handle the tablets with dry hands, and hands should then be thoroughly washed.
Instructions for patient self-administration of Mavenclad® tablets
- Prepare a glass of water and ensure your hands are dry and clean.
- Take the medication box and orient it towards you with the side showing the instructions.

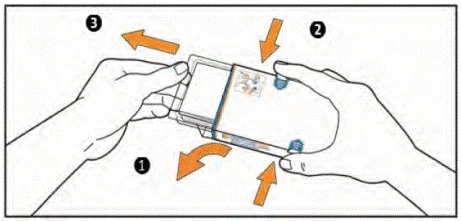
- (1) Open the flap on the left side of the box.
(2) Simultaneously press the tabs on both sides of the box with your index and thumb fingers and hold them in this position.
(3) Pull out the push-through blister pack until it moves. WARNING: Do not remove the push-through blister pack from the box.
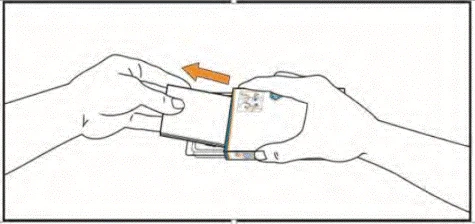
- Remove the package leaflet from the box. Ensure you have carefully read its contents, including the instructions for self-administration of the tablets.
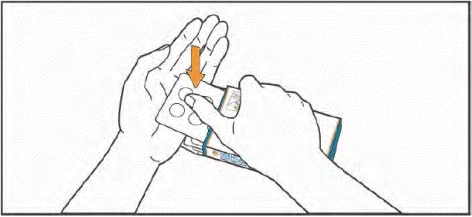
- Lift the blister by pressing your fingers through the opening in the push-through blister pack. Place your hand under the blister and push 1 or 2 tablets into your hand, as prescribed by your doctor.
- Take the tablets with water. The tablets must be swallowed whole, without chewing and without allowing them to dissolve in the mouth. Contact between the tablets and the skin should be minimized. During tablet administration, do not touch your nose, eyes, or other body parts with your hands.
- After taking the tablets, wash your hands thoroughly with water and soap.
- Return the push-through blister pack into the box. Store it in the original packaging to protect from moisture.
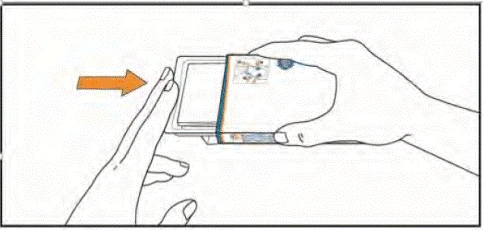
Store the tablets in the blister until the next dose is due. Do not push tablets out of the blister or store them in another container.
Children.
The safety and efficacy of Mavenclad® in children (under 18 years of age) have not been established. Appropriate data are lacking.
Overdose.
There is limited experience with cladribine overdose following oral administration. It is known that lymphopenia developing as a result of overdose is dose-dependent.
In patients with cladribine overdose, particularly careful monitoring of hematological parameters is recommended.
There is no specific antidote for cladribine. Management consists of careful observation of the patient and appropriate supportive measures. Consideration should be given to the potential need to discontinue Mavenclad®. Due to the rapid and extensive intracellular and tissue distribution, it is unlikely that hemodialysis would significantly eliminate cladribine.
Adverse Reactions
Brief Summary of Safety Profile
The most clinically significant adverse reactions are lymphopenia (25.6%) and herpes zoster (3.0%). The incidence of herpes zoster was higher during periods when grade 3 or 4 lymphopenia (from < 500 to 200 cells/mm³ or < 200 cells/mm³) was observed, compared to periods when patients did not have grade 3 or 4 lymphopenia.
List of Adverse Reactions
The information on adverse reactions described below is based on pooled data from clinical studies of relapsing-remitting multiple sclerosis (MS) treatment with oral cladribine used as monotherapy at a cumulative dose of 3.5 mg/kg. The safety database from these studies includes data from 923 patients. Adverse reactions identified during the post-marketing surveillance period are marked with a footnote (*).
The following classification was used to define the frequency of adverse reactions: very common (≥ 1/10), common (≥ 1/100 to < 1/10), uncommon (≥ 1/1,000 to < 1/100), rare (≥ 1/10,000 to < 1/1,000), very rare (< 1/10,000), frequency not known (cannot be estimated from available data).
Infections and infestations
Common: oral herpes, dermatomal herpes zoster.
Rare: tuberculosis.
Blood and lymphatic system disorders
Very common: lymphopenia.
Common: decreased neutrophil count.
Immune system disorders
Common: hypersensitivity reactions*, including pruritus, urticaria, rash, and isolated cases of angioedema.
Hepatobiliary disorders
Uncommon: liver injury*.
Skin and subcutaneous tissue disorders
Common: rash, alopecia.
Description of Selected Adverse Reactions
Lymphopenia
In clinical studies, transient grade 3 or 4 lymphopenia developed in 20–25% of patients receiving a cumulative dose of cladribine 3.5 mg/kg over 2 years as monotherapy. Grade 4 lymphopenia was observed in less than 1% of patients. The highest proportion of patients with grade 3 or 4 lymphopenia was observed 2 months after the first dose of cladribine each year (4.0% and 11.3% of patients with grade 3 lymphopenia in year 1 and year 2, respectively; 0% and 0.4% with grade 4 lymphopenia in year 1 and year 2, respectively). In most patients, lymphocyte counts are expected to recover to normal levels or to grade 1 lymphopenia within 9 months.
To reduce the risk of severe lymphopenia, lymphocyte counts should be monitored before, during, and after treatment with cladribine, and strict criteria for initiating and continuing cladribine therapy must be followed.
Malignant neoplasms
In clinical studies and during long-term follow-up, malignancies were observed more frequently in patients treated with oral cladribine at a cumulative dose of 3.5 mg/kg (10 cases in 3,414 patient-years [0.29 cases per 100 patient-years]) compared to placebo-treated patients (3 cases in 2,022 patient-years [0.15 cases per 100 patient-years]).
Hypersensitivity
In clinical studies, hypersensitivity reactions occurred more frequently in patients receiving oral cladribine at a cumulative dose of 3.5 mg/kg (11.8%) compared to placebo-treated patients (8.4%). Serious hypersensitivity reactions were observed in 0.3% of cladribine-treated patients and were not observed in the placebo group. Hypersensitivity reactions led to discontinuation of treatment in 0.4% of cladribine-treated patients and in 0.3% of placebo-treated patients.
Liver injury
During post-marketing surveillance, uncommon cases of liver injury, including serious cases and cases leading to discontinuation of treatment, temporally associated with the use of Mavencles® have been reported. Transient elevations in serum transaminases typically exceeded the upper limit of normal (ULN) by more than 5 times. Isolated cases of transient increases in serum transaminase levels up to 40 times above ULN and/or symptomatic hepatitis with transient increases in bilirubin and jaundice have been observed. These events occurred at various time intervals after treatment initiation, but most cases were observed within 8 weeks after the first treatment course (see section "Special precautions").
Reporting suspected adverse reactions after medicine authorization is important. It allows continued monitoring of the benefit-risk balance of the medicine. Healthcare professionals and patients, or their legal representatives, are encouraged to report all suspected adverse reactions and lack of efficacy through the Automated Pharmacovigilance Information System at: https://aisf.dec.gov.ua.
Shelf life. 4 years.
Do not use after the expiry date stated on the packaging.
Storage conditions.
Store in the original packaging to protect from moisture.
Keep out of the reach and sight of children.
Packaging. 1, 4, or 6 tablets in an aluminum blister pack, sealed in a cardboard sleeve, placed in a contour cell packaging, and inserted into a cardboard box with child-resistant protection.
Prescription category. Prescription only.
Manufacturer. NerPharMa S.R.L. / NerPharMa S.R.L.
Manufacturer's address and location of its business activities.
Viale Pasteur 10 (loc. Nerviano), 20014 Milan (MI), Italy /
Viale Pasteur 10 (loc. Nerviano), 20014 Milan (MI), Italy.