Lipobon
Ukraina
Spis treści
INSTRUKCJA DOTYCZĄCA STOSOWANIA LEKU Lipobon (LIPOBON)
Skład:
substancja czynna: ezetymib;
1 tabletka zawiera ezetymibu 10 mg;
substancje pomocnicze: celuloza mikrokryształowa, manitol, sodowa kroskarboksymetyloceluloza, hydroksypropyloceluloza o niskim stopniu zastępczości, povidon-K25, sodowy laurylosiarczan, stearyna magnezu.
Postać leku. Tabletki.
Główne właściwości fizykochemiczne: wydłużone tabletki białego lub prawie białego koloru z oznaczeniem „E 611” po jednej stronie i bez żadnych oznaczeń po drugiej stronie.
Grupa farmakoterapeutyczna.
Inne leki modyfikujące poziom lipidów. Kod ATC C10AX09.
Właściwości farmakologiczne.
Farmakodynamika.
Mechanizm działania
Ezetymib to przedstawiciel nowej klasy leków obniżających stężenie lipidów, które selektywnie hamują jelitową absorpcję cholesterolu i pokrewnych fitosteroli. Ezetymib jest doustnie aktywny i ma mechanizm działania różny od innych klas leków obniżających stężenie cholesterolu (np. statyn, wiązarek kwasów żółciowych (żywic), kwasowych pochodnych fibranów i fitostanolów). Cząsteczkowym celem ezetymibu jest transporter steroli Niemann-Pick C1-Like 1 (NPC1L1), odpowiedzialny za wchłanianie cholesterolu i fitosteroli w jelitach.
Ezetymib lokalizuje się na powierzchni szczoteczkowej jelita cienkiego i hamuje absorpcję cholesterolu, zmniejszając dostarczanie jelitowego cholesterolu do wątroby; statyny natomiast zmniejszają synteze cholesterolu w wątrobie, a razem te mechanizmy zapewniają dodatkowe obniżenie stężenia cholesterolu. Po dwutygodniowym zastosowaniu klinicznym u 18 pacjentów z hipercholesterolemia ezetymib obniżał absorpcję cholesterolu o 54% w porównaniu z placebo.
Właściwości farmakodynamiczne
Przeprowadzono serię badań przedklinicznych w celu określenia selektywności ezetymibu w zakresie hamowania absorpcji cholesterolu. Ezetymib hamował absorpcję [14C]-cholesterolu bez wpływu na absorpcję trójglicerydów, kwasów tłuszczowych, kwasów żółciowych, progesteronu, etynylowego estradiolu czy witamin rozpuszczalnych w tłuszczach A i D.
Badania epidemiologiczne wykazały, że choroby sercowo-naczyniowe i śmiertelność zmieniają się wprost proporcjonalnie do poziomu całkowitego cholesterolu i cholesterolu lipoprotein o niskiej gęstości (Ch-LDL) oraz odwrotnie proporcjonalnie do poziomu cholesterolu lipoprotein o wysokiej gęstości (Ch-HDL).
Zastosowanie ezetymibu w połączeniu ze statyną skutecznie zmniejsza ryzyko zdarzeń sercowo-naczyniowych u pacjentów z chorobą wieńcową i ostrym zespołem wieńcowym (OZW) w wywiadzie.
Skuteczność kliniczna i bezpieczeństwo
W kontrolowanych badaniach klinicznych ezetymib stosowany jako monoterapia, jak również w połączeniu ze statyną, znacząco obniżał całkowity cholesterol (całkowity Ch), cholesterol lipoprotein o niskiej gęstości (Ch-LDL), apolipoproteinę B (Apo-B) i trójglicerydy (TG) oraz zwiększał cholesterol lipoprotein o wysokiej gęstości (Ch-HDL) u pacjentów z hipercholesterolemią.
Pierwotna hipercholesterolemia
W podwójnie ślepej, placebo-kontrolowanej 8-tygodniowej próbie 769 pacjentów z hipercholesterolemią, którzy otrzymywali już monoterapię statyną i nie osiągali celu Ch-LDL zgodnie z Narodowym Programem Edukacyjnym dotyczącym cholesterolu (NCEP) (od 2,6 do 4,1 mmol/l [od 100 do 160 mg/dl], w zależności od wyjściowych cech), losowo przydzielono do przyjmowania ezetymibu 10 mg lub placebo jako dodatek do obecnej terapii statyną.
Wśród pacjentów otrzymujących statyny, którzy nie osiągali celu Ch-LDL na początku badania (~82%), znacznie więcej pacjentów przydzielonych do przyjmowania ezetymibu osiągnęło cel Ch-LDL w końcowym punkcie badania w porównaniu z pacjentami przydzielonymi do przyjmowania placebo, odpowiednio 72% i 19%. Odpowiednie obniżenie Ch-LDL istotnie różniło się (25% i 4% dla ezetymibu w porównaniu z placebo, odpowiednio). Ponadto ezetymib dodany do obecnej terapii statyną istotnie obniżył całkowity Ch, Apo-B, TG i zwiększył Ch-HDL w porównaniu z placebo. Ezetymib lub placebo dodane do terapii statyną zmniejszyły średni poziom białka C-reaktywnego odpowiednio o 10% lub 0% w stosunku do wartości wyjściowej.
W dwóch podwójnie ślepych, randomizowanych, placebo-kontrolowanych badaniach trwających 12 tygodni, w których wzięło udział 1719 pacjentów z pierwotną hipercholesterolemią, ezetymib w dawce 10 mg istotnie obniżył całkowity Ch (13%), LDL (19%), Apo-B (14%) i TG (8%) oraz zwiększył Ch-HDL (3%) w porównaniu z placebo. Ponadto ezetymib nie wpływał na stężenie witamin rozpuszczalnych w tłuszczach A, D i E, nie wpływał na czas protrombinowy i, podobnie jak inne leki obniżające stężenie lipidów, nie zakłócał produkcji sterydowego hormonu kory nadnerczy.
W wieloośrodkowym, podwójnie ślepych, kontrolowanym badaniu klinicznym (ENHANCE) 720 pacjentów z heterozygotyczną hipercholesterolemią rodzinną zostało losowo przydzielonych do przyjmowania ezetymibu 10 mg w połączeniu z symwastatyną 80 mg (n = 357) lub samej symwastatyny 80 mg (n = 363) przez 2 lata. Głównym celem badania była ocena wpływu terapii kombinowanej ezetymibem/symwastatyną na grubość intima-media (TIM) tętnic szyjnych w porównaniu z monoterapią symwastatyną. Wpływ tego markera zastępczego na chorobę sercowo-naczyniową i śmiertelność nie został jeszcze wykazany. Pierwotny punkt końcowy, zmiana średniej TIM we wszystkich sześciu segmentach tętnic szyjnych, nie różnił się istotnie (p = 0,29) między dwiema grupami leczenia, jak określono za pomocą ultrasonografii w trybie B. W trakcie dwuletniego badania grubość intima-media zwiększyła się odpowiednio o 0,0111 mm i 0,0058 mm przy stosowaniu ezetymibu 10 mg w połączeniu z symwastatyną 80 mg lub samej symwastatyny 80 mg (początkowa grubość TIM tętnic szyjnych wynosiła odpowiednio 0,68 mm i 0,69 mm).
Ezetymib 10 mg w połączeniu z symwastatyną 80 mg istotnie bardziej obniżył poziom Ch-LDL, całkowitego Ch, Apo-B i TG niż symwastatyna 80 mg. Procentowy wzrost Ch-HDL był podobny w obu grupach leczenia. Reakcje niepożądane zgłaszane przy stosowaniu ezetymibu 10 mg w połączeniu z symwastatyną 80 mg odpowiadały jego znanemu profilowi bezpieczeństwa.
Dzieci
W wieloośrodkowym, podwójnie ślepych, kontrolowanym badaniu 138 pacjentów (59 chłopców i 79 dziewcząt) w wieku od 6 do 10 lat (średni wiek 8,3 roku) z heterozygotyczną hipercholesterolemią rodzinną lub nie-rodzinną (HHR) z wyjściowymi poziomami Ch-LDL między 3,74 a 9,92 mmol/l, losowo przydzielono do przyjmowania ezetymibu 10 mg lub placebo przez 12 tygodni.
W 12. tygodniu ezetymib istotnie obniżył całkowity Ch (-21% vs. 0%), Ch-LDL (-28% vs. -1%), Apo-B (-22% vs. -1%), Ch-LDL (-26% vs. 0%) w porównaniu z placebo. Wyniki dla dwóch grup leczenia były podobne dla TG i Ch-HDL (-6% vs. +8% i +2% vs. +1%, odpowiednio).
W wieloośrodkowym, podwójnie ślepych, kontrolowanym badaniu 142 chłopców (stopień dojrzewania wg Tanner’a II lub wyższy) i 106 dziewcząt po menarche w wieku od 10 do 17 lat (średni wiek 14,2 roku) z heterozygotyczną hipercholesterolemią rodzinną (HHR) z wyjściowymi poziomami Ch-LDL między 4,1 a 10,4 mmol/l, losowo przydzielono do przyjmowania ezetymibu 10 mg w połączeniu z symwastatyną (10, 20 lub 40 mg) lub samej symwastatyny (10, 20 lub 40 mg) przez 6 tygodni, następnie w połączeniu z ezetymibem i 40 mg symwastatyny lub samą 40 mg symwastatyną przez kolejne 27 tygodni, a następnie z otwartym dodatkowym przyjmowaniem ezetymibu i symwastatyny (10 mg, 20 mg lub 40 mg) przez 20 tygodni.
W 6. tygodniu ezetymib w połączeniu ze statyną (we wszystkich dawkach) istotnie zmniejszył całkowity Ch (38% vs. 26%), Ch-LDL (49% vs. 34%), Apo-B (39% vs. 27%), Ch-LDL (47% vs. 33%) w porównaniu z monoterapią statyną (we wszystkich dawkach). Wyniki dla dwóch grup leczenia były podobne dla TG i Ch-HDL (-17% vs. -12% i +7% vs. +6%, odpowiednio). W 33. tygodniu wyniki były zgodne z wynikami w 6. tygodniu, a znacznie większa liczba pacjentów przyjmujących ezetymib i 40 mg symwastatyny (62%) osiągnęła optymalny cel zgodnie z NCEP AAP (< 2,8 mmol/l [110 mg/dl]) dla Ch-LDL w porównaniu z pacjentami przyjmującymi 40 mg symwastatyny (25%). W 53. tygodniu, na końcu otwartego badania dodatkowego, wpływ na parametry lipidowe utrzymywał się.
Bezpieczeństwo i skuteczność ezetymibu stosowanego w połączeniu z symwastatyną w dawce przekraczającej 40 mg dziennie u dzieci w wieku od 10 do 17 lat nie były badane. Bezpieczeństwo i skuteczność ezetymibu stosowanego w połączeniu z symwastatyną u dzieci poniżej 10 roku życia nie były badane. Długoterminowa skuteczność terapii ezetymibem u pacjentów poniżej 17 roku życia w celu zmniejszenia zachorowalności i śmiertelności w dorosłości nie była badana.
Profilaktyka zdarzeń sercowo-naczyniowych
Poprawione skrócenie wyników: międzynarodowe badanie efektywności Witorynu (IMPROVE-IT) – to wieloośrodkowe, randomizowane, podwójnie ślepe badanie z aktywnym kontrolowaniem, w którym wzięło udział 18144 pacjentów, którzy zostali włączeni w ciągu 10 dni po hospitalizacji z powodu ostrego zespołu wieńcowego (OZW, ostry zawał mięśnia sercowego [ZM] lub niestabilna dławica piersiowa [ND]). W momencie wystąpienia OZW poziom Ch-LDL u pacjentów wynosił ≤ 125 mg/dl (≤ 3,2 mmol/l), jeśli nie otrzymywali terapii lipidotnej, lub ≤ 100 mg/dl (≤ 2,6 mmol/l), jeśli otrzymywali terapię lipidotną. Wszyscy pacjenci zostali losowo przydzieleni w stosunku 1:1 do przyjmowania ezetymibu/symwastatyny 10/40 mg (n = 9067) lub symwastatyny 40 mg (n = 9077) i byli obserwowani średnio przez 6 lat.
Średni wiek pacjentów wynosił 63,6 roku; 76% stanowili mężczyźni, 84% miało pochodzenie kaukaskie, a 27% miało cukrzycę. Średni poziom Ch-LDL w momencie wystąpienia kwalifikującego zdarzenia wynosił 80 mg/dl (2,1 mmol/l) u pacjentów otrzymujących terapię lipidotną (n = 6390) i 101 mg/dl (2,6 mmol/l) u tych, którzy nie otrzymywali wcześniejszej terapii lipidotnej (n = 11 594). Przed hospitalizacją z powodu OZW 34% pacjentów otrzymywało terapię statynami. W ciągu 1 roku średni poziom Ch-LDL u pacjentów kontynuujących terapię wynosił 53,2 mg/dl (1,4 mmol/l) w grupie ezetymibu/symwastatyny i 69,9 mg/dl (1,8 mmol/l) w grupie monoterapii symwastatyną. Wartości lipidów ogólnie były uzyskiwane dla pacjentów, którzy pozostali w badanej terapii.
Pierwotnym punktem końcowym była kombinacja obejmująca zgon z powodu choroby sercowo-naczyniowej, ostry zespół wieńcowy (OZW, definiowany jako nielętny zawał mięśnia sercowego, udokumentowana niestabilna dławica piersiowa wymagająca hospitalizacji lub jakakolwiek procedura rewaskularyzacji wieńcowej przeprowadzona co najmniej 30 dni po przydzieleniu randomizowanej terapii) oraz nielętny udar. Badanie wykazało, że leczenie ezetymibem dodanym do symwastatyny zapewnia stopniową korzyść w redukcji pierwotnego punktu końcowego obejmującego zgon z powodu choroby sercowo-naczyniowej, OZW i nielętny udar w porównaniu z samą symwastatyną (względne ryzyko zmniejsza się o 6,4%, p = 0,016). Pierwotny punkt końcowy zaobserwowano u 2572 z 9067 pacjentów (7-letni wskaźnik oparty na metodzie Kaplana-Meiera [KM] 32,72%) w grupie ezetymibu/symwastatyny i u 2742 z 9077 pacjentów (7-letni wskaźnik oparty na metodzie KM 34,67%) w grupie symwastatyny. (patrz Rys.1 i Tabela 1). Oczekuje się, że ta dodatkowa korzyść będzie podobna przy jednoczesnym stosowaniu innych statyn, które wykazały skuteczność w zmniejszaniu ryzyka zdarzeń sercowo-naczyniowych. Ogólna śmiertelność w tej grupie wysokiego ryzyka nie uległa zmianie (patrz Tabela 1).
Dla wszystkich udarów istniała ogólna korzyść; jednak zaobserwowano niewielki, nieistotny wzrost udarów krwotocznych w grupie ezetymibu-symwastatyny w porównaniu z grupą symwastatyny (patrz Tabela 1). Ryzyko udaru krwotocznego przy stosowaniu ezetymibu wraz z wysoko aktywnymi statynami w długoterminowych badaniach wyników nie zostało ocenione.
Efekt terapeutyczny ezetymibu/symwastatyny ogólnie odpowiadał ogólnym wynikom w wielu podgrupach, w tym płci, wieku, rasie, obecności cukrzycy w wywiadzie, wyjściowym poziomie lipidów, poprzedniej terapii statynami, poprzednim udarze i nadciśnieniu.
Rys. 1. Wpływ ezetymibu/symwastatyny na pierwotny punkt końcowy obejmujący zgon z powodu choroby sercowo-naczyniowej, ostry zespół wieńcowy i nielętny udar.
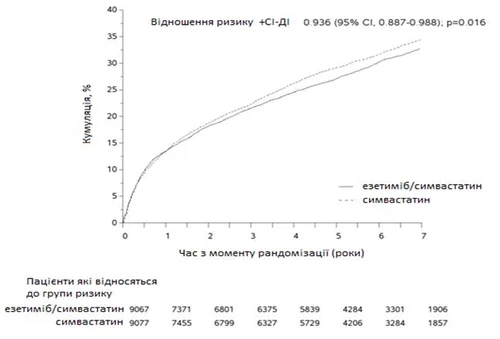
Główne zdarzenia sercowo-naczyniowe według grup leczenia u wszystkich pacjentów losowo przydzielonych w badaniu IMPROVE-IT.
Tabela 1.
| Wynik |
Lipobon 10/40 mga (N = 9067) |
Simwastatyna (N = 9077) |
Stosunek ryzyka (95 % CI) |
Wartość p |
||
| n |
K-M %c |
n |
K-M %c |
|||
| Pierwotny punkt końcowy złożony |
||||||
| (śmiertelny skutek z powodu choroby sercowo-naczyniowej, ostry zespół wieńcowy i udar nieśmiertelny) |
2572 |
32,72 % |
2742 |
34,67 % |
0,936 (0,887, 0,988) |
0,016 |
| Punkty końcowe wtórne złożone |
||||||
| Śmiertelny skutek z powodu choroby niedokrwiennej serca (CZS), nieśmiertelny zawał mięśnia sercowego (ZMS), pilna rewaskularyzacja wieńcowa po 30 dniach |
1322 |
17,52 % |
1448 |
18,88 % |
0,912 (0,847, 0,983) |
0,016 |
| OZW, nieśmiertelny udar, śmiertelny skutek (wszystkie przyczyny) |
3089 |
38,65 % |
3246 |
40,25 % |
0,948 (0,903, 0,996) |
0,035 |
| Śmiertelny skutek z powodu choroby sercowo-naczyniowej, nieśmiertelny ZMS, niestabilna dławica piersiowa wymagająca hospitalizacji, wszelkie rewaskularyzacje, nieśmiertelny udar |
2716 |
34,49 % |
2869 |
36,20 % |
0,945 (0,897, 0,996) |
0,035 |
| Składniki pierwotnego punktu końcowego złożonego oraz wybrane punkty końcowe skuteczności (pierwsze wystąpienie danego zdarzenia w dowolnym czasie) |
||||||
| Śmiertelny skutek z powodu choroby sercowo-naczyniowej |
537 |
6,89 % |
538 |
6,84 % |
1,000 (0,887, 1,127) |
0,997 |
| Ostry zespół wieńcowy: |
||||||
| Nieśmiertelny ZMS |
945 |
12,77 % |
1083 |
14,41 % |
0,871 (0,798, 0,950) |
0,002 |
| Niestabilna dławica piersiowa wymagająca hospitalizacji |
156 |
2,06 % |
148 |
1,92 % |
1,059 (0,846, 1,326) |
0,618 |
| Pilna rewaskularyzacja wieńcowa po 30 dniach |
1690 |
21,84 % |
1793 |
23,36 % |
0,947 (0,886, 1,012) |
0,107 |
| Nieśmiertelny udar |
245 |
3,49 % |
305 |
4,24 % |
0,802 (0,678, 0,949) |
0,010 |
| Wszystkie ZMS (śmiertelne i nieśmiertelne) |
977 |
13,13 % |
1118 |
14,82 % |
0,872 (0,800, 0,950) |
0,002 |
| Wszystkie udary (śmiertelne i nieśmiertelne) |
296 |
4,16 % |
345 |
4,77 % |
0,857 (0,734, 1,001) |
0,052 |
| Niekrwotoczny udard |
242 |
3,48 % |
305 |
4,23 % |
0,793 (0,670, 0,939) |
0,007 |
| Udar krwotoczny |
59 |
0,77 % |
43 |
0,59 % |
1,377 (0,930, 2,040) |
0,110 |
| Śmiertelny skutek z dowolnej przyczyny |
1215 |
15,36 % |
1231 |
15,28 % |
0,989 (0,914, 1,070) |
0,782 |
u 6 % pacjentów dawkę ezetymibu/symwastatyny zwiększono do 10/80 mg.
u 27 % pacjentów dawkę symwastatyny zwiększono do 80 mg.
c wartość 7-letnia oszacowana metodą Kaplana-Meiera.
d obejmuje udar niedokrwienny lub udar o nieokreślonym typie.
Profilaktyka głównych zdarzeń naczyniowych przy przewlekłej chorobie nerek (PChN)
Badanie dotyczące ochrony serca i nerek (SHARP) to wielonarodowe, randomizowane, placebo kontrolowane, podwójnie ślepe badanie, w którym wzięło udział 9438 pacjentów z przewlekłą chorobą nerek, z których jedna trzecia na początku była na dializie. Ogółem 4650 pacjentom przyznaczono stałą dawkę kombinacji ezetymibu 10 mg ze symwastatyną 20 mg, a 4620 – placebo; pacjentów obserwowano średnio przez 4,9 roku. Średni wiek pacjentów wynosił 62 lata; 63 % stanowili mężczyźni; 72 % miało pochodzenie kaukaskie; 23 % miało cukrzycę, a u tych, którzy nie byli na dializie, średnie oszacowane tempo filtracji kłębuszkowej (eGFR) wynosiło 26,5 ml/min/1,73 m². Nie stawiano kryteriów wprowadzania lipidów. Średni poziom cholesterolu LDL na początku wynosił 108 mg/dl. Po roku, w tym u pacjentów, którzy już nie przyjmowali badanych leków, cholesterol LDL zmniejszył się o 26 % przy monoterapii symwastatyną w dawce 20 mg oraz o 38 % przy stosowaniu ezetymibu w dawce 10 mg w połączeniu ze symwastatyną w dawce 20 mg w porównaniu z placebo.
Pierwotne porównanie zdefiniowane w protokole SHARP to analiza zgodnie z początkowo przydzielonym leczeniem „głównych zdarzeń naczyniowych” (GZN, definiowanych jako niemortalny zawał serca lub zatrzymanie krążenia, udar lub jakakolwiek procedura rewaskularyzacji) wyłącznie u tych pacjentów, którzy na początku zostali zrandomizowani do grupy ezetymibu w połączeniu ze symwastatyną (n = 4193) lub do grupy placebo (n = 4191). Analizy wtórne obejmowały analogiczną kombinację, przeanalizowaną dla całej kohorty zrandomizowanej (na początku badania lub po 1 roku) do przyjmowania ezetymibu w połączeniu ze symwastatyną (n = 4650) lub placebo (n = 4620), a także składniki tej kombinacji. Analiza pierwotnego punktu końcowego wykazała, że ezetymib w połączeniu ze symwastatyną istotnie zmniejszył ryzyko wystąpienia zdarzeń naczyniowych (749 pacjentów z zdarzeniami w grupie placebo i 639 w grupie ezetymibu w połączeniu ze symwastatyną) z względnym zmniejszeniem ryzyka o 16 % (p = 0,001). Jednakże ten projekt badania nie pozwolił na oddzielenie wkładu samodzielnego składnika ezetymibu w skuteczność, aby istotnie zmniejszyć ryzyko wystąpienia zdarzeń naczyniowych u pacjentów z PChN. Poszczególne składniki GZN u wszystkich zrandomizowanych pacjentów przedstawiono w Tabeli 2. Ezetymib w połączeniu ze symwastatyną istotnie zmniejszył ryzyko udaru oraz jakiejś rewaskularyzacji, z niewielkimi różnicami liczbowymi na korzyść ezetymibu w połączeniu ze symwastatyną w przypadku niemortalnego zawału serca i zatrzymania krążenia.
Główne zdarzenia naczyniowe według grup leczenia u wszystkich zrandomizowanych pacjentów w badaniu SHARPa.
Tabela 2.
| Wynik |
ezetymib 10 mg w kombinacji z symwastatyną 20 mg (N = 4650) |
Placebo (N = 4620) |
Stosunek ryzyka (95 % CI) |
Wartość p |
| Podstawowe zdarzenia waskularne |
701 (15,1 %) |
814 (17,6 %) |
0,85 (0,77 – 0,94) |
0,001 |
| Nieśmiertelny ZI |
134 (2,9 %) |
159 (3,4 %) |
0,84 (0,66 – 1,05) |
0,12 |
| Zatrzymanie serca |
253 (5,4 %) |
272 (5,9 %) |
0,93 (0,78 – 1,10) |
0,38 |
| Udar niedokrwienny |
171 (3,7 %) |
210 (4,5 %) |
0,81 (0,66 – 0,99) |
0,038 |
| Udar niedokrwienny mózgu |
131 (2,8 %) |
174 (3,8 %) |
0,75 (0,60 – 0,94) |
0,011 |
| Udar krwotoczny |
45 (1,0 %) |
37 (0,8 %) |
1,21 (0,78 – 1,86) |
0,40 |
| Wszelkie rewaskularyzacje |
284 (6,1 %) |
352 (7,6 %) |
0,79 (0,68 – 0,93) |
0,004 |
| Podstawowe zdarzenia przeciwmiażdżycowe (PZP)b |
526 (11,3 %) |
619 (13,4 %) |
0,83 (0,74 – 0,94) |
0,002 |
aAnaliza według pierwotnie przypisanego leczenia dla wszystkich pacjentów SHARP, randomizowanych do przyjmowania ezetymibu w połączeniu z symwastatyną lub placebo zarówno na etapie początkowym, jak i po 1 roku.
bZdarzenia związane z chorobą tętnic wieńcowych (ZCTW); definiowane jako kombinacja niezgonnego zawału mięśnia serca, śmierci wieńcowej, udaru niehemoragicznego lub jakiejkolwiek rewaskularyzacji.
Bezwzględne obniżenie stężenia cholesterolu LPNŻ osiągnięte przez ezetymib w połączeniu z symwastatyną było niższe u pacjentów z niższym początkowym stężeniem cholesterolu LPNŻ (< 2,5 mmol/l) oraz u pacjentów poddawanych dializę na etapie początkowym w porównaniu z innymi pacjentami, a odpowiadające temu zmniejszenie ryzyka w tych dwóch grupach było osłabione.
Homozygotyczna hipercholesterolemia rodzinna (HHR)
W podwójnie ślepej, randomizowanej 12-tygodniowej próbie wzięło udział 50 pacjentów z kliniczną i/lub genotypową diagnozą HHR, którzy otrzymywali atorwastatynę lub symwastatynę (40 mg) z lub bez wspomagającego aferezy LPNŻ. Ezetymib w połączeniu z atorwastatyną (40 lub 80 mg) lub symwastatyną (40 lub 80 mg) znacząco obniżył stężenie cholesterolu LPNŻ o 15% w porównaniu ze zwiększeniem dawki symwastatyny lub monoterapią atorwastatyną od 40 do 80 mg.
Homozygotyczna sytosterolemia (fitosterolemia)
W podwójnie ślepej, placebo-kontrolowanej 8-tygodniowej próbie 37 pacjentów z homozygotyczną sytosterolemią zostało zrandomizowanych do przyjmowania ezetymibu 10 mg (n = 30) lub placebo (n = 7). Niektórzy pacjenci otrzymywali inne leczenie (np. statyny, żywice). Ezetymib znacząco obniżył oba główne roślinne sterydy: sytosterol i kampesterol, odpowiednio o 21% i 24% w porównaniu z poziomem wyjściowym. Wpływ obniżenia stężenia sytosterolu na zachorowalność i śmiertelność w tej populacji jest nieznany.
Zawał aortalny
Badanie Symwastatynu i Ezetymibu w Leczeniu Zawału Aortalnego (SEAS) to wieloośrodkowe, podwójnie ślepe, placebo-kontrolowane badanie trwające średnio 4,4 roku z udziałem 1873 pacjentów z bezobjawowym zawałem aortalnym (ZA) potwierdzonym wynikami echokardiografii Dopplerowskiej, w której prędkość przepływu szczytowego aorty była w zakresie od 2,5 do 4,0 m/s. Do badania zakwalifikowano tylko tych pacjentów, którzy według opinii lekarzy nie wymagali leczenia statynami w celu zmniejszenia ryzyka miażdżycowych chorób sercowo-naczyniowych. Pacjenci zostali zrandomizowani w stosunku 1:1 do przyjmowania placebo lub wspomagającego podawania ezetymibu 10 mg i symwastatyny 40 mg dziennie.
Pierwotnym punktem końcowym była kombinacja głównych zdarzeń sercowo-naczyniowych (GZSN), obejmująca śmiertelność z przyczyn sercowo-naczyniowych, wymianę zastawki aortalnej (WZA), niewydolność serca (NWS) spowodowaną postępem ZA, niezgonny zawał mięśnia serca, tętnicze sztuczne pomostowanie (TSP), interwencję wieńcową (ICW), hospitalizację z powodu niestabilnej dławicy piersiowej oraz udar niehemoragiczny. Kluczowymi wtórnymi punktami końcowymi były kombinacje podkategorii zdarzeń pierwotnych punktów końcowych.
W porównaniu z placebo, ezetymib/symwastatyna 10/40 mg nieistotnie zmniejszyła ryzyko GZSN. Pierwotny wynik zaobserwowano u 333 pacjentów (35,3%) w grupie ezetymibu/symwastatyny i u 355 pacjentów (38,2%) w grupie placebo (stosunek ryzyka w grupie ezetymibu/symwastatyny 0,96; 95% przedział ufności, od 0,83 do 1,12; p = 0,59). Wymiana zastawki aortalnej została przeprowadzona u 267 pacjentów (28,3%) w grupie ezetymibu/symwastatyny i u 278 pacjentów (29,9%) w grupie placebo (stosunek ryzyka 1,00; 95% PU, od 0,84 do 1,18; p = 0,97). Mniej pacjentów miało zdarzenia sercowo-naczyniowe w grupie ezetymibu/symwastatyny (n = 148) niż w grupie placebo (n = 187) (stosunek ryzyka, 0,78; 95% PU, od 0,63 do 0,97; p = 0,02), głównie z powodu mniejszej liczby pacjentów, którzy przeszli sztuczne pomostowanie tętnicy wieńcowej.
Nowotwory występowały częściej w grupie ezetymibu/symwastatyny (105 vs. 70, p = 0,01). Kliniczna istotność tego obserwowania jest nieokreślona, ponieważ w badaniu SHARP większa liczba pacjentów z dowolnym nowym nowotworem (438 w grupie ezetymibu/symwastatyny i 439 w grupie placebo) nie różniła się. Ponadto, w badaniu IMPROVE-IT ogólna liczba pacjentów z nową chorobą nowotworową (853 w grupie ezetymibu/symwastatyny i 863 w grupie symwastatyny) istotnie się nie różniła, a zatem wynik badania SEAS nie może być potwierdzony przez badania SHARP lub IMPROVE-IT.
Właściwości farmakokinetyczne
Wchłanianie. Po podaniu doustnie ezetymib jest szybko wchłaniany i aktywnie ulega koniugacji, tworząc farmakologicznie aktywny fenolowy glukuronid (ezetymib-glukuronid). Średnie stężenie maksymalne (Cmax) w osoczu osiągane jest po 1–2 godzinach dla ezetymibu-glukuronidu i po 4–12 godzinach dla ezetymibu. Bezwzględną biodostępność ezetymibu nie można określić, ponieważ ten związek jest nierozpuszczalny w środowisku wodnym odpowiednim do wstrzykiwań.
Jednoczesne spożycie pokarmu (o niskiej lub wysokiej zawartości tłuszczu) nie wpływa na biodostępność doustną ezetymibu. Ezetymib można przyjmować niezależnie od posiłku.
Rozkład. Ezetymib i ezetymib-glukuronid wiążą się z białkami osocza krwi człowieka w 99,7% i 88–92% odpowiednio.
Metabolizm. Główny metabolizm ezetymibu zachodzi w jelicie cienkim i wątrobie poprzez koniugację z glukuronidem (reakcja fazy II) z późniejszym wydaleniem z żółcią. Minimalny metabolizm utleniający (reakcja fazy I) obserwowano na wszystkich etapach transformacji. Ezetymib i ezetymib-glukuronid są głównymi substancjami wykrywalnymi w osoczu krwi i stanowią odpowiednio około 10–20% i 80–90% całkowitej zawartości leku w osoczu. Ezetymib i ezetymib-glukuronid są powoli wydalane z osocza krwi w procesie krążenia jelitowo-wątrobowego. Okres półtrwania ezetymibu i ezetymibu-glukuronidu wynosi około 22 godziny.
Wydalanie. Po podaniu ochotnikom doustnie 20 mg 14C-ezetymibu w osoczu krwi wykryto około 93% całkowitego ezetymibu z całkowitej radioaktywności osocza. W ciągu 10 dni około 78% i 11% przyjętej dawki radioaktywnej zostało wydalone odpowiednio z kałem i moczem. Po 48 godzinach ślady radioaktywności w osoczu krwi nie były wykrywalne.
Osobliwe grupy pacjentów
Dzieci
Farmakokinetyka ezetymibu jest podobna u dzieci w wieku od 6 lat i dorosłych. Brak danych farmakokinetycznych dotyczących stosowania u dzieci poniżej 6 roku życia. Doświadczenie kliniczne u dzieci i młodzieży obejmuje pacjentów z HHR, heterozygotyczną hipercholesterolemią rodzinną (HeHR) lub sytosterinemią.
Pacjenci w podeszłym wieku
U pacjentów w podeszłym wieku (powyżej 65 lat) stężenie w osoczu krwi całkowitego ezetymibu jest około dwukrotnie wyższe niż u młodszych pacjentów (18–45 lat). Obniżenie cholesterolu LPNŻ i profil bezpieczeństwa są zbliżone u pacjentów w podeszłym wieku i młodszych pacjentów przyjmujących ezetymib. Dlatego nie ma potrzeby dostosowywania dawki u pacjentów w podeszłym wieku.
Niewydolność wątroby
Po jednorazowym podaniu 10 mg ezetymibu wartość średniej powierzchni pod krzywą „stężenie–czas” (AUC) dla całkowitego ezetymibu była 1,7 razy wyższa u pacjentów z niewydolnością wątroby lekkiego stopnia (5–6 punktów według skali Childa-Pugha) niż u zdrowych ochotników. W 14-dniowym badaniu z wielokrotnym podawaniem ezetymibu (10 mg dziennie) u pacjentów z niewydolnością wątroby umiarkowanego stopnia (7–9 punktów według skali Childa-Pugha) wartość AUC dla całkowitego ezetymibu wzrosła około 4 razy w 1. i 14. dniu w porównaniu z tym wskaźnikiem u zdrowych ochotników. Pacjentom z niewydolnością wątroby lekkiego stopnia nie wymaga się dostosowania dawki. Ponieważ efekty zwiększonego stężenia ezetymibu u pacjentów z niewydolnością wątroby umiarkowanego lub ciężkiego stopnia (powyżej 9 punktów według skali Childa-Pugha) są nieznane, ezetymib nie jest zalecany do stosowania u tej grupy pacjentów (patrz sekcja „Szczególne ostrzeżenia i środki ostrożności”).
Niewydolność nerek
Po jednorazowym podaniu 10 mg ezetymibu u pacjentów z ciężką niewydolnością nerek (n = 8; klirens kreatyniny ≤ 30 ml/min/1,73 m²) średnia wartość AUC dla całkowitego ezetymibu wzrosła około 1,5 razy w porównaniu z tym wskaźnikiem u zdrowych ochotników (n = 9). Wynik ten nie jest uważany za klinicznie istotny. U pacjentów z zaburzeniem funkcji nerek nie ma potrzeby dostosowywania dawki.
U jednego pacjenta w tym badaniu (który miał przeszczep nerki i otrzymywał leczenie wielolekowe, w tym cyklosporynę) stężenie całkowitego ezetymibu było 12 razy wyższe.
Płeć
Stężenie całkowitego ezetymibu w osoczu krwi jest nieco wyższe (około 20%) u kobiet niż u mężczyzn. Obniżenie stężenia cholesterolu LPNŻ i profil bezpieczeństwa są zbliżone u mężczyzn i kobiet przyjmujących ezetymib. Dlatego nie ma potrzeby dostosowywania dawki w zależności od płci.
Charakterystyka kliniczna.
Wskazania.
Pierwotna hipercholesterolemia
Ezetymiba w połączeniu z inhibitorem reduktazy hydroksymetyloglutarylo-koenzymu A (HMG-CoA) (statyną) wskazana jest jako terapia wspomagająca dietę u pacjentów z pierwotną (heterozygotyczną rodzinną i nieryzyczną) hipercholesterolemią, gdy terapia wyłącznie statyną jest niewystarczająca.
Monoterapia ezetymibą wskazana jest jako terapia wspomagająca dietę u pacjentów z pierwotną (heterozygotyczną rodzinną i nieryzyczną) hipercholesterolemią, u których stosowanie statyny jest nieuzasadnione lub występuje nietolerancja wobec statyn.
Profilaktyka zdarzeń sercowo-naczyniowych
Ezetymiba jest wskazana do zmniejszenia ryzyka zdarzeń sercowo-naczyniowych (patrz sekcja „Farmakodynamika”) u pacjentów z chorobą niedokrwienną serca (CNS) i zespołem wieńcowym ostrym (ZWO) w wywiadzie, w połączeniu z aktualnie stosowaną terapią statynami lub w terapii jednoczesnej ze statyną.
Homozygotyczna hipercholesterolemia rodzinna (HoHR)
Ezetymiba w połączeniu ze statyną jest wskazana jako terapia wspomagająca dietę u pacjentów z HoHR. Pacjenci mogą również otrzymywać leczenie wspomagające (np. afereza LDL).
Homozygotyczna sitosterolemia (fitosterolemia)
Ezetymiba jest wskazana jako terapia wspomagająca dietę u pacjentów z homozygotyczną rodzinną sitosterolemią.
Przeciwwskazania.
Nadwrażliwość na substancję czynną lub na którąkolwiek z substancji pomocniczych.
W przypadku jednoczesnego stosowania ezetymiby z którąkolwiek statyną należy zapoznać się z instrukcją stosowania dla tego konkretnego leku.
Łączna terapia ezetymiby z którąkolwiek statyną jest przeciwwskazana w okresie ciąży lub karmienia piersią.
Stosowanie ezetymiby w połączeniu z którąkolwiek statyną jest przeciwwskazane u pacjentów z chorobą wątroby w fazie zaostrzenia lub niejasnymi, trwałymi podwyższeniami stężenia aminotransferaz surowicy.
Interakcje z innymi lekami i inne rodzaje interakcji.
Badania przedkliniczne wykazały, że ezetymiba nie indukuje enzymów układu cytochromu P450 metabolizujących lek. Nie zaobserwowano żadnych klinicznie istotnych interakcji farmakokinetycznych między ezetymibą a lekami metabolizowanymi przez enzymy cytochromu P450 1A2, 2D6, 2C8, 2C9 i 3A4 lub przez N-acetylotransferazę.
W badaniach klinicznych interakcji lekowej ezetymiba w terapii skojarzonej nie wpływała na farmakokinetykę dapsonu, dextrometorfanu, digoxyny, doustnych środków antykoncepcyjnych (etynyloestradiolu i lewonorgestrelu), glipezydu, tolbutamidu ani midazolamu. Cymetydyna w terapii skojarzonej z ezetymibą nie wpływała na biodostępność ezetymiby.
Antacida. Jednoczesne stosowanie antacidy prowadzi do zmniejszenia stopnia absorpcji ezetymiby, ale nie wpływa na jej biodostępność. Takie zmniejszenie stopnia absorpcji nie jest uważane za klinicznie istotne.
Cholestyramina. W przypadku stosowania łącznie z cholestyraminą średnia wartość pola pod krzywą „stężenie-czas” (AUC) sumarycznej ezetymiby (ezetymiba i ezetymiba-glukuronid) zmniejszała się o około 55%. W przypadku dodania ezetymiby do cholestyraminy może zachodzić opóźnione zmniejszanie stężenia cholesterolu lipoprotein o niskiej gęstości (cholesterol LDL). Fibraty. Jeżeli pacjenci otrzymują fenofibrat i ezetymibę, lekarze powinni być poinformowani o możliwym ryzyku wystąpienia kamicy żółciowej i choroby pęcherza żółciowego. W przypadku podejrzenia kamicy żółciowej u pacjenta otrzymującego ezetymibę i fenofibrat, należy przeprowadzić badania pęcherza żółciowego, a taką terapię należy przerwać (patrz sekcja „Reakcje niepożądane”).
Jednoczesne stosowanie fenofibratu lub gemfibrozylu nieznacznie zwiększało całkowite stężenie ezetymiby (odpowiednio około 1,5 i 1,7 raza).
Jednoczesnego stosowania ezetymiby z innymi fibratami nie badano.
Fibraty mogą zwiększać wydzielanie cholesterolu do żółci, co prowadzi do kamicy żółciowej. W badaniach na zwierzętach ezetymiba czasem zwiększała stężenie cholesterolu w żółci, ale nie u wszystkich gatunków. Ryzyko litogenne związane z terapeutycznym stosowaniem ezetymiby nie może być wykluczone.
Statyny. Nie wykryto żadnych klinicznie istotnych interakcji farmakokinetycznych przy jednoczesnym stosowaniu ezetymiby z atorwastatyną, simwastatyną, prawastatyną, lowastatyną, flawastatyną ani rosuwastatyną.
Cyklosporyna. W badaniu z udziałem 8 pacjentów po przeszczepie nerki z klirensem kreatyniny > 50 ml/min przy stabilnej dawce cyklosporyny, jedna dawka ezetymiby 10 mg prowadziła do 3,4-krotnego wzrostu (zakres od 2,3 do 7,9 razy) średniego AUC sumarycznej ezetymiby w porównaniu z odpowiednim wskaźnikiem w populacji kontrolnej zdrowych pacjentów otrzymujących wyłącznie ezetymibę, w innym badaniu (n = 17). W jeszcze jednym badaniu u pacjenta z przeszczepioną nerką i ciężką niewydolnością nerek, otrzymującego cyklosporynę i wiele innych leków, zaobserwowano 12-krotne zwiększenie ekspozycji na sumaryczną ezetymibę w porównaniu z pacjentami kontrolnymi otrzymującymi wyłącznie ezetymibę. W badaniu krzyżowym z dwoma okresami z udziałem 12 zdrowych ochotników, codzienne podawanie 20 mg ezetymiby przez 8 dni z jedną dawką 100 mg cyklosporyny w 7. dniu prowadziło do wzrostu AUC cyklosporyny średnio o 15% (zakres od zmniejszenia o 10% do wzrostu o 51%) w porównaniu z odpowiednim wskaźnikiem przy podaniu jednej dawki 100 mg cyklosporyny. Kontrolowanego badania wpływu jednoczesnego przyjmowania ezetymiby na ekspozycję na cyklosporynę u pacjentów z przeszczepioną nerką nie przeprowadzono. Leczenie ezetymibą u pacjentów przyjmujących cyklosporynę należy rozpoczynać ostrożnie. U pacjentów przyjmujących ezetymibę i cyklosporynę należy kontrolować stężenia cyklosporyny.
Antykoagulanta. Jednoczesne stosowanie ezetymiby (10 mg raz dziennie) nie miało istotnego wpływu na biodostępność warfaryny i czas protrombinowy w badaniu z udziałem 12 zdrowych dorosłych mężczyzn. Jednak po rejestracji zgłaszano przypadki zwiększenia międzynarodowego wskaźnika znormalizowanego (INR) u pacjentów, którym dodawano ezetymibę do warfaryny lub fluindionu. Jeśli ezetymiba jest stosowana w połączeniu z warfaryną, innym antykoagulantem z grupy kumaryn lub fluindionem, należy odpowiednio kontrolować INR.
Dzieci. Badania interakcji przeprowadzono u dorosłych.
Szczególne środki ostrożności dotyczące stosowania.
Przy jednoczesnym stosowaniu ezetymiby z dowolnym statyną należy zapoznać się z instrukcją do konkretnego leku.
Enzymy wątrobowe
W kontrolowanych badaniach terapii skojarzonej u pacjentów przyjmujących ezetymibę ze statynami obserwowano kolejne wzrosty poziomu transaminaz (3 lub więcej razy wyższe niż górna granica normy [GGN]). Podczas stosowania kombinacji ezetymiby ze statyną należy wykonać badania funkcji wątroby na początku terapii oraz zgodnie z zaleceniami dotyczącymi statyny.
W dużym randomizowanym badaniu: Międzynarodowym Badaniu Efektywności Vytorina (IMPROVE-IT), 18144 pacjentów z chorobą niedokrwienną serca i przypadłościami OZW w wywiadzie zostało randomizowanych do przyjmowania ezetymiby/symwastatyny 10/40 mg dziennie (n = 9067) lub symwastatyny 40 mg dziennie (n = 9077). W okresie średniego obserwowania 6 lat częstość kolejnego wzrostu transaminaz (3 lub więcej razy wyższa niż GGN) wynosiła 2,5% dla ezetymiby/symwastatyny oraz 2,3% dla symwastatyny.
W kontrolowanym badaniu klinicznym, w którym ponad 9000 pacjentów z przewlekłą chorobą nerek zostało randomizowanych do przyjmowania ezetymiby 10 mg w kombinacji z symwastatyną 20 mg dziennie (n = 4650) lub placebo (n = 4620), (średni okres obserwacji 4,9 roku), częstość kolejnego wzrostu transaminaz (3 lub więcej razy wyższa niż GGN) wynosiła 0,7% dla ezetymiby w kombinacji z symwastatyną oraz 0,6% dla placebo.
Mięśnie szkieletowe
W okresie pozarejestrowym zgłaszano przypadki miopatii i rabdomiolizy podczas stosowania ezetymiby. Większość pacjentów, u których rozwinęła się rabdomioliza, przyjmowała statynę jednocześnie z ezetymibą. Jednakże bardzo rzadko zgłaszano przypadki rabdomiolizy przy monoterapii ezetymibą oraz w przypadku dodania ezetymiby do innych leków, z którymi wiąże się zwiększony ryzyko wystąpienia rabdomiolizy. W przypadku podejrzenia miopatii, objawiającej się dolegliwościami ze strony mięśni lub wzrostem poziomu kinazy kreatynowej ponad 10 razy w stosunku do górnej granicy normy, należy natychmiast przerwać przyjmowanie ezetymiby, każdej statyny oraz każdego innego leku, który pacjent przyjmuje równocześnie. Wszystkich pacjentów rozpoczynających leczenie ezetymibą należy poinformować o ryzyku wystąpienia miopatii oraz konieczności natychmiastowego zgłaszania bólu mięśni, osłabienia lub słabości o nieznanej etiologii.
W badaniu IMPROVE-IT 18144 pacjentów z chorobą niedokrwienną serca i przypadłościami OZW w wywiadzie zostało randomizowanych do przyjmowania ezetymiby/symwastatyny 10/40 mg dziennie (n = 9067) lub symwastatyny 40 mg dziennie (n = 9077). W okresie średniego obserwowania 6 lat częstość miopatii wynosiła 0,2% dla ezetymiby/symwastatyny oraz 0,1% dla symwastatyny, przy czym miopatię definiowano jako osłabienie mięśni lub ból o nieznanej etiologii z poziomem kinazy kreatynowej w surowicy krwi 10 lub więcej razy wyższym niż GGN lub z dwoma kolejnymi wynikami kinazy kreatynowej 5 lub więcej razy i 10 razy niższe niż GGN. Częstość rabdomiolizy wynosiła 0,1% dla ezetymiby/symwastatyny oraz 0,2% dla symwastatyny, przy czym rabdomiolizę definiowano jako osłabienie mięśni lub ból o nieznanej etiologii z poziomem kinazy kreatynowej w surowicy krwi 10 lub więcej razy wyższym niż GGN z objawami uszkodzenia nerek, 5 lub więcej razy wyższym i 10 razy niższym niż GGN dwukrotnie z objawami uszkodzenia nerek lub poziomem kinazy kreatynowej ≥ 10000 IU/l bez objawów uszkodzenia nerek.
W badaniu klinicznym, w którym ponad 9000 pacjentów z przewlekłą chorobą nerek zostało randomizowanych do przyjmowania ezetymiby 10 mg w kombinacji z symwastatyną 20 mg dziennie (n = 4650) lub placebo (n = 4620), (średni okres obserwacji 4,9 roku), częstość miopatii/rabdomiolizy wynosiła 0,2% dla ezetymiby w kombinacji z symwastatyną oraz 0,1% dla placebo.
Niewydolność wątroby
Ponieważ skutki zwiększonego stężenia ezetymiby u pacjentów z umiarkowanym i ciężkim uszkodzeniem wątroby są nieznane, ezetymibę nie zaleca się stosować tej grupie pacjentów.
Dzieci
Wiadomo, że skuteczność i bezpieczeństwo ezetymiby u pacjentów w wieku od 6 do 10 lat z heterozygotyczną rodzinną lub nieryczynną hipercholesterolemią oceniano w badaniu klinicznym kontrolowanym placebo przez 12 tygodni. Wpływ ezetymiby w okresie leczenia dłuższym niż 12 tygodni w tej grupie wiekowej nie był badany.
Wpływ ezetymiby nie był badany u pacjentów poniżej 6 roku życia.
Wiadomo, że skuteczność i bezpieczeństwo ezetymiby stosowanej w kombinacji z symwastatyną u pacjentów w wieku od 10 do 17 lat z heterozygotyczną rodzinną hipercholesterolemią oceniano w kontrolowanym badaniu klinicznym z udziałem chłopców (stopień Tanner II lub wyższy) i dziewcząt (co najmniej rok po menarche).
W trakcie tego ograniczonego kontrolowanego badania nie zaobserwowano żadnego istotnego wpływu na wzrost i dojrzewanie płciowe chłopców i dziewcząt w wieku nastoletnim ani żadnego wpływu na długość cyklu menstruacyjnego u dziewcząt. Jednakże wpływ ezetymiby na wzrost i dojrzewanie płciowe w okresie leczenia dłuższym niż 33 tygodnie nie był badany.
Bezpieczeństwa i skuteczności ezetymiby stosowanej w kombinacji z symwastatyną w dawce przekraczającej 40 mg dziennie u dzieci w wieku od 10 do 17 lat nie badano.
Bezpieczeństwa i skuteczności ezetymiby stosowanej w kombinacji z symwastatyną u dzieci poniżej 10 roku życia nie badano.
Długoterminowej skuteczności terapii ezetymibą u pacjentów poniżej 17 roku życia w celu zmniejszenia zachorowalności i śmiertelności w dorosłym życiu nie badano.
Fibraty
Bezpieczeństwo i skuteczność ezetymiby przy stosowaniu z fibratami nie są ustalone.
W przypadku podejrzenia kamicy żółciowej u pacjenta przyjmującego ezetymibę i fenofibrat, należy wykonać badanie pęcherza żółciowego, a taką terapię należy przerwać.
Cyklosporyna
Należy ostrożnie rozpoczynać leczenie ezetymibą u pacjentów przyjmujących cyklosporynę. U pacjentów przyjmujących ezetymibę i cyklosporynę należy kontrolować stężenia cyklosporyny.
Antykoagulancy
Jeśli ezetymibę stosuje się w połączeniu z warfaryną, innym antykoagulacyjnym lekiem z grupy kumaryn lub fluindonem, należy odpowiednio kontrolować międzynarodowe znormalizowane stężenie (INR).
Stosowanie w czasie ciąży lub karmienia piersią.
Terapia ezetymibą w kombinacji ze statyną jest przeciwwskazana w czasie ciąży lub karmienia piersią (patrz sekcja „Przeciwwskazania”); należy zapoznać się z instrukcją do stosowania dla tej statyny.
Ciąża
Ezetymibę należy przepisywać kobietom w ciąży tylko wtedy, gdy jest to wyraźnie konieczne. Brak danych klinicznych dotyczących stosowania ezetymiby w czasie ciąży. Badania na zwierzętach z wykorzystaniem ezetymiby jako monoterapii wykazały brak bezpośredniego lub pośredniego szkodliwego wpływu na ciążę, rozwój płodowo-embrionalny, poród lub rozwój poporodowy.
Karmienie piersią
Ezetymiby nie należy stosować w czasie karmienia piersią. Badania na szczurach wykazały, że ezetymiba przenika do mleka karmiących zwierząt. Nie wiadomo, czy ezetymiba przenika do mleka matki.
Plodność
Brak danych klinicznych dotyczących wpływu ezetymiby na płodność człowieka. Ezetymiba nie wykazała żadnego wpływu.
Wpływ na zdolność prowadzenia pojazdów i obsługi urządzeń mechanicznych.
Nie przeprowadzono badań dotyczących wpływu na zdolność prowadzenia samochodu i innych urządzeń mechanicznych. Należy jednak wziąć pod uwagę, że zgłaszano zawroty głowy podczas prowadzenia samochodu i innych urządzeń.
Sposób stosowania i dawki
Podczas całego okresu leczenia ezetymibem pacjent powinien przestrzegać standardowej diety obniżającej poziom lipidów.
Dawkowanie
Zalecana dawka ezetymibu wynosi 10 mg (1 tabletka) na dobę.
W przypadku dodania ezetymibu do statyny należy kontynuować przyjmowanie początkowej dawki tej statyny lub wyższej dawki statyny, która została już wcześniej przepisana. W takim przypadku należy zapoznać się z instrukcją do stosowania tej statyny.
Stosowanie u pacjentów z chorobą niedokrwienną serca i przypadkami zawału serca w wywiadzie
W celu stopniowego obniżenia ryzyka wystąpienia zdarzeń sercowo-naczyniowych pacjentom z chorobą niedokrwienną serca i przypadkami zawału serca w wywiadzie ezetymib (10 mg) może być stosowany w połączeniu ze statyną, której korzystny wpływ na układ sercowo-naczyniowy został potwierdzony.
Stosowanie równoczesne z wiązaczami kwasów żółciowych
Ezetymib należy przyjmować nie później niż 2 godziny przed lub nie wcześniej niż 4 godziny po podaniu wiązacza kwasu żółciowego.
Pacjenci w podeszłym wieku
Pacjenci w podeszłym wieku nie wymagają korekty dawki.
Niewydolność wątroby
U pacjentów z niewydolnością wątroby w stopniu lekkim (5–6 punktów według skali Childa-Pugha) korekta dawki nie jest wymagana. Leczenie ezetymibem nie jest zalecane u pacjentów z niewydolnością wątroby w stopniu umiarkowanym (7–9 punktów według skali Childa-Pugha) lub ciężkim (powyżej 9 punktów według skali Childa-Pugha).
Niewydolność nerek
Pacjenci z niewydolnością nerek nie wymagają korekty dawki.
Sposób stosowania
Lek należy stosować doustnie. Ezetymib można przyjmować o dowolnej porze dnia niezależnie od posiłków.
Dzieci
Na początku leczenia wymagana jest opieka specjalisty.
Dzieci i młodzież w wieku od 6 lat: wiadomo, że bezpieczeństwo i skuteczność stosowania ezetymibu u dzieci w wieku od 6 do 17 lat nie zostały ustalone. Dane dostępne obecnie zawarte są w punktach „Farmakodynamika”, „Właściwości farmakokinetyczne”, „Szczególne ostrzeżenia i środki ostrożności”, „Działania niepożądane”, jednak nie można podać rekomendacji dotyczących dawkowania.
W przypadku stosowania ezetymibu w połączeniu ze statyną należy zapoznać się z instrukcją do stosowania dotyczącą dawkowania u dzieci.
Dzieci poniżej 6 roku życia: wiadomo, że bezpieczeństwo i skuteczność stosowania ezetymibu u dzieci poniżej 6 roku życia nie zostały ustalone. Brak informacji.
Przedawkowanie
W badaniach, w których 15 zdrowym uczestnikom podawano ezetymib w dawce 50 mg/dobę przez 14 dni lub 18 pacjentom z pierwotną hipercholesterolemią w dawce 40 mg/dobę przez do 56 dni, lek był ogólnie dobrze tolerowany. U zwierząt nie zaobserwowano toksyczności po pojedynczych dawkach doustnych 5000 mg/kg ezetymibu u szczurów i myszy oraz 3000 mg/kg u psów.
Zgłoszono kilka przypadków przedawkowania ezetymibu; w większości przypadków przedawkowanie nie powodowało niepożądanych zjawisk. Obserwowane działania niepożądane nie były poważne. W przypadku przedawkowania należy podjąć działania objawowe i wspierające.
Efekty uboczne
Poniżej przedstawiono listę efektów ubocznych (badania kliniczne i okres po rejestracji). W badaniach trwających do 112 tygodni ezetymib w dawce 10 mg na dobę podawano samodzielnie 2396 pacjentom lub w kombinacji ze statyną 11308 pacjentom, albo z fenofybratem 185 pacjentom. Efekty uboczne były zazwyczaj niewielkie i krótkotrwałe. Ogólna częstość występowania efektów ubocznych była podobna między ezetymibem a placebo. Podobnie częstość przerwania leczenia z powodu działań niepożądanych była porównywalna między ezetymibem a placebo.
Ezetymib jako monoterapia lub w połączeniu ze statyną
Poniżej wymienione efekty uboczne obserwowano w badaniach klinicznych u pacjentów leczonych ezetymibem (N = 2396), które występowały częściej niż w grupie placebo (N = 1159), lub u pacjentów leczonych ezetymibem w połączeniu ze statyną (N = 11308), które występowały częściej niż w grupie przyjmującej wyłącznie statynę (N = 9361). Efekty uboczne z okresu po rejestracji pochodzą z doniesień, w których ezetymib podawano samodzielnie lub w połączeniu ze statyną.
Częstość występowania działań niepożądanych sklasyfikowano następująco: bardzo często (≥1/10), często (≥1/100 do <1/10), nieczęsto (≥1/1000 do <1/100), rzadko (≥1/10000 do <1/1000), bardzo rzadko (<1/10 000) oraz nieznane (niemożliwe do oszacowania na podstawie dostępnych danych).
| Monoterapia ezetymibem |
||
| Klasy układów narządów |
Reakcje niepożądane |
Częstotliwość |
| Zaburzenia metabolizmu i odżywiania |
spadek apetytu |
Nieczęsto |
| Zaburzenia układu naczyniowego |
napływy gorąca; nadciśnienie |
Nieczęsto |
| Zaburzenia układu oddechowego, jamy piersiowej i śródpiersia |
kaszel |
Nieczęsto |
| Zaburzenia ze strony przewodu pokarmowego |
ból brzucha; biegunka; wzdęcia |
Często |
| dyspepsja; refluks żołądkowo-jelitowy; nudności |
Nieczęsto |
|
| Zaburzenia ze strony tkanki mięśniowej i tkanki łącznej |
artralgia; skurcze mięśni; ból szyi |
Nieczęsto |
| Zaburzenia ogólne i miejsce podania |
zmęczenie |
Często |
| ból w klatce piersiowej, ból |
Nieczęsto |
|
| Badania laboratoryjne |
Podwyższenie ALAT i/lub ASAT; podwyższenie stężenia kinazy kreatynowej w surowicy; podwyższenie stężenia gamma-glutamylotransferazy; wyniki badań funkcji wątroby poza normą |
Nieczęsto |
| Dodatkowe reakcje niepożądane przy jednoczesnym stosowaniu ezetymibu ze statyną |
||
| Klasy układów narządów |
Reakcje niepożądane |
Częstotliwość |
| Zaburzenia ze strony układu nerwowego |
ból głowy |
Często |
| parestezja |
Nieczęsto |
|
| Zaburzenia ze strony przewodu pokarmowego |
sucha jamy ustnej; zapalenie żołądka |
Nieczęsto |
| Zaburzenia ze strony skóry i tkanek podskórnych |
świerdzenie; wysypka; pokrzywka |
Nieczęsto |
| Zaburzenia ze strony tkanki mięśniowej i tkanki łącznej |
mialgia |
Często |
| ból pleców; osłabienie mięśni; ból kończyn |
Nieczęsto |
|
| Zaburzenia ogólne i miejsce podania |
astenia; obrzęki obwodowe |
Nieczęsto |
| Badania laboratoryjne |
Podwyższenie ALAT i/lub ASAT |
Często |
| Okres po rejestracji (ze statyną lub bez) |
||
| Klasy układów narządów |
Reakcje niepożądane |
Częstotliwość |
| Zaburzenia układu krwiotwórczego i limfatycznego |
zespół zakrzepowo-zaropnowy |
Nieznane |
| Zaburzenia układu odpornościowego |
podatność na alergię, w tym wysypkę, świerdzenie, anafilaksję i obrzęk naczynioruchowy |
Nieznane |
| Zaburzenia psychiczne |
depresja |
Nieznane |
| Zaburzenia ze strony układu nerwowego |
zawroty głowy; parestezja |
Nieznane |
| Zaburzenia układu oddechowego, jamy piersiowej i śródpiersia |
dyspneja |
Nieznane |
| Zaburzenia ze strony przewodu pokarmowego |
zapalenie trzustki; zaparcia |
Nieznane |
| Zaburzenia ze strony wątroby i dróg żółciowych |
zapalenie wątroby; kamica żółciowa; zapalenie pęcherza żółciowego |
Nieznane |
| Zaburzenia ze strony skóry i tkanek podskórnych |
rumień wielopostaciowy |
Nieznane |
| Zaburzenia ze strony tkanki mięśniowej i tkanki łącznej |
mialgia; miopatia/rabdomyoliza |
Nieznane |
| Zaburzenia ogólne i miejsce podania |
astenia |
Nieznane |
Stosowanie równoległe ezetymiby i fenofibratu
Zaburzenia ze strony przewodu pokarmowego: ból brzucha (często).
W wieloośrodkowym, podwójnym, ślepym, z randomizacją i kontrolą placebo badaniu klinicznym u pacjentów z hiperlipidemią mieszaną, 625 pacjentów otrzymywało leczenie przez okres do 12 tygodni, a 576 pacjentów przez okres do 1 roku. W tym badaniu 172 pacjentów, którzy otrzymywali leczenie ezetymibą i fenofibratem, ukończyło 12-tygodniową terapię, a 230 pacjentów, którzy otrzymywali leczenie ezetymibą i fenofibratem (w tym 109 pacjentów, którzy otrzymywali ezetymibę jako monoterapię przez pierwsze 12 tygodni), ukończyło roczne leczenie. Badanie to nie było zaprojektowane w celu porównania grup leczenia pod względem występowania rzadkich zdarzeń. Częstości występowania (95 % CI) klinicznie istotnych wzrostów (trzykrotnie wyższe niż górna granica normy, kolejne) stężeń transaminaz osocza wynosiły odpowiednio 4,5 % (1,9 i 8,8) i 2,7 % (1,2 i 5,4) dla monoterapii fenofibratem oraz jednoczesnego stosowania ezetymiby z fenofibratem, skorygowane względem ekspozycji na leczenie. Odpowiednie wskaźniki częstości występowania dla cholecystektomii wynosiły odpowiednio 0,6 % (0,0 i 3,1) i 1,7 % (0,6 i 4,0) dla monoterapii fenofibratem oraz jednoczesnego stosowania ezetymiby z fenofibratem, skorygowane względem ekspozycji na leczenie.
Dzieci (w wieku od 6 do 17 lat)
Wiadomo, że w trakcie badania z udziałem dzieci w wieku od 6 do 10 lat z heterozygotyczną hipercholesterolemią rodzinną lub nieryzyczną (n = 138) obserwowano wzrost stężeń ALAT i/lub ASPAT (trzykrotnie lub więcej wyższe niż górna granica normy, kolejne) u 1,1 % (1 pacjent) pacjentów otrzymujących leczenie ezetymibą w porównaniu z 0 % w grupie placebo. Nie obserwowano wzrostów stężeń CK (10 lub więcej razy wyższe niż górna granica normy). Nie zgłaszano przypadków miopatii.
Wiadomo, że w innym badaniu z udziałem nastolatków (w wieku od 10 do 17 lat) z heterozygotyczną hipercholesterolemią rodzinną (N = 248) obserwowano wzrosty ALAT i/lub ASPAT (trzykrotnie lub więcej wyższe niż górna granica normy, kolejne) u 3 % (4 pacjenci) w grupie ezetymiba/symwastatyny w porównaniu z 2 % (2 pacjenci) w grupie monoterapii symwastatyną; odpowiednie wskaźniki dla wzrostu kinazy kreatynowej (CK) 10 lub więcej razy wyższego niż górna granica normy wynosiły odpowiednio 2 % (2 pacjenci) i 0 %. Nie zgłaszano przypadków miopatii.
W tym badaniu nie porównywano rzadkich działań niepożądanych leku.
Pacjenci z chorobą niedokrwienną serca i wywiadem zawału serca
W badaniu IMPROVE-IT (patrz sekcja „Farmakodynamika”) z udziałem 18144 pacjentów, którzy otrzymywali ezetymibę/symwastatynę 10/40 mg (n = 9067; z czego 6 % pacjentom podwyższono dawkę ezetymiby/symwastatyny do 10/80 mg) lub symwastatynę 40 mg (n = 9077, z czego 27 % pacjentom podwyższono dawkę symwastatyny do 80 mg), profile bezpieczeństwa były podobne przez średni okres obserwacji wynoszący 6 lat. Częstość przerwania leczenia z powodu działań niepożądanych wynosiła 10,6 % u pacjentów otrzymujących ezetymibę/symwastatynę i 10,1 % u pacjentów otrzymujących symwastatynę. Częstość wystąpienia miopatii wynosiła 0,2 % dla ezetymiby/symwastatyny i 0,1 % dla symwastatyny, gdzie miopatię definiowano jako osłabienie mięśni lub ból o niejasnej etiologii oraz poziom kinazy kreatynowej we krwi 10 lub więcej razy wyższy niż górna granica normy lub dwa kolejne wyniki kinazy kreatynowej 5 lub więcej razy wyższe i 10 razy niższe niż górna granica normy. Częstość wystąpienia rabdomiolizy wynosiła 0,1 % dla ezetymiby/symwastatyny i 0,2 % dla symwastatyny, gdzie rabdomiolizę definiowano jako osłabienie mięśni lub ból o niejasnej etiologii oraz poziom kinazy kreatynowej we krwi 10 lub więcej razy wyższy niż górna granica normy z objawami uszkodzenia nerek, 5 lub więcej razy wyższy i 10 razy niższy niż górna granica normy dwa razy z rzędu z objawami uszkodzenia nerek lub poziom kinazy kreatynowej ≥ 10000 U/l bez objawów uszkodzenia nerek. Częstość wystąpienia kolejnych wzrostów transaminaz (3 lub więcej razy wyższe niż górna granica normy) wynosiła 2,5 % dla ezetymiby/symwastatyny i 2,3 % dla symwastatyny (patrz sekcja „Szczególne ostrzeżenia i środki ostrożności stosowania”). Działania niepożądane związane z pęcherzem żółciowym stwierdzono u 3,1 % i 3,5 % pacjentów otrzymujących odpowiednio ezetymibę/symwastatynę i symwastatynę. Częstość hospitalizacji z powodu cholecystektomii wynosiła 1,5 % w obu grupach leczenia. Nowotwór (definiowany jako każdy nowy nowotwór złośliwy) został zdiagnozowany podczas badania u 9,4 % i 9,5 % odpowiednio.
Pacjenci z przewlekłą chorobą nerek
W badaniu dotyczącego ochrony serca i nerek (SHARP) (patrz sekcja „Farmakodynamika”) z udziałem ponad 9000 pacjentów, którzy otrzymywali stałą kombinację ezetymiby 10 mg z symwastatyną 20 mg dziennie (n = 4650) lub placebo (n = 4620), profile bezpieczeństwa były porównywalne przez średni okres obserwacji wynoszący 4,9 roku. W tym badaniu rejestrowano jedynie poważne działania niepożądane oraz przerwanie leczenia z powodu jakichkolwiek działań niepożądanych. Częstość przerwania leczenia z powodu działań niepożądanych była porównywalna (10,4 % u pacjentów otrzymujących ezetymibę w kombinacji z symwastatyną i 9,8 % u pacjentów otrzymujących placebo). Częstość wystąpienia miopatii/rabdomiolizy wynosiła 0,2 % u pacjentów otrzymujących ezetymibę w kombinacji z symwastatyną i 0,1 % u pacjentów otrzymujących placebo. Kolejne wzrosty transaminaz (3 razy wyższe niż górna granica normy) obserwowano u 0,7 % pacjentów otrzymujących ezetymibę w kombinacji z symwastatyną w porównaniu z 0,6 % pacjentów otrzymujących placebo (patrz sekcja „Szczególne ostrzeżenia i środki ostrożności stosowania”). W tym badaniu nie stwierdzono statystycznie istotnego wzrostu częstości wcześniej zdefiniowanych działań niepożądanych, w tym nowotworów (9,4 % dla ezetymiby w kombinacji z symwastatyną, 9,5 % dla placebo), zapalenia wątroby, cholecystektomii ani powikłań kamieni żółciowych lub zapalenia trzustki.
Dane badań laboratoryjnych
W kontrolowanych badaniach klinicznych monoterapii klinicznie istotne wzrosty transaminaz osocza (ALAT i/lub ASPAT 3 lub więcej razy wyższe niż górna granica normy) były podobne dla ezetymiby (0,5 %) i placebo (0,3 %). W badaniach terapii skojarzonej częstość występowania wynosiła 1,3 % u pacjentów przyjmujących równolegle ezetymibę i statynę oraz 0,4 % u pacjentów przyjmujących tylko statynę. Takie wzrosty były zazwyczaj bezobjawowe, nie związane z cholestazą, a wartości wracały do poziomu wyjściowego po odstawieniu leczenia lub przy kontynuacji terapii.
W badaniach klinicznych obserwowano wzrost CK 10 lub więcej razy wyższy niż górna granica normy u 4 z 1674 pacjentów (0,2 %) przyjmujących tylko ezetymibę, w porównaniu z 1 z 786 pacjentów (0,1 %) przyjmujących placebo oraz u 1 z 917 pacjentów (0,1 %) przyjmujących ezetymibę równolegle ze statyną, w porównaniu z 4 z 929 pacjentów (0,4 %) przyjmujących tylko statynę. Nie stwierdzono przewagi w występowaniu miopatii lub rabdomiolizy przy stosowaniu ezetymiby w porównaniu z odpowiednią grupą kontrolną (placebo lub tylko statyna).
Zgłaszanie podejrzewanych działań niepożądanych
Bardzo ważne jest zgłaszanie podejrzewanych działań niepożądanych, które wystąpiły po rejestracji leku. Pozwala to na ciągłe monitorowanie stosunku korzyści do ryzyka stosowania tego leku. Zwracamy się do pracowników systemu opieki zdrowotnej z prośbą o zgłaszanie wszelkich działań niepożądanych za pomocą krajowego systemu zgłaszania.
Okres ważności. 3 lata.
Warunki przechowywania.
Przechowywać w temperaturze nie wyższej niż 30 °C. Przechowywać w oryginalnym opakowaniu w miejscu chronionym przed wilgocią.
Opakowanie.
Po 10 tabletek w blistrze; po 3, 6 lub 9 blisterów w tece kartonowej.
Kategoria dystrybucji. Na receptę.
Producent.
Spółka Akcyjna Zakład Farmaceutyczny Egis.
Adres siedziby producenta i miejsca prowadzenia działalności.
1165, miasto Budapeszt, ul. Bekényfeldi 118-120, Węgry.