Lipobon
Ukraine
Table of Contents
INSTRUCTIONS FOR MEDICAL USE OF THE MEDICINAL PRODUCT LIPIDBON (LIPOBON)
Composition:
Active substance: ezetimibe;
1 tablet contains 10 mg of ezetimibe;
Excipients: microcrystalline cellulose, mannitol, sodium croscarmellose, low-substituted hydroxypropylcellulose, povidone-K25, sodium lauryl sulfate, magnesium stearate.
Pharmaceutical form. Tablets.
Main physicochemical properties: white or almost white, elongated tablets with the imprint "E 611" on one side and no markings on the other side.
Pharmacotherapeutic group.
Other lipid-modifying agents. ATC code: C10AX09.
Pharmacological Properties.
Pharmacodynamics.
Mechanism of Action
Ezetimibe is a representative of a new class of lipid-lowering agents that selectively inhibit intestinal cholesterol and related phytosterol absorption. Ezetimibe is orally active and has a mechanism of action distinct from other classes of cholesterol-lowering drugs (e.g., statins, bile acid sequestrants (resins), fibrate acid derivatives, and plant stanols). The molecular target of ezetimibe is the Niemann-Pick C1-Like 1 (NPC1L1) sterol transporter, which mediates cholesterol and phytosterol absorption in the intestine.
Ezetimibe localizes to the brush border of the small intestine and inhibits cholesterol absorption, thereby reducing delivery of intestinal cholesterol to the liver; statins reduce cholesterol synthesis in the liver, and together these mechanisms provide additive cholesterol reduction. After two weeks of clinical use in 18 patients with hypercholesterolemia, ezetimibe reduced cholesterol absorption by 54% compared to placebo.
Pharmacodynamic Effects
A series of preclinical studies were conducted to determine the selectivity of ezetimibe in inhibiting cholesterol absorption. Ezetimibe inhibited the absorption of [14C]-cholesterol without affecting the absorption of triglycerides, fatty acids, bile acids, progesterone, ethinyl estradiol, or fat-soluble vitamins A and D.
Epidemiological studies have established that cardiovascular disease and mortality are directly related to levels of total cholesterol and LDL-C and inversely related to HDL-C levels.
The use of ezetimibe with a statin effectively reduces the risk of cardiovascular events in patients with ischemic heart disease and a history of acute coronary syndrome (ACS).
Clinical Efficacy and Safety
In controlled clinical trials, ezetimibe as monotherapy, as well as when used concomitantly with a statin, significantly reduced total cholesterol (total-C), low-density lipoprotein cholesterol (LDL-C), apolipoprotein B (Apo-B), and triglycerides (TG), and increased high-density lipoprotein cholesterol (HDL-C) in patients with hypercholesterolemia.
Primary Hypercholesterolemia
In a double-blind, placebo-controlled 8-week study, 769 patients with hypercholesterolemia who were already receiving statin monotherapy and not achieving LDL-C goals according to the National Cholesterol Education Program (NCEP) (ranging from 2.6 to 4.1 mmol/L [100 to 160 mg/dL], depending on baseline characteristics) were randomized to receive either ezetimibe 10 mg or placebo in addition to ongoing statin therapy.
Among patients receiving statins who did not achieve LDL-C goals at baseline (~82%), significantly more patients randomized to ezetimibe achieved LDL-C goals at study endpoint compared to those randomized to placebo (72% vs. 19%, respectively). Corresponding LDL-C reductions were significantly different (25% vs. 4% for ezetimibe vs. placebo, respectively). Additionally, ezetimibe added to ongoing statin therapy significantly reduced total-C, Apo-B, and TG and increased HDL-C compared to placebo. Ezetimibe or placebo added to statin therapy reduced mean C-reactive protein by 10% or 0%, respectively, from baseline.
In two double-blind, randomized, placebo-controlled 12-week studies involving 1719 patients with primary hypercholesterolemia, ezetimibe 10 mg significantly reduced total-C (13%), LDL-C (19%), Apo-B (14%), and TG (8%) and increased HDL-C (3%) compared to placebo. Furthermore, ezetimibe did not affect concentrations of fat-soluble vitamins A, D, and E, did not affect prothrombin time, and, like other lipid-lowering agents, did not interfere with adrenal corticosteroid hormone production.
In a multicenter, double-blind, controlled clinical trial (ENHANCE), 720 patients with heterozygous familial hypercholesterolemia were randomized to receive either ezetimibe 10 mg in combination with simvastatin 80 mg (n = 357) or simvastatin 80 mg alone (n = 363) for 2 years. The primary objective of the study was to evaluate the effect of combined ezetimibe/simvastatin therapy on carotid intima-media thickness (CIMT) compared to simvastatin monotherapy. The impact of this surrogate marker on cardiovascular disease and mortality has not yet been demonstrated. The primary endpoint, change in mean CIMT across all six carotid artery segments, did not differ significantly (p = 0.29) between the two treatment groups as assessed by B-mode ultrasound. With ezetimibe 10 mg in combination with simvastatin 80 mg or simvastatin 80 mg alone, CIMT increased by 0.0111 mm and 0.0058 mm, respectively, over the two-year study (baseline CIMT was 0.68 mm and 0.69 mm, respectively).
Ezetimibe 10 mg in combination with simvastatin 80 mg reduced LDL-C, total-C, Apo-B, and TG levels significantly more than simvastatin 80 mg alone. The percentage increase in HDL-C was similar in both treatment groups. Adverse reactions reported with ezetimibe 10 mg in combination with simvastatin 80 mg were consistent with its known safety profile.
Children
In a multicenter, double-blind, controlled trial, 138 patients (59 boys and 79 girls) aged 6 to 10 years (mean age 8.3 years) with heterozygous familial or non-familial hypercholesterolemia (HeFH) and baseline LDL-C levels between 3.74 and 9.92 mmol/L were randomized to receive ezetimibe 10 mg or placebo for 12 weeks.
At week 12, ezetimibe significantly reduced total-C (-21% vs. 0%), LDL-C (-28% vs. -1%), Apo-B (-22% vs. -1%), and non-HDL-C (-26% vs. 0%) compared to placebo. Results for TG and HDL-C were similar between the two treatment groups (-6% vs. +8% and +2% vs. +1%, respectively).
In a multicenter, double-blind, controlled trial, 142 boys (Tanner stage II or higher) and 106 post-menarchal girls aged 10 to 17 years (mean age 14.2 years) with heterozygous familial hypercholesterolemia (HeFH) and baseline LDL-C levels between 4.1 and 10.4 mmol/L were randomized to receive ezetimibe 10 mg plus simvastatin (10, 20, or 40 mg) or simvastatin alone (10, 20, or 40 mg) for 6 weeks, followed by coadministration of ezetimibe and 40 mg simvastatin or simvastatin 40 mg alone for the next 27 weeks, then open-label coadministration of ezetimibe and simvastatin (10 mg, 20 mg, or 40 mg) for 20 weeks.
At week 6, ezetimibe coadministered with simvastatin (all doses) significantly reduced total-C (38% vs. 26%), LDL-C (49% vs. 34%), Apo-B (39% vs. 27%), and non-HDL-C (47% vs. 33%) compared to simvastatin monotherapy (all doses). Results for TG and HDL-C were similar between the two treatment groups (-17% vs. -12% and +7% vs. +6%, respectively). At week 33, results were consistent with those at week 6, and significantly more patients receiving ezetimibe plus 40 mg simvastatin (62%) achieved the NCEP ATP-III LDL-C goal (< 2.8 mmol/L [110 mg/dL]) compared to those receiving 40 mg simvastatin alone (25%). At week 53, at the end of the open-label extension phase, the lipid-lowering effect was maintained.
The safety and efficacy of ezetimibe administered in combination with simvastatin at doses greater than 40 mg daily in children aged 10 to 17 years have not been studied. The safety and efficacy of ezetimibe administered in combination with simvastatin in children under 10 years of age have not been studied. The long-term efficacy of ezetimibe therapy in patients under 17 years of age for reducing morbidity and mortality in adulthood has not been investigated.
Prevention of Cardiovascular Events
Improved Reduction of Outcomes: Vytorin Efficacy International Trial (IMPROVE-IT) was a multicenter, randomized, double-blind, active-controlled study involving 18,144 patients enrolled within 10 days of hospitalization for acute coronary syndrome (ACS, acute myocardial infarction [MI] or unstable angina [UA]). At the time of ACS presentation, patients had LDL-C ≤ 125 mg/dL (≤ 3.2 mmol/L) if not receiving lipid-lowering therapy, or ≤ 100 mg/dL (≤ 2.6 mmol/L) if already on lipid-lowering therapy. All patients were randomized in a 1:1 ratio to receive either ezetimibe/simvastatin 10/40 mg (n = 9067) or simvastatin 40 mg (n = 9077) and were followed for a median of 6 years.
The mean age of patients was 63.6 years; 76% were male, 84% were Caucasian, and 27% had diabetes. The mean LDL-C level at qualifying event was 80 mg/dL (2.1 mmol/L) for those on lipid-lowering therapy (n = 6390) and 101 mg/dL (2.6 mmol/L) for those not on prior lipid-lowering therapy (n = 11,594). Prior to hospitalization for ACS, 34% of patients were receiving statin therapy. Over 1 year, the mean LDL-C level among patients continuing therapy was 53.2 mg/dL (1.4 mmol/L) in the ezetimibe/simvastatin group and 69.9 mg/dL (1.8 mmol/L) in the simvastatin monotherapy group. Lipid values were generally obtained from patients who remained on study therapy.
The primary endpoint was a composite of cardiovascular death, acute coronary syndrome (ACS, defined as non-fatal MI, documented unstable angina requiring hospitalization, or any coronary revascularization procedure occurring at least 30 days after randomization), and non-fatal stroke. The study demonstrated that adding ezetimibe to simvastatin provided a gradual benefit in reducing the primary composite endpoint of cardiovascular death, ACS, and non-fatal stroke compared to simvastatin alone (relative risk reduction of 6.4%, p = 0.016). The primary endpoint occurred in 2572 of 9067 patients (7-year Kaplan-Meier [KM] rate 32.72%) in the ezetimibe/simvastatin group and in 2742 of 9077 patients (7-year KM rate 32.72%) in the simvastatin group. (See Fig. 1 and Table 1). This additional benefit is expected to be similar when ezetimibe is coadministered with other statins proven effective in reducing cardiovascular risk. Overall mortality in this high-risk group was unchanged (see Table 1).
A general benefit was observed for all strokes; however, a small, non-significant increase in hemorrhagic stroke was observed in the ezetimibe/simvastatin group compared to simvastatin alone (see Table 1). The risk of hemorrhagic stroke with ezetimibe coadministered with high-potency statins in long-term outcomes trials has not been evaluated.
The treatment effect of ezetimibe/simvastatin was generally consistent across multiple subgroups, including sex, age, race, history of diabetes, baseline lipid levels, prior statin therapy, prior stroke, and hypertension.
Fig. 1. Effect of ezetimibe/simvastatin on the primary composite endpoint including cardiovascular death, acute coronary syndrome, and non-fatal stroke.
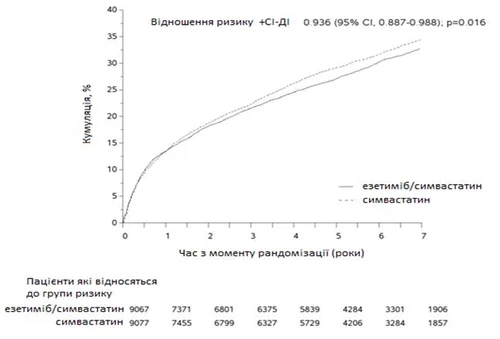
Major cardiovascular events by treatment group in all randomized patients in the IMPROVE-IT study.
Table 1.
| Result |
Ezetimibe/simvastatin 10/40 mga (N = 9067) |
Simvastatin (N = 9077) |
Relative risk (95 % CI) |
p-value |
||
| n |
K-M % c |
n |
K-M % c |
|||
| Primary composite endpoint |
||||||
| (Cardiovascular death, acute coronary syndrome, and non-fatal stroke) |
2572 |
32.72 % |
2742 |
34.67 % |
0.936 (0.887, 0.988) |
0.016 |
| Secondary composite endpoints |
||||||
| Death due to ischemic heart disease (IHD), non-fatal myocardial infarction (MI), urgent coronary revascularization after 30 days |
1322 |
17.52 % |
1448 |
18.88 % |
0.912 (0.847, 0.983) |
0.016 |
| Acute coronary syndrome, non-fatal stroke, death (all causes) |
3089 |
38.65 % |
3246 |
40.25 % |
0.948 (0.903, 0.996) |
0.035 |
| Cardiovascular death, non-fatal MI, unstable angina requiring hospitalization, any revascularization, non-fatal stroke |
2716 |
34.49 % |
2869 |
36.20 % |
0.945 (0.897, 0.996) |
0.035 |
| Components of the primary composite endpoint and selected efficacy endpoints (first occurrence of specified event at any time) |
||||||
| Cardiovascular death |
537 |
6.89 % |
538 |
6.84 % |
1.000 (0.887, 1.127) |
0.997 |
| Acute coronary syndrome: |
||||||
| Non-fatal MI |
945 |
12.77 % |
1083 |
14.41 % |
0.871 (0.798, 0.950) |
0.002 |
| Unstable angina requiring hospitalization |
156 |
2.06 % |
148 |
1.92 % |
1.059 (0.846, 1.326) |
0.618 |
| Urgent coronary revascularization after 30 days |
1690 |
21.84 % |
1793 |
23.36 % |
0.947 (0.886, 1.012) |
0.107 |
| Non-fatal stroke |
245 |
3.49 % |
305 |
4.24 % |
0.802 (0.678, 0.949) |
0.010 |
| All MI (fatal and non-fatal) |
977 |
13.13 % |
1118 |
14.82 % |
0.872 (0.800, 0.950) |
0.002 |
| All strokes (fatal and non-fatal) |
296 |
4.16 % |
345 |
4.77 % |
0.857 (0.734, 1.001) |
0.052 |
| Non-hemorrhagic stroked |
242 |
3.48 % |
305 |
4.23 % |
0.793 (0.670, 0.939) |
0.007 |
| Hemorrhagic stroke |
59 |
0.77 % |
43 |
0.59 % |
1.377 (0.930, 2.040) |
0.110 |
| Death from any cause |
1215 |
15.36 % |
1231 |
15.28 % |
0.989 (0.914, 1.070) |
0.782 |
a 6 % of patients, the dose of ezetimibe/simvastatin was increased to 10/80 mg.
a 27 % of patients, the dose of simvastatin was increased to 80 mg.
c 7-year rate based on the Kaplan-Meier method.
d includes ischemic stroke or stroke of undefined type.
Prevention of major vascular events in chronic kidney disease (CKD)
The Study of Heart and Renal Protection (SHARP) was a multinational, randomized, placebo-controlled, double-blind study involving 9438 patients with chronic kidney disease, one-third of whom were on dialysis at baseline. A total of 4650 patients were assigned to receive fixed-dose combination of ezetimibe 10 mg with simvastatin 20 mg, and 4620 to placebo; patients were followed for a median of 4.9 years. The mean patient age was 62 years; 63 % were male; 72 % were Caucasian; 23 % had diabetes, and for those not on dialysis, the mean estimated glomerular filtration rate (eGFR) was 26.5 mL/min/1.73 m². There were no lipid entry criteria. The mean baseline LDL-C level was 108 mg/dL. After one year, including in patients who were no longer taking the study medication, LDL-C was reduced by 26 % with simvastatin monotherapy 20 mg and by 38 % with ezetimibe 10 mg in combination with simvastatin 20 mg compared to placebo.
The primary comparison prespecified in the SHARP protocol was the intention-to-treat analysis of "major vascular events" (MVEs, defined as non-fatal myocardial infarction or cardiac arrest, stroke, or any revascularization procedure) only in those patients initially randomized to ezetimibe with simvastatin (n = 4193) or placebo (n = 4191). Secondary analyses included the same composite analyzed across the entire cohort randomized (at baseline or after 1 year) to receive ezetimibe with simvastatin (n = 4650) or placebo (n = 4620), as well as components of this composite. Analysis of the primary endpoint showed that ezetimibe in combination with simvastatin significantly reduced the risk of vascular events (749 patients with events in the placebo group and 639 in the ezetimibe with simvastatin group), representing a 16 % relative risk reduction (p = 0.001). However, this study design did not allow for determination of the individual contribution of ezetimibe alone to efficacy in significantly reducing the risk of vascular events in patients with CKD. Individual components of MVEs in all randomized patients are presented in Table 2. Ezetimibe in combination with simvastatin significantly reduced the risk of stroke and any revascularization, with numerically favorable but non-significant differences in non-fatal myocardial infarction and cardiac arrest.
Major vascular events by treatment group in all randomized patients in the SHARP studya.
Table 2.
| Result |
ezetimibe 10 mg in combination with simvastatin 20 mg (N = 4650) |
Placebo (N = 4620) |
Hazard ratio (95 % CI) |
P-value |
| Major vascular events |
701 (15.1 %) |
814 (17.6 %) |
0.85 (0.77 – 0.94) |
0.001 |
| Non-fatal MI |
134 (2.9 %) |
159 (3.4 %) |
0.84 (0.66 – 1.05) |
0.12 |
| Cardiac arrest |
253 (5.4 %) |
272 (5.9 %) |
0.93 (0.78 – 1.10) |
0.38 |
| Any stroke |
171 (3.7 %) |
210 (4.5 %) |
0.81 (0.66 – 0.99) |
0.038 |
| Non-hemorrhagic stroke |
131 (2.8 %) |
174 (3.8 %) |
0.75 (0.60 – 0.94) |
0.011 |
| Hemorrhagic stroke |
45 (1.0 %) |
37 (0.8 %) |
1.21 (0.78 – 1.86) |
0.40 |
| Any revascularization |
284 (6.1 %) |
352 (7.6 %) |
0.79 (0.68 – 0.93) |
0.004 |
| Major atherosclerotic events (MAE)b |
526 (11.3 %) |
619 (13.4 %) |
0.83 (0.74 – 0.94) |
0.002 |
aAnalysis based on the initial treatment assignment for all SHARP patients randomized to receive ezetimibe in combination with simvastatin or placebo both at baseline and after 1 year.
bMajor atherosclerotic events (MAE); defined as a composite of non-fatal myocardial infarction, coronary death, non-hemorrhagic stroke, or any revascularization procedure.
The absolute reduction in LDL-C levels achieved with ezetimibe in combination with simvastatin was lower in patients with lower baseline LDL-C levels (< 2.5 mmol/L) and in patients undergoing dialysis at baseline compared to other patients, and the corresponding risk reduction in these two groups was attenuated.
Homozygous familial hypercholesterolemia (HoFH)
In a double-blind, randomized, 12-week study, 50 patients with a clinical and/or genotypic diagnosis of HoFH who were receiving atorvastatin or simvastatin (40 mg) with or without concomitant LDL apheresis were enrolled. Ezetimibe co-administered with atorvastatin (40 or 80 mg) or simvastatin (40 or 80 mg) significantly reduced LDL-C levels by 15% compared to increasing the dose of simvastatin or switching from atorvastatin 40 mg to 80 mg monotherapy.
Homozygous sitosterolemia (phytosterolemia)
In a double-blind, placebo-controlled, 8-week study, 37 patients with homozygous sitosterolemia were randomized to receive ezetimibe 10 mg (n = 30) or placebo (n = 7). Some patients were receiving other treatments (e.g., statins, resins). Ezetimibe significantly reduced both primary plant sterols—sitosterol and campesterol—by 21% and 24%, respectively, compared to baseline levels. The impact of reducing sitosterol levels on morbidity and mortality in this population is unknown.
Aortic stenosis
The Simvastatin and Ezetimibe in Aortic Stenosis (SEAS) study was a multicenter, double-blind, placebo-controlled trial with a mean duration of 4.4 years involving 1873 patients with asymptomatic aortic stenosis (AS) confirmed by Doppler echocardiography showing peak aortic flow velocity between 2.5 and 4.0 m/s. Only patients whom physicians judged not to require statin therapy for reducing the risk of atherosclerotic cardiovascular disease were enrolled. Patients were randomized 1:1 to receive either placebo or combination ezetimibe 10 mg and simvastatin 40 mg daily.
The primary endpoint was a composite of major atherosclerotic events (MAE), including cardiovascular death, aortic valve replacement (AVR), heart failure due to progression of AS, non-fatal myocardial infarction, coronary artery bypass grafting (CABG), percutaneous coronary intervention (PCI), hospitalization for unstable angina, and non-hemorrhagic stroke. Key secondary endpoints were combinations of subclasses of the primary endpoint categories.
Compared with placebo, ezetimibe/simvastatin 10/40 mg did not significantly reduce the risk of MAE. The primary endpoint occurred in 333 patients (35.3%) in the ezetimibe/simvastatin group and in 355 patients (38.2%) in the placebo group (hazard ratio for the ezetimibe/simvastatin group, 0.96; 95% confidence interval [CI], 0.83 to 1.12; p = 0.59). Aortic valve replacement was performed in 267 patients (28.3%) in the ezetimibe/simvastatin group and in 278 patients (29.9%) in the placebo group (hazard ratio, 1.00; 95% CI, 0.84 to 1.18; p = 0.97). Fewer patients experienced ischemic cardiovascular events in the ezetimibe/simvastatin group (n = 148) than in the placebo group (n = 187) (hazard ratio, 0.78; 95% CI, 0.63 to 0.97; p = 0.02), primarily due to fewer patients undergoing coronary artery bypass grafting.
Cancer was observed more frequently in the ezetimibe/simvastatin group (105 vs. 70, p = 0.01). The clinical significance of this observation is uncertain, as in the SHARP study, the number of patients with any newly diagnosed cancer was not significantly different between groups (438 in the ezetimibe/simvastatin group and 439 in the placebo group). Furthermore, in the IMPROVE-IT study, the total number of patients with any new malignancy (853 in the ezetimibe/simvastatin group and 863 in the simvastatin group) did not differ significantly. Therefore, the findings from the SEAS study cannot be confirmed by the SHARP or IMPROVE-IT studies.
Pharmacokinetic properties
Absorption. After oral administration, ezetimibe is rapidly absorbed and actively conjugated to form the pharmacologically active phenolic glucuronide (ezetimibe-glucuronide). The mean peak plasma concentration (Cmax) is reached within 1–2 hours for ezetimibe-glucuronide and within 4–12 hours for ezetimibe. Absolute bioavailability of ezetimibe cannot be determined because the compound is insoluble in aqueous media suitable for injection.
Concomitant food intake (low- or high-fat meal) does not affect the oral bioavailability of ezetimibe. Ezetimibe can be administered independently of meals.
Distribution. Ezetimibe and ezetimibe-glucuronide are bound to human plasma proteins by 99.7% and 88–92%, respectively.
Metabolism. The primary metabolism of ezetimibe occurs in the small intestine and liver via glucuronidation (Phase II reaction), followed by biliary excretion. Minimal oxidative metabolism (Phase I reaction) is observed throughout the transformation process. Ezetimibe and ezetimibe-glucuronide are the major components detected in plasma, accounting for approximately 10–20% and 80–90% of total drug-related material in plasma, respectively. Ezetimibe and ezetimibe-glucuronide are slowly eliminated from plasma through enterohepatic recirculation. The elimination half-life of ezetimibe and ezetimibe-glucuronide is approximately 22 hours.
Excretion. After oral administration of 20 mg of 14C-ezetimibe to healthy volunteers, approximately 93% of total ezetimibe-related radioactivity was recovered in plasma. Approximately 78% and 11% of the administered radioactive dose were excreted in feces and urine, respectively, within 10 days. No detectable traces of radioactivity were found in plasma 48 hours after administration.
Special patient populations
Children
The pharmacokinetics of ezetimibe are similar in children aged 6 years and older and in adults. Pharmacokinetic data in children under 6 years of age are lacking. Clinical experience in pediatric and adolescent patients includes those with HoFH, heterozygous familial hypercholesterolemia (HeFH), or sitosterolemia.
Elderly patients
In elderly patients (over 65 years of age), plasma concentrations of total ezetimibe are approximately twice as high as in younger patients (18–45 years). However, the LDL-C reduction and safety profile are approximately similar between elderly and younger patients receiving ezetimibe. Therefore, no dose adjustment is required for elderly patients.
Hepatic impairment
After a single 10 mg dose of ezetimibe, the mean area under the concentration-time curve (AUC) for total ezetimibe was 1.7 times higher in patients with mild hepatic impairment (Child-Pugh score 5–6) compared to healthy volunteers. In a 14-day multiple-dose study (10 mg daily) in patients with moderate hepatic impairment (Child-Pugh score 7–9), the AUC for total ezetimibe increased approximately 4-fold on both Day 1 and Day 14 compared to healthy volunteers. Dose adjustment is not required for patients with mild hepatic impairment. Since the effects of increased ezetimibe exposure in patients with moderate or severe hepatic impairment (Child-Pugh score >9) are unknown, ezetimibe is not recommended for use in these patients (see section "Special precautions for use").
Renal impairment
After a single 10 mg dose of ezetimibe, the mean AUC for total ezetimibe increased approximately 1.5-fold in patients with severe renal impairment (n = 8; creatinine clearance ≤30 mL/min/1.73 m²) compared to healthy volunteers (n = 9). This increase is not considered clinically significant. No dose adjustment is required for patients with renal impairment.
In one patient in this study (who had received a kidney transplant and was on multiple therapies, including cyclosporine), total ezetimibe levels were 12 times higher.
Gender
Plasma concentrations of total ezetimibe are slightly higher (approximately 20%) in women than in men. However, LDL-C reduction and safety profile are approximately similar between men and women receiving ezetimibe. Therefore, no dose adjustment is required based on gender.
Clinical characteristics.
Indications.
Primary hypercholesterolemia
Ezetimibe in combination with a hydroxymethylglutaryl-coenzyme A (HMG-CoA) reductase inhibitor (statin) is indicated as adjunctive therapy to diet in patients with primary (heterozygous familial and non-familial) hypercholesterolemia when treatment with statins alone is insufficient.
Monotherapy with ezetimibe is indicated as adjunctive therapy to diet in patients with primary (heterozygous familial and non-familial) hypercholesterolemia for whom statin therapy is inappropriate or not tolerated.
Prevention of cardiovascular events
Ezetimibe is indicated to reduce the risk of cardiovascular events (see section "Pharmacodynamics") in patients with ischemic heart disease (IHD) and a history of acute coronary syndrome (ACS) when added to ongoing statin therapy or used concomitantly with a statin.
Homozygous familial hypercholesterolemia (HoFH)
Ezetimibe in combination with a statin is indicated as adjunctive therapy to diet in patients with HoFH. Patients may also receive additional treatment (e.g., LDL apheresis).
Homozygous sitosterolemia (phytosterolemia)
Ezetimibe is indicated as adjunctive therapy to diet in patients with homozygous familial sitosterolemia.
Contraindications.
Hypersensitivity to the active substance or to any of the excipients.
When co-administering ezetimibe with any statin, refer to the prescribing information for that specific medicinal product.
Combination therapy with ezetimibe and any statin is contraindicated during pregnancy or breastfeeding.
Use of ezetimibe in combination with any statin is contraindicated in patients with active liver disease or unexplained persistent elevations in serum transaminases.
Interaction with other medicinal products and other types of interactions.
Preclinical studies have shown that ezetimibe does not induce cytochrome P450 enzymes involved in drug metabolism. No clinically significant pharmacokinetic interactions have been observed between ezetimibe and drugs metabolized by cytochrome P450 enzymes 1A2, 2D6, 2C8, 2C9, and 3A4 or by N-acetyltransferase.
In clinical drug interaction studies, ezetimibe did not affect the pharmacokinetics of dapsone, dextromethorphan, digoxin, oral contraceptives (ethinylestradiol and levonorgestrel), glipizide, tolbutamide, or midazolam when used in combination therapy. Cimetidine did not affect the bioavailability of ezetimibe when co-administered.
Antacids. Concomitant administration of antacids results in reduced extent of absorption of ezetimibe but does not affect its bioavailability. This reduction in absorption is not considered clinically significant.
Cholestyramine. When used concomitantly with cholestyramine, the mean area under the concentration-time curve (AUC) of total ezetimibe (ezetimibe and ezetimibe-glucuronide) is reduced by approximately 55%. When adding ezetimibe to cholestyramine, the gradual reduction in low-density lipoprotein cholesterol (LDL-C) may be slowed.
Fibrates. When patients are receiving fenofibrate and ezetimibe, physicians should be aware of the potential risk of gallstone formation and gallbladder disease. In patients suspected of having gallstones while receiving ezetimibe and fenofibrate, gallbladder investigations are indicated and such therapy should be discontinued (see section "Adverse reactions").
Concomitant use of fenofibrate or gemfibrozil slightly increased the total concentration of ezetimibe (approximately 1.5 and 1.7 times, respectively).
The concomitant use of ezetimibe with other fibrates has not been studied.
Fibrates may increase biliary cholesterol excretion, leading to gallstone formation. In animal studies, ezetimibe sometimes increased cholesterol levels in bile, but not in all species. The lithogenic risk associated with therapeutic use of ezetimibe cannot be excluded.
Statins. No clinically significant pharmacokinetic interactions were observed when ezetimibe was co-administered with atorvastatin, simvastatin, pravastatin, lovastatin, fluvastatin, or rosuvastatin.
Cyclosporine. In a study involving 8 kidney transplant patients with creatinine clearance > 50 ml/min on stable doses of cyclosporine, a single 10 mg dose of ezetimibe resulted in a 3.4-fold increase (range: 2.3 to 7.9-fold) in the mean AUC of total ezetimibe compared to control healthy subjects receiving ezetimibe alone, in another study (n = 17). In another case involving a kidney transplant patient with severe renal insufficiency receiving cyclosporine and multiple other drugs, a 12-fold increase in exposure to total ezetimibe was observed compared to control subjects receiving ezetimibe alone. In a two-period crossover study involving 12 healthy volunteers, daily administration of 20 mg ezetimibe for 8 days with a single 100 mg dose of cyclosporine on day 7 resulted in a mean 15% increase (range: 10% decrease to 51% increase) in cyclosporine AUC compared to administration of a single 100 mg dose of cyclosporine alone. A controlled study evaluating the effect of concomitant ezetimibe administration on cyclosporine exposure in kidney transplant patients has not been conducted. Ezetimibe therapy should be initiated with caution in patients taking cyclosporine. Cyclosporine concentrations should be monitored in patients receiving both ezetimibe and cyclosporine.
Anticoagulants. Concomitant administration of ezetimibe (10 mg once daily) had no significant effect on the bioavailability of warfarin or prothrombin time in a study involving 12 healthy adult males. However, post-marketing reports have documented increases in international normalized ratio (INR) in patients receiving ezetimibe added to warfarin or fluindione. If ezetimibe is used concomitantly with warfarin, another coumarin anticoagulant, or fluindione, INR should be appropriately monitored.
Pediatric population. Interaction studies were performed in adults.
Special precautions for use.
When co-administering ezetimibe with any statin, refer to the package leaflet for that specific medicinal product.
Liver enzymes
In controlled combination therapy studies of patients receiving ezetimibe with statins, consistent increases in transaminase levels (≥3 times the upper limit of normal [ULN]) were observed. Liver function tests should be performed at the start of combination therapy with ezetimibe and a statin, and periodically thereafter in accordance with recommendations for statins.
In the Improved Reduction of Outcomes: Vytorin Efficacy International Trial (IMPROVE-IT), 18,144 patients with ischemic heart disease and a history of cardiovascular events were randomized to receive ezetimibe/simvastatin 10/40 mg daily (n = 9,067) or simvastatin 40 mg daily (n = 9,077). Over a median follow-up period of 6 years, the incidence of consecutive transaminase elevations (≥3 times ULN) was 2.5% for ezetimibe/simvastatin and 2.3% for simvastatin.
In a controlled clinical trial in which over 9,000 patients with chronic kidney disease were randomized to receive ezetimibe 10 mg in combination with simvastatin 20 mg daily (n = 4,650) or placebo (n = 4,620) (median follow-up 4.9 years), the incidence of consecutive transaminase elevations (≥3 times ULN) was 0.7% for ezetimibe plus simvastatin and 0.6% for placebo.
Skeletal muscle
Post-marketing reports have described cases of myopathy and rhabdomyolysis with ezetimibe use. Most patients who developed rhabdomyolysis were taking a statin concomitantly with ezetimibe. However, very rare cases of rhabdomyolysis have been reported during ezetimibe monotherapy and when ezetimibe was added to other agents known to be associated with an increased risk of rhabdomyolysis. If myopathy is suspected, manifested by muscle symptoms or creatine kinase levels >10 times the upper limit of normal, ezetimibe, any statin, and any other concurrently administered medications should be discontinued immediately. All patients initiating ezetimibe therapy should be informed about the risk of myopathy and the need to promptly report any unexplained muscle pain, tenderness, or weakness.
In the IMPROVE-IT study, 18,144 patients with ischemic heart disease and a history of cardiovascular events were randomized to receive ezetimibe/simvastatin 10/40 mg daily (n = 9,067) or simvastatin 40 mg daily (n = 9,077). Over a median follow-up of 6 years, the incidence of myopathy was 0.2% for ezetimibe/simvastatin and 0.1% for simvastatin. Myopathy was defined as unexplained muscle weakness or pain with serum creatine kinase levels ≥10 times ULN or two consecutive creatine kinase results ≥5 times and <10 times ULN. The incidence of rhabdomyolysis was 0.1% for ezetimibe/simvastatin and 0.2% for simvastatin. Rhabdomyolysis was defined as unexplained muscle weakness or pain with serum creatine kinase levels ≥10 times ULN and evidence of renal impairment, ≥5 times ULN on two consecutive occasions with evidence of renal impairment, or creatine kinase ≥10,000 IU/L without evidence of renal impairment.
In a clinical trial in which over 9,000 patients with chronic kidney disease were randomized to receive ezetimibe 10 mg in combination with simvastatin 20 mg daily (n = 4,650) or placebo (n = 4,620) (median follow-up 4.9 years), the incidence of myopathy/rhabdomyolysis was 0.2% for ezetimibe plus simvastatin and 0.1% for placebo.
Hepatic impairment
Since the effects of increased ezetimibe exposure in patients with moderate to severe hepatic impairment are unknown, ezetimibe is not recommended for use in these patients.
Children
The efficacy and safety of ezetimibe in patients aged 6 to 10 years with heterozygous familial or non-familial hypercholesterolemia were evaluated in a placebo-controlled clinical trial over 12 weeks. The effect of ezetimibe beyond 12 weeks of treatment in this age group has not been studied.
The effect of ezetimibe has not been studied in patients under 6 years of age.
The efficacy and safety of ezetimibe in combination with simvastatin in patients aged 10 to 17 years with heterozygous familial hypercholesterolemia were evaluated in a controlled clinical trial involving boys (Tanner stage II or higher) and girls (at least one year post-menarche).
During this limited controlled study, no clinically significant effects on growth or sexual maturation in adolescent boys and girls were observed, nor any effect on menstrual cycle length in girls. However, the effect of ezetimibe on growth and sexual maturation beyond 33 weeks of treatment has not been studied.
The safety and efficacy of ezetimibe in combination with simvastatin at doses exceeding 40 mg daily in children aged 10 to 17 years have not been studied.
The safety and efficacy of ezetimibe in combination with simvastatin in children under 10 years of age have not been studied.
The long-term efficacy of ezetimibe therapy in patients under 17 years of age for reducing morbidity and mortality in adulthood has not been established.
Fibrates
The safety and efficacy of ezetimibe when used in combination with fibrates have not been established.
If gallstones are suspected in a patient receiving ezetimibe and fenofibrate, gallbladder imaging is indicated, and this combination therapy should be discontinued.
Cyclosporine
Ezetimibe therapy should be initiated with caution in patients receiving cyclosporine. In patients taking both ezetimibe and cyclosporine, cyclosporine concentrations should be monitored.
Anticoagulants
If ezetimibe is used concomitantly with warfarin, other coumarin anticoagulants, or fluindione, the international normalized ratio (INR) should be monitored appropriately.
Use during pregnancy or breastfeeding.
Ezetimibe therapy in combination with a statin is contraindicated during pregnancy or breastfeeding (see section "Contraindications"); refer to the package leaflet for the specific statin.
Pregnancy
Ezetimibe should be administered to pregnant women only if clearly needed. There are no clinical data on the use of ezetimibe during pregnancy. Animal studies using ezetimibe as monotherapy showed no direct or indirect harmful effects on pregnancy, fetal or embryonic development, parturition, or postnatal development.
Breastfeeding
Ezetimibe should not be used during breastfeeding. Animal studies in rats have shown that ezetimibe is excreted in the milk of lactating animals. It is not known whether ezetimibe is excreted in human breast milk.
Fertility
Clinical data on the effect of ezetimibe on human fertility are lacking. Ezetimibe showed no effect in animal studies.
Ability to affect driving and use of machines.
No studies on the effect on the ability to drive and use machines have been conducted. However, dizziness has been reported during driving and operating machinery.
Dosage and Administration
Throughout the entire course of treatment with ezetimibe, the patient should adhere to a standard lipid-lowering diet.
Dosage
The recommended dose of ezetimibe is 10 mg (1 tablet) once daily.
When adding ezetimibe to a statin, the initial dose of that statin or a higher dose already prescribed should be continued. In such cases, refer to the product information for that statin.
Use in patients with ischemic heart disease and history of acute coronary syndrome (ACS)
To gradually reduce the risk of cardiovascular events in patients with ischemic heart disease and a history of ACS, ezetimibe (10 mg) may be used in combination with a statin proven to have beneficial cardiovascular effects.
Concomitant use with bile acid sequestrants
Ezetimibe should be administered no later than 2 hours before or no earlier than 4 hours after administration of a bile acid sequestrant.
Elderly patients
Elderly patients do not require dose adjustment.
Hepatic impairment
No dose adjustment is required in patients with mild hepatic impairment (Child-Pugh score of 5–6). Treatment with ezetimibe is not recommended in patients with moderate (Child-Pugh score of 7–9) or severe (Child-Pugh score >9) hepatic impairment.
Renal impairment
Patients with renal impairment do not require dose adjustment.
Administration method
The drug is administered orally. Ezetimibe can be taken at any time of the day, regardless of food intake.
Children
Specialist supervision is required at the beginning of treatment.
Children and adolescents aged 6 years and older: Safety and efficacy of ezetimibe in children aged 6 to 17 years have not been established. Available data are provided in the sections “Pharmacodynamics”, “Pharmacokinetic properties”, “Special precautions”, and “Adverse reactions”, but dosage recommendations cannot be made.
When ezetimibe is used concomitantly with a statin, refer to the statin’s product information for pediatric dosage recommendations.
Children under 6 years of age: Safety and efficacy of ezetimibe in children under 6 years of age have not been established. Information is lacking.
Overdose
In studies where 50 mg/day of ezetimibe was administered to 15 healthy subjects for 14 days or 40 mg/day to 18 patients with primary hypercholesterolemia for up to 56 days, the drug was generally well tolerated. In animal studies, no toxicity was observed after single oral doses of 5000 mg/kg in rats and mice, and 3000 mg/kg in dogs.
Several cases of ezetimibe overdose have been reported; in most cases, overdose did not result in adverse events. The adverse events observed were not serious. In case of overdose, symptomatic and supportive measures should be implemented.
Adverse Reactions
Below is a list of adverse reactions (from clinical studies and post-marketing experience). In studies lasting up to 112 weeks, ezetimibe 10 mg once daily was administered as monotherapy to 2396 patients or in combination with a statin to 11,308 patients or with fenofibrate to 185 patients. Adverse reactions were generally mild and transient. The overall incidence of adverse reactions was similar between ezetimibe and placebo. Similarly, the rate of discontinuation due to adverse events was comparable between ezetimibe and placebo.
Ezetimibe as monotherapy or in combination with a statin
The adverse reactions listed below were observed in clinical trials in patients treated with ezetimibe (N = 2396) and occurred more frequently than in the placebo group (N = 1159), or were observed in patients treated with ezetimibe in combination with a statin (N = 11,308) and occurred more frequently than in the group receiving statin alone (N = 9361). Adverse reactions in the post-marketing period were derived from reports where ezetimibe was administered either alone or concomitantly with a statin.
The frequency of adverse reactions is classified as follows: very common (≥1/10), common (≥1/100 to <1/10), uncommon (≥1/1000 to <1/100), rare (≥1/10,000 to <1/1000), very rare (<1/10,000), and not known (cannot be estimated from the available data).
| Monotherapy with ezetimibe |
||
| System organ classes |
Adverse reactions |
Frequency |
| Metabolism and nutrition disorders |
decreased appetite |
Uncommon |
| Vascular disorders |
flushing; hypertension |
Uncommon |
| Respiratory, thoracic and mediastinal disorders |
cough |
Uncommon |
| Gastrointestinal disorders |
abdominal pain; diarrhea; flatulence |
Common |
| dyspepsia; gastroesophageal reflux; nausea |
Uncommon |
|
| Musculoskeletal and connective tissue disorders |
arthralgia; muscle spasms; neck pain |
Uncommon |
| General disorders and administration site conditions |
fatigue |
Common |
| chest pain, pain |
Uncommon |
|
| Laboratory investigations |
Elevated ALT and/or AST; elevated serum creatine phosphokinase levels; elevated gamma-glutamyl transferase levels; abnormal liver function tests |
Uncommon |
| Additional adverse reactions with concomitant use of ezetimibe with a statin |
||
| System organ classes |
Adverse reactions |
Frequency |
| Nervous system disorders |
headache |
Common |
| paraesthesia |
Uncommon |
|
| Gastrointestinal disorders |
dry mouth; gastritis |
Uncommon |
| Skin and subcutaneous tissue disorders |
pruritus; rash; urticaria |
Uncommon |
| Musculoskeletal and connective tissue disorders |
myalgia |
Common |
| back pain; muscle weakness; limb pain |
Uncommon |
|
| General disorders and administration site conditions |
asthenia; peripheral oedema |
Uncommon |
| Laboratory investigations |
Elevated ALT and/or AST |
Common |
| Post-marketing period (with or without statin) |
||
| System organ classes |
Adverse reactions |
Frequency |
| Blood and lymphatic system disorders |
thrombocytopenia |
Not known |
| Immune system disorders |
hypersensitivity, including rash, urticaria, anaphylaxis and angioneurotic oedema |
Not known |
| Psychiatric disorders |
depression |
Not known |
| Nervous system disorders |
dizziness; paraesthesia |
Not known |
| Respiratory, thoracic and mediastinal disorders |
dyspnoea |
Not known |
| Gastrointestinal disorders |
pancreatitis; constipation |
Not known |
| Hepatobiliary disorders |
hepatitis; cholelithiasis; cholecystitis |
Not known |
| Skin and subcutaneous tissue disorders |
erythema multiforme |
Not known |
| Musculoskeletal and connective tissue disorders |
myalgia; myopathy/rhabdomyolysis |
Not known |
| General disorders and administration site conditions |
asthenia |
Not known |
Concomitant use of ezetimibe and fenofibrate
Gastrointestinal disorders: abdominal pain (common).
In a multicenter, double-blind, placebo-controlled clinical trial in patients with mixed hyperlipidemia, 625 patients were treated for up to 12 weeks and 576 patients for up to 1 year. In this study, 172 patients receiving treatment with ezetimibe and fenofibrate completed the 12-week treatment period, and 230 patients receiving treatment with ezetimibe and fenofibrate (including 109 patients who received ezetimibe as monotherapy during the first 12 weeks) completed one year of treatment. This study was not designed to compare treatment groups regarding the occurrence of infrequent events. The exposure-adjusted incidence rates (95 % CI) for clinically significant elevations (≥3 times the upper limit of normal, confirmed) of serum transaminases were 4.5 % (1.9 and 8.8) and 2.7 % (1.2 and 5.4) for fenofibrate monotherapy and concomitant use of ezetimibe with fenofibrate, respectively. The corresponding incidence rates for cholecystectomy were 0.6 % (0.0 and 3.1) and 1.7 % (0.6 and 4.0) for fenofibrate monotherapy and concomitant use of ezetimibe with fenofibrate, respectively, adjusted for treatment exposure.
Children (aged 6 to 17 years)
In a study involving children aged 6 to 10 years with heterozygous familial or non-familial hypercholesterolemia (n = 138), elevations in ALT and/or AST levels (≥3 times the upper limit of normal, confirmed) occurred in 1.1 % (1 patient) of patients receiving ezetimibe compared to 0 % in the placebo group. Elevations in CK levels (≥10 times the upper limit of normal) were not observed. No cases of myopathy were reported.
In another study involving adolescents (aged 10 to 17 years) with heterozygous familial hypercholesterolemia (N = 248), elevations in ALT and/or AST (≥3 times the upper limit of normal, confirmed) occurred in 3 % (4 patients) in the ezetimibe/simvastatin group compared to 2 % (2 patients) in the simvastatin monotherapy group; the corresponding rates for creatine kinase (CK) elevations (≥10 times the upper limit of normal) were 2 % (2 patients) and 0 %, respectively. No cases of myopathy were reported.
This study did not compare rare adverse drug reactions.
Patients with ischemic heart disease and history of acute coronary syndrome
In the IMPROVE-IT study (see section "Pharmacodynamics") involving 18,144 patients who received ezetimibe/simvastatin 10/40 mg (n = 9,067; of whom 6 % had their ezetimibe/simvastatin dose increased to 10/80 mg) or simvastatin 40 mg (n = 9,077; of whom 27 % had their simvastatin dose increased to 80 mg), the safety profiles were similar over a mean follow-up period of 6 years. The rate of discontinuation due to adverse events was 10.6 % in patients receiving ezetimibe/simvastatin and 10.1 % in those receiving simvastatin. The incidence of myopathy was 0.2 % for ezetimibe/simvastatin and 0.1 % for simvastatin, where myopathy was defined as muscle weakness or pain of unknown etiology with serum creatine kinase levels ≥10 times the upper limit of normal or with two consecutive results ≥5 times and <10 times the upper limit of normal. The incidence of rhabdomyolysis was 0.1 % for ezetimibe/simvastatin and 0.2 % for simvastatin, where rhabdomyolysis was defined as muscle weakness or pain of unknown etiology with serum creatine kinase levels ≥10 times the upper limit of normal with evidence of renal impairment, ≥5 times and <10 times the upper limit of normal on two consecutive occasions with evidence of renal impairment, or creatine kinase ≥10,000 IU/L without evidence of renal impairment. The incidence of confirmed transaminase elevations (≥3 times the upper limit of normal) was 2.5 % for ezetimibe/simvastatin and 2.3 % for simvastatin (see section "Special precautions for use"). Gallbladder-related adverse events occurred in 3.1 % versus 3.5 % of patients receiving ezetimibe/simvastatin and simvastatin, respectively. The rate of hospitalization due to cholecystectomy was 1.5 % in both treatment groups. Cancer (defined as any new malignancy) was diagnosed during the study in 9.4 % versus 9.5 %, respectively.
Patients with chronic kidney disease
In the Study of Heart and Renal Protection (SHARP) (see section "Pharmacodynamics") involving over 9,000 patients receiving a fixed combination of ezetimibe 10 mg with simvastatin 20 mg daily (n = 4,650) or placebo (n = 4,620), the safety profiles were comparable over a mean follow-up period of 4.9 years. In this study, only serious adverse reactions and discontinuations due to any adverse reactions were recorded. The rate of discontinuation due to adverse reactions was comparable (10.4 % in patients receiving ezetimibe in combination with simvastatin and 9.8 % in those receiving placebo). The incidence of myopathy/rhabdomyolysis was 0.2 % in patients receiving ezetimibe in combination with simvastatin and 0.1 % in those receiving placebo. Confirmed transaminase elevations (≥3 times the upper limit of normal) were observed in 0.7 % of patients receiving ezetimibe in combination with simvastatin compared to 0.6 % of patients receiving placebo (see section "Special precautions for use"). In this study, there was no statistically significant increase in the incidence of pre-specified adverse reactions, including cancer (9.4 % for ezetimibe in combination with simvastatin, 9.5 % for placebo), hepatitis, cholecystectomy, or complications related to gallstones or pancreatitis.
Laboratory test data
In controlled clinical trials of monotherapy, clinically significant elevations in serum transaminases (ALT and/or AST ≥3 times the upper limit of normal) were similar with ezetimibe (0.5 %) and placebo (0.3 %). In combination therapy studies, the incidence was 1.3 % in patients receiving concomitant ezetimibe and statin compared to 0.4 % in patients receiving statin alone. Such elevations were typically asymptomatic, not associated with cholestasis, and returned to baseline levels either after discontinuation of therapy or with continued treatment.
In clinical trials, CK elevations ≥10 times the upper limit of normal occurred in 4 of 1,674 patients (0.2 %) receiving ezetimibe alone, compared to 1 of 786 patients (0.1 %) receiving placebo, and in 1 of 917 patients (0.1 %) receiving ezetimibe concomitantly with a statin, compared to 4 of 929 patients (0.4 %) receiving statin alone. There was no increased risk of myopathy or rhabdomyolysis with ezetimibe compared to the respective control groups (placebo or statin alone).
Reporting suspected adverse reactions
It is very important to report suspected adverse reactions after marketing authorization of the medicinal product. This allows continuous monitoring of the benefit-risk balance of the medicinal product. Healthcare professionals are requested to report any adverse reactions via the national reporting system.
Shelf life. 3 years.
Storage conditions.
Store at temperatures not exceeding 30 °C. Store in the original packaging in a moisture-protected place.
Packaging.
10 tablets in a blister; 3, 6, or 9 blisters in a cardboard box.
Prescription category. Prescription only.
Manufacturer.
EGIS Pharmaceuticals Ltd.
Manufacturer's address and place of business.
1165 Budapest, Bekenyfeldi u. 118-120, Hungary.