Kiskali
Ukraina
Spis treści
INSTRUKCJA DOTYCZĄCA STOSOWANIA LEKU KISKALI (KISQALI)
Skład:
substancja czynna: ribociclib;
1 tabletka zawiera ribociclibu succynian w ilości odpowiadającej ribociclibowi 200 mg;
substancje pomocnicze: celuloza mikrokryształowa, crospovidon (typ A), hydroksypropyloceluloza niskoestryfikowana, stearynian magnezu, dwutlenek krzemu koloidalny bezwodny, tlenek żelaza czarny (E 172), tlenek żelaza czerwony (E 172), lecytyna z soi, alkohol polowinylowy częściowo zhydrolizowany, talk, dwutlenek tytanu (E 171), gumę ksantanową.
Postać leku. Tabletki powlekane o powłoce filmowej.
Główne właściwości fizykochemiczne: okrągłe, wypukłe tabletki z pochylonymi krawędziami, jasnoszaro-fioletowe, bez zarysowania, z tłoczonym napisem „RIC” po jednej stronie i „NVR” po drugiej.
Grupa farmakoterapeutyczna. Środki przeciwnowotworowe. Inhibitory proteinkinaz. Inhibitory kinazy zależnej od cykliny (CDK). Ribociclib.
Kod ATX L01E F02.
Właściwości farmakologiczne
Farmakodynamika
Mechanizm działania.
Ribocyklib to selektywny inhibitor cyklinozależnych kinaz (CDK) 4 i 6, powodujący ich 50-procentową inhibicję (IC50) w testach biochemicznych przy stężeniach odpowiednio 0,01 (4,3 ng/ml) i 0,039 μM (16,9 ng/ml). Kinazy te aktywują się poprzez wiązanie z cyklinami D i odgrywają kluczową rolę w szlakach sygnałowych regulujących cykl komórkowy i proliferację komórek. Kompleks cyklina D-CDK4/6 reguluje postęp cyklu komórkowego poprzez fosforylację białka retinoblastomy (pRb).
In vitro ribocyklib zmniejszał fosforylację pRb, prowadząc do zatrzymania fazy G1 cyklu komórkowego, zmniejszenia proliferacji oraz fenotypu starzenia się w modelach raka piersi. In vivo monoterapia ribocyklibem prowadziła do regresji guza, co odpowiadało inhibicji fosforylacji pRb.
W badaniach in vivo na modelu ksenotransplantatu pacjenta z rakiem piersi pozytywnym pod względem receptorów estrogenowych (ER+), stosowanie kombinacji ribocyklibu i antyestrogenów (np. letrozolu) prowadziło do silniejszego hamowania wzrostu guza, trwałej regresji guza oraz opóźnionego wznowienia wzrostu guza po zakończeniu leczenia w porównaniu z zastosowaniem każdego leku oddzielnie.
U pacjentów przyjmujących ribocyklib możliwe jest również działanie immunomodulacyjne poprzez zmniejszenie liczby regulatorowych komórek T oraz względnego poziomu komórek T CD3+.
Ponadto, w warunkach in vivo oceniano aktywność przeciwnowotworową ribocyklibu w kombinacji z fulvestrantem u myszy z niedoborem odporności, które były nosicielami ksenotransplantatów ludzkiego ER+ raka piersi ZR751; zastosowanie tej kombinacji prowadziło do całkowitego hamowania wzrostu guza.
Analiza panelu linii komórkowych raka piersi znanego pod względem statusu ER wykazała wyższą skuteczność ribocyklibu w liniach komórkowych raka piersi z statusem ER+ niż w tych z statusem ER-. W badanych modelach przedklinicznych do aktywności ribocyklibu wymagany był nietknięty pRb.
Elektrofizjologia serca
W celu oceny wpływu ribocyklibu na interwał QTc u pacjentów z zaawansowanym nowotworem wykonano serie EKG, trzykrotnie po podaniu dawki pojedynczej w stanie równowagi. Do analizy farmakokinetyki i farmakodynamiki włączono dane ogółem 997 pacjentów otrzymujących leczenie ribocyklibem w zakresie dawek od 50 do 1200 mg. Analiza wykazała, że ribocyklib powoduje zależne od stężenia wydłużenie interwału QTc.
U pacjentów z lokalnie zaawansowanym lub przerzutowym rakiem piersi obliczono średnią zmianę QTcF od wartości wyjściowej przy stosowaniu leku Kiskali w dawce 600 mg w kombinacji z nie-steroidowym inhibitorem aromatazy (NSIA) lub fulvestrantem, która wynosiła odpowiednio 22,0 ms (90 % CI: 20,56; 23,44) i 23,7 ms (90 % CI: 22,31; 25,08) przy geometrycznej średniej Cmax w stanie równowagi, w porównaniu z 34,7 ms (90 % CI: 31,64; 37,78) przy stosowaniu w kombinacji z tamoksyfenem (patrz sekcja „Szczególne ostrzeżenia i środki ostrożności”).
U pacjentów z rakiem piersi w wczesnym stadium występuje podobne, zależne od stężenia wydłużenie interwału QTc. Szacunkowa średnia zmiana QTcF od wartości wyjściowej jest niższa u pacjentów z rakiem piersi w wczesnym stadium otrzymujących 400 mg leku Kiskali w porównaniu z pacjentami z lokalnie zaawansowanym lub przerzutowym rakiem piersi otrzymującymi 600 mg leku Kiskali.
Skuteczność i bezpieczeństwo kliniczne
Rak piersi w wczesnym stadium
Badanie CLEE011O12301C (NATALEE)
Kiskali badano w randomizowanym, otwartym, wieloośrodkowym badaniu klinicznym fazy III, w którym ten lek stosowano w leczeniu kobiet w okresie przed- i po menopauzie oraz mężczyzn z rakiem piersi HR+ i HER2- w wczesnym stadium anatomicznym II lub III niezależnie od statusu węzłów chłonnych, z wysokim ryzykiem nawrotu, w kombinacji z inhibitorem aromatazy (IA) letrozolem lub anastrozolem, w porównaniu z leczeniem tylko IA:
- Grupa stadium IIB–III lub
- Grupa stadium IIA w połączeniu z następującymi kryteriami:
- Węzeł chłonny pachowy pozytywny lub
- Węzeł chłonny pachowy negatywny:
- guz G3 lub
- guz G2 w połączeniu z dowolnym z poniższych kryteriów:
- Ki67 ≥ 20 %;
- wysokie ryzyko nawrotu na podstawie wyników testu genetycznego.
Kobiety w przedmenopauzie oraz mężczyźni otrzymywali również agonistę LH-RH gozerelinę. Według kryteriów TNM do badania NATALEE zakwalifikowano pacjentów z dowolnym zaangażowaniem węzłów chłonnych lub bez zaangażowania węzłów chłonnych z rozmiarem guza > 5 cm lub z rozmiarem guza 2–5 cm stopnia G2 (i wysokim ryzykiem genetycznym nawrotu lub Ki67 ≥ 20 %) lub stopnia G3.
Ogółem 5101 pacjentów, w tym 20 mężczyzn, zostało zrandomizowanych w stosunku 1:1 do otrzymywania leku Kiskali w dawce 400 mg oraz IA (n = 2549) lub tylko IA (n = 2552). Randomizacja do otrzymywania leczenia była uwarstwiona według stadium anatomicznego (grupa II [n = 2154 (42,2 %)] vs grupa III [n = 2947 (57,8 %)]), wcześniejszego leczenia (chemioterapia neoadiuwantowa/adjuwantowa – tak [n = 4432 (86,9 %)] lub nie [n = 669 (13,1 %)]), statusu menopauzy (kobiety i mężczyźni w przedmenopauzie [n = 2253 (44,2 %)] vs kobiety w po menopauzie [n = 2848 (55,8 %)]) oraz regionu (Ameryka Północna/Zachodnia Europa/Oceanie [n = 3128 (61,3 %)] vs inne kraje świata [n = 1973 (38,7 %)]). Lek Kiskali podawano doustnie w dawce 400 mg raz dziennie przez 21 dni z rzędu z następującą 7-dniową przerwą w leczeniu w kombinacji z letrozolem 2,5 mg lub anastrozolem 1 mg doustnie raz dziennie przez 28 dni. Gozerelin podawano w dawce 3,6 mg w postaci podskórnej iniekcji implantu, podawanej w 1. dniu każdego 28-dniowego cyklu. Leczenie lekiem Kiskali trwało do ukończenia 3-letniego cyklu leczenia od daty randomizacji (około 39 cykli).
Średni wiek pacjentów włączonych do tego badania wynosił 52 lata (zakres od 24 do 90 lat). 15,2 % pacjentów miało 65 lat i więcej, w tym 123 pacjentów (2,4 %) w wieku 75 lat i więcej. Wśród pacjentów byli reprezentanci rasy europejskiej (73,4 %), azjatyckiej (13,2 %) oraz czarnoskórej lub Afroamerykanów (1,7 %). Wszyscy pacjenci mieli status funkcjonalny wg skali ECOG 0 lub 1. Ogółem 88,2 % pacjentów otrzymało chemioterapię w trybie neoadiuwantowym lub adiuwantowym, a 71,6 % otrzymało terapię hormonalną w trybie neoadiuwantowym lub adiuwantowym przez 12 miesięcy przed włączeniem do badania.
Pierwotnym punktem końcowym badania była przeżycia wolne od choroby inwazyjnej (PFS), zdefiniowana jako czas od randomizacji do pierwszego przypadku: lokalnego nawrotu inwazyjnego raka piersi, regionalnego nawrotu inwazyjnego raka, odległego nawrotu, śmierci (z dowolnej przyczyny), nowotworu kontralateralnego inwazyjnego raka piersi lub innego pierwotnego inwazyjnego raka (z wyłączeniem raka podstawnokomórkowego i grzybicy płaskokomórkowej skóry).
Pierwotny punkt końcowy badania został osiągnięty podczas analizy pierwotnej (data zakończenia zbierania danych: 11.01.2023). Statystycznie istotna poprawa PFS (HR: 0,748; 95 % CI: 0,618; 0,906; wartość p uzyskana z jednostronnego uwarstwionego testu log-rank, 0,0014) została wykazana u pacjentów przyjmujących lek Kiskali i IA w porównaniu z pacjentami otrzymującymi tylko IA. Spójne wyniki obserwowano w podgrupach według stadium anatomicznego, statusu menopauzy, regionu, stanu węzłów chłonnych, wieku, rasy oraz wcześniejszej terapii adiuwantowej/neoadiuwantowej lub terapii hormonalnej.
Dane analizy uzupełniającej (data zakończenia zbierania danych: 21.07.2023) podsumowano w tabeli 1, a wykres Kaplana-Meiera dla PFS przedstawiono na rysunku 1. Mediana czasu trwania leczenia w momencie końcowej analizy PFS wynosiła około 30 miesięcy, a mediana czasu obserwacji dla PFS – 33,3 miesiąca w obu badanych grupach. Dane dotyczące przeżycia całkowitego (OS) są obecnie niewystarczające. Ogółem 172 pacjentów (3,5 %) zmarło (83/2525 w grupie ribocyklibu vs 89/2442 w grupie leczenia tylko IA, HR 0,892; 95 % CI: 0,661; 1,203).
Tabela 1. NATALEE – Wyniki oceny skuteczności (PFS) według danych badacza (data zakończenia zbierania danych: 21.07.2023)
| Kiskali i IA* N = 2549 |
IA N = 2552 |
||
| Przeżycie wolne od choroby inwazyjnej (PWIa) |
|||
| Liczba pacjentów z zdarzeniem (n, %) |
226 (8,9 %) |
283 (11,1 %) |
|
| Stosunek ryzyka (95 % DI) |
0,749 (0,628; 0,892) |
||
| Wartość pb |
0,0006 |
||
| PWI po 36 miesiącach (%, 95 % DI) |
90,7 (89,3; 91,8) |
87,6 (86,1; 88,9) |
|
| DI – przedział ufności; N – liczba pacjentów. a PWI definiuje się jako czas od chwili randomizacji do pierwszego wystąpienia: nawrotu miejscowego inwazyjnego raka piersi, nawrotu regionalnego inwazyjnego raka, nawrotu odległego, śmierci (z dowolnej przyczyny), kontralateralnego inwazyjnego raka piersi lub innego pierwotnego raka inwazyjnego, różnego od raka piersi (z wyłączeniem raków podstawnokomórkowego i płaskokomórkowego skóry). b Nominalna wartość p uzyskana przy użyciu zstratifikowanego jednostronnego testu log-rank. * Letrozol lub anastrozol. |
|||
IА – inhibitor aromatazy (letrozol lub anastrozol)
Wartość p na podstawie skategoryzowanego testu log-rankowego jest jednostronna.
Rys. 1. NATALEE – krzywa przeżycia Kaplana-Meiera dla DFS według oceny badacza (data zamknięcia zbierania danych: 21.07.2023)
Wskaźnik przeżycia bez odległych przerzutów (DDFS) w grupie leczonych lekiem Kiskali i IA wyniósł 204 (8,0 %) w porównaniu z 256 (10 %) w grupie leczonej wyłącznie IA (HR: 0,749; 95 % CI: 0,623; 0,900).
Uogólniony rak piersi
Badanie CLEE011A2301 (MONALEESA-2)
Lek Kiskali oceniano w randomizowanym, podwójnie ślepym, placebo-kontrolowanym, wieloośrodkowym badaniu klinicznym fazy III u kobiet w okresie menopauzy z hormonozależnym, HER2 (czynnikiem wzrostu ludzkiego typu 2) – negatywnym uogólnionym rakiem piersi, które wcześniej nie otrzymywały terapii ze względu na uogólnienie choroby, w połączeniu z letrozolem w porównaniu z samym letrozolem.
Ogółem 668 pacjentek zostało randomizowanych w stosunku 1:1 w celu otrzymania leku Kiskali w dawce 600 mg i letrozolu (n = 334) lub placebo i letrozolu (n = 334) oraz podzielonych według występowania przerzutów do wątroby i/lub płuc (tak [n = 292 (44 %)] lub nie [n = 376 (56 %)]). Dane demograficzne i pierwotne cechy choroby były zrównoważone i porównywalne w grupach badawczych. Lek Kiskali podawano doustnie w dawce 600 mg na dobę przez 21 kolejnych dni z 7-dniową przerwą w leczeniu, w połączeniu z letrozolem w dawce 2,5 mg raz na dobę przez 28 dni. Pacjentkom nie umożliwiono przejścia z placebo na lek Kiskali w trakcie badania ani po postępie choroby.
Średni wiek pacjentek włączonych do tego badania wyniósł 62 lata (zakres od 23 do 91). 44,2 % pacjentek miało 65 lat i więcej, w tym 69 pacjentek powyżej 75 roku życia. Spośród pacjentek byli reprezentanci rasy europejskiej (82,2 %), mongolskiej (7,6 %) i czarnej (2,5 %). Wszystkie pacjentki miały status czynnościowy wg skali ECOG 0 lub 1. W grupie leczonej lekiem Kiskali 46,6 % pacjentek otrzymywało chemioterapię w trybie neoadiuwantowym lub adiuwantowym, a 51,3 % otrzymywało terapię antyhormonalną w trybie neoadiuwantowym lub adiuwantowym przed włączeniem do badania. 34,1 % pacjentek miało nowotwór de novo (pierwszy raz zdiagnozowany rak). U 22,0 % pacjentek stwierdzono przerzuty wyłącznie do kości, a u 58,8 % – przerzuty do narządów wewnętrznych. Pacjentki, które wcześniej otrzymywały terapię (neo)adiuvantową anastrozolem lub letrozolem, musiały zakończyć tę terapię co najmniej 12 miesięcy przed randomizacją do badania.
Analiza pierwotna
Pierwotny punkt końcowy w badaniu osiągnięto podczas planowanego analizy pośredniej, przeprowadzonej po zaobserwowaniu 80 % zaplanowanych przypadków przeżycia bez postępu choroby (PFS) z wykorzystaniem kryteriów oceny odpowiedzi w nowotworach litych (RECIST, wersja 1.1) na podstawie oceny badacza w całej populacji (wszyscy randomizowani pacjenci) i potwierdzono niezależną oceną radiologiczną centralną danych w sposób zamaskowany.
Wyniki oceny skuteczności wykazały istotne statystycznie poprawienie PFS u pacjentek otrzymujących lek Kiskali i letrozol w porównaniu z pacjentkami otrzymującymi placebo i letrozol, przy analizie całej populacji (stosunek ryzyka 0,556; 95 % CI: 0,429; 0,720; wartość p według jednostronnego skategoryzowanego testu log-rankowego = 0,00000329), co wskazuje na klinicznie istotny efekt leczenia.
Ogólne dane dotyczące stanu zdrowia/jakości życia nie wykazały różnic między grupą stosującą lek Kiskali i letrozol a grupą stosującą placebo i letrozol.
Późniejsze aktualizacje danych dotyczących skuteczności (data zamknięcia zbierania danych: 2 stycznia 2017 r.) przedstawiono w tabelach 2 i 3 (MONALEESA-2: wyniki oceny skuteczności (PFS) na podstawie radiologicznej oceny badacza oraz MONALEESA-2: wyniki oceny skuteczności (OS, PFS) na podstawie oceny badacza).
Mediana PFS wyniosła 25,3 miesiąca (95 % CI: 23,0; 30,3) u pacjentek otrzymujących rybocyklib i letrozol oraz 16,0 miesiąca (95 % CI: 13,4; 18,2) u pacjentek otrzymujących placebo i letrozol. U 54,7 % pacjentek otrzymujących rybocyklib i letrozol nie zaobserwowano postępu choroby po 24 miesiącach, w porównaniu z 35,9 % w grupie placebo i letrozol.
Tabela 2. MONALEESA-2: wyniki oceny skuteczności (PFS) na podstawie radiologicznej oceny badacza (data zamknięcia zbierania danych: 2 stycznia 2017 r.)
| Zaktualizowana analiza |
||
| Wskaźniki |
Kiskali i letrozol N = 334 |
Placebo i letrozol N = 334 |
| Przeżycie wolne od progresji |
||
| Mediana PFS [miesiące] (95 % CI) |
25,3 (23,0; 30,3) |
16,0 (13,4; 18,2) |
| Stosunek ryzyka (95 % CI) |
0,568 (0,457; 0,704) |
|
| Wartość pa |
9,63 × 10-8 |
|
| CI – przedział ufności; N – liczba pacjentów a Wartość p uzyskano jednostronnym warstwowym testem log-rank. |
||
 |
| Chwile oceny Rybocyklib (N = 334) Placebo (N = 334) |
| Liczba zdarzeń: rybocyklif – 140, placebo – 205 Stosunek ryzyka 0,568; 95 % przedział ufności [0,457; 0,704] Mediana Kaplana-Meiera: rybocyklif – 25,3 miesiąca; placebo – 16,0 miesiąca Wartość p testu log-rank 9,63*10^(-8) |
Czas (miesiące)
| Liczba pacjentów, którzy wciąż są narażeni na ryzyko |
||||||||||||||||||
| Czas |
0 |
2 |
4 |
6 |
8 |
10 |
12 |
14 |
16 |
18 |
20 |
22 |
24 |
26 |
28 |
30 |
32 |
34 |
| Ribocyklib |
334 |
294 |
277 |
257 |
240 |
227 |
207 |
196 |
188 |
176 |
164 |
132 |
97 |
46 |
17 |
11 |
1 |
0 |
| Placebo |
334 |
279 |
265 |
239 |
219 |
196 |
179 |
156 |
138 |
124 |
110 |
93 |
63 |
34 |
10 |
7 |
2 |
0 |
Rys. 2. MONALEESA-2: wykres Kaplana-Meiera dla PFS według oceny badacza (data zakończenia zbierania danych: 2 stycznia 2017 r.)
Przeprowadzono serię analiz PFS w wcześniej określonych podgrupach na podstawie czynników prognostycznych i wyjściowych cech pacjentów w celu zbadania wewnętrznej stabilności działania leczenia. Obniżenie ryzyka postępowania choroby lub śmierci na korzyść grupy leczonej lekiem Kiskali i letrozolem obserwowano we wszystkich pojedynczych podgrupach pacjentów ze względu na wiek, przynależność rasową, poprzednią terapię adiuwantową lub neoadiuwantową (chemioterapię lub terapię hormonalną), zaangażowanie wątroby i/lub płuc oraz przerzuty wyłącznie do kości. Efekt był widoczny u pacjentów z przerzutami do wątroby i/lub płuc (HR 0,561 [95 % CI: 0,424; 0,743], mediana przeżycia bez postępowania choroby [mPFS] wyniosła 24,8 miesiąca przy stosowaniu kombinacji leku Kiskali i letrozolu w porównaniu do 13,4 miesiąca przy samym letrozolu) oraz u pacjentów bez przerzutów do wątroby i/lub płuc (HR 0,597 [95 % CI: 0,426; 0,837], mPFS 27,6 miesiąca w porównaniu do 18,2 miesiąca).
Zaktualizowane wyniki dotyczące całkowitej odpowiedzi i częstości skuteczności klinicznej przedstawiono w tabeli 3.
Tabela 3. MONALEESA-2: wyniki oceny skuteczności (ORR, CBR) według oceny badacza (data zakończenia zbierania danych: 2 stycznia 2017 r.)
| Analiza |
Kiskali i letrozol (%, 95 % CI) |
Placebo i letrozol (%, 95 % CI) |
Wartość pb |
| Cała populacja analityczna |
N = 334 |
N = 334 |
|
| Częstość całkowitej odpowiedzia |
42,5 (37,2; 47,8) |
28,7 (23,9; 33,6) |
9,18 × 10-5 |
| Częstość skuteczności klinicznejb |
79,9 (75,6; 84,2) |
73,1 (68,3; 77,8) |
0,018 |
| Pacjenci z mierzalną chorobą |
n = 257 |
n = 245 |
|
| Częstość całkowitej odpowiedzia |
54,5 (48,4; 60,6) |
38,8 (32,7; 44,9) |
2,54 × 10-4 |
| Częstość skuteczności klinicznejb |
80,2 (75,3; 85,0) |
71,8 (66,2; 77,5) |
0,018 |
| a CZO: częstość całkowitej odpowiedzi = odsetek pacjentów z pełną odpowiedzią + częściową odpowiedzią. b CSK: częstość skuteczności klinicznej = odsetek pacjentów z pełną odpowiedzią + częściową odpowiedzią (+ stan stabilny lub niepełna odpowiedź / brak postępu choroby przez ≥ 24 tygodnie). b Wartość p uzyskano przy użyciu jednostronnego testu chi-kwadrat Cochrana-Mantela-Haenszela. |
|||
Podsumowanie analizy ORR
Wyniki analizy całkowitej odpowiedzi (ORR) w ogólnej populacji badanej przedstawiono w tabeli 4 i na rysunku 3.
Tabela 4. MONALEESA-2: wyniki oceny skuteczności (ORR) (data zakończenia zbierania danych – 10 czerwca 2021 r.)
| Całkowite przeżycie, populacja badana ogólna |
Kiskali i letrozol N = 334 |
Placebo i letrozol N = 334 |
| Liczba przypadków, n [%] |
181 (54,2) |
219 (65,6) |
| Mediana CZW [miesiące] (95% CI) |
63,9 (52,4; 71,0) |
51,4 (47,2; 59,7) |
| Stosunek ryzykaa (95 % CI) |
0,765 (0,628, 0,932) |
|
| Wartość p-b |
0,004 |
|
| CZW bez powikłań, (%) (95 % CI) |
||
| 24 miesiące |
86,6 (82,3, 89,9) |
85,0 (80,5, 88,4) |
| 60 miesięcy |
52,3 (46,5, 57,7) |
43,9 (38,3, 49,4) |
| 72 miesiące |
44,2 (38,5, 49,8) |
32,0 (26,8, 37,3) |
| CI – przedział ufności. a Stosunek ryzyka uzyskano za pomocą skategoryzowanego modelu Coxa. b Wartość p uzyskano za pomocą jednostronnego skategoryzowanego testu log-rank (p < 0,0219 dla stwierdzenia większej skuteczności). Kategoryzacja przeprowadzona według statusu przerzutów w płucach i/lub wątrobie według interaktywnej technologii odpowiedzi (IRT). |
||
| Momenty cenzurowania Rybozycylib (N = 334) Placbo (N = 334) |
| Liczba zdarzeń: rybocyklib: 181, placebo: 219 Stosunek ryzyka = 0,765 Mediana Kaplana-Meiera: rybocyklib: 63,9 miesiąca placebo: 51,4 miesiąca Wartość p testu log-rank = 0,004 |
| Czas (miesiące) |
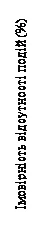 |
| Liczba pacjentów, którzy wciąż są narażeni na ryzyko |
| Czas Rybocyklib Placebo |
Rys. 3. MONALEESA-2: wykres Kaplana-Meiera dla PFS w populacji ogólnej (data zakończenia zbierania danych – 10 czerwca 2021 r.)
Test log-rankowy i model HR Coxa ze stratyfikacją według statusu przerzutów w płuca lub wątrobę według danych IRT.
Jednostronna wartość p uzyskana za pomocą stratyfikowanego testu log-rankowego.
Badanie CLEE011E2301 (MONALEESA-7)
Preparat Kiskali oceniano w randomizowanym, podwójnie ślepych, kontrolowanym placebo wieloośrodkowym badaniu klinicznym fazy III leczenia kobiet w okresie przed- i okołomenopauzalnym z pozytywnym pod względem receptora hormonu, HER2-negatywnym zaawansowanym rakiem piersi w połączeniu z AISS lub tamoksyfenem i gozerelina w porównaniu z placebo w połączeniu z AISS lub tamoksyfenem i gozerelina. Pacjentki w badaniu MONALEESA-7 nie otrzymywały wcześniejszej terapii endokrynnej z powodu zaawansowanego raka piersi.
Łącznie 672 pacjentki zostało zrandomizowanych w stosunku 1:1 w celu otrzymania preparatu Kiskali w dawce 600 mg oraz AISS/tamoksyfenu i gozereliny (n = 335) lub placebo i AISS/tamoksyfenu i gozereliny (n = 337) oraz ze stratyfikacją według obecności przerzutów w wątrobie i/lub płucach (tak [n = 344 (51,2 %)] lub nie [n = 328 (48,8 %)]), wcześniejszej chemioterapii z powodu zaawansowanej choroby (tak [n = 120 (17,9 %)] lub nie [n = 552 (82,1 %)]) oraz dodatkowego leku w ramach kombinowanej terapii endokrynnej (AISS i gozerelina [n = 493 (73,4 %)] lub tamoksyfen i gozerelina [n = 179 (26,6 %)]). Dane demograficzne i wyjściowe cechy choroby były zrównoważone i porównywalne w grupach badawczych. Preparat Kiskali podawano doustnie w dawce 600 mg na dobę przez 21 kolejnych dni z 7-dniową przerwą w leczeniu w połączeniu z AISS (letrozol w dawce 2,5 mg lub anastrozol w dawce 1 mg) lub tamoksyfenem (20 mg) doustnie raz na dobę przez 28 dni oraz gozereliną (3,6 mg) podskórnie co 28 dni aż do postępu choroby lub rozwoju nieakceptowalnej toksyczności. Pacjentkom nie umożliwiano przejścia z placebo na preparat Kiskali w trakcie badania ani po postępie choroby. Zmiana dodatkowego leku w ramach kombinowanej terapii endokrynnej również nie była dozwolona.
Średni wiek pacjentek włączonych do tego badania wynosił 44 lata (zakres od 25 do 58), a 27,7 % pacjentek miało poniżej 40 roku życia. Większość pacjentek była rasy europejskiej (57,7 %), mongolskiej (29,5 %) lub negroidalnej (2,8 %), niemal wszystkie pacjentki (99,0 %) miały wyjściowy status czynnościowy ECOG 0 lub 1. Przed włączeniem do badania spośród tych 672 pacjentek 14 % otrzymywało chemioterapię z powodu choroby przerzutowej, 32,6 % otrzymywało chemioterapię w trybie adiuwantnym, a 18,0 % – w trybie neoadiuwantnym; 39,6 % otrzymywało terapię endokrynną w trybie adiuwantnym, a 0,7 % – w trybie neoadiuwantnym. W badaniu E2301 40,2 % pacjentek miało chorobę przerzutową de novo (pierwotnie zdiagnozowaną), u 23,7 % pacjentek stwierdzono przerzuty wyłącznie w kościach, a u 56,7 % – przerzuty w narządach wewnętrznych.
Pierwotny punkt końcowy w badaniu został osiągnięty w analizie pierwotnej przeprowadzonej po zaobserwowaniu 318 przypadków przeżycia bez postępu choroby (PFS) na podstawie oceny badacza z wykorzystaniem kryteriów RECIST wersja 1.1 w całej populacji (wszystkie zrandomizowane pacjentki). Pierwotne wyniki oceny skuteczności potwierdzono wynikami PFS na podstawie niezależnej centralnej oceny radiologicznej zamaskowanych danych. Średni czas dalszej obserwacji w momencie pierwotnej analizy PFS wyniósł 19,2 miesiąca.
W populacji ogólnej badania wyniki oceny skuteczności wykazały statystycznie istotne poprawy PFS u pacjentek otrzymujących preparat Kiskali oraz AISS/tamoksyfen i gozerelinę w porównaniu z pacjentkami otrzymującymi placebo oraz AISS/tamoksyfen i gozerelinę (stosunek ryzyka 0,553; 95 % CI: 0,441; 0,694; wartość p z jednostronnego stratyfikowanego testu log-rankowego 9,83 × 10-8) z klinicznie istotnym efektem leczenia.
Mediana PFS wyniosła 23,8 miesiąca (95 % CI: 19,2; nie ocenianej (NO)) u pacjentek otrzymujących preparat Kiskali oraz AISS/tamoksyfen i gozerelinę oraz 13,0 miesiąca (95 % CI: 11,0; 16,4) u pacjentek otrzymujących placebo oraz AISS/tamoksyfen i gozerelinę.
Rozkład PFS podsumowano na krzywej Kaplana-Meiera dla PFS, przedstawionej na rysunku 4.
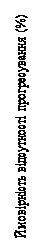
| Momenty cenzurowania Rybocyklіb (N = 335) Placbo (N = 337) |
| Liczba zdarzeń Rybofiklib – 131, placebo – 187 Stosunek ryzyka 0,553 95 % CI [0,441; 0,694] Mediana Kaplana-Meiera Rybofiklib – 23,8 miesiąca Placebo – 13,0 miesiąca Wartość p testu log-rank 9,83*10^(-8) |
Czas (miesiące)
| Liczba pacjentów, którzy wciąż są narażeni na ryzyko |
||||||||||||||||
| Czas (miesiące) |
0 |
2 |
4 |
6 |
8 |
10 |
12 |
14 |
16 |
18 |
20 |
22 |
24 |
26 |
28 |
30 |
| Ryocyklib |
335 |
301 |
284 |
264 |
245 |
235 |
219 |
178 |
136 |
90 |
54 |
40 |
20 |
3 |
1 |
0 |
| Placebo |
337 |
273 |
248 |
230 |
207 |
183 |
165 |
124 |
94 |
62 |
31 |
24 |
13 |
3 |
1 |
0 |
Rys. 4. MONALEESA-7: wykres Kaplana-Meiera dla PFS w populacji ogólnej na podstawie oceny badacza
Wyniki PFS na podstawie niezależnej, centralnej oceny radiologicznej zamaskowanych danych podgrupy losowo wybranych pacjentów (około 40 % wszystkich pacjentów zrandomizowanych) potwierdziły pierwotne wyniki dotyczące skuteczności na podstawie oceny badacza (szacunek HR 0,427; 95 % CI: 0,288; 0,633).
W momencie pierwotnej analizy PFS dane dotyczące przeżycia ogólnego były niekompletne, odnotowano 89 (13 %) przypadków śmierci (HR 0,916 [95 % CI: 0,601; 1,396]).
Wskaźnik odpowiedzi całkowitej (ORR) według oceny badacza, na podstawie kryteriów RECIST w wersji 1.1, był wyższy w grupie leczonej lekiem Kiskali (40,9 %; 95 % CI: 35,6; 46,2) w porównaniu z grupą placebo (29,7 %; 95 % CI: 24,8; 34,6; p = 0,00098). Wskaźnik skuteczności klinicznej (CBR) był wyższy w grupie leczonej lekiem Kiskali (79,1 %; 95 % CI: 74,8; 83,5) w porównaniu z grupą placebo (69,7 %; 95 % CI: 64,8; 74,6; p = 0,002).
W analizie wcześniej zdefiniowanych podgrup u 495 pacjentek, które otrzymywały lek Kiskali lub placebo w połączeniu z AI i gozareliną, mediana PFS wynosiła 27,5 miesiąca (95 % CI: 19,1; NR) w podgrupie leczonej lekiem Kiskali i AI oraz 13,8 miesiąca (95 % CI: 12,6; 17,4) w podgrupie otrzymującej placebo i AI [HR: 0,569; 95 % CI: 0,436; 0,743]. Wyniki oceny skuteczności przedstawiono w tabeli 5, a krzywe Kaplana-Meiera dla PFS na rysunku 5.
| Tabela 5. MONALEESA-7: wyniki oceny skuteczności (PFS) u pacjentów leczonych AI |
||
| Wskaźniki |
Kiskali i AI i goserelina N = 248 |
Placebo i AI i goserelina N = 247 |
| Przeżycie wolne od postępu chorobya |
||
| Mediana PFS [miesiące] (95 % CI) |
27,5 (19,1; nie oszacowano) |
13,8 (12,6; 17,4) |
| Stosunek ryzyka (95 % CI) |
0,569 (0,436; 0,743) |
|
| CI – przedział ufności; N – liczba pacjentów; nie oszacowano – nie oszacowano. a PFS na podstawie radiologicznej oceny badacza. |
||
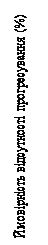 |
| Momenty cenzurowania Rybocyklib (N = 248) Placebo (N = 247) |
| Liczba zdarzeń Rybozyl – 92, placebo – 132 Stosunek ryzyka 0,569 95 % przedział ufności [0,436; 0,743] Mediana Kaplana-Meiera Rybozyl – 27,5 miesiąca Placebo – 13,8 miesiąca |
Czas (miesiące)
| Liczba pacjentów, którzy wciąż są narażeni na ryzyko |
||||||||||||||||
| Czas (miesiące) |
0 |
2 |
4 |
6 |
8 |
10 |
12 |
14 |
16 |
18 |
20 |
22 |
24 |
26 |
28 |
30 |
| Rybowcylib |
248 |
223 |
212 |
199 |
183 |
175 |
163 |
132 |
100 |
66 |
38 |
27 |
15 |
2 |
1 |
0 |
| Placebo |
247 |
195 |
177 |
163 |
149 |
138 |
126 |
95 |
72 |
48 |
25 |
19 |
9 |
2 |
1 |
0 |
Ryc. 5. MONALEESA-7: wykres Kaplana-Meiera dla PFS na podstawie oceny badacza u pacjentów leczonych CDT
Wyniki oceny skuteczności dotyczące częstotliwości odpowiedzi ogólnej (ORR) i częstotliwości skuteczności klinicznej (CBR) według oceny badacza na podstawie kryteriów RECIST w wersji 1.1 przedstawiono w tabeli 6.
| Tabela 6. MONALEESA-7: wyniki oceny skuteczności (ORR, CBR) na podstawie oceny badacza u pacjentów otrzymujących AISS |
||
| Analiza |
Kiskali i AISS i goserelina (%, 95 % CI) |
Placebo i AISS i goserelina (%, 95 % CI) |
| Cała populacja do analizy |
N = 248 |
N = 247 |
| Częstość odpowiedzi ogólnej (ORR)a |
39,1 (33,0; 45,2) |
29,1 (23,5; 34,8) |
| Częstość korzyści klinicznej (CBR)b |
80,2 (75,3; 85,2) |
67,2 (61,4; 73,1) |
| Pacjenci z mierzalną chorobą |
n = 192 |
n = 199 |
| Częstość odpowiedzi ogólneja |
50,5 (43,4; 57,6) |
36,2 (29,5; 42,9) |
| Częstość korzyści klinicznego b |
81,8 (76,3; 87,2) |
63,8 (57,1; 70,5) |
| a ORR: odsetek pacjentów z pełną odpowiedzią + częściową odpowiedzią. b CBR: odsetek pacjentów z pełną odpowiedzią + częściową odpowiedzią + (stan stabilny lub niepełna odpowiedź/brak postępu choroby przez ≥ 24 tygodnie). |
||
Wyniki w podgrupie leczonej lekiem Kiskali w połączeniu z AI były zgodne między podgrupami pod względem wieku, pochodzenia rasowego, wcześniejszej chemioterapii adjuwantowej/neoadjuwantowej lub leczenia hormonalnego, zaangażowania wątroby i/lub płuc oraz przerzutów wyłącznie do kości.
Późniejsza aktualizacja danych dotyczących przeżycia całkowitego (data zakończenia zbierania danych: 30 listopada 2018 r.) przedstawiona jest w tabeli 7 oraz na rysunkach 6 i 7.
W drugiej analizie OS badanie osiągnęło swój kluczowy wtórny punkt końcowy, wykazując istotne statystycznie poprawy przeżycia całkowitego.
Tabela 7. MONALEESA-7: wyniki oceny skuteczności (OS) (data zakończenia zbierania danych – 30 listopada 2018 r.)
| Zaktualizowany analiz |
||
| Przeżycie ogólne, populacja ogólna badania |
Kiskali 600 mg N = 335 |
Placebo N = 337 |
| Liczba przypadków, n [%] |
83 (24,8) |
109 (32,3) |
| Mediana PO [miesiące] (95 % CI) |
NR (NR, NR) |
40,9 (37,8; NR) |
| Stosunek ryzyka (95 % CI) |
0,712 (0,535; 0,948) |
|
| Wartość pa |
0,00973 |
|
| Przeżycie ogólne, podgrupa NSIA |
Kiskali 600 mg n = 248 |
Placebo n = 247 |
| Liczba przypadków, n [%] |
61 (24,6) |
80 (32,4) |
| Mediana PO [miesiące] (95 % CI) |
NR (NR, NR) |
40,7 (37,4; NR) |
| Stosunek ryzyka (95 % CI) |
0,699 (0,501; 0,976) |
|
| CI – przedział ufności; NR – nie oszacowano; N – liczba pacjentów. a Wartość p uzyskano przy użyciu jednostronnego testu log-rank, ze stratyfikacją według obecności przerzutów w wątrobie i/lub płucach, wcześniejszej chemioterapii z powodu zaawansowanego schorzenia oraz dodatkowego leku w ramach kombinowanej terapii endokrynnej według interaktywnej technologii odpowiedzi (IRT). |
||
| Chwile oceny Rybocyklib (N = 335) Placebo (N = 337) |
| Liczba przypadków Rybozycylib – 83, placebo – 109 Stosunek ryzyka 0,712 95 % CI [0,535; 0,948] Mediana Kaplana-Meiera Rybozycylib – NV Placebo – 40,9 miesiąca Wartość p testu log-rank 0,00973 |
Czas (miesiące)
Liczba pacjentów, którzy wciąż podlegają ryzyku
Czas (miesiące)
Ribociklib
Placebo
Rys. 6. MONALEESA-7: wykres Kaplana-Meiera dla ostatecznej analizy WSW (data zakończenia zbierania danych: 30 listopada 2018 r.)
Test log-rank oraz model Coxa uwarstwione według obecności przerzutów do płuc i/lub wątroby, wcześniejszej chemioterapii z powodu zaawansowanej choroby oraz dodatkowego leku w ramach kombinowanej terapii endokrynnej według interaktywnej technologii odpowiedzi (IRT).
| Momenty cenzurowania Rybozyclib (N = 248) Placebo (N = 247) |
| Liczba przypadków Rybozylib – 61, placebo – 80 Stosunek ryzyka 0,699 95 % przedział ufności [0,501; 0,976] Mediana Kaplana-Meiera Rybozylib – NV Placebo – 40,7 miesiąca |
Czas (miesiące)
Liczba pacjentów nadal narażonych na ryzyko
Czas (miesiące)
Ribocyklib
Placebo
Rys. 7. MONALEESA-7: wykres Kaplana-Meiera dla ostatecznej analizy OS u pacjentek otrzymujących terapię endokrynną pierwszego rzutu (data zakończenia zbierania danych: 30 listopada 2018 r.)
Stosunek ryzyka oparty na niestratyfikowanym modelu Coxa.
Ponadto prawdopodobieństwo progresji podczas leczenia kolejnej linii lub śmierci (PFS2) u pacjentek, które wcześniej otrzymywały ribocyklib w badaniu, było niższe w porównaniu z pacjentkami w grupie placebo, z HR wynoszącym 0,692 (95 % CI: 0,548; 0,875) w populacji ogólnej badania. Mediana PFS2 wyniosła 32,3 miesiąca (95 % CI: 27,6; 38,3) w grupie placebo i nie została osiągnięta (95 % CI: 39,4; NR) w grupie ribocyklibu. Podobne wyniki zaobserwowano w podgrupie terapii endokrynnej pierwszego rzutu, przy czym HR wynosiło 0,660 (95 % CI: 0,503; 0,868), a mediana PFS2 wyniosła 32,3 miesiąca (95 % CI: 26,9; 38,3) w grupie placebo w porównaniu z nieosiągniętą (95 % CI: 39,4; NR) w grupie ribocyklibu.
Badanie CLEE011F2301 (MONALEESA-3)
Lek Kiskali oceniano w randomizowanym, podwójnie ślepy, kontrolowanym placebo, wieloośrodkowym badaniu klinicznym fazy III z udziałem 726 kobiet w okresie postmenopauzalnym z pozytywnym pod względem receptora hormonu, HER2-negatywnym zaawansowanym rakiem piersi, które wcześniej nie otrzymywały lub otrzymały tylko jeden cykl poprzedniej terapii endokrynnej w połączeniu z fulwestrantem w porównaniu z fulwestrantem samym.
Średni wiek pacjentek włączonych do tego badania wynosił 63 lata (zakres od 31 do 89). 46,7 % pacjentek miało co najmniej 65 lat, w tym 13,8 % pacjentek w wieku co najmniej 75 lat. Pacjenci włączeni do badania byli reprezentantami rasy europejskiej (85,3 %), mongolskiej (8,7 %) i negroidalnej (0,7 %), a niemal wszystkie pacjentki (99,7 %) miały stan funkcjonalny według skali ECOG 0 lub 1. Do badania włączono pacjentki leczone w pierwszej i drugiej linii (w tym 19,1 % z przerzutami de novo). Przed włączeniem do badania 42,7 % pacjentek otrzymywało chemioterapię w trybie adiuwantnym, a 13,1 % – w trybie neoadiuwantnym, podczas gdy 58,5 % pacjentek otrzymywało terapię endokrynną w trybie adiuwantnym, 1,4 % – w trybie neoadiuwantnym, a 21 % otrzymało poprzednią terapię endokrynną z powodu zaawansowanego raka piersi. W badaniu F2301 u 21,2 % pacjentek stwierdzono przerzuty wyłącznie do kości, a u 60,5 % pacjentek stwierdzono przerzuty do narządów wewnętrznych.
Analiza pierwotna
Pierwotny punkt końcowy w badaniu został osiągnięty w analizie pierwotnej przeprowadzonej po ocenie 361 przypadków przeżycia bez progresji (PFS) na podstawie oceny badacza z wykorzystaniem kryteriów RECIST w wersji 1.1, w populacji ogólnej (wszystkie pacjentki zrandomizowane, data zakończenia zbierania danych: 03.11.2017). Średni czas dalszej obserwacji w momencie pierwotnej analizy PFS wyniósł 20,4 miesiąca.
Wyniki oceny pierwotnej skuteczności wykazały statystycznie istotne poprawy PFS u pacjentek otrzymujących lek Kiskali i fulwestrant w porównaniu z pacjentkami otrzymującymi placebo i fulwestrant w populacji ogólnej (stosunek ryzyka 0,593; 95 % CI: 0,480; 0,732; wartość p dla jednostronnego, stratyfikowanego testu log-rank 4,1 × 10–7), co odpowiada oszacowanemu zmniejszeniu względnego ryzyka progresji lub śmierci o 41 % na korzyść grupy leczonej lekiem Kiskali i fulwestrantem.
Wyniki PFS na podstawie niezależnej, centralnej oceny radiologicznej zamaskowanych danych przypadkowo wybranej podgrupy około 40 % pacjentek zrandomizowanych potwierdziły pierwotne wyniki dotyczące skuteczności na podstawie oceny badacza (stosunek ryzyka 0,492; 95 % CI: 0,345; 0,703).
Opisowe aktualizacje PFS przeprowadzono w momencie drugiej pośredniej analizy OS; zaktualizowane wyniki PFS dla populacji ogólnej oraz podgrup na podstawie poprzedniej terapii endokrynnej podsumowano w tabeli 8, a krzywą Kaplana-Meiera przedstawiono na rysunku 8.
| Tabela 8. MONALEESA-3 (F2301): zaktualizowane wyniki (PFS) na podstawie oceny badacza (data zakończenia zbierania danych – 03.06.2019) |
||
| Wskaźniki |
Kiskali i fulvestrant N = 484 |
Placebo i fulvestrant N = 242 |
| Przeżycie bez postępu choroby, populacja ogólna badania |
||
| Liczba przypadków, n [%] |
283 (58,5) |
193 (79,8) |
| Mediana PFS [miesiące] (95 % CI) |
20,6 (18,6; 24,0) |
12,8 (10,9; 16,3) |
| Stosunek ryzyka (95 % CI) |
0,587 (0,488; 0,705) |
|
| Podgrupa leczenia w pierwszej linii a |
Kiskali i fulvestrant n = 237 |
Placebo i fulvestrant n = 128 |
| Liczba przypadków, n [%] |
112 (47,3) |
95 (74,2) |
| Mediana PFS [miesięcy] (95 % CI) |
33,6 (27,1; 41,3) |
19,2 (14,9; 23,6) |
| Stosunek ryzyka (95 % CI) |
0,546 (0,415–0,718) |
|
| Podgrupa leczenia w drugiej linii lub wczesnego nawrotu b |
Kiskali i fulvestrant n = 237 |
Placebo i fulvestrant n = 109 |
| Liczba przypadków, n [%] |
167 (70,5) |
95 (87,2) |
| Mediana PFS [miesięcy] (95 % CI) |
14,6 (12,5; 18,6) |
9,1 (5,8; 11,0) |
| Stosunek ryzyka (95 % CI) |
0,571 (0,443; 0,737) |
|
| CI – przedział ufności. a Pacjenci z nowotworem przerzutowym piersi de novo bez wcześniejszej terapii hormonalnej oraz pacjenci z nawrotem po zakończeniu 12-miesięcznej terapii (neo)adjuwantowej. b Pacjenci z nawrotem podczas terapii adjuwantowej lub w ciągu 12 miesięcy po zakończeniu terapii (neo)adjuwantowej oraz pacjenci z postępem choroby po jednej linii terapii hormonalnej zaawansowanego schorzenia. |
||
| Liczba zdarzeń Rybozycylib + fulwestrant: 283, placebo + fulwestrant: 193 Stosunek ryzyka = 0,587 95% CI [0,488, 0,705] Mediana Kaplana-Meiera Rybozycylib + fulwestrant: 20,6 mies. Placebo + fulwestrant: 12,8 mies. |
| Momenty cenzurowania Rybozycylib + fulwestrant (N = 484) Placebo + fulwestrant (N = 242) |
| 100 |
| 80 |
| 60 |
| 40 |
| 20 |
| 0 |
| 0 |
| 2 |
| 4 |
| 6 |
| 8 |
| 10 |
| 12 |
| 14 |
| 16 |
| 18 |
| 20 |
| 22 |
| 24 |
| 26 |
| 28 |
| 30 |
| 32 |
| 34 |
| 36 |
| 38 |
| 40 |
| 42 |
| 44 |
| 46 |
| Czas (miesiące) |
| Placebo |
| Ribociklib |
| 242 |
| 484 |
| 168 |
| 156 |
| 144 |
| 364 |
| 346 |
| 323 |
| 106 |
| 98 |
| 88 |
| 258 |
| 239 |
| 225 |
| 68 |
| 62 |
| 198 |
| 181 |
| 47 |
| 156 |
| 41 |
| 127 |
| 13 |
| 65 |
| 2 |
| 11 |
| 0 |
| 0 |
| 1 |
| 4 |
| 6 |
| 29 |
| 21 |
| 92 |
| 45 |
| 149 |
| 59 |
| 51 |
| 174 |
| 159 |
| 82 |
| 205 |
| 134 |
| 116 |
| 305 |
| 282 |
| 195 |
| 403 |
Rys. 8. MONALEESA-3 (F2301): wykres Kaplana-Meiera dla PFS według oceny badacza (niezależna komisja ds. oceny skuteczności) (data zakończenia zbierania danych – 03.06.2019)
Wyniki oceny skuteczności dotyczące wskaźnika odpowiedzi całkowitej (ORR) oraz wskaźnika korzyści klinicznej (CBR) według oceny badacza na podstawie kryteriów RECIST w wersji 1.1 przedstawiono w tabeli 9.
| Tabela 9. MONALEESA-3: wyniki oceny skuteczności (ORR, CBR) na podstawie oceny badacza (data zakończenia zbierania danych – 03.11.2017) |
||
| Analiza |
Kiskali i fulvestrant (%, 95 % CI) |
Placebo i fulvestrant (%, 95 % CI) |
| Cała populacja do analizy |
N = 484 |
N = 242 |
| Częstość odpowiedzi ogólnej (ORR)a |
32,4 (28,3; 36,6) |
21,5 (16,3; 26,7) |
| Częstość korzyści klinicznej (CBR)b |
70,2 (66,2; 74,3) |
62,8 (56,7; 68,9) |
| Pacjenci z mierzalną chorobą |
n = 379 |
n = 181 |
| Częstość odpowiedzi ogólneja |
40,9 (35,9; 45,8) |
28,7 (22,1; 35,3) |
| Częstość korzyści klinicznego b |
69,4 (64,8; 74,0) |
59,7 (52,5; 66,8) |
| a ORR: odsetek pacjentów z odpowiedzią pełną + częściową. b CBR: odsetek pacjentów z odpowiedzią pełną + częściową + (choroba stabilna lub brak pełnej odpowiedzi/brak postępu choroby przez ≥ 24 tygodnie). |
||
Stosunek ryzyka na podstawie analizy wcześniej zdefiniowanych podgrup pacjentów otrzymujących leczenie lekiem Kiskali i fulvestrantem wykazał stabilną skuteczność w różnych podgrupach, w tym pod względem wieku, przynależności rasowej, wcześniejszej terapii (na wczesnym etapie lub zaawansowanego raka), wcześniejszej terapii adjuwantowej/neoadjuwantowej za pomocą chemioterapii lub terapii hormonalnej, zaangażowania wątroby i/lub płuc oraz przerzutów wyłącznie do kości.
Analiza OS
W drugiej analizie OS osiągnięto wtórną punktową końcową badania, wykazując istotne statystycznie poprawy OS.
Wyniki końcowej analizy OS w ogólnej populacji badawczej oraz analizy podgrup przedstawiono w tabeli 10 i na rysunku 9.
| Tabela 10. MONALEESA-3 (F2301): wyniki skuteczności (PFS) (data zakończenia zbierania danych – 03.06.2019) |
||
| Wskaźniki |
Kiskali i fulvestrant |
Placebo i fulvestrant |
| Cała populacja badawcza |
N = 484 |
N = 242 |
| Liczba przypadków, n [%] |
167 (34,5) |
108 (44,6) |
| Mediana PFS [miesiące] (95 % CI) |
NR, (NR, NR) |
40 (37, NR) |
| Stosunek ryzyka (95 % CI)a |
0,724 (0,568; 0,924) |
|
| Wartość p b |
0,00455 |
|
| Podgrupa leczenia w I linii |
n = 237 |
n = 128 |
| Liczba przypadków, n [%] |
63 (26,6) |
47 (36,7) |
| Stosunek ryzyka (95 % CI)c |
0,700 (0,479; 1,021) |
|
| Podgrupa leczenia w II linii lub wczesna nawroty |
n = 237 |
n = 109 |
| Liczba przypadków, n [%] |
102 (43,0) |
60 (55,0) |
| Stosunek ryzyka (95 % CI)c |
0,730 (0,530; 1,004) |
|
| NR – nie oszacowano. a Stosunek ryzyka uzyskany z modelu Coxa PH, wyodrębniony według obecności przerzutów w płucach i/lub wątrobie oraz poprzedniej terapii endokrynnej. b Jednostronna wartość p uzyskana z testu log-rank, wyodrębniona według obecności przerzutów w płucach i/lub wątrobie oraz poprzedniej terapii endokrynnej zgodnie z IRT. Wartość p jest jednostronna i porównywana z wartością progową 0,01129, określoną na podstawie funkcji zależności błędów I rodzaju od uzyskanej frakcji niezbędnych informacji Lan-DeMetsa (O’Bryana-Fleminga) dla ogólnego poziomu istotności 0,025. c Stosunek ryzyka uzyskany z niestratyfikowanego modelu Coxa PH. |
||
| Liczba zdarzeń Rybozyclib + fulwestrant: 167; placebo + fulwestrant: 108 Stosunek ryzyka = 0,724 95% przedział ufności [0,568; 0,924] Mediana Kaplana-Meiera Rybozyclib + fulwestrant: brak danych Placebo + fulwestrant: 40,0 mies. Wartość p testu log-rank = 0,00455 |
| Momenty cenzurowania Rybozcyklib + fulwestrant (N = 484) Placbo + fulwestrant (N = 242) |
| 242 |
| 233 |
| 227 |
| 223 |
| 218 |
| 213 |
| 207 |
| 199 |
| 194 |
| 187 |
| 184 |
| 174 |
| 169 |
| 159 |
| 155 |
| 147 |
| 141 |
| 134 |
| 107 |
| 64 |
| 37 |
| 14 |
| 3 |
| 0 |
| 100 |
| 80 |
| 60 |
| 40 |
| 20 |
| 0 |
| 0 |
| 2 |
| 4 |
| 6 |
| 8 |
| 10 |
| 12 |
| 14 |
| 16 |
| 18 |
| 20 |
| 22 |
| 24 |
| 26 |
| 28 |
| 30 |
| 32 |
| 34 |
| 36 |
| 38 |
| 40 |
| 42 |
| 44 |
| 46 |
| 48 |
| 484 |
| 470 |
| 454 |
| 444 |
| 436 |
| 428 |
| 414 |
| 402 |
| 397 |
| 389 |
| 374 |
| 365 |
| 348 |
| 334 |
| 326 |
| 309 |
| Czas (miesiące) |
| Placebo |
| Ribociclib |
| 300 |
| 287 |
| 237 |
| 159 |
| 92 |
| 41 |
| 14 |
| 2 |
| 0 |
| 0 |
Rys. 9. MONALEESA-3 (F2301): wykres Kaplana-Meiera dla PFS (zestaw danych do pełnej analizy [ZDPA]) (data zakończenia zbierania danych – 03.06.2019)
Test log-rank i model Coxa zostały ustratyfikowane według obecności przerzutów w płucach i/lub wątrobie, wcześniejszej chemioterapii w chorobie uogólnionej oraz składnika kombinacji endokrynnej według IRT.
Czas do progresji podczas terapii kolejnej linii lub śmierci (PFS2) u pacjentów w grupie Kiskali był dłuższy niż u pacjentów w grupie placebo (HR (szansa ryzyka): 0,670 [95 % CI: 0,542; 0,830]) w populacji ogólnej badania. Mediana PFS2 wyniosła 39,8 miesiąca (95 % CI: 32,5; ND) dla grupy Kiskali oraz 29,4 miesiąca (95 % CI: 24,1; 33,1) dla grupy placebo.
Pacjenci starsi
Wśród wszystkich pacjentów otrzymujących lek Kiskali w badaniach MONALEESA-2 i MONALEESA-3, do tej grupy wiekowej należeli pacjenci w wieku ≥ 65 lat i ≥ 75 lat. Nie zaobserwowano ogólnych różnic w zakresie bezpieczeństwa lub skuteczności stosowania leku Kiskali pomiędzy tymi pacjentami a pacjentami młodszymi (patrz sekcja „Sposób stosowania i dawki”).
Pacjenci z niewydolnością nerek
W trzech badaniach podstawowych (MONALEESA-2, MONALEESA-3 i MONALEESA-7) leczenie rybocyklibem otrzymało 510 (53,8 %) pacjentów z normalną funkcją nerek, 341 (36 %) pacjentów z łagodną niewydolnością nerek oraz 97 (10,2 %) pacjentów z umiarkowaną niewydolnością nerek. Żaden pacjent z ciężką niewydolnością nerek nie został włączony do badań. Dane dotyczące PFS były podobne u pacjentów z łagodną i umiarkowaną niewydolnością nerek otrzymujących rybocyklib w dawce początkowej 600 mg w porównaniu z pacjentami z normalną funkcją nerek. Profil bezpieczeństwa u pacjentów z zaburzeniami funkcji nerek był ogólnie porównywalny z takim u pacjentów bez zaburzeń funkcji nerek (patrz sekcja „Reakcje niepożądane”).
Dzieci
Europejska Agencja Leków odroczyła obowiązek przedłożenia wyników badań leku Kiskali w odniesieniu do wszystkich podgrup dziecięcych w raku piersi (patrz informacje dotyczące stosowania u dzieci w sekcji „Sposób stosowania i dawki”).
Farmakokinetyka
Farmakokinetyka rybocyklibu została zbadana u pacjentów z chorobą uogólnioną po doustnym podaniu leku w dawce dobowej od 50 do 1200 mg. Zdrowi wolontariusze otrzymywali doustnie dawki jednorazowe w zakresie od 400 mg do 600 mg lub dawki powtarzane po 400 mg dziennie (8 dni).
Wchłanianie
Średnia geometryczna wartość całkowitej biodostępności rybocyklibu po jednorazowym doustnym podaniu dawki 600 mg wyniosła 65,8 % u zdrowych wolontariuszy.
Czas osiągnięcia Cmax (Tmax) po doustnym podaniu rybocyklibu wynosił 1–4 godziny. Obserwowano nieznaczne nadproporcjonalne zwiększenie ekspozycji (Cmax i AUC) rybocyklibu w badanym zakresie dawek (od 50 do 1200 mg). Po wielokrotnym podawaniu 1 raz dziennie stan równowagi był osiągany zazwyczaj po 8 dniach, a kumulacja rybocyklibu zachodziła ze średnią geometryczną wartością współczynnika kumulacji wynoszącą 2,51 (zakres: od 0,97 do 6,40).
Wpływ posiłku
W porównaniu z podawaniem na czczo, doustne podanie tabletek rybocyklibu w jednorazowej dawce 600 mg z posiłkiem o wysokiej zawartości kalorii i tłuszczu nie wpływało na szybkość i stopień wchłaniania rybocyklibu.
Rozkład
Wiązanie rybocyklibu z białkami osocza krwi człowieka in vitro wynosiło około 70 % i nie zależało od stężenia leku (od 10 do 10 000 ng/ml). Rybocyklib równomiernie rozkładał się między erytrocytami a osoczem krwi ze średnią wartością stosunku krew/osocze in vivo wynoszącą 1,04. Według danych analizy farmakokinetycznej populacyjnej pozorny objętość rozkładu w stanie równowagi (Vss/F) wynosiła 1090 l.
Biotransformacja
Badania in vitro i in vivo wykazały, że u człowieka rybocyklib jest metabolizowany głównie w wątrobie, głównie za udziałem CYP3A4. Po doustnym podaniu [14C] rybocyklibu w jednorazowej dawce 600 mg u człowieka głównymi drogami metabolizmu rybocyklibu były oksydacja (dealkilacja, oksygenacja C i/lub N, oksydacja (-2H)) oraz ich kombinacje. Koniugaty II fazy metabolitów I fazy rybocyklibu poddawane były N-acetylacji, sulfatacji, koniugacji z cysteiną, glikozylozylacji i glukuronidacji. Rybocyklib był główną krążącą pochodną substancji leczniczej. Głównymi krążącymi metabolitami były metabolit M13 (CCI284, N-hydroksylacja), M4 (LEQ803, N-demetylacja) i M1 (wtórny glukuronid). Aktywność kliniczna (właściwości farmakologiczne i bezpieczeństwo) rybocyklibu była głównie uzależniona od substancji czynnej i tylko nieznacznie od krążących metabolitów.
Rybocyklib był intensywnie metabolizowany, przy czym ilość niezmienionego leku w kale i moczu wynosiła odpowiednio 17,3 % i 12,1 % dawki. Metabolit LEQ803 występował w dużych ilościach w stolcu i stanowił około 13,9 % i 3,74 % podanej dawki w kale i moczu odpowiednio. Wykryto wiele innych metabolitów w kale i moczu w niewielkich ilościach (≤ 2,78 % podanej dawki).
Wydalanie
W stanie równowagi przy podawaniu dawki 600 mg pacjentom z chorobą uogólnioną średnia geometryczna wartość efektywnego okresu półtrwania w osoczu (na podstawie współczynnika kumulacji) wynosiła 32,0 godziny (63 % CV), a średnia geometryczna wartość pozornego klirensu (CL/F) po podaniu doustnym wynosiła 25,5 l/h (66 % CV). Na podstawie danych analizy farmakokinetycznej populacyjnej uważa się, że ekspozycja na rybocyklib u pacjentek z rakiem piersi w stadium wczesnym będzie nieco niższa niż u pacjentek z rakiem piersi uogólnionym otrzymujących tę samą dawkę. Średnia geometryczna wartość pozornego końcowego okresu półtrwania w osoczu (T1/2) rybocyklibu wynosiła od 29,7 do 54,7 godziny, a średnia geometryczna wartość CL/F rybocyklibu wynosiła od 39,9 do 77,5 l/h przy podawaniu dawki 600 mg zdrowym wolontariuszom we wszystkich badaniach.
Rybocyklib i jego metabolity są wydalane głównie przez przewód pokarmowy, a jedynie niewielka ilość – przez nerki. U 6 zdrowych mężczyzn po doustnym podaniu jednorazowej dawki [14C] rybocyklibu 91,7 % całkowitej podanej dawki promieniotwórczej zostało wydalone w ciągu 22 dni; główną drogą wydalania był stolec (69,1 %), a 22,6 % dawki zostało wydalone z moczem.
Liniowość/nieliniowość
Obserwowano nieznaczne nadproporcjonalne zwiększenie ekspozycji (Cmax i AUC) rybocyklibu w badanym zakresie dawek (od 50 do 1200 mg) po podaniu zarówno jednorazowych, jak i powtarzanych dawek. Ten analiz został ograniczony małą liczbą próby dla większości kohort otrzymujących określone dawki; największa ilość danych została uzyskana w kohorcie otrzymującej dawkę 600 mg.
Specjalne grupy pacjentów
Zaburzenia funkcji nerek
Wpływ zaburzeń funkcji nerek na farmakokinetykę rybocyklibu oceniano w badaniu obejmującym 14 zdrowych wolontariuszy z normalną funkcją nerek (absolutna szybkość filtracji kłębuszkowej [aSKF] ≥ 90 ml/min), 8 osób z łagodną niewydolnością nerek (aSKF od 60 do < 90 ml/min), 6 osób z umiarkowaną niewydolnością nerek (aSKF od 30 do < 60 ml/min), 7 osób z ciężką niewydolnością nerek (aSKF od 15 do < 30 ml/min) oraz 3 osoby z końcowym stadium niewydolności nerek (aSKF < 15 ml/min), którym podawano rybocyklib jednorazowo w dawce 400 mg.
AUCinf zwiększała się odpowiednio 1,6, 1,9 i 2,7 razy, a Cmax – 1,8, 1,8 i 2,3 razy u osób z łagodną, umiarkowaną i ciężką niewydolnością nerek w porównaniu z ekspozycją u osób z normalną funkcją nerek. Ponieważ badania skuteczności i bezpieczeństwa rybocyklibu obejmowały dużą część pacjentów z łagodną niewydolnością nerek (patrz sekcja „Farmakodynamika”), dane dotyczące pacjentów z umiarkowaną lub ciężką niewydolnością nerek w badaniu z zaburzeniami funkcji nerek porównywano również z połączonymi danymi dotyczącymi osób z normalną funkcją nerek i łagodną niewydolnością nerek. W porównaniu z połączonymi danymi dotyczącymi osób z normalną funkcją nerek i łagodną niewydolnością nerek, AUCinf zwiększała się 1,6 i 2,2 razy, a Cmax – 1,5 i 1,9 razy u osób z umiarkowaną i ciężką niewydolnością nerek odpowiednio. Wielokrotna różnica dla osób z końcowym stadium niewydolności nerek nie została obliczona z powodu niewielkiej liczby takich osób, ale wyniki wskazują na podobne lub nieco większe zwiększenie ekspozycji na rybocyklib w porównaniu z osobami z ciężką niewydolnością nerek.
Wpływ zaburzeń funkcji nerek na farmakokinetykę rybocyklibu oceniano również u chorych na raka piersi miejscowo zaawansowanego lub przerzutowego, włączonych do badań skuteczności i bezpieczeństwa, gdzie pacjenci otrzymywali dawkę początkową 600 mg (patrz sekcja „Farmakodynamika”). Analiza danych farmakokinetycznych w podgrupach pacjentów z rakiem piersi miejscowo zaawansowanym lub przerzutowym po doustnym przyjęciu 600 mg rybocyklibu w formie dawki jednorazowej lub powtarzanych dawek wykazała, że AUCinf i Cmax rybocyklibu u pacjentów z łagodną (n = 57) lub umiarkowaną (n = 14) niewydolnością nerek były porównywalne z takimi u pacjentów z normalną funkcją nerek (n = 86), co wskazuje na brak klinicznie istotnego wpływu łagodnej lub umiarkowanej niewydolności nerek na ekspozycję na rybocyklib.
Zaburzenia funkcji wątroby
Na podstawie danych z badania farmakokinetycznego u osób bez nowotworu, zaburzenia funkcji wątroby w stopniu łagodnym nie wpływały na ekspozycję na rybocyklib (patrz sekcja „Sposób stosowania i dawki”). Średnia ekspozycja na rybocyklib u pacjentów z zaburzeniami funkcji wątroby w stopniu umiarkowanym (stosunek średnich geometrycznych [SSG]: 1,44 dla Cmax; 1,28 dla AUCinf) i ciężkim (SSG: 1,32 dla Cmax; 1,29 dla AUCinf) była zwiększona mniej niż 2 razy (patrz sekcja „Sposób stosowania i dawki”).
Dane analizy farmakokinetycznej populacyjnej obejmującej 160 pacjentów z rakiem piersi miejscowo zaawansowanym lub przerzutowym i normalną funkcją wątroby oraz 47 pacjentów z łagodnym zaburzeniem funkcji wątroby również wykazały, że łagodne zaburzenia funkcji wątroby nie wpływały na ekspozycję na rybocyklib. Nie badano stosowania rybocyklibu u pacjentek z rakiem piersi z umiarkowanym lub ciężkim zaburzeniem funkcji wątroby.
Wpływ wieku, masy ciała, płci i pochodzenia rasowego pacjenta
Analiza farmakokinetyczna populacyjna wykazała, że wiek, masa ciała i płeć pacjenta nie miały klinicznie istotnego wpływu na ogólnoustrojową ekspozycję na rybocyklib, który mógłby wymagać korekty dawki. Dane dotyczące różnic farmakokinetyki związanej z pochodzeniem rasowym są zbyt ograniczone, aby wyciągnąć wnioski.
Dane interakcji in vitro
Wpływ rybocyklibu na enzymy cytochromu P450
W warunkach in vitro rybocyklib jest odwracalnym inhibitorem CYP1A2, CYP2E1 i CYP3A4/5 oraz inhibitorem zależnym od czasu CYP3A4/5 w klinicznie istotnych stężeniach. Badania in vitro wykazały, że rybocyklib nie hamuje aktywności CYP2A6, CYP2B6, CYP2C8, CYP2C9, CYP2C19 i CYP2D6 w klinicznie istotnych stężeniach. Rybocyklib nie ma potencjału do hamowania zależnego od czasu CYP1A2, CYP2C9 i CYP2D6.
Dane in vitro wskazują, że rybocyklib nie ma potencjału do indukcji enzymów UGT ani enzymów CYP, takich jak CYP2C9, CYP2C19 i CYP3A4, poprzez receptor PXR (pregnan-X). W związku z tym mało prawdopodobne jest, że lek Kiskali będzie wpływać na substraty tych enzymów. Dane in vitro są niewystarczające, aby wykluczyć potencjał rybocyklibu do indukcji CYP2B6 poprzez receptor CAR (constitutive androstane receptor).
Wpływ transporterów na rybocyklib
W warunkach in vitro rybocyklib jest substratem P-gp, ale na podstawie danych bilansu masy mało prawdopodobne jest, że hamowanie P-gp lub BCRP będzie wpływać na ekspozycję na rybocyklib w dawkach terapeutycznych. Rybocyklib nie jest substratem transporterów wychwytu wątrobowego OATP1B1, OATP1B3 ani OCT-1 w warunkach in vitro.
Wpływ rybocyklibu na transportery
Badania in vitro wykazały, że rybocyklib ma potencjał do hamowania aktywności transporterów leków P-gp, BCRP, OATP1B1/1B3, OCT1, OCT2, MATE1 i BSEP. Rybocyklib nie hamował OAT1, OAT3 ani MRP2 w klinicznie istotnych stężeniach w warunkach in vitro.
Dane przedkliniczne dotyczące bezpieczeństwa
Bezpieczeństwo farmakologiczne
Badania kardiologicznej bezpieczeństwa in vivo na psach wykazały zależne od dawki i stężenia wydłużenie interwału QTc przy ekspozycji osiągalnej u pacjentów po podaniu zalecanej dawki 600 mg. Ponadto przy podwyższonych poziomach ekspozycji (około 5 razy wyższych niż oczekiwane kliniczne Cmax) istnieje możliwość indukcji przedwczesnych skurczów komorowych.
Toxiczność powtarzanych dawek
W badaniach toksyczności powtarzanych dawek (schemat leczenia: 3 tygodnie leczenia / 1 tydzień przerwy) trwających do 27 tygodni u szczurów i do 39 tygodni u psów stwierdzono, że głównym organem, na który rybocyklib działa toksycznie, jest układ wątrobowo-żółciowy (zmiany proliferacyjne, cholestaza, podobne do piasku konkrementy w pęcherzu żółciowym i gęsta żółć). Do organów docelowych związanych z działaniem farmakologicznym rybocyklibu w badaniach zastosowania powtarzanych dawek należą szpik kostny (hipocelularność), układ limfatyczny (wyczerpanie tkanki limfatycznej), błona śluzowa jelita (atropia), skóra (atropia), kości (zmniejszenie formowania tkanki kostnej), nerki (jednoczesna degeneracja i regeneracja nabłonka kanalików nerkowych) oraz jądra (atropia). Oprócz zmian atroficznych obserwowanych w jądrach, które miały tendencję do odwracalności, wszystkie inne zmiany były całkowicie odwracalne po 4-tygodniowym okresie bez leczenia. Ekspozycja na rybocyklib u zwierząt w badaniach toksyczności była zazwyczaj niższa lub porównywalna z ekspozycją u pacjentów otrzymujących wielokrotne dawki 600 mg/dobę przy raku piersi miejscowo zaawansowanym lub przerzutowym (na podstawie AUC).
Toxyczność reprodukcyjna / wpływ na płodność
Rybocyklib wykazywał fetotoksyczność i teratogenność u szczurów i królików w dawkach, które nie wywierały toksycznego wpływu na ciężarną samicę. Po prenatalnej ekspozycji u szczurów obserwowano zwiększenie przypadków poimplantacyjnej śmierci płodu i zmniejszenie masy ciała płodu, a u królików rybocyklib wykazywał działanie teratogenne przy ekspozycji do 1,5 razy wyższej niż u człowieka przy stosowaniu w najwyższej zalecanej dawce 600 mg/dobę (na podstawie AUC).
U szczurów stwierdzono zmniejszenie masy ciała płodów, które towarzyszyły zmiany szkieletowe uznawane za tymczasowe i/lub związane z mniejszą masą ciała płodu. U królików obserwowano niekorzystny wpływ na rozwój embrionalny/fetalny, co wyrażało się zwiększoną częstością anomalii rozwojowych płodu (wady rozwoju i anomalie zewnętrzne, wisceralne i szkieletowe) oraz wzrostu płodu (zmniejszenie masy ciała płodów). Do tych anomalii należały zmniejszone/male płaty płuc, dodatkowa tętnica na łuku aorty i przepuklina przeponowa, brak dodatkowego płata lub (częściowo) zrośnięte płaty płuc i zmniejszone/male dodatkowe płaty płuc (30 i 60 mg/kg), dodatkowe/rudymentarne trzynaste żebra, zdeformowana kość podjęzykowa i zmniejszona liczba paliczków pierwszego palca. Nie odnotowano przypadków śmierci embrionalnej/fetalnej.
W badaniu płodności u samic szczurów rybocyklib nie wpływał na funkcję rozrodczą, płodność ani wczesne stadia embriogenezy przy jakiejkolwiek dawce do 300 mg/kg/dobę (prawdopodobnie przy ekspozycji niższej lub równej ekspozycji klinicznej u pacjentów przy najwyższej zalecanej dawce rybocyklibu 600 mg/dobę na podstawie AUC).
Rybocyklib nie był oceniany w badaniach płodności mężczyzn. Jednak w badaniach toksyczności u szczurów i psów raportowano o zmianach atroficznych w jądrach przy ekspozycjach niższych lub porównywalnych z ekspozycją u człowieka przy najwyższej zalecanej dawce dobowej 600 mg/dobę na podstawie AUC. Te efekty mogą być związane z bezpośrednim działaniem antyproliferacyjnym na komórki zarodkowe jąder, prowadzącym do atrofii kanalików nasieniowych.
Rybocyklib i jego metabolity łatwo przenikają do mleka u szczurów. Ekspozycja na rybocyklib w mleku była wyższa niż w osoczu.
Genotoksyczność
W badaniach genotoksyczności w bakteryjnych systemach in vitro oraz w systemach in vitro i in vivo u ssaków z aktywacją metaboliczną i bez niej nie stwierdzono żadnych objawów potencjału genotoksycznego rybocyklibu.
Karcinogenność
Karcinogenność rybocyklibu oceniano w 2-letnim badaniu na szczurach.
Doustne podawanie rybocyklibu przez 2 lata prowadziło do zwiększenia częstości guzów nabłonkowych endometrium, hiperplazji gruczołowej i płaskonabłonkowej macicy/szyjki macicy u samic szczurów w dawkach ≥ 300 mg/kg/dobę, a także do zwiększenia częstości guzów folikularnych tarczycy u samców szczurów w dawce 50 mg/kg/d. Średnia ekspozycja w stanie równowagi (AUC0–24 h) u samic i samców szczurów, u których obserwowano zmiany nowotworowe, była odpowiednio 1,2 i 1,4 razy wyższa niż u pacjentów przy zalecanej dawce 600 mg/d. Średnia ekspozycja w stanie równowagi (AUC0–24 h) u samic i samców szczurów, u których obserwowano zmiany nowotworowe, była odpowiednio 2,2 i 2,5 razy wyższa niż u pacjentów przy dawce 400 mg/d.
Dodatkowe nie-nowotworowe zmiany proliferacyjne obejmowały zwiększenie liczby ogniskowych zmian w wątrobie (komórki zasadochłonne i przejrzyste) oraz hiperplazję komórek międzypłytkowych jąder (komórki Leydiga) u samców szczurów w dawkach ≥ 5 mg/kg/d i 50 mg/kg/d odpowiednio.
Potencjalne mechanizmy zmian tarczycy u samców szczurów obejmują specyficzne dla gryzoni indukcję enzymów mikrosomalnych w wątrobie, które uważa się za nieistotne dla człowieka. Wpływ na macicę/szyjkę macicy i komórki międzystykowe jąder (komórki Leydiga) może być związany z długotrwałą hipoprolaktynemią wynikającą z hamowania funkcji komórek laktotropowych przysadki przez CDK4, co zmienia oś podwzgórze-przysadka-gonady.
Każde potencjalne zwiększenie stosunku estrogen/progesteron u człowieka pod wpływem tego mechanizmu jest kompensowane działaniem inhibitora terapii przeciwestrogenowej towarzyszącej syntezie estrogenów, ponieważ lek Kiskali jest wskazany u ludzi w połączeniu z lekami obniżającymi poziom estrogenów.
Biorąc pod uwagę istotne różnice między gryzoniami a człowiekiem pod względem syntezy i roli prolaktyny, oczekuje się, że ten mechanizm nie będzie miał konsekwencji dla człowieka.
Charakterystyki kliniczne
Wskazania
Rak piersi wczesny
Kiskali jest wskazany w połączeniu z inhibitorem aromatazy w leczeniu uzupełniającym kobiet z rakiem piersi wczesnym z dodatnim statusem HR (receptorów hormonów) i ujemnym statusem HER2 (czynnika wzrostu ludzkiego 2 typu) z wysokim ryzykiem nawrotu (kryteria doboru pacjentek patrz w sekcji „Właściwości farmakologiczne”).
U kobiet w okresie przedmenopauzalnym lub w okresie przejściowym do menopauzy lub u mężczyzn terapię inhibitorem aromatazy należy prowadzić w połączeniu z agonistą hormonu uwalniającego hormon uwalniający gonadotropinę (GnRH).
Rak piersi lokalnie zaawansowany lub przerzutowy
Kiskali jest wskazany w połączeniu z inhibitorem aromatazy lub fulwestrantem w leczeniu kobiet z lokalnie zaawansowanym lub przerzutowym rakiem piersi z dodatnim statusem HR i ujemnym statusem HER2 jako wstępną terapię hormonalną lub w leczeniu kobiet, które wcześniej otrzymywały terapię hormonalną.
U kobiet w okresie przedmenopauzalnym lub w okresie przejściowym do menopauzy terapię hormonalną należy prowadzić w połączeniu z agonistą GnRH.
Przeciwwskazania
Podwyższona wrażliwość na substancję czynną lub na orzechy ziemne, soję lub dowolny składnik pomocniczy produktu leczniczego.
Interakcje z innymi lekami i inne formy interakcji
Leki mogące zwiększać stężenie rybocyklibu w osoczu
Rybocyklib jest metabolizowany głównie przez CYP3A4. Dlatego leki mogące wpływać na aktywność enzymu CYP3A4 mogą również wpływać na farmakokinetykę rybocyklibu. Jednoczesne stosowanie mocnego inhibitora CYP3A4 rytonawiru (100 mg 2 razy dziennie przez 14 dni) z rybocyklibem w dawce pojedynczej 400 mg u zdrowych ochotników prowadziło do zwiększenia ekspozycji (AUCinf) i maksymalnego stężenia (Cmax) rybocyklibu odpowiednio 3,2 i 1,7 razy w porównaniu ze stosowaniem rybocyklibu w dawce pojedynczej 400 mg oddzielnie. Cmax i AUClast dla LEQ803 (główny metabolit rybocyklibu, stanowiący mniej niż 10 % ekspozycji substancji pierwotnej) zmniejszały się odpowiednio o 96 % i 98 %. Farmakokinetyczne modelowanie oparte na fizjologii (physiologically based pharmacokinetic modelling (PBPK)) przy jednoczesnym przyjmowaniu rytonawiru (100 mg 2 razy dziennie) wykazało, że Cmax i AUC0-24h rybocyklibu w stanie stacjonarnym (400 mg raz dziennie) zwiększyły się odpowiednio 1,5 i 1,8 razy.
Należy unikać jednoczesnego stosowania mocnych inhibitorów CYP3A4, takich jak np. klaritromycyna, indynawir, itrakonazol, ketokonazol, lopinawir, rytonawir, nefazodon, nelfinawir, posakonazol, sakwinawir, telaprewir, telitromycyna, werapamil i worykonazol (patrz sekcja „Szczególne ostrzeżenia i środki ostrożności postępowania”). Należy rozważyć możliwość stosowania leków alternatywnych, które są słabszymi inhibitorami CYP3A4, oraz prowadzić monitorowanie stanu pacjentów pod kątem reakcji niepożądanych (RN) związanych z rybocyklibem (patrz sekcje „Sposób stosowania i dawki”, „Szczególne ostrzeżenia i środki ostrożności postępowania” i „Farmakokinetyka”).
Jeśli nie można uniknąć jednoczesnego stosowania produktu Kiskali z mocnym inhibitorem CYP3A4, należy zmienić dawkę produktu Kiskali, jak opisano w sekcji „Sposób stosowania i dawki”. Dane kliniczne dotyczące takiej korekty dawki jednak brakują. Z uwagi na międzyindywidualną zmienność zalecana korekta dawki może nie być optymalna dla wszystkich pacjentów, dlatego zaleca się staranne monitorowanie pod kątem RN związanych z rybocyklibem. W przypadku wystąpienia efektów toksycznych rybocyklibu należy skorygować dawkę leku lub tymczasowo przerwać jego stosowanie do ustąpienia objawów toksyczności (patrz sekcje „Sposób stosowania i dawki” i „Farmakokinetyka”). W przypadku przerwania stosowania mocnego inhibitora CYP3A4 co najmniej po upływie 5 okresów półtrwania jego wydalania (patrz instrukcja do stosowania medycznego inhibitora CYP3A4 w razie pytań) stosowanie produktu Kiskali należy wznowić w tej samej dawce, co przed rozpoczęciem stosowania mocnego inhibitora CYP3A4.
PBPK wykazało, że przy stosowaniu rybocyklibu w dawce 600 mg umiarkowany inhibitor CYP3A4 (erytromycyna) może zwiększać Cmax i AUC rybocyklibu w stanie stacjonarnym odpowiednio 1,1 i 1,1 razy. PBPK wykazało, że przy stosowaniu rybocyklibu w dawce 400 mg umiarkowany inhibitor CYP3A4 może zwiększać Cmax i AUC rybocyklibu w stanie stacjonarnym odpowiednio 1,1 i 1,2 razy. Przewiduje się, że przy stosowaniu rybocyklibu w dawce 200 mg 1 raz dziennie obserwowane będzie zwiększenie Cmax i AUC odpowiednio 1,3 i 1,5 razy w stanie stacjonarnym. Na początku leczenia słabymi lub umiarkowanymi inhibitorami CYP3A4 korekta dawki rybocyklibu nie jest wymagana. Jednak zaleca się prowadzenie monitorowania RN związanych z rybocyklibem.
Pacjentów należy poinformować o konieczności unikania spożycia grejpfrutów i soku grejpfrutowego. Wiadomo, że te produkty hamują enzymy cytochromu CYP3A4 i mogą zwiększać ekspozycję na rybocyklib.
Leki mogące obniżać stężenie rybocyklibu w osoczu
Jednoczesne stosowanie mocnego induktora CYP3A4 ryfampicyny (600 mg dziennie przez 14 dni) z rybocyklibem w dawce pojedynczej 600 mg u zdrowych ochotników obniżało AUCinf i Cmax rybocyklibu odpowiednio o 89 % i 81 % w porównaniu ze stosowaniem rybocyklibu w dawce pojedynczej 600 mg oddzielnie. Cmax LEQ803 zwiększało się 1,7 razy, a AUCinf zmniejszało się o 27 % odpowiednio. W związku z tym jednoczesne stosowanie mocnych induktorów CYP3A4 może prowadzić do obniżenia ekspozycji i, jako konsekwencja, do ryzyka utraty skuteczności. Należy unikać jednoczesnego stosowania mocnych induktorów CYP3A4, takich jak np. fenytoina, ryfampicyna, karbamazepina i dziurawiec (Hypericum perforatum). Należy rozważyć możliwość jednoczesnego stosowania leku alternatywnego, który nie indukuje lub ma minimalną zdolność indukowania CYP3A4.
Wpływ umiarkowanych induktorów CYP3A4 na ekspozycję rybocyklibu nie był badany. PBPK wykazało, że umiarkowany induktor CYP3A4 (efawirenz) może obniżać Cmax i AUC rybocyklibu w stanie stacjonarnym odpowiednio o 55 % i 74 %, a przy stosowaniu rybocyklibu w dawce 600 mg – odpowiednio o 52 % i 71 %. Jednoczesne stosowanie umiarkowanych induktorów CYP3A4 może zatem prowadzić do obniżenia ekspozycji i, jako konsekwencja, do ryzyka zmniejszenia skuteczności, szczególnie u pacjentów otrzymujących rybocyklib w dawce 400 mg lub 200 mg 1 raz dziennie.
Leki, na stężenie których w osoczu może wpływać Kiskali
Rybocyklib jest umiarkowanym/mocnym inhibitorem CYP3A4 i może oddziaływać z substancjami leczniczymi metabolizowanymi za pośrednictwem CYP3A4, co może prowadzić do zwiększenia stężenia w surowicy krwi współstosowanego leku.
Jednoczesne stosowanie midazolamu (substrat CYP3A4) z wielokrotnymi dawkami produktu Kiskali (400 mg) u zdrowych ochotników zwiększało ekspozycję na midazolam o 280 % (3,8 razy) w porównaniu ze stosowaniem midazolamu oddzielnie. Modelowanie z wykorzystaniem PBPK wykazało, że przy stosowaniu produktu Kiskali w dawce 600 mg oczekuje się zwiększenia AUC midazolamu 5,2 razy. Dlatego ogólnie przy jednoczesnym stosowaniu rybocyklibu z innymi lekami należy zapoznać się z zaleceniami dotyczącymi jednoczesnego stosowania z inhibitorami CYP3A4 w instrukcjach do stosowania medycznego tych leków. Zaleca się zachowanie ostrożności przy jednoczesnym stosowaniu z wrażliwymi substratami CYP3A4 o wąskim indeksie terapeutycznym (patrz sekcja „Szczególne ostrzeżenia i środki ostrożności postępowania”). Może być wymagane zmniejszenie dawki wrażliwych substratów CYP3A4 o wąskim indeksie terapeutycznym, takich jak np. alfentanil, cyklosporyna, ewerolimus, fentanil, sirolimus i tacrolius, ponieważ rybocyklib może zwiększać ich ekspozycję.
Należy unikać jednoczesnego stosowania rybocyklibu z następującymi substratami CYP3A4: alfuzozyna, amiodaron, cizapryda, pimozyd, chinidyna, ergotamina, dihydroergotamina, kwietyapina, lowastatyna, simwastatyna, syldenafil, midazolam, triazolam.
Jednoczesne stosowanie kofeiny (substrat CYP1A2) z wielokrotnymi dawkami produktu Kiskali (400 mg) u zdrowych ochotników zwiększało ekspozycję na kofeinę o 20 % (1,2 razy) w porównaniu ze stosowaniem kofeiny oddzielnie. Przy stosowaniu klinicznie istotnej dawki 600 mg modelowanie z wykorzystaniem modeli farmakokinetycznych opartych na fizjologii prognozowało jedynie słaby wpływ hamujący rybocyklibu na substraty CYP1A2 (zwiększenie AUC o < 2 razy).
Leki będące substratami transporterów
Badania in vitro wykazały, że rybocyklib ma potencjał do hamowania aktywności transporterów leków P-gp, BCRP, OATP1B1/1B3, OCT1, OCT2, MATE1 i BSEP. Zaleca się zachowanie ostrożności i prowadzenie monitorowania pod kątem objawów toksyczności podczas jednoczesnego leczenia wrażliwymi substratami tych transporterów, które mają wąski indeks terapeutyczny, takimi jak np. digoksyna, pitawastatyna, prawastatyna, rosuwastatyna i metformina.
Interakcja leku z pożywieniem
Produkt Kiskali można stosować niezależnie od przyjmowania pokarmu (patrz sekcje „Sposób stosowania i dawki” i „Farmakokinetyka”).
Leki zwiększające pH żołądka
Rybocyklib charakteryzuje się wysoką rozpuszczalnością przy pH 4,5 lub niższym i w środowisku biologicznym (przy pH 5,0 i 6,5). Jednoczesne stosowanie rybocyklibu z lekami zwiększającymi pH żołądka nie było oceniane w badaniach klinicznych; jednak w analizie farmakokinetycznej populacyjnej ani w analizie farmakokinetycznej niekompartementowej nie zaobserwowano zaburzeń wchłaniania rybocyklibu.
Interakcja między rybocyklibem a letrozolem
Dane badania klinicznego z udziałem pacjentek z rakiem piersi oraz analizy farmakokinetycznej populacyjnej wykazały brak interakcji między rybocyklibem a letrozolem po jednoczesnym stosowaniu tych leków.
Interakcja między rybocyklibem a anastrozolem
Dane badania klinicznego z udziałem pacjentek z rakiem piersi wykazały brak klinicznie istotnej interakcji między rybocyklibem a anastrozolem po jednoczesnym stosowaniu tych leków.
Interakcja między rybocyklibem a fulwestrantem
Dane badania klinicznego z udziałem pacjentek z rakiem piersi wykazały brak klinicznie istotnego wpływu fulwestrantu na ekspozycję rybocyklibu po jednoczesnym stosowaniu tych leków.
Interakcja między rybocyklibem a tamoksyfenem
Dane badania klinicznego z udziałem pacjentek z rakiem piersi wykazały, że po jednoczesnym stosowaniu rybocyklibu i tamoksyfenu ekspozycja tamoksyfenu zwiększała się około 2 razy.
Interakcja między rybocyklibem a doustnymi środkami antykoncepcyjnymi
Badania interakcji między rybocyklibem a doustnymi środkami antykoncepcyjnymi nie przeprowadzono (patrz sekcja „Stosowanie w okresie ciąży lub karmienia piersią”).
Przewidywane interakcje
Leki przeciwarhythmiczne i inne leki mogące wydłużać odstęp QT
Należy unikać jednoczesnego stosowania produktu Kiskali z lekami mogące wydłużać odstęp QT, takimi jak leki przeciwarhythmiczne (np. amiodaron, dysopyramida, prokaina, chinidyna i sotalol) i inne leki (np. chlorochina, halofantryna, klaritromycyna, ciprofloksacyna, lewofloksacyna, azitromycyna, haloperidol, metadon, moxifloksacyna, beprydyl, pimozyd i ondansetron do wstrzykiwania dożylnej) (patrz sekcja „Szczególne ostrzeżenia i środki ostrożności postępowania”). Produkt Kiskali nie powinien być również stosowany w połączeniu z tamoksyfenem (patrz sekcje „Wskazania”, „Szczególne ostrzeżenia i środki ostrożności postępowania” i „Farmakodynamika”).
Szczególne ostrzeżenia i środki ostrożności dotyczące stosowania
Choroba układowa zagrażająca życiu
Skuteczność i bezpieczeństwo stosowania rybocyklibu u pacjentów z chorobą układową zagrażającą życiu nie zostały ocenione.
Neutropenia
W zależności od stopnia nasilenia neutropenii leczenie lekiem Kiskali może wymagać tymczasowego wstrzymania, zmniejszenia dawki lub przerwania, zgodnie z opisem w sekcjach „Sposób stosowania i dawki” oraz „Działania niepożądane”.
Toxiczność wątrobowo-żółciowa
Przed rozpoczęciem leczenia lekiem Kiskali należy wykonać oznaczenie funkcji wątroby. Funkcję wątroby należy monitorować po rozpoczęciu leczenia (patrz sekcje „Sposób stosowania i dawki” oraz „Działania niepożądane”).
W zależności od stopnia podwyższenia poziomu aminotransferaz leczenie lekiem Kiskali może wymagać tymczasowego wstrzymania, zmniejszenia dawki lub przerwania, zgodnie z opisem w sekcjach „Sposób stosowania i dawki” oraz „Działania niepożądane”. Brak zaleceń dotyczących pacjentów z podwyższeniem poziomu aspартaminotransferazy (AST)/alaninaminotransferazy (ALT) ≥ 3 razy w stosunku do wartości wyjściowej.
<u wydłużenie interwału QT
Należy unikać stosowania leku Kiskali u pacjentów z wydłużeniem interwału QTc lub z istotnym ryzykiem jego wystąpienia. Obejmuje to pacjentów z:
- zespołem wydłużonego QT;
- niekontrolowaną lub znaczącą chorobą serca, w tym niedawnym zawałem mięśnia sercowego, przewlekłą niewydolnością serca, niestabilną dławicą piersiową i bradyarytmią;
- zaburzeniami równowagi elektrolitowej.
Należy unikać stosowania leku Kiskali z lekami, które mogą wydłużać interwał QTc, oraz/lub z silnymi inhibitorami CYP3A4, ponieważ może to prowadzić do klinicznie istotnego wydłużenia interwału QTcF (patrz sekcje „Sposób stosowania i dawki”, „Interakcje z innymi lekami i inne formy interakcji” oraz „Farmakodynamika”). Jeśli leczenie lekiem Kiskali i silnym inhibitorem CYP3A4 nie może być uniknięte, dawkę leku Kiskali należy dostosować, zgodnie z opisem w sekcji „Sposób stosowania i dawki”.
Na podstawie wyników badania E2301 (MONALEESA-7) leczenie lekiem Kiskali w połączeniu z tamoksyfenem nie jest zalecane (patrz sekcje „Działania niepożądane” oraz „Farmakodynamika”).
Rak piersi w stadium wczesnym
W badaniu O12301C (NATALEE) wydłużenie interwału QTcF > 60 ms od wartości wyjściowej obserwowano u 19 (0,8%) pacjentów otrzymujących Kiskali w połączeniu z inhibitorem aromatazy.
Przed rozpoczęciem leczenia należy wykonać EKG. Leczenie lekiem Kiskali należy rozpoczynać wyłącznie u pacjentów z wartością QTcF poniżej 450 ms. EKG należy powtórzyć około 14. dnia pierwszego cyklu, a następnie – zgodnie z wskazaniami klinicznymi (patrz sekcje „Sposób stosowania i dawki” oraz „Działania niepożądane”).
Pacjentom z rakiem piersi w stadium wczesnym należy odpowiednio monitorować poziom elektrolitów w surowicy krwi (w tym potasu, wapnia, fosforu i magnezu) przed rozpoczęciem leczenia, na początku pierwszych 6 cykli, a następnie – zgodnie z wskazaniami klinicznymi. Wszelkie odchylenia od normy należy skorygować przed rozpoczęciem leczenia lekiem Kiskali i podczas trwania leczenia.
W zależności od zaobserwowanego wydłużenia QT leczenie lekiem Kiskali może wymagać tymczasowego wstrzymania, zmniejszenia dawki lub przerwania, zgodnie z opisem w tabeli 14 (patrz sekcje „Sposób stosowania i dawki”, „Działania niepożądane” oraz „Farmakokinetyka”).
Lokalnie zaawansowany lub przerzutowy rak piersi
W badaniu E2301 (MONALEESA-7) wydłużenie interwału QTcF > 60 ms od wartości wyjściowej obserwowano u 14/87 (16,1%) pacjentów otrzymujących Kiskali w połączeniu z tamoksyfenem oraz u 18/245 (7,3%) pacjentów otrzymujących Kiskali w połączeniu z niesteroidowym inhibitorem aromatazy (NSIA).
Przed rozpoczęciem leczenia należy wykonać EKG. Leczenie lekiem Kiskali należy rozpoczynać wyłącznie u pacjentów z wartością QTcF poniżej 450 ms. EKG należy powtórzyć około 14. dnia pierwszego cyklu, a następnie – zgodnie z wskazaniami klinicznymi (patrz sekcje „Sposób stosowania i dawki” oraz „Działania niepożądane”).
Pacjentom z lokalnie zaawansowanym lub przerzutowym rakiem piersi należy odpowiednio monitorować poziom elektrolitów w surowicy krwi (w tym potasu, wapnia, fosforu i magnezu) przed rozpoczęciem leczenia, na początku pierwszych 6 cykli, a następnie – zgodnie z wskazaniami klinicznymi. Wszelkie odchylenia od normy należy skorygować przed rozpoczęciem leczenia lekiem Kiskali i podczas trwania leczenia.
W zależności od zaobserwowanego podczas leczenia wydłużenia QT leczenie lekiem Kiskali może wymagać tymczasowego wstrzymania, zmniejszenia dawki lub przerwania, zgodnie z opisem w sekcjach „Sposób stosowania i dawki”, „Działania niepożądane” oraz „Farmakokinetyka”.
Ciężkie reakcje skórne
Podczas leczenia lekiem Kiskali zgłaszano toksyczny epidermalny nekrolyz (TEN). W przypadku wystąpienia objawów i objawów sugerujących ciężkie reakcje skórne (np. postępujące, rozległe wysypki skórne, często z pęcherzami lub zaangażowaniem błon śluzowych) leczenie lekiem Kiskali należy natychmiast przerwać.
Choroba międzybłoniowa płuc / zapalenie płuc
Chorobę międzybłoniową płuc (CMP) / zapalenie płuc obserwowano podczas stosowania Kiskali. Należy obserwować pacjentów pod kątem objawów płucnych wskazujących na CMP/zapalenie płuc, które mogą obejmować hipoksję, kaszel i duszność, oraz należy dostosować dawkę (patrz sekcja „Sposób stosowania i dawki”, tabela 15).
W zależności od stopnia nasilenia CMP/zapalenia płuc, które może być śmiertelne, może być konieczne tymczasowe wstrzymanie przyjmowania, zmniejszenie dawki lub całkowite przerwanie stosowania leku Kiskali (patrz sekcja „Sposób stosowania i dawki”, tabela 15).
Zwiększenie stężenia kreatyniny we krwi
RyboCyklib może powodować wzrost stężenia kreatyniny we krwi jako inhibitor transporterów nerkowych, organicznego kationowego transportera 2 (OCT2) i białka 1 wielolekowej ekstruzji i ekstruzji toksyn (MATE1), zaangażowanych w aktywną sekrecję kreatyniny z kanalików nerkowych (patrz sekcja „Interakcje z innymi lekami i inne formy interakcji”). W przypadku wzrostu stężenia kreatyniny we krwi podczas leczenia zaleca się dalszą ocenę funkcji nerek w celu wykluczenia niewydolności nerek.
Substraty CYP3A4
RyboCyklib jest silnym inhibitorem CYP3A4 w dawce 600 mg i umiarkowanym inhibitorem CYP3A4 w dawce 400 mg. W związku z tym ryboCyklib może oddziaływać na leki metabolizowane przez CYP3A4, co może prowadzić do zwiększenia stężenia substratów CYP3A4 w surowicy krwi (patrz sekcja „Interakcje z innymi lekami i inne formy interakcji”). Zaleca się zachowanie ostrożności przy jednoczesnym stosowaniu z wrażliwymi substratami CYP3A4 o wąskim indeksie terapeutycznym oraz zapoznanie się z instrukcjami do tych leków dotyczącymi jednoczesnego stosowania z inhibitorami CYP3A4.
Niewydolność nerek
Szacuje się, że zalecana dawka początkowa 200 mg u pacjentów z ciężką niewydolnością nerek prowadzi do około 45% niższej ekspozycji w porównaniu ze standardową dawką początkową 600 mg u pacjentów z lokalnie zaawansowanym lub przerzutowym rakiem piersi i prawidłową funkcją nerek. Skuteczność przy tej dawce początkowej nie była badana. Lek należy stosować z ostrożnością u pacjentów z ciężką niewydolnością nerek i dokładnie monitorować objawy toksyczności (patrz sekcje „Sposób stosowania i dawki” oraz „Farmakokinetyka”).
Kobiety w wieku rozrodczym
Kobietom w wieku rozrodczym należy zalecać stosowanie skutecznych metod antykoncepcji podczas leczenia lekiem Kiskali oraz co najmniej 21 dni po podaniu ostatniej dawki (patrz sekcja „Stosowanie w czasie ciąży lub karmienia piersią”).
Lecytyna z soi
Lek Kiskali zawiera lecytynę z soi. Pacjentom z podwyższoną wrażliwością na orzechy ziemne lub soję nie należy przyjmować Kiskali (patrz sekcja „Przeciwwskazania”).
Stosowanie w czasie ciąży lub karmienia piersią
Kobiety w wieku rozrodczym / antykoncepcja
Przed rozpoczęciem leczenia lekiem Kiskali należy wykonać test ciążowy.
Kobietom w wieku rozrodczym otrzymującym lek Kiskali należy zalecać stosowanie skutecznych metod antykoncepcji (np. podwójna metoda barierowa) podczas leczenia oraz co najmniej 21 dni po zakończeniu leczenia lekiem Kiskali.
Ciąża
Nie ma odpowiednich i dobrze kontrolowanych badań u ciężarnych kobiet. Ze względu na dane uzyskane na zwierzętach ryboCyklib stosowany u ciężarnej kobiety może powodować szkodliwy wpływ na płód (patrz sekcja „Dane przedkliniczne dotyczące bezpieczeństwa”). Leku Kiskali nie zaleca się stosować ciężarnym ani kobietom w wieku rozrodczym, które nie stosują środków antykoncepcyjnych.
Karmienie piersią
Nie wiadomo, czy ryboCyklib przenika do mleka matki. Brak danych dotyczących wpływu ryboCyklibu na niemowlę karmione piersią lub wpływu ryboCyklibu na produkcję mleka. RyboCyklib i jego metabolity łatwo przenikały do mleka ssących szczurów. Pacjentkom otrzymującym lek Kiskali nie należy karmić piersią przez co najmniej 21 dni po podaniu ostatniej dawki.
Fertylność
Brak danych klinicznych dotyczących wpływu ryboCyklibu na płodność. Ze względu na badania na zwierzętach ryboCyklib może wpływać na płodność mężczyzn z potencjałem rozrodczym (patrz sekcja „Dane przedkliniczne dotyczące bezpieczeństwa”).
Wpływ na zdolność do kierowania pojazdami i obsługi urządzeń
Lek Kiskali może mieć nieznaczny wpływ na zdolność do kierowania pojazdami i obsługi urządzeń. Pacjentom należy zalecać zachowanie ostrożności podczas kierowania pojazdami i pracy z innymi urządzeniami w przypadku wystąpienia zmęczenia, zawrotów głowy lub zawrotów podczas leczenia lekiem Kiskali (patrz sekcja „Działania niepożądane”).
Sposób stosowania i dawki
Leczenie lekiem Kiskali należy rozpoczynać pod nadzorem lekarza posiadającego doświadczenie w stosowaniu leków przeciwnowotworowych.
Test na pozytywny status HR i negatywny status HER2
Wybór pacjentów do leczenia lekiem Kiskali na podstawie ekspresji HR i HER2 w guzie należy przeprowadzać za pomocą urządzenia diagnostycznego in vitro (in vitro diagnostic (IVD)) posiadającego oznakowanie CE i przeznaczonego do tego celu. Jeśli test IVD z oznakowaniem CE nie jest dostępny, należy użyć alternatywnego, zwalidowanego testu.
Dawkowanie
Rak piersi w wczesnym etapie
Zalecana dawka to 400 mg (dwie tabletki powlekane o opłaszczeniu błonowym po 200 mg) rybocyklibu 1 raz dziennie przez 21 kolejnych dni, po czym następuje 7-dniowa przerwa, co stanowi pełen cykl 28-dniowy. Pacjentki z rakiem piersi w wczesnym etapie powinny przyjmować lek Kiskali do ukończenia 3-letniego cyklu leczenia lub do wystąpienia nawrotu choroby lub nieznośnej toksyczności.
W przypadku stosowania leku Kiskali w połączeniu z inhibitorem aromatazy, inhibitor aromatazy należy stosować doustnie 1 raz dziennie bez przerwy przez cały 28-dniowy cykl. Aby uzyskać bardziej szczegółowe informacje, zobacz instrukcję do stosowania medycznego (ISM) inhibitora aromatazy.
W leczeniu kobiet w okresie przedmenopauzalnym lub w okresie okołomenopauzalnym oraz mężczyzn inhibitor aromatazy należy łączyć z agonistą LH-RH.
Lokalnie zaawansowany lub rak piersi z przerzutami
Zalecana dawka to 600 mg (trzy tabletki powlekane o opłaszczeniu błonowym po 200 mg) rybocyklibu 1 raz dziennie przez 21 kolejnych dni, po czym następuje 7-dniowa przerwa, co stanowi pełny cykl 28-dniowy. U pacjentów z lokalnie zaawansowanym lub przerzutowym rakiem piersi leczenie należy kontynuować tak długo, jak długo utrzymuje się kliniczna skuteczność terapii lub do wystąpienia nieznośnej toksyczności.
W przypadku stosowania leku Kiskali w połączeniu z inhibitorem aromatazy, inhibitor aromatazy należy stosować doustnie 1 raz dziennie bez przerwy przez cały 28-dniowy cykl. Aby uzyskać bardziej szczegółowe informacje, zobacz ISM inhibitora aromatazy.
W przypadku stosowania leku Kiskali w połączeniu z fulwestrantem, fulwestrant podaje się w sposób dożylny w 1., 15. i 29. dniu, a następnie 1 raz miesięcznie. Aby uzyskać bardziej szczegółowe informacje, zobacz instrukcję do stosowania medycznego fulwestrantu.
Leczenie kobiet w okresie przed- i okołomenopauzalnym z wykorzystaniem zatwierdzonych kombinacji z lekiem Kiskali powinno również obejmować agonisty LH-RH zgodnie z lokalnymi standardami praktyki klinicznej.
Korekta dawki
Leczenie ciężkich lub nieznośnych działań niepożądanych może wymagać tymczasowego przerwania leczenia, zmniejszenia dawki lub całkowitego zaprzestania stosowania leku Kiskali. Wskazówki dotyczące zmniejszenia zalecanej dawki przedstawiono w tabeli 11.
Tabela 11. Wskazówki dotyczące korekty zalecanej dawki
| Kiskali |
||
| Dawka |
Liczba tabletek 200 mg |
|
| Rak piersi wczesny |
||
| Dawka początkowa |
400 mg/dobę |
2 |
| Redukcja dawki |
200 mg*/dobę |
1 |
| Rak piersi uogólniony miejscowo lub przerzutowy |
||
| Dawka początkowa |
600 mg/dobę |
3 |
| Pierwsza redukcja dawki |
400 mg/dobę |
2 |
| Druga redukcja dawki |
200 mg*/dobę |
1 |
| * Jeśli konieczne jest dalsze zmniejszenie dawki poniżej 200 mg/dobę, leczenie należy ostatecznie przerwać. |
||
W tabelach 12–16 podsumowano zalecenia dotyczące przerwania leczenia, zmniejszenia dawki lub zaprzestania przyjmowania leku Kiskali w przypadku leczenia poszczególnych działań niepożądanych. W ocenie klinicznej lekarz powinien kierować się planem postępowania wobec każdego pacjenta, biorąc pod uwagę ocenę stosunku korzyści do ryzyka w każdym przypadku (patrz rozdział „Szczególne ostrzeżenia i środki ostrożności stosowania”).
Przed rozpoczęciem leczenia lekiem Kiskali należy wykonać ogólny morfologiczny obraz krwi (OMK). Po rozpoczęciu leczenia OMK należy wykonywać co 2 tygodnie przez pierwsze 2 cykle, na początku każdego z kolejnych 4 cykli, a następnie w dalszym ciągu – zgodnie z wskazaniami klinicznymi.
Tabela 12. Korekta dawki i leczenie w przypadku neutropenii
| Neutropenia |
Stopień 1 lub 2* (AKN 1000/mm3 – ≤ NMN) |
Stopień 3* (AKN 500 – < 1000/mm3) |
Stopień 3* nieutropenia febrilna** |
Stopień 4* (AKN < 500/mm3) |
| Korekta dawki nie jest wymagana. |
Tymczasowo przerwać przyjmowanie do odzyskania poziomu ≤ stopnia 2. Wznowić przyjmowanie leku Kiskali w tej samej dawce. W przypadku ponownego wystąpienia toksyczności stopnia 3: tymczasowo przerwać przyjmowanie do odzyskania poziomu ≤ stopnia 2, następnie wznowić przyjmowanie leku Kiskali w dawce obniżonej o 1 poziom dawki. |
Tymczasowo przerwać przyjmowanie do odzyskania poziomu ≤ stopnia 2. Wznowić przyjmowanie leku Kiskali w dawce obniżonej o 1 poziom dawki. |
Tymczasowo przerwać przyjmowanie do odzyskania poziomu ≤ stopnia 2. Wznowić przyjmowanie leku Kiskali w dawce obniżonej o 1 poziom dawki. |
|
| * Określenie stopnia zgodnie z CTCAE, wersja 4.03 (CTCAE – Common Terminology Criteria for Adverse Events – ogólne kryteria terminologiczne oceny działań niepożądanych). ** Neutropenia stopnia 3 z pojedynczym epizodem gorączki > 38,3°C (lub ≥ 38°C przez więcej niż 1 godzinę i/lub współistniejące zakażenie). AKN – absolutna liczba neutrofili; NMN – niższa granica normy. |
||||
Przed rozpoczęciem leczenia lekiem Kiskali należy wykonać testy funkcji wątroby (LFT). Po rozpoczęciu leczenia LFT należy wykonywać co 2 tygodnie przez pierwsze 2 cykle, na początku każdego z kolejnych 4 cykli, a następnie zgodnie z wskazaniami klinicznymi. W przypadku zaburzeń o stopniu ≥ 2 zaleca się częstsze monitorowanie.
Tabela 13. Korekta dawki i leczenie w przypadku toksyczności hepatobilijarnej
| Wskaźniki |
Stopień 1* (> ULN – 3 × ULN) |
Stopień 2 * (> 3–5 × ULN) |
Stopień 3* (> 5–20 × ULN) |
Stopień 4* (> 20 × ULN) |
| Zwiększenie AST i/lub ALT w porównaniu do poziomu wyjściowego** bez jednoczesnego wzrostu całkowitego bilirubiny powyżej 2 × ULN |
Korekta dawki nie jest wymagana. |
Poziom wyjściowy < 2: czasowo wstrzymać przyjmowanie do odzyskania poziomu ≤ poziomu wyjściowego, następnie wznowić przyjmowanie leku Kiskali w tej samej dawce. W przypadku ponownego wystąpienia toksyczności stopnia 2 wznowić przyjmowanie leku Kiskali w dawce zmniejszonej do najbliższego poziomu dawkowania. |
Czasowo wstrzymać przyjmowanie leku Kiskali do odzyskania poziomu ≤ poziomu wyjściowego, następnie wznowić przyjmowanie leku w dawce zmniejszonej do najbliższego poziomu dawkowania. W przypadku ponownego wystąpienia toksyczności stopnia 3 całkowicie przerwać przyjmowanie leku Kiskali. |
Całkowicie przerwać przyjmowanie leku Kiskali. |
| Poziom wyjściowy = 2: przerwanie przyjmowania nie jest wymagane. |
||||
| Jednoczesne zwiększenie AST i/lub ALT wraz ze wzrostem całkowitego bilirubiny bez cech cholestazy |
Jeśli u pacjenta występuje wzrost ALT i/lub AST > 3 × ULN wraz ze wzrostem całkowitego bilirubiny > 2 × ULN, niezależnie od poziomu wyjściowego, całkowicie przerwać przyjmowanie leku Kiskali. |
|||
| * Klasyfikacja stopnia zgodnie z CTCAE, wersja 4.03. ** Poziom wyjściowy – przed rozpoczęciem leczenia. ULN – górna granica normy. |
||||
Przed rozpoczęciem leczenia lekiem Kiskali należy wykonać wszystkim pacjentom EKG.
Leczenie lekiem Kiskali należy rozpoczynać wyłącznie u pacjentów z wartościami QTcF mniejszymi niż 450 ms.
Po rozpoczęciu leczenia EKG należy powtarzać około w 14. dniu pierwszego cyklu, a następnie – zgodnie z wskazaniami klinicznymi.
W przypadku wydłużenia QTcF podczas leczenia zaleca się częstsze monitorowanie stanu pacjentek z rakiem piersi wczesnym oraz lokalnie zaawansowanym lub przerzutowym.
Tabela 14. Korekta dawki i leczenie przy wydłużeniu QT
| Wydłużenie QTcF* |
Rak piersi wczesny |
Lokalnie zaawansowany lub przerzutowy rak piersi |
| > 480 ms – ≤ 500 ms |
Należy tymczasowo przerwać stosowanie leku Kiskali do przywrócenia interwału QTcF do < 481 ms. |
|
| Wznowić leczenie lekiem w tym samym poziomie dawki. |
Zmniejszyć dawkę do następnego niższego poziomu dawki. |
|
| Jeśli interwał QTcF ponownie wzrośnie do ≥ 481 ms, należy tymczasowo przerwać przyjmowanie leku Kiskali do przywrócenia interwału QTcF do < 481 ms, a następnie wznowić przyjmowanie leku Kiskali w dawce zmniejszonej do najbliższego poziomu dawkowania. |
||
| > 500 ms |
Należy tymczasowo przerwać przyjmowanie leku Kiskali do przywrócenia interwału QTcF do < 481 ms, a następnie wznowić przyjmowanie leku Kiskali w dawce zmniejszonej do najbliższego poziomu dawkowania. Jeśli interwał QTcF ponownie wzrośnie do > 500 ms, należy przerwać przyjmowanie leku Kiskali. |
|
| Jeśli interwał QTcF przekracza 500 ms lub występuje jego zmiana o więcej niż 60 ms w porównaniu z wartością wyjściową w połączeniu z tachyarytmią komorową typu torsade de pointes lub polimorficzną tachyarytmią komorową lub objawami/objawami ciężkiej arytmii, należy ostatecznie przerwać przyjmowanie leku Kiskali. |
||
| Uwaga. Jeśli konieczne jest dalsze zmniejszenie dawki poniżej 200 mg/dzień, leczenie lekiem Kiskali należy ostatecznie przerwać. *QTcF – skorygowany interwał QT z zastosowaniem wzoru Fridericia. |
||
Tabela 15. Dostosowanie dawki i leczenie w przypadku ICHL/pneumonitis
| 1 stopień* (bezobjawowy) |
2 stopień* (objawowy) |
3 lub 4 stopień* (ciężki) |
|
| ChL/pneumonit |
Korekty dawki nie wymaga się. Rozpocząć odpowiednią terapię lekową i prowadzić monitorowanie kliniczne wskazane klinicznie. |
Chwilowo przerwać przyjmowanie leku Kiskali do ustabilizowania stanu do ≤ 1 stopnia, następnie wznowić przyjmowanie w dawce zmniejszonej do najbliższego poziomu dawki**. |
Ostatecznie przerwać przyjmowanie leku Kiskali. |
| * Stopień zgodnie z CTCAE, wersja 4.03. ** Przy rozważaniu możliwości wznowienia przyjmowania Kiskali należy przeprowadzić indywidualną ocenę korzyści i ryzyka. ChL — choroba międzywistowata płuc. |
|||
Tabela 16. Korekta dawki i leczenie w przypadku innych skutków toksycznych*
| Inne efekty toksyczne |
Stopień 1 lub 2** |
Stopień 3** |
Stopień 4** |
| Korekta dawki nie jest wymagana. Rozpocząć odpowiednią terapię lekową i prowadzić monitorowanie zgodnie z wskazaniami klinicznymi. |
Chwilowo przerwać przyjmowanie leku do ustąpienia do poziomu ≤ stopień 1, następnie wznowić przyjmowanie leku Kiskali w tej samej dawce. W przypadku ponownego wystąpienia toksyczności stopnia 3 wznowić przyjmowanie leku Kiskali w dawce zmniejszonej do najbliższego poziomu dawkowania. |
Trwale przerwać przyjmowanie leku Kiskali. |
|
| * Z wyjątkiem neutropenii, hepatotoksyczności, wydłużenia przedziału QT oraz RL/Pneumonitis. ** Stopnie oceny zgodnie z CTCAE, wersja 4.03. |
|||
Wskazówki dotyczące dostosowania dawki oraz inne odpowiednie informacje dotyczące bezpieczeństwa w przypadku wystąpienia efektów toksycznych należy znaleźć w instrukcji dołączanej do inhibitora aromatazy, fulwestrantu lub agonisty GnRH stosowanego współbieżnie.
Dostosowanie dawki przy stosowaniu leku Kiskali z silnymi inhibitorami CYP3A4
Należy unikać jednoczesnego stosowania silnych inhibitorów CYP3A4; należy rozważyć możliwość zastosowania leków alternatywnych, które są słabszymi inhibitorami CYP3A4. W przypadku konieczności jednoczesnego stosowania silnego inhibitora CYP3A4 z rybocyklibem, dawkę leku Kiskali należy zmniejszyć (patrz sekcja „Interakcje z innymi lekami i inne formy interakcji”).
Pacjentom przyjmującym 600 mg rybocyklibu na dobę, u których nie można uniknąć jednoczesnego stosowania silnego inhibitora CYP3A4, należy dodatkowo zmniejszyć dawkę do 400 mg.
Pacjentom przyjmującym 400 mg rybocyklibu na dobę, u których nie można uniknąć jednoczesnego stosowania silnego inhibitora CYP3A4, należy dodatkowo zmniejszyć dawkę do 200 mg.
Pacjentom, u których dawkę rybocyklibu zmniejszono już do 200 mg na dobę i u których nie można uniknąć jednoczesnego stosowania silnego inhibitora CYP3A4, należy tymczasowo przerwać leczenie lekiem Kiskali.
Z powodu międzyosobniczej zmienności zalecane dostosowanie dawki może nie być optymalne dla wszystkich pacjentów, dlatego zaleca się staranne monitorowanie pod kątem objawów toksyczności. Po zakończeniu stosowania silnego inhibitora CYP3A4 dawkę leku Kiskali należy przywrócić do dawki stosowanej przed rozpoczęciem przyjmowania silnego inhibitora CYP3A4, po upływie co najmniej 5 okresów półtrwania silnego inhibitora CYP3A4 (patrz sekcje „Szczególne ostrzeżenia i środki ostrożności dotyczące stosowania”, „Interakcje z innymi lekami i inne formy interakcji” oraz „Farmakokinetyka”).
Grupy specjalne
Zaburzenia funkcji nerek
Nie jest wymagane dostosowanie dawki u pacjentów z zaburzeniami funkcji nerek o lekkim lub umiarkowanym nasileniu. U pacjentów z ciężkim zaburzeniem funkcji nerek zaleca się dawkę początkową 200 mg. Nie przeprowadzono badań dotyczących stosowania leku Kiskali u pacjentek z rakiem piersi z ciężką niewydolnością nerek (patrz sekcje „Szczególne ostrzeżenia i środki ostrożności dotyczące stosowania”, „Farmakodynamika” oraz „Farmakokinetyka”).
Zaburzenia funkcji wątroby
Nie jest wymagane dostosowanie dawki u pacjentek z wczesnym etapem raka piersi i zaburzeniami funkcji wątroby (patrz sekcja „Farmakokinetyka”). U pacjentów z lokalnie zaawansowanym lub przerzutowym rakiem piersi i zaburzeniami funkcji wątroby o lekkim nasileniu (klasa A wg klasyfikacji Childa-Pugha) nie jest wymagane dostosowanie dawki; u pacjentów z zaburzeniami funkcji wątroby o umiarkowanym (klasa B wg klasyfikacji Childa-Pugha) i ciężkim nasileniu (klasa C wg klasyfikacji Childa-Pugha) może występować zwiększona (mniej niż 2-krotnie) ekspozycja na rybocyklib, dlatego zalecana dawka początkowa leku Kiskali wynosi 400 mg raz na dobę (patrz sekcja „Farmakokinetyka”).
Pacjenci w wieku podeszłym
Nie jest wymagane dostosowanie dawki u pacjentów w wieku 65 lat i starszych (patrz sekcja „Farmakokinetyka”).
Sposób stosowania
Lek Kiskali przeznaczony jest do doustnego stosowania raz na dobę niezależnie od posiłku (patrz sekcja „Interakcje z innymi lekami i inne formy interakcji”). Pacjentom zaleca się przyjmowanie dawki w przybliżenie o tej samej porze każdego dnia, najlepiej rano. W przypadku wystąpienia u pacjenta wymiotów po przyjęciu leku lub pominięcia dawki, nie należy przyjmować dodatkowej dawki w tym dniu. Następną przepisaną dawkę należy przyjąć w ustalonym czasie. Tabletki należy połykać całkowite, nie żując, nie łamiąc ani nie dzieląc przed połknięciem. Nie należy przyjmować tabletki, która jest złamana, popękana lub inaczej uszkodzona.
Każdy niewykorzystany lek lub jego odpady należy utylizować zgodnie z lokalnymi przepisami.
Dzieci
Bezpieczeństwo i skuteczność stosowania leku Kiskali u dzieci (do 18. roku życia) nie zostały ustalone. Brak danych.
Przedawkowanie
Doniesienia o przypadkach przedawkowania leku Kiskali są ograniczone. W przypadku przedawkowania mogą wystąpić takie objawy jak nudności i wymioty. Ponadto może występować toksyczność hematologiczna (np. neutropenia, trombocytopenia) oraz wydłużenie odcinka QT. We wszystkich przypadkach przedawkowania należy w razie potrzeby stosować ogólną terapię wspierającą.
Efekty uboczne
Podsumowanie profilu bezpieczeństwa
Rak piersi wczesny
W połączonych bazach danych najczęściej występującymi reakcjami niepożadanymi (RN) (≥ 20 %), których częstość przy stosowaniu leku Kiskali i inhibitora aromatazy (IA) była wyższa niż przy stosowaniu samego IA, były: neutropenia, infekcje, nudności, ból głowy, zmęczenie, leukopenia oraz odchylenia wyników czynnościowych prób wątrobowych.
W połączonych bazach danych najczęściej występującymi RN stopnia 3/4 (> 2 %), których częstość przy stosowaniu leku Kiskali i IA była wyższa niż przy stosowaniu samego IA, były: neutropenia, odchylenia wyników czynnościowych prób wątrobowych oraz leukopenia.
Zmniejszenie dawki z powodu wystąpienia działań niepożądanych niezależnie od przyczyny odnotowano u 22,8 % pacjentów otrzymujących lek Kiskali i IA w ramach badania klinicznego fazy III. Leczenie zostało ostatecznie zakończone u 19,7 % pacjentów otrzymujących lek Kiskali i IA w ramach badania klinicznego fazy III.
Rak piersi uogólniony lokalnie lub przerzutowy
W połączonych bazach danych najczęściej występującymi RN (≥ 20 %), których częstość przy stosowaniu leku Kiskali i dowolnej kombinacji była wyższa niż przy stosowaniu placebo i dowolnej kombinacji, były: neutropenia, infekcje, nudności, zmęczenie, biegunka, leukopenia, wymioty, ból głowy, zaparcia, alopecia, kaszel, wysypka, ból pleców, anemia oraz odchylenia wyników czynnościowych prób wątrobowych.
W połączonych bazach danych najczęściej występującymi RN stopnia 3/4 (> 2 %), których częstość przy stosowaniu leku Kiskali i dowolnej kombinacji była wyższa niż przy stosowaniu placebo i dowolnej kombinacji, były: neutropenia, leukopenia, odchylenia wyników czynnościowych prób wątrobowych, limfopenia, infekcje, ból pleców, anemia, zmęczenie, hipofosfatemia oraz wymioty.
Zmniejszenie dawki z powodu wystąpienia działań niepożądanych niezależnie od przyczyny odnotowano u 39,5 % pacjentów otrzymujących lek Kiskali w ramach badań klinicznych fazy III niezależnie od kombinacji. Leczenie zostało ostatecznie zakończone u 8,7 % pacjentów otrzymujących lek Kiskali i dowolną kombinację w ramach badań klinicznych fazy III.
Lista reakcji niepożądanych na lek
Rak piersi wczesny
Ogólny profil bezpieczeństwa leku Kiskali oparto na danych uzyskanych od 2525 pacjentów, którzy przyjmowali lek Kiskali w kombinacji z IA i zostali włączeni do randomizowanego otwartego badania klinicznego fazy III NATALEE.
Średnia długość leczenia rybocyklibem w badaniu wyniosła 33,0 miesiąca, przy czym 69,4 % pacjentów przyjmowało rybocyklib ponad 24 miesiące, a 42,8 % pacjentów ukończyło 36-miesięczny schemat przyjmowania rybocyklibu.
Rak piersi uogólniony lokalnie lub przerzutowy
Ogólny profil bezpieczeństwa leku Kiskali oparto na połączonych danych uzyskanych od 1065 pacjentów, którzy otrzymywali Kiskali w kombinacji z terapią endokrynną (582 pacjentów – w kombinacji z inhibitory aromatazy oraz 483 pacjentów – w kombinacji z fulwestrantem) i zostali włączeni do randomizowanych, podwójnie ślepych, kontrolowanych placebo badań klinicznych fazy III MONALEESA-2, MONALEESA-7 podgrupa NSIA oraz MONALEESA-3.
Średnia długość leczenia lekiem Kiskali w ramach badań fazy III wyniosła 19,2 miesiąca, przy czym 61,7 % pacjentów otrzymywało lek ≥ 12 miesięcy.
RN na lek obserwowane w trakcie badań klinicznych fazy III (tabela 16) u pacjentek z rakiem piersi wczesnym oraz z rakiem piersi uogólnionym lokalnie lub przerzutowym przedstawiono według klas układów narządów zgodnie z MedDRA [Medical Dictionary for Regulatory Activities – Słownik medyczny dla działalności regulacyjnej]. W każdej grupie według klas układów narządów reakcje niepożądane na lek wymieniono w kolejności malejącej częstości występowania. W każdej grupie według częstości reakcje niepożądane na lek wymieniono w kolejności malejącej ich powagi. Ponadto odpowiednia kategoria częstości dla każdej reakcji niepożądanej na lek określona jest następująco (CIOMS III): bardzo często (≥ 1/10); często (od ≥ 1/100 do < 1/10); nieczęsto (od ≥ 1/1000 do < 1/100); rzadko (od ≥ 1/10 000 do < 1/1000); bardzo rzadko (< 1/10 000); częstość nieznana (niemożliwe oszacowanie na podstawie dostępnych danych).
Tabela 17. Reakcje niepożądane na lek, które obserwowano w badaniach klinicznych fazy III oraz w okresie obserwacji popożyciowej
| Częstotliwość |
Pacjentki z rakiem piersi wczesnym otrzymujące rybocyklib w dawce początkowej 400 mg |
Pacjentki z rakiem piersi lokalnie zaawansowanym lub przerzutowym otrzymujące rybocyklib w dawce początkowej 600 mg |
| Infekcje i inwazje |
||
| Bardzo często |
Infekcje1 |
Infekcje1 |
| Zaburzenia układu krwi i chłonnego |
||
| Bardzo często |
Neutropenia, leukopenia |
Neutropenia, leukopenia, anemia, limfopenia |
| Często |
Anemia, trombocytopenia, limfopenia |
Trombocytopenia, febrilna neutropenia |
| Rzadko |
Febrilna neutropenia |
- |
| Zaburzenia przemiany materii i odżywiania |
||
| Bardzo często |
- |
Spadek apetytu |
| Często |
Hypokalcemia, hypokaliemia, spadek apetytu |
Hypokalcemia, hypokaliemia, hypofosfatemia |
| Zaburzenia układu nerwowego |
||
| Bardzo często |
Bóle głowy |
Bóle głowy, zawroty głowy |
| Często |
Zawroty głowy |
Vertigo |
| Zaburzenia oczu |
||
| Często |
- |
Zwiększone łzawienie, suchość oczu |
| Zaburzenia serca |
||
| Często |
- |
Syncope |
| Zaburzenia oddechowe, klatki piersiowej i jamy błędnika |
||
| Bardzo często |
Kaszel |
Utrudnione oddychanie, kaszel |
| Często |
Utrudnione oddychanie, choroba śródmiąższowa płuc (ILD)/zapalenie płuc |
Choroba śródmiąższowa płuc (ILD)/zapalenie płuc |
| Zaburzenia układu pokarmowego |
||
| Bardzo często |
Światłość, biegunka, zaparcia, ból brzucha2 |
Światłość, biegunka, wymioty, zaparcia, ból brzucha2, stomatyt, dyspepsja |
| Często |
Wymioty, stomatyt3 |
Dysgezja |
| Zaburzenia układu wątrobowo-żółciowego |
||
| Często |
Heptotoksyczność4 |
Heptotoksyczność4 |
| Zaburzenia skóry i tkanek podskórnych |
||
| Bardzo często |
Alodpecia |
Alodpecia, wysypka5, świąd |
| Często |
Wysypka5, świąd |
Susza skóry, zaczerwienienie, witiligo |
| Rzadko |
- |
Erythema multiforme |
| Nieznana częstotliwość |
- |
Toxyczny zespół martwiczy nabłonka (TEN) |
| Zaburzenia układu mięśniowo-szkieletowego i tkanki łącznej |
||
| Bardzo często |
- |
Ból pleców |
| Zaburzenia ogólne i w miejscu podania |
||
| Bardzo często |
Zmęczenie, osłabienie, podwyższenie temperatury ciała |
Zmęczenie, obrzęki obwodowe, podwyższenie temperatury ciała, osłabienie |
| Często |
Obrzęki obwodowe, ból gardła i jamy ustnej |
Ból gardła i jamy ustnej, suchość w ustach |
| Badania laboratoryjne i instrumentalne |
||
| Bardzo często |
Odchylenia wyników badań czynności wątroby6 |
Odchylenia wyników badań czynności wątroby6 |
| Często |
Zwiększenie stężenia kreatyniny we krwi, wydłużenie odstępu QT w zapisie EKG |
Zwiększenie stężenia kreatyniny we krwi, wydłużenie odstępu QT w zapisie EKG |
| 1 Infekcje: infekcje dróg moczowych, infekcje dróg oddechowych, gastroenteritis (tylko u pacjentek z rakiem piersi lokalnie zaawansowanym lub przerzutowym), sepsa (tylko u < 1 % pacjentek z rakiem piersi lokalnie zaawansowanym lub przerzutowym). 2 Ból brzucha: ból brzucha, ból w górnej części brzucha. 3 Stomatyt przy raku piersi wczesnym: stomatyt, mukityt. 4 Hepatotoksyczność: cytoliza wątrobowo-komórkowa, uszkodzenie komórek wątroby (tylko u pacjentek z rakiem piersi lokalnie zaawansowanym lub przerzutowym), uszkodzenie wątroby lekowe (tylko u < 1 % pacjentów z rakiem piersi wczesnym i pacjentów z rakiem piersi lokalnie zaawansowanym lub przerzutowym), hepatotoksyczność, niewydolność wątroby (tylko u pacjentek z rakiem piersi lokalnie zaawansowanym lub przerzutowym), autoimmunologiczny zapalenie wątroby (jeden przypadek u pacjentki z rakiem piersi wczesnym i jeden przypadek u pacjentki z rakiem piersi lokalnie zaawansowanym lub przerzutowym). 5 Wysypka: wysypka, wysypka makulopapularna, wysypka z świądem. 6 Odchylenia wyników badań czynności wątroby: zwiększenie stężenia ALT, zwiększenie stężenia AST, zwiększenie stężenia bilirubiny we krwi. |
||
Opis poszczególnych działań niepożądanych
Neutropenia
W badaniu fazy III u pacjentów z rakiem piersi w stadium wczesnym najczęstszym działaniem niepożądanym był neutropenia (62,5%), a zmniejszenie liczby neutrofili stopnia 3 lub 4 (na podstawie danych laboratoryjnych) zaobserwowano u 45,1% pacjentów otrzymujących lek Kiskali i IA.
U pacjentów z rakiem piersi w stadium wczesnym, u których wystąpiła neutropenia stopnia 2, 3 lub 4, średni czas do wystąpienia zdarzenia wynosił 0,6 miesiąca. Średni czas odzyskania neutropenii ≥ stopnia 3 (do normalizacji lub < stopnia 3) wynosił 0,3 miesiąca w grupie leczonej lekiem Kiskali i IA po przerwaniu leczenia i/lub zmniejszeniu dawki i/lub zaprzestaniu leczenia. Febrylne neutropenie zaobserwowano u 0,3% pacjentów przyjmujących lek Kiskali i IA. Częstość zaprzestania leczenia z powodu neutropenii była niska (1,1%) u pacjentów otrzymujących lek Kiskali i IA (patrz sekcje „Sposób stosowania i dawki” oraz „Szczególne wskazania”).
W badaniach fazy III u pacjentek z lokalnie zaawansowanym lub przerzutowym rakiem piersi najczęstszym działaniem niepożądanym był neutropenia (75,4%), a zmniejszenie liczby neutrofili stopnia 3 lub 4 (na podstawie danych laboratoryjnych) zaobserwowano u 62,0% pacjentów otrzymujących lek Kiskali i dowolną kombinację.
U pacjentów z lokalnie zaawansowanym lub przerzutowym rakiem piersi, u których wystąpiła neutropenia stopnia 2, 3 lub 4, średni czas do wystąpienia zjawiska wynosił 17 dni. Średni czas odzyskania wskaźnika przy stopniu ≥ 3 (do normalizacji lub stopnia < 3) wynosił 12 dni w grupach leczonych lekiem Kiskali i dowolnymi kombinacjami po przerwaniu leczenia i/lub zmniejszeniu dawki i/lub zaprzestaniu leczenia. Febrylne neutropenie obserwowano u około 1,7% pacjentów otrzymujących lek Kiskali w trakcie badań fazy III.
Częstość zaprzestania leczenia z powodu neutropenii była niska (0,8%) (patrz sekcje „Sposób stosowania i dawki” oraz „Szczególne wskazania”).
Wszystkich pacjentów należy poinstruować o konieczności natychmiastowego zgłaszania wszelkiego podwyższenia temperatury ciała.
Toksykożółciowość wątroby
W ramach badań klinicznych fazy III u pacjentów z rakiem piersi w stadium wczesnym oraz lokalnie zaawansowanym lub przerzutowym rakiem piersi obserwowano podwyższenie poziomu transaminaz.
W badaniu fazy III u pacjentów z rakiem piersi w stadium wczesnym przypadki toksykożółciowości wątroby obserwowano częściej w grupie leczonej lekiem Kiskali i IA w porównaniu z grupą IA samą (26,4% vs 11,2%), przy czym działania niepożądane stopnia 3/4 występowały częściej u pacjentów otrzymujących leczenie lekiem Kiskali i IA (8,6% vs 1,7%). Jednoczesne podwyższenie poziomu ALAT lub ASPAT powyżej trzech razy wyższego od górnej granicy normy i bilirubiny całkowitej powyżej dwóch razy wyższego od górnej granicy normy przy normalnym poziomie fosfatazy alkalicznej zaobserwowano u 8 pacjentów przyjmujących lek Kiskali i IA (u 6 pacjentów wartości ALAT lub ASPAT normalizowały się w ciągu 65 i 303 dni po zaprzestaniu leczenia lekiem Kiskali).
Tymczasowe przerwanie leczenia z powodu toksykożółciowości wątroby miało miejsce u 12,4% pacjentek z rakiem piersi w stadium wczesnym, które przyjmowały lek Kiskali i IA, głównie z powodu podwyższenia poziomu ALAT (10,1%) i/lub podwyższenia poziomu ASPAT (6,8%). Korekta dawki z powodu toksykożółciowości wątroby występowała u 2,6% pacjentów przyjmujących lek Kiskali i IA, głównie z powodu podwyższenia poziomu ALAT (1,9%) i/lub podwyższenia poziomu ASPAT (0,6%). Ostateczne zaprzestanie leczenia lekiem Kiskali z powodu odchyleń wskaźników funkcji wątroby lub hepatotoksyczności odnotowano u 8,9% i 0,1% pacjentów odpowiednio (patrz sekcje „Sposób stosowania i dawki” oraz „Szczególne wskazania”).
W ramach badania klinicznego fazy III u pacjentów z rakiem piersi w stadium wczesnym 80,9% (165/204) przypadków podwyższenia poziomu ALAT lub ASPAT stopnia 3 lub 4 zaobserwowano w ciągu pierwszych 6 miesięcy leczenia. U pacjentów z podwyższeniem poziomu ALAT/ASPAT stopnia 3 lub 4 średni czas do wystąpienia tych zjawisk wynosił 2,8 miesiąca w grupie leczonej lekiem Kiskali i IA. Średni czas do odzyskania wskaźnika (do normalizacji lub stopnia ≤ 2) wynosił 0,7 miesiąca w grupie leczonej lekiem Kiskali i IA.
W badaniach klinicznych fazy III u pacjentek z lokalnie zaawansowanym lub przerzutowym rakiem piersi przypadki toksykożółciowości wątroby obserwowano częściej u pacjentów w grupach leczonych lekiem Kiskali i dowolną kombinacją w porównaniu z grupami otrzymującymi placebo i dowolną kombinację (27,3% i 19,6% odpowiednio), przy czym działania niepożądane stopnia 3/4 występowały częściej u pacjentów otrzymujących leczenie lekiem Kiskali i dowolną kombinacją (13,2% i 6,1% odpowiednio). Podwyższenie poziomu ALAT (11,2% i 1,7%) oraz ASPAT (7,8% i 2,1%) stopnia 3 lub 4 zaobserwowano w grupach stosowania leku Kiskali i placebo odpowiednio. Jednoczesne podwyższenie ALAT lub ASPAT powyżej trzech razy wyższego od górnej granicy normy i bilirubiny całkowitej powyżej dwóch razy wyższego od górnej granicy normy przy normalnym poziomie fosfatazy alkalicznej i przy braku cholestazy zaobserwowano u 6 pacjentów (4 pacjenci w badaniu A2301 [MONALEESA-2], u których wartości te normalizowały się w ciągu 154 dni, i 2 pacjenci w badaniu F2301 [MONALEESA-3], u których wartości te normalizowały się w ciągu 121 i 532 dni odpowiednio po zaprzestaniu leczenia lekiem Kiskali). W badaniu E2301 (MONALEESA-7) nie zgłaszano takich przypadków.
Tymczasowe przerwanie leczenia i/lub korekta dawki z powodu toksykożółciowości wątroby były konieczne u 12,3% pacjentów z lokalnie zaawansowanym lub przerzutowym rakiem piersi, którzy otrzymywali leczenie lekiem Kiskali i dowolną kombinacją, głównie z powodu podwyższenia poziomu ALAT (7,9%) i/lub podwyższenia poziomu ASPAT (7,3%). Ostateczne zaprzestanie leczenia lekiem Kiskali i dowolną kombinacją z powodu odchyleń wskaźników funkcji wątroby lub hepatotoksyczności odnotowano u 2,4% i 0,3% pacjentów odpowiednio (patrz sekcje „Sposób stosowania i dawki” oraz „Szczególne wskazania”).
W badaniach klinicznych fazy III u pacjentów z lokalnie zaawansowanym lub przerzutowym rakiem piersi 70,9% (90/127) przypadków podwyższenia poziomu ALAT lub ASPAT stopnia 3 lub 4 zaobserwowano w ciągu pierwszych 6 miesięcy leczenia. U pacjentów z podwyższeniem poziomu ALAT/ASPAT stopnia 3 lub 4 średni czas do wystąpienia tych zjawisk wynosił 92 dni w grupach leczonych lekiem Kiskali i dowolną kombinacją. Średni czas do odzyskania wskaźnika (do normalizacji lub stopnia ≤ 2) wynosił 21 dni w grupach leczonych lekiem Kiskali z dowolną kombinacją.
Wydłużenie interwału QT
W badaniu fazy III u pacjentów z rakiem piersi w stadium wczesnym wydłużenie interwału QT zaobserwowano u 5,3% pacjentów w grupie leczonej lekiem Kiskali i IA oraz u 1,4% pacjentów w grupie leczonej tylko IA. W grupie leczonej lekiem Kiskali i IA przypadki wydłużenia interwału QT były reprezentowane głównie przez wydłużenie interwału QT w EKG (4,3%), co było jedyną potwierdzoną reakcją niepożądaną stosowania leku Kiskali. Przerwanie leczenia z powodu wydłużenia interwału QT w EKG i omdlenia było konieczne u 1,1% pacjentów przyjmujących lek Kiskali. Korekta dawki z powodu wydłużenia interwału QT w EKG była konieczna u 0,1% pacjentów przyjmujących lek Kiskali.
Analiza centralna danych EKG wykazała, że u 10 pacjentów (0,4%) i 4 pacjentów (0,2%) zaobserwowano co najmniej jeden przypadek wydłużenia interwału QTcF > 480 ms po rozpoczęciu leczenia w grupach leczonych lekiem Kiskali i IA oraz grupie leczonej tylko IA odpowiednio. U pacjentów z wydłużeniem QTcF > 480 ms w grupie leczonej lekiem Kiskali i IA średni czas do wystąpienia tych zjawisk wynosił 15 dni i zmiany te były odwracalne po tymczasowym przerwaniu leczenia i/lub korekcie dawki. Zmiana interwału QTcF > 60 ms w porównaniu z wartością wyjściową odnotowana została u 19 pacjentów (0,8%) w grupie leczonej lekiem Kiskali i IA, a interwał QTcF > 500 ms po rozpoczęciu leczenia zaobserwowano u 3 pacjentów (0,1%) w grupie leczonej lekiem Kiskali i IA.
W badaniu E2301 (MONALEESA-7) u pacjentów z lokalnie zaawansowanym lub przerzutowym rakiem piersi obserwowane średnie wydłużenie interwału QTcF od wartości wyjściowej było o około 10 ms większe w podgrupie stosującej tamoksyfen i placebo w porównaniu z podgrupą stosującą NSA i placebo, co wskazuje, że tamoksyfen sam w sobie powodował wydłużenie interwału QTcF i może wpływać na wartości QTcF obserwowane w grupie stosującej lek Kiskali i tamoksyfen. W grupie placebo wydłużenie interwału QTcF o > 60 ms od wartości wyjściowej zaobserwowano u 6/90 (6,7%) pacjentów otrzymujących tamoksyfen i nie zaobserwowano u żadnego z pacjentów otrzymujących NSA (patrz sekcja „Farmakokinetyka”). Wydłużenie interwału QTcF o > 60 ms od wartości wyjściowej zaobserwowano u 14/87 (16,1%) pacjentów otrzymujących lek Kiskali i tamoksyfen oraz u 18/245 (7,3%) pacjentów otrzymujących lek Kiskali i NSA. Kiskali nie zaleca się stosować w połączeniu z tamoksyfenem (patrz sekcja „Farmakodynamika”).
W badaniach klinicznych fazy III u 9,3% pacjentów z lokalnie zaawansowanym lub przerzutowym rakiem piersi w grupach leczonych lekiem Kiskali i IA lub fulwestrantem oraz u 3,5% pacjentów w grupach stosujących placebo i IA lub fulwestrant zaobserwowano co najmniej jeden przypadek wydłużenia interwału QT (w tym wydłużenie interwału QT w EKG i omdlenia). Przegląd danych EKG wykazał, że u 15 pacjentów (1,4%) wartość QTcF po rozpoczęciu leczenia wynosiła > 500 ms, a u 61 pacjentów (5,8%) zaobserwowano zwiększenie interwału QTcF o > 60 ms od wartości wyjściowej. O przypadkach tachykardii komorowej typu „torsade de pointes” nie zgłaszano. Przerwanie leczenia / korekta dawki z powodu wydłużenia interwału QT w EKG i omdleń wymagane były u 2,9% pacjentów otrzymujących lek Kiskali i IA lub fulwestrant.
Analiza danych EKG wykazała, że u 55 pacjentów (5,2%) i 12 pacjentów (1,5%) zaobserwowano co najmniej jeden przypadek wydłużenia interwału QTcF > 480 ms po rozpoczęciu leczenia w grupach stosowania leku Kiskali i inhibitora aromatazy lub fulwestrantu oraz grupach stosowania placebo i inhibitora aromatazy lub fulwestrantu odpowiednio. U pacjentów z wydłużeniem QTcF > 480 ms średni czas do wystąpienia tego zjawiska wynosił 15 dni niezależnie od kombinacji i zmiany te były odwracalne po tymczasowym przerwaniu leczenia i/lub zmniejszeniu dawki (patrz sekcje „Sposób stosowania i dawki”, „Szczególne wskazania” oraz „Farmakokinetyka”).
Pacjenci z niewydolnością nerek
W ramach badania klinicznego fazy III u pacjentów z rakiem piersi w stadium wczesnym 983 pacjentów z łagodnym zaburzeniem funkcji nerek oraz 71 pacjentów z umiarkowanym zaburzeniem funkcji nerek otrzymywało rybocyklib. Żaden pacjent z ciężkim zaburzeniem funkcji nerek nie został włączony do badania (patrz sekcja „Farmakodynamika”).
W trzech badaniach podstawowych 341 pacjentów z lokalnie zaawansowanym lub przerzutowym rakiem piersi i łagodną niewydolnością nerek oraz 97 pacjentów z umiarkowaną niewydolnością nerek otrzymywało leczenie rybocyklibem. Żaden pacjent z ciężką niewydolnością nerek nie został włączony do badania (patrz sekcja „Farmakodynamika”). Zaobserwowano korelację między stopniem niewydolności nerek na poziomie wyjściowym a wartościami kreatyniny we krwi podczas leczenia. U pacjentów z łagodną lub umiarkowaną niewydolnością nerek zaobserwowano nieznaczne zwiększenie częstości występowania wydłużenia interwału QT i trombocytopenii. Rekomendacje dotyczące monitorowania i korekty dawki w przypadku takich toksycznych efektów patrz w sekcjach „Sposób stosowania i dawki” i „Szczególne wskazania”.
Zgłaszanie podejrzewanych działań niepożądanych
Zgłaszanie działań niepożądanych po rejestracji leku ma duże znaczenie. Umożliwia to monitorowanie stosunku korzyści do ryzyka stosowania tego leku. Lekarze, pracownicy farmaceutyczni, pacjenci oraz ich ustawieni przedstawiciele powinni zgłaszać wszystkie przypadki podejrzewanych działań niepożądanych i braku skuteczności leku poprzez Automatyczny System Informacyjny nadzoru farmakologicznego pod adresem: https://aisf.dec.gov.ua
Okres ważności. 1 rok.
Warunki przechowywania
Przechowywać w lodówce (od 2 do 8 °C) przez 10 miesięcy w strefie magazynowej.
Przechowywać w temperaturze nie wyższej niż 25 °C przez 2 miesiące w czasie stosowania leku przez pacjenta. Przechowywać w oryginalnym opakowaniu. Przechowywać w miejscu niedostępnym dla dzieci.
Opakowanie
- Po 21 tabletek w blisterze, po 3 blisterach w pudełku kartonowym.
- Po 21 tabletek w blisterze, po 3 blisterach w pudełku kartonowym, po 3 pudełka w pudełku kartonowym.
Kategoria wydania. Na receptę.
Producent
Novartis Pharmaceuticals Manufacturing LLC.
Miejsce produkcji i adres miejsca prowadzenia działalności
ul. Verovškova 57, Lublana, 1000, Słowenia.