Kisqali
Ucraina
Indice
ISTRUZIONI PER L'USO MEDICINALE DEL MEDICINALE KISQALI (KISQALI)
Composizione:
principio attivo: ribociclib;
1 compressa contiene ribociclib succinato in quantità tale da corrispondere a 200 mg di ribociclib;
eccipienti: cellulosa microcristallina, crospovidone (tipo A), idrossipropilcellulosa a basso grado di sostituzione, magnesio stearato, biossido di silicio colloidale anidro, ossido di ferro nero (E 172), ossido di ferro rosso (E 172), lecitina di soia, alcool polivinilico parzialmente idrolizzato, talco, biossido di titanio (E 171), gomma xantana.
Forma farmaceutica. Compresse rivestite con film.
Principali caratteristiche fisico-chimiche: compresse rotonde biconvesse con bordi smussati, di colore grigio chiaro tendente al viola, senza riga di incisione, con impresso «RIC» su un lato e «NVR» sull'altro.
Gruppo farmacoterapeutico. Agenti antineoplastici. Inibitori delle proteina chinasi. Inibitori della chinasi dipendente dai ciclini (CDK). Ribociclib.
Codice ATC L01E F02.
Proprietà farmacologiche
Farmacodinamica
Meccanismo d'azione
Ribociclib è un inibitore selettivo delle chinasi dipendenti dal ciclo (CDK) 4 e 6 e determina la loro inibizione al 50% (IC50) in saggi biochimici alle concentrazioni di 0,01 (4,3 ng/ml) e 0,039 µM (16,9 ng/ml), rispettivamente. Queste chinasi vengono attivate dal legame con le cicline D e svolgono un ruolo fondamentale nelle vie di segnalazione che regolano il ciclo cellulare e la proliferazione cellulare. Il complesso ciclina D-CDK4/6 regola il progresso del ciclo cellulare attraverso la fosforilazione della proteina della retinoblastoma (pRb).
In vitro, ribociclib riduce la fosforilazione di pRb, determinando l'arresto della fase G1 del ciclo cellulare, riduzione della proliferazione e fenotipo di senescenza nei modelli di cancro al seno. In vivo, la monoterapia con ribociclib ha indotto regressione tumorale, correlata all'inibizione della fosforilazione di pRb.
Negli studi in vivo su modelli di xeno-trapianto derivati da pazienti con carcinoma mammario positivo ai recettori degli estrogeni (ER+), l'uso combinato di ribociclib e antiestrogeni (ad esempio letrozolo) ha determinato un'inibizione più marcata della crescita tumorale, con regressione sostenuta del tumore e ritardo nella ripresa della crescita tumorale dopo l'interruzione del trattamento, rispetto all'uso di ciascun farmaco singolarmente.
Quando somministrato ai pazienti, ribociclib può esercitare anche un'azione immunomodulante, riducendo il numero di linfociti T regolatori e aumentando il rapporto relativo di linfociti T CD3+.
Inoltre, in condizioni in vivo, è stata valutata l'attività antitumorale di ribociclib in combinazione con fulvestrant in topi immunodeficienti portatori di xeno-trapianti umani ER+ di carcinoma mammario ZR751; tale combinazione ha determinato l'inibizione completa della crescita tumorale.
L'analisi di un pannello di linee cellulari di carcinoma mammario con noto stato ER ha dimostrato una maggiore efficacia di ribociclib nelle linee cellulari ER+ rispetto a quelle ER-. Nei modelli preclinici studiati, per l'attività di ribociclib era necessario un pRb integro.
Elettrofisiologia cardiaca
Per valutare l'effetto di ribociclib sull'intervallo QTc in pazienti con cancro avanzato, sono state effettuate serie di ECG ripetuti tre volte dopo una dose singola allo stato stazionario. Nell'analisi farmacocinetica e farmacodinamica sono stati inclusi dati complessivi di 997 pazienti che ricevevano ribociclib in un intervallo di dosi da 50 a 1200 mg. L'analisi ha mostrato che ribociclib induce un prolungamento dell'intervallo QTc dipendente dalla concentrazione.
In pazienti con carcinoma mammario localmente avanzato o metastatico, la variazione media calcolata di QTcF rispetto al basale con l'assunzione di Kisqali alla dose di 600 mg in combinazione con un inibitore non steroideo dell'aromatasi (NSAI) o fulvestrant è stata di 22,0 ms (IC 90%: 20,56; 23,44) e 23,7 ms (IC 90%: 22,31; 25,08), rispettivamente, in corrispondenza della Cmax geometrica media allo stato stazionario, rispetto a 34,7 ms (IC 90%: 31,64; 37,78) con la combinazione con tamoxifene (vedere sezione «Informazioni importanti sulla somministrazione»).
In pazienti con carcinoma mammario in stadio precoce si osserva un aumento simile e dipendente dalla concentrazione dell'intervallo QTc. Secondo le stime, la variazione media calcolata di QTcF rispetto al basale è inferiore in pazienti con carcinoma mammario in stadio precoce che ricevono 400 mg di Kisqali rispetto ai pazienti con carcinoma mammario localmente avanzato o metastatico che ricevono 600 mg di Kisqali.
Efficacia e sicurezza clinica
Carcinoma mammario in stadio precoce
Studio CLEE011O12301C (NATALEE)
Kisqali è stato studiato in uno studio clinico di fase III randomizzato, aperto, multicentrico, in cui il farmaco è stato utilizzato per il trattamento di donne in pre-/post-menopausa e uomini con carcinoma mammario HR-positivo e HER2-negativo in stadio anatomico precoce II o III indipendentemente dallo stato linfonodale, ad alto rischio di recidiva, in combinazione con un inibitore dell'aromatasi (IA) come letrozolo o anastrozolo, rispetto al trattamento con IA da solo:
- Gruppo stadio IIB–III oppure
- Gruppo stadio IIA in combinazione con i seguenti criteri:
- Linfonodo ascellare positivo oppure
- Linfonodo ascellare negativo:
- tumore di grado G3 oppure
- tumore di grado G2, in combinazione con uno qualsiasi dei seguenti criteri:
- Ki67 ≥ 20%;
- alto rischio di recidiva secondo i profili genetici.
Donne in pre-menopausa e uomini hanno ricevuto anche il goserelina, agonista del GnRH. Secondo i criteri TNM, nello studio NATALEE sono stati inclusi pazienti con qualsiasi coinvolgimento linfonodale o senza coinvolgimento linfonodale con tumore di dimensioni > 5 cm oppure con tumore di dimensioni 2–5 cm di grado G2 (e alto rischio genomica di recidiva o Ki67 ≥ 20%) o grado G3.
Complessivamente, 5101 pazienti, inclusi 20 uomini, sono stati randomizzati in rapporto 1:1 per ricevere Kisqali 400 mg più IA (n = 2549) oppure solo IA (n = 2552). La randomizzazione per il trattamento è stata stratificata per stadio anatomico (gruppo II [n = 2154 (42,2%)] vs gruppo III [n = 2947 (57,8%)]), trattamento precedente (chemioterapia neoadiuvante/adjuvante – sì [n = 4432 (86,9%)] o no [n = 669 (13,1%)]), stato menopausico (donne e uomini in pre-menopausa [n = 2253 (44,2%)] vs donne in post-menopausa [n = 2848 (55,8%)]) e regione (Nord America/Europa Occidentale/Oceania [n = 3128 (61,3%)] vs altri paesi del mondo [n = 1973 (38,7%)]). Kisqali è stato somministrato per via orale alla dose di 400 mg una volta al giorno per 21 giorni consecutivi seguiti da una pausa di 7 giorni nel trattamento, in combinazione con letrozolo 2,5 mg o anastrozolo 1 mg per via orale una volta al giorno per 28 giorni. Goserelina è stata somministrata alla dose di 3,6 mg come impianto sottocutaneo iniettabile, somministrato il 1° giorno di ogni ciclo di 28 giorni. Il trattamento con Kisqali è proseguito fino al completamento di un periodo di trattamento di 3 anni dalla data di randomizzazione (circa 39 cicli).
L'età media dei pazienti inclusi nello studio era di 52 anni (range da 24 a 90 anni). Il 15,2% dei pazienti aveva un'età ≥ 65 anni, di cui 123 pazienti (2,4%) avevano un'età ≥ 75 anni. Tra i pazienti erano rappresentate le razze caucasiche (73,4%), asiatiche (13,2%) e afroamericane o nere (1,7%). Tutti i pazienti avevano uno stato funzionale ECOG 0 o 1. Complessivamente, l'88,2% dei pazienti aveva ricevuto chemioterapia in regime neoadiuvante o adiuvante e il 71,6% aveva ricevuto terapia endocrina in regime neoadiuvante o adiuvante nei 12 mesi precedenti l'inclusione nello studio.
Il punto primario dello studio era la sopravvivenza libera da malattia invasiva (iDFS), definita come il tempo tra la randomizzazione e il primo evento di: recidiva locale di carcinoma mammario invasivo, recidiva regionale di carcinoma invasivo, recidiva a distanza, morte (per qualsiasi causa), carcinoma mammario controlaterale invasivo o altro cancro primario invasivo (esclusi il carcinoma basocellulare e il carcinoma a cellule squamose della cute).
Il punto primario dello studio è stato raggiunto durante l'analisi primaria (data di chiusura dei dati: 11.01.2023). È stato dimostrato un miglioramento statisticamente significativo dell'iDFS (HR: 0,748; IC 95%: 0,618; 0,906; valore p ottenuto dal test log-rank stratificato unilaterale, 0,0014) nei pazienti che assumevano Kisqali più IA rispetto ai pazienti che ricevevano solo IA. Risultati coerenti sono stati osservati in sottogruppi definiti da stadio anatomico, stato menopausico, regione, stato linfonodale, età, razza e chemioterapia o terapia ormonale adiuvante/neoadiuvante precedente.
I dati di un'analisi aggiuntiva (data di chiusura dei dati: 21.07.2023) sono riassunti nella Tabella 1 e il grafico di Kaplan-Meier per l'iDFS è riportato in Figura 1. La mediana della durata del trattamento al momento dell'analisi finale dell'iDFS era di circa 30 mesi e la mediana del tempo di follow-up per l'iDFS era di 33,3 mesi in entrambi i gruppi di studio. Attualmente non sono disponibili dati sufficienti per determinare la sopravvivenza globale (OS). Complessivamente, 172 pazienti (3,5%) sono deceduti (83/2525 nel gruppo ribociclib vs 89/2442 nel gruppo IA da solo, HR 0,892; IC 95%: 0,661; 1,203).
Tabella 1. NATALEE – Risultati di efficacia (iDFS) valutati dall'investigatore (data di chiusura dei dati: 21.07.2023)
| Kisqali e IA* N = 2549 |
IA N = 2552 |
||
| Sopravvivenza libera da malattia invasiva (SLLMIa) |
|||
| Numero di pazienti con evento (n, %) |
226 (8,9 %) |
283 (11,1 %) |
|
| Rapporto dei rischi (IC 95 %) |
0,749 (0,628; 0,892) |
||
| Valore pb |
0,0006 |
||
| SLLMI a 36 mesi (%, IC 95 %) |
90,7 (89,3; 91,8) |
87,6 (86,1; 88,9) |
|
| IC – intervallo di confidenza; N – numero di pazienti. a La SLLMI è definita come il tempo dalla randomizzazione al primo evento: recidiva locale di carcinoma mammario invasivo, recidiva regionale di carcinoma invasivo, recidiva a distanza, morte (per qualsiasi causa), carcinoma mammario invasivo controlaterale o altro carcinoma primario invasivo diverso dal carcinoma mammario (esclusi il carcinoma basocellulare e il carcinoma a cellule squamose della pelle). b Il valore p nominale è stato ottenuto mediante il test log-rank stratificato unilaterale. * Letrozolo o anastrozolo. |
|||
IA – inibitore dell’aromatasi (letrozolo o anastrozolo)
Il valore p è basato sul test log-rank stratificato unidirezionale.
Fig. 1. NATALEE – Curva di Kaplan-Meier per la SLD (Sopravvivenza libera da malattia distante) basata sulla valutazione dell’investigatore (data di chiusura raccolta dati: 21 luglio 2023).
Il tasso di sopravvivenza libera da metastasi a distanza (SLMD) nel gruppo trattato con Kisqali e IA era di 204 (8,0 %) rispetto a 256 (10 %) nel gruppo trattato solo con IA (HR: 0,749; IC 95 %: 0,623; 0,900).
Carcinoma mammario avanzato
Studio CLEE011A2301 (MONALEESA-2)
Kisqali è stato valutato in uno studio clinico multicentrico di Fase III, randomizzato, in doppio cieco, controllato con placebo, che ha coinvolto donne in post-menopausa con carcinoma mammario ormono-recettore-positivo e HER2 (recettore del fattore di crescita epidermico umano di tipo 2)-negativo in stadio avanzato, precedentemente non trattate per la malattia avanzata, in combinazione con letrozolo rispetto a letrozolo da solo.
Un totale di 668 pazienti sono state randomizzate in rapporto 1:1 per ricevere Kisqali 600 mg più letrozolo (n = 334) o placebo più letrozolo (n = 334), stratificate in base alla presenza di metastasi epatiche e/o polmonari (sì [n = 292 (44 %)] o no [n = 376 (56 %)]). Le caratteristiche demografiche e basali della malattia erano bilanciate e comparabili tra i gruppi di studio. Kisqali è stato somministrato per via orale alla dose di 600 mg al giorno per 21 giorni consecutivi, seguiti da una pausa di 7 giorni nel ciclo di trattamento, in combinazione con letrozolo alla dose di 2,5 mg una volta al giorno per 28 giorni. Non era consentito il passaggio da placebo a Kisqali durante lo studio o dopo il progressione della malattia.
L’età media dei pazienti inclusi in questo studio era di 62 anni (range da 23 a 91). Il 44,2 % dei pazienti aveva un’età ≥ 65 anni, compresi 69 pazienti con età superiore a 75 anni. Tra i pazienti erano rappresentate le razze caucasica (82,2 %), mongolica (7,6 %) e nera (2,5 %). Tutti i pazienti avevano uno stato funzionale ECOG pari a 0 o 1. Nel gruppo trattato con Kisqali, il 46,6 % dei pazienti aveva ricevuto chemioterapia in regime neoadiuvante o adiuvante, e il 51,3 % aveva ricevuto terapia antiormonale in regime neoadiuvante o adiuvante prima dell’inclusione nello studio. Il 34,1 % dei pazienti era de novo (diagnosi di cancro alla mammella per la prima volta). Nel 22,0 % dei pazienti le metastasi erano localizzate esclusivamente alle ossa, mentre nel 58,8 % erano presenti metastasi viscerali. I pazienti che avevano ricevuto in precedenza terapia (neo)adiuvante con anastrozolo o letrozolo dovevano aver concluso tale trattamento almeno 12 mesi prima della randomizzazione nello studio.
Analisi primaria
Il punto finale primario dello studio è stato raggiunto nell’analisi intermedia pianificata, eseguita dopo aver osservato l’80 % degli eventi pianificati di sopravvivenza libera da progressione (SLP), utilizzando i criteri RECIST (Response Evaluation Criteria in Solid Tumors, versione 1.1), basata sulla valutazione dell’investigatore nell’intera popolazione (tutti i pazienti randomizzati), e confermata da una valutazione radiologica centrale indipendente e in cieco.
I risultati dell’efficacia hanno dimostrato un miglioramento statisticamente significativo della SLP nei pazienti trattati con Kisqali e letrozolo rispetto ai pazienti trattati con placebo e letrozolo, nell’analisi dell’intera popolazione (hazard ratio 0,556; IC 95 %: 0,429; 0,720; valore p unidirezionale stratificato con test log-rank = 0,00000329), con un effetto clinico rilevante del trattamento.
I dati complessivi relativi allo stato di salute/guarigione non hanno mostrato differenze tra il gruppo trattato con Kisqali e letrozolo e il gruppo trattato con placebo e letrozolo.
Un aggiornamento successivo dei dati di efficacia (data di chiusura raccolta dati: 2 gennaio 2017) è riportato nelle tabelle 2 e 3 (MONALEESA-2: risultati della valutazione dell’efficacia (SLP) basati sulla valutazione radiologica dell’investigatore e MONALEESA-2: risultati della valutazione dell’efficacia (SOSa, TTPb) basati sulla valutazione dell’investigatore).
La mediana della SLP è stata di 25,3 mesi (IC 95 %: 23,0; 30,3) nei pazienti trattati con ribociclib e letrozolo e di 16,0 mesi (IC 95 %: 13,4; 18,2) nei pazienti trattati con placebo e letrozolo. Il 54,7 % dei pazienti trattati con ribociclib e letrozolo non ha mostrato progressione della malattia a 24 mesi, rispetto al 35,9 % nel gruppo trattato con placebo e letrozolo.
Tabella 2. MONALEESA-2: risultati della valutazione dell’efficacia (SLP) basati sulla valutazione radiologica dell’investigatore (data di chiusura raccolta dati: 2 gennaio 2017)
| Analisi aggiornata |
||
| Parametri |
Kisqali e letrozolo N = 334 |
Placebo e letrozolo N = 334 |
| Sopravvivenza libera da progressione |
||
| Mediana PFS [mesi] (IC 95 %) |
25,3 (23,0; 30,3) |
16,0 (13,4; 18,2) |
| Rapporto dei rischi (IC 95 %) |
0,568 (0,457; 0,704) |
|
| Valore di pa |
9,63 × 10-8 |
|
| IC – intervallo di confidenza; N – numero di pazienti aIl valore di p è stato ottenuto mediante il test log-rank stratificato unilaterale. |
||
 |
| Moments of data cutoff Ribociclib (N = 334) Placebo (N = 334) |
| Numero di eventi: ribociclib – 140, placebo – 205 Rapporto di rischio 0,568; IC 95% [0,457; 0,704] Mediana di Kaplan-Meier: ribociclib – 25,3 mesi; placebo – 16,0 mesi Valore p del log-rank 9,63*10^(-8) |
Tempo (mesi)
| Numero di pazienti ancora a rischio |
||||||||||||||||||
| Tempo |
0 |
2 |
4 |
6 |
8 |
10 |
12 |
14 |
16 |
18 |
20 |
22 |
24 |
26 |
28 |
30 |
32 |
34 |
| Kisqali |
334 |
294 |
277 |
257 |
240 |
227 |
207 |
196 |
188 |
176 |
164 |
132 |
97 |
46 |
17 |
11 |
1 |
0 |
| Placebo |
334 |
279 |
265 |
239 |
219 |
196 |
179 |
156 |
138 |
124 |
110 |
93 |
63 |
34 |
10 |
7 |
2 |
0 |
Fig. 2. MONALEESA-2: curva di Kaplan-Meier per la PFS secondo la valutazione dello sperimentatore (data di chiusura del database 2 gennaio 2017)
Sono state condotte analisi di PFS in sottogruppi predefiniti basati su fattori prognostici e caratteristiche basali per valutare la coerenza interna dell'effetto del trattamento. Una riduzione del rischio di progressione della malattia o di morte a favore del gruppo trattato con Kisqali e letrozolo è stata osservata in tutti i singoli sottogruppi di pazienti in base all'età, all'etnia, alla chemioterapia adiuvante o neoadiuvante o alla terapia ormonale precedente, all'interessamento epatico e/o polmonare e alle metastasi ossee esclusive. L'effetto è stato evidente nei pazienti con metastasi epatiche e/o polmonari (HR 0,561 [95 % CI: 0,424; 0,743], la mediana della PFS [mPFS] era di 24,8 mesi con la combinazione di Kisqali e letrozolo rispetto a 13,4 mesi con letrozolo da solo) o senza metastasi epatiche e/o polmonari (HR 0,597 [95 % CI: 0,426; 0,837], mPFS di 27,6 mesi rispetto a 18,2 mesi).
I risultati aggiornati relativi alla risposta globale e alla frequenza di efficacia clinica sono riportati nella Tabella 3.
Tabella 3. MONALEESA-2: risultati di efficacia (ORRa, CBRb) secondo la valutazione dello sperimentatore (data di chiusura del database 2 gennaio 2017)
| Analisi |
Kisqali e letrozolo (%, 95 % CI) |
Placebo e letrozolo (%, 95 % CI) |
valore pb |
| Popolazione analizzabile |
N = 334 |
N = 334 |
|
| Tasso di risposta globalea |
42,5 (37,2; 47,8) |
28,7 (23,9; 33,6) |
9,18 × 10-5 |
| Tasso di efficacia clinicab |
79,9 (75,6; 84,2) |
73,1 (68,3; 77,8) |
0,018 |
| Pazienti con malattia misurabile |
n = 257 |
n = 245 |
|
| Tasso di risposta globalea |
54,5 (48,4; 60,6) |
38,8 (32,7; 44,9) |
2,54 × 10-4 |
| Tasso di efficacia clinicab |
80,2 (75,3; 85,0) |
71,8 (66,2; 77,5) |
0,018 |
| a TGR: tasso di risposta globale = frazione di pazienti con risposta completa + risposta parziale. b CBR: tasso di efficacia clinica = frazione di pazienti con risposta completa + risposta parziale (+ malattia stabile o risposta incompleta/assenza di progressione della malattia per ≥ 24 settimane). b Il valore p è stato ottenuto mediante il test chi-quadro unilaterale di Cochran-Mantel-Haenszel. |
|||
Analisi finale della RFS
I risultati dell'analisi finale della risposta globale (RFS) nell'intera popolazione studiata sono riportati nella Tabella 4 e nella Figura 3.
Tabella 4. MONALEESA-2: risultati della valutazione dell'efficacia (RFS) (data di chiusura della raccolta dati – 10 giugno 2021)
| Sopravvivenza globale, popolazione analizzata complessivamente |
Kisqali e letrozolo N = 334 |
Placebo e letrozolo N = 334 |
| Numero di eventi, n [%] |
181 (54,2) |
219 (65,6) |
| Mediana SG [mesi] (IC 95%) |
63,9 (52,4; 71,0) |
51,4 (47,2; 59,7) |
| Rapporto dei rischia (IC 95%) |
0,765 (0,628; 0,932) |
|
| Valore p b |
0,004 |
|
| SG senza complicazioni, (%) (IC 95%) |
||
| 24 mesi |
86,6 (82,3; 89,9) |
85,0 (80,5; 88,4) |
| 60 mesi |
52,3 (46,5; 57,7) |
43,9 (38,3; 49,4) |
| 72 mesi |
44,2 (38,5; 49,8) |
32,0 (26,8; 37,3) |
| IC – intervallo di confidenza. a Il rapporto dei rischi è stato ottenuto mediante il modello di Cox stratificato. b Il valore p è stato ottenuto mediante il test log-rank stratificato unilaterale (p < 0,0219 per affermare una maggiore efficacia). La stratificazione è stata effettuata in base allo stato di metastasi nei polmoni e/o nel fegato mediante tecnologia interattiva di risposta (IRT). |
||
| Moments of censorship Ribociclib (N = 334) Placebo (N = 334) |
| Numero di eventi: ribociclib: 181, placebo: 219 Rapporto di rischio = 0,765 Mediana di Kaplan-Meier: ribociclib: 63,9 mesi placebo: 51,4 mesi Valore p del log-rank = 0,004 |
| Tempo (mesi) |
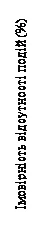 |
| Numero di pazienti ancora a rischio |
| Tempo Ribociclib Placebo |
Fig. 3. MONALEESA-2: curva di Kaplan-Meier per la SFP nella popolazione complessiva (data di chiusura del database 10 giugno 2021)
Test log-rank e modello di Cox stratificato per lo stato di metastasi epatiche e/o polmonari secondo IRT.
Valore p unilaterale ottenuto dal test log-rank stratificato.
Studio CLEE011E2301 (MONALEESA-7)
Kisqali è stato valutato in uno studio clinico randomizzato, in doppio cieco, controllato con placebo, multicentrico di Fase III, che ha coinvolto donne in pre- o perimenopausa con carcinoma mammario ormono-recettore-positivo, HER2-negativo e in stadio avanzato, trattate in combinazione con un inibitore dell’aromatasi (IA) o tamoxifene e goserelina, rispetto a placebo in combinazione con un IA o tamoxifene e goserelina. I pazienti nello studio MONALEESA-7 non avevano ricevuto precedente terapia endocrina per il carcinoma mammario in stadio avanzato.
Complessivamente, 672 pazienti sono stati randomizzati in rapporto 1:1 a ricevere Kisqali alla dose di 600 mg più un IA/tamoxifene e goserelina (n = 335) oppure placebo più un IA/tamoxifene e goserelina (n = 337), stratificati in base alla presenza di metastasi epatiche e/o polmonari (sì [n = 344 (51,2 %)] o no [n = 328 (48,8 %)]), alla chemioterapia precedente per malattia avanzata (sì [n = 120 (17,9 %)] o no [n = 552 (82,1 %)]) e al farmaco aggiuntivo nella terapia endocrina combinata (IA e goserelina [n = 493 (73,4 %)] o tamoxifene e goserelina [n = 179 (26,6 %)]). Le caratteristiche demografiche e basali della malattia erano bilanciate e comparabili tra i gruppi di studio. Kisqali è stato somministrato per via orale alla dose di 600 mg al giorno per 21 giorni consecutivi seguiti da una pausa terapeutica di 7 giorni, in combinazione con un IA (letrozolo 2,5 mg oppure anastrozolo 1 mg) o tamoxifene (20 mg) per via orale una volta al giorno per 28 giorni e goserelina (3,6 mg) per via sottocutanea ogni 28 giorni, fino a progressione della malattia o insorgenza di tossicità inaccettabile. Non era consentito il passaggio da placebo a Kisqali durante lo studio o dopo la progressione della malattia. Non era consentita neppure la modifica del farmaco aggiuntivo nella terapia endocrina combinata.
L’età media dei pazienti inclusi nello studio era di 44 anni (range da 25 a 58), e il 27,7 % dei pazienti aveva meno di 40 anni. La maggior parte dei pazienti apparteneva alla razza caucasica (57,7 %), asiatica (29,5 %) o afrodiscendente (2,8 %); quasi tutti i pazienti (99,0 %) avevano uno stato funzionale basale ECOG pari a 0 o 1. Alla randomizzazione, tra questi 672 pazienti, il 14 % aveva ricevuto chemioterapia per malattia metastatica, il 32,6 % chemioterapia in regime adiuvante e il 18,0 % in regime neoadiuvante; il 39,6 % aveva ricevuto terapia endocrina in regime adiuvante e lo 0,7 % in regime neoadiuvante. Nello studio E2301, il 40,2 % dei pazienti aveva una malattia metastatica de novo (diagnosticata per la prima volta), nel 23,7 % dei pazienti le metastasi erano localizzate solo a livello osseo e nel 56,7 % erano presenti metastasi in organi viscerali.
Il punto finale primario dello studio è stato raggiunto con l’analisi primaria, effettuata dopo aver osservato 318 eventi di sopravvivenza libera da progressione (SLP) secondo la valutazione dell’investigatore, utilizzando i criteri RECIST versione 1.1, nell’intera popolazione (tutti i pazienti randomizzati). I risultati primari di efficacia sono stati confermati dai risultati di SLP ottenuti tramite valutazione radiologica centrale indipendente sui dati mascherati. Il tempo mediano di follow-up al momento dell’analisi primaria della SLP era di 19,2 mesi.
Nella popolazione complessiva dello studio, i risultati di efficacia hanno dimostrato un miglioramento statisticamente significativo della SLP nei pazienti trattati con Kisqali più IA/tamoxifene e goserelina, rispetto ai pazienti trattati con placebo più IA/tamoxifene e goserelina (hazard ratio 0,553; IC 95 %: 0,441; 0,694; valore p unilaterale dal test log-rank stratificato 9,83 × 10-8), con un effetto terapeutico clinicamente rilevante.
La mediana della SLP è stata di 23,8 mesi (IC 95 %: 19,2; non stimabile (NS)) nei pazienti trattati con Kisqali più IA/tamoxifene e goserelina e di 13,0 mesi (IC 95 %: 11,0; 16,4) nei pazienti trattati con placebo più IA/tamoxifene e goserelina.
La distribuzione della SLP è riassunta nella curva di Kaplan-Meier per la SLP riportata nella Figura 4.
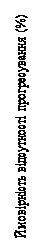
| Momenti di censura Ribociclib (N = 335) Placebo (N = 337) |
| Numero di eventi Ribociclib – 131, placebo – 187 Rapporto di rischio 0,553 IC 95% [0,441; 0,694] Mediana di Kaplan-Meier Ribociclib – 23,8 mesi Placebo – 13,0 mesi Valore p del log-rank 9,83*10^(-8) |
Tempo (mesi)
| Numero di pazienti ancora a rischio |
||||||||||||||||
| Tempo (mesi) |
0 |
2 |
4 |
6 |
8 |
10 |
12 |
14 |
16 |
18 |
20 |
22 |
24 |
26 |
28 |
30 |
| Kisqali |
335 |
301 |
284 |
264 |
245 |
235 |
219 |
178 |
136 |
90 |
54 |
40 |
20 |
3 |
1 |
0 |
| Placebo |
337 |
273 |
248 |
230 |
207 |
183 |
165 |
124 |
94 |
62 |
31 |
24 |
13 |
3 |
1 |
0 |
Fig. 4. MONALEESA-7: curva di Kaplan-Meier per PFS nella popolazione generale basata sulla valutazione dell’investigatore
I risultati di PFS basati sulla valutazione radiologica centrale indipendente e in cieco dei dati di un sottogruppo casualmente selezionato di circa il 40 % dei pazienti randomizzati hanno confermato i risultati primari sull’efficacia basati sulla valutazione dell’investigatore (hazard ratio 0,427; IC 95 %: 0,288; 0,633).
Al momento dell’analisi primaria di PFS, i dati sulla sopravvivenza globale erano ancora incompleti, con 89 (13 %) eventi di morte osservati (HR 0,916 [IC 95 %: 0,601; 1,396]).
La percentuale di risposta globale (ORR), secondo la valutazione dell’investigatore basata sui criteri RECIST versione 1.1, è risultata più alta nel gruppo trattato con Kisqali (40,9 %; IC 95 %: 35,6; 46,2) rispetto al gruppo placebo (29,7 %; IC 95 %: 24,8; 34,6; p = 0,00098). La percentuale di efficacia clinica (CBR) è risultata più alta nel gruppo trattato con Kisqali (79,1 %; IC 95 %: 74,8; 83,5) rispetto al gruppo placebo (69,7 %; IC 95 %: 64,8; 74,6; p = 0,002).
Nell’analisi di un sottogruppo precedentemente definito di 495 pazienti trattati con Kisqali o placebo in combinazione con un inibitore dell’aromatasi (AI) e goserelina, la mediana di PFS è stata di 27,5 mesi (IC 95 %: 19,1; NE) nel sottogruppo trattato con Kisqali e AI e di 13,8 mesi (IC 95 %: 12,6; 17,4) nel sottogruppo trattato con placebo e AI [HR: 0,569; IC 95 %: 0,436; 0,743]. I risultati dell’efficacia sono riportati nella Tabella 5 e le curve di Kaplan-Meier per PFS sono mostrate in Figura 5.
| Tabella 5. MONALEESA-7: risultati della valutazione di efficacia (PFS) nei pazienti trattati con terapia ormonale inibitrice delle gonadotropine |
||
| Parametri |
Kisqali e terapia ormonale inibitrice delle gonadotropine e goserelina N = 248 |
Placebo e terapia ormonale inibitrice delle gonadotropine e goserelina N = 247 |
| Survival libera da progressionea |
||
| Mediana PFS [mesi] (IC 95%) |
27,5 (19,1; NE) |
13,8 (12,6; 17,4) |
| Rapporto dei rischi (IC 95%) |
0,569 (0,436; 0,743) |
|
| IC – intervallo di confidenza; N – numero di pazienti; NE – non stimato. a PFS basato sulla valutazione radiologica dello sperimentatore. |
||
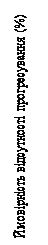 |
| Momenti di censura Ribociclib (N = 248) Placebo (N = 247) |
| Numero di eventi Ribociclib – 92, placebo – 132 Rapporto di rischio 0,569 IC 95% [0,436; 0,743] Mediana di Kaplan-Meier Ribociclib – 27,5 mesi Placebo – 13,8 mesi |
Tempo (mesi)
| Numero di pazienti ancora a rischio |
||||||||||||||||
| Tempo (mesi) |
0 |
2 |
4 |
6 |
8 |
10 |
12 |
14 |
16 |
18 |
20 |
22 |
24 |
26 |
28 |
30 |
| Ribociclib |
248 |
223 |
212 |
199 |
183 |
175 |
163 |
132 |
100 |
66 |
38 |
27 |
15 |
2 |
1 |
0 |
| Placebo |
247 |
195 |
177 |
163 |
149 |
138 |
126 |
95 |
72 |
48 |
25 |
19 |
9 |
2 |
1 |
0 |
Fig. 5. MONALEESA-7: curva di Kaplan-Meier per PFS basata sulla valutazione dello sperimentatore nei pazienti trattati con CDK4/6
I risultati di efficacia relativi alla percentuale di risposta globale (ORR) e alla percentuale di efficacia clinica (CBR), secondo la valutazione dello sperimentatore basata sui criteri RECIST versione 1.1, sono riportati nella tabella 6.
| Tabella 6. MONALEESA-7: risultati della valutazione dell'efficacia (ORR, CBR) basati sulla valutazione dello sperimentatore nei pazienti trattati con inibitori del CYP450 |
||
| Analisi |
Kisqali e inibitori del CYP450 e goserelina (%, 95 % CI) |
Placebo e inibitori del CYP450 e goserelina (%, 95 % CI) |
| Popolazione per l'analisi |
N = 248 |
N = 247 |
| Tasso di risposta globale (ORR)a |
39,1 (33,0; 45,2) |
29,1 (23,5; 34,8) |
| Tasso di beneficio clinico (CBR)b |
80,2 (75,3; 85,2) |
67,2 (61,4; 73,1) |
| Pazienti con malattia misurabile |
n = 192 |
n = 199 |
| Tasso di risposta globalea |
50,5 (43,4; 57,6) |
36,2 (29,5; 42,9) |
| Tasso di beneficio clinico b |
81,8 (76,3; 87,2) |
63,8 (57,1; 70,5) |
| a ORR: proporzione di pazienti con risposta completa + risposta parziale. b CBR: proporzione di pazienti con risposta completa + risposta parziale + (malattia stabile o risposta incompleta/assenza di progressione della malattia per ≥ 24 settimane). |
||
I risultati nel sottogruppo trattato con Kisqali e ISNA sono stati coerenti tra i sottogruppi per età, razza, chemioterapia adiuvante/neoadiuvante o terapia ormonale precedente, coinvolgimento epatico e/o polmonare e metastasi solo ossee.
Un aggiornamento successivo dei dati sulla sopravvivenza globale (data di chiusura del database: 30 novembre 2018) è riportato nella Tabella 7 e nelle Figure 6 e 7.
Nella seconda analisi OS, lo studio ha raggiunto il suo principale obiettivo secondario, dimostrando un miglioramento statisticamente significativo della OS.
Tabella 7. MONALEESA-7: risultati della valutazione dell'efficacia (OS) (data di chiusura del database: 30 novembre 2018)
| Analisi aggiornata |
||
| Sopravvivenza globale, popolazione totale dello studio |
Kisqali 600 mg N = 335 |
Placebo N = 337 |
| Numero di eventi, n [%] |
83 (24,8) |
109 (32,3) |
| Mediana SG [mesi] (95 % CI) |
NE (NE, NE) |
40,9 (37,8; NE) |
| Rapporto dei rischi (95 % CI) |
0,712 (0,535; 0,948) |
|
| Valore pa |
0,00973 |
|
| Sopravvivenza globale, sottogruppo NSAID |
Kisqali 600 mg n = 248 |
Placebo n = 247 |
| Numero di eventi, n [%] |
61 (24,6) |
80 (32,4) |
| Mediana SG [mesi] (95 % CI) |
NE (NE, NE) |
40,7 (37,4; NE) |
| Rapporto dei rischi (95 % CI) |
0,699 (0,501; 0,976) |
|
| CI – intervallo di confidenza; NE – non stimato; N – numero di pazienti. a Il valore p è stato ottenuto mediante il test log-rank unilaterale, stratificato per presenza di metastasi polmonari e/o epatiche, chemioterapia precedente per malattia avanzata e trattamento aggiuntivo nell’ambito della terapia endocrina combinata, mediante tecnologia di risposta interattiva (IRT). |
||
| Momenti di censura Ribociclib (N = 335) Placebo (N = 337) |
| Numero di casi Ribociclib – 83, placebo – 109 Rapporto di rischio 0,712 IC 95% [0,535; 0,948] Mediana di Kaplan-Meier Ribociclib – NV Placebo – 40,9 mesi Valore p del log-rank 0,00973 |
Tempo (mesi)
Numero di pazienti ancora a rischio
Tempo (mesi)
Ribociclib
Placebo
Fig. 6. MONALEESA-7: curva di Kaplan-Meier per l'analisi finale di OS (data di chiusura del database 30 novembre 2018)
Il test log-rank e il modello di Cox sono stratificati per la presenza di metastasi polmonari e/o epatiche, chemioterapia precedente per malattia avanzata e trattamento aggiuntivo nell'ambito della terapia endocrina combinata, in base alla tecnologia interattiva di randomizzazione (IRT).
| Momenti di censura Ribociclib (N = 248) Placebo (N = 247) |
| Numero di casi Ribociclib – 61, placebo – 80 Rapporto di rischio 0,699 IC 95% [0,501; 0,976] Mediana di Kaplan-Meier Ribociclib – NV Placebo – 40,7 mesi |
Tempo (mesi)
Numero di pazienti ancora a rischio
Tempo (mesi)
Ribociclib
Placebo
Fig. 7. MONALEESA-7: curva di Kaplan-Meier per l'analisi finale di OS nei pazienti trattati con terapia endocrina in prima linea (data di chiusura della raccolta dati: 30 novembre 2018)
Il rapporto dei rischi si basa su un modello di Cox non stratificato.
Inoltre, la probabilità di progressione al trattamento di linea successiva o morte (PFS2) nei pazienti precedentemente trattati con ribociclib nello studio era inferiore rispetto ai pazienti nel gruppo placebo, con un HR pari a 0,692 (IC 95%: 0,548; 0,875) nell'intera popolazione dello studio. La mediana del PFS2 era di 32,3 mesi (IC 95%: 27,6; 38,3) nel gruppo placebo e non raggiunta (IC 95%: 39,4; NE) nel gruppo ribociclib. Risultati simili sono stati osservati nel sottogruppo con terapia endocrina in prima linea, con un HR pari a 0,660 (IC 95%: 0,503; 0,868) e una mediana del PFS2 di 32,3 mesi (IC 95%: 26,9; 38,3) nel gruppo placebo rispetto a non raggiunta (IC 95%: 39,4; NE) nel gruppo ribociclib.
Studio CLEE011F2301 (MONALEESA-3)
Kisqali è stato valutato in uno studio clinico di Fase III, randomizzato, in doppio cieco, controllato con placebo, multicentrico, che ha coinvolto 726 donne in postmenopausa con carcinoma mammario avanzato ormono-recettore-positivo e HER2-negativo, precedentemente non trattate o trattate con un solo ciclo di terapia endocrina precedente in combinazione con fulvestrant rispetto a fulvestrant da solo.
L'età media dei pazienti inclusi in questo studio era di 63 anni (range da 31 a 89). Il 46,7% dei pazienti aveva un'età ≥65 anni, di cui il 13,8% aveva un'età ≥75 anni. I pazienti inclusi nello studio erano di razza caucasica (85,3%), mongoloide (8,7%) e negra (0,7%), e quasi tutti i pazienti (99,7%) avevano uno stato funzionale basale ECOG 0 o 1. Lo studio ha incluso pazienti di prima e seconda linea (dei quali il 19,1% presentava metastasi de novo). Prima dell'inclusione nello studio, il 42,7% dei pazienti aveva ricevuto chemioterapia in regime adiuvante e il 13,1% in regime neoadiuvante, mentre il 58,5% aveva ricevuto terapia endocrina in regime adiuvante e l'1,4% in regime neoadiuvante, e il 21% aveva ricevuto una terapia endocrina precedente per carcinoma mammario avanzato. Nello studio F2301, il 21,2% dei pazienti aveva metastasi solo ossee, e il 60,5% aveva metastasi in organi viscerali.
Analisi primaria
L'endpoint primario dello studio è stato raggiunto nell'analisi primaria, condotta dopo la valutazione di 361 eventi di sopravvivenza libera da progressione (PFS) secondo la valutazione dello sperimentatore basata sui criteri RECIST, versione 1.1, nell'intera popolazione (tutti i pazienti randomizzati; data di chiusura della raccolta dati: 03.11.2017). Il tempo medio di ulteriore follow-up al momento dell'analisi primaria del PFS era di 20,4 mesi.
I risultati della valutazione dell'efficacia primaria hanno dimostrato un miglioramento statisticamente significativo del PFS nei pazienti trattati con Kisqali e fulvestrant rispetto ai pazienti trattati con placebo e fulvestrant, nell'intera popolazione (hazard ratio 0,593; IC 95%: 0,480; 0,732; valore p dal test log-rank stratificato unilaterale 4,1 × 10–7), con una riduzione stimata del rischio relativo di progressione o morte del 41% a favore del gruppo trattato con Kisqali e fulvestrant.
I risultati del PFS basati sulla valutazione radiologica centrale indipendente e in cieco di un sottogruppo casuale di circa il 40% dei pazienti randomizzati hanno confermato i risultati primari di efficacia basati sulla valutazione dello sperimentatore (hazard ratio 0,492; IC 95%: 0,345; 0,703).
Un aggiornamento descrittivo del PFS è stato effettuato in concomitanza con la seconda analisi intermedia di OS; i risultati aggiornati del PFS per l'intera popolazione e per i sottogruppi in base alla terapia endocrina precedente sono riportati nella Tabella 8, mentre la curva di Kaplan-Meier è mostrata nella Figura 8.
| Tabella 8. MONALEESA-3 (F2301): risultati aggiornati (PFS) basati sulla valutazione dello sperimentatore (data di chiusura del data cut – 03.06.2019) |
||
| Parametri |
Kisqali e fulvestrant N = 484 |
Placebo e fulvestrant N = 242 |
| Sopravvivenza libera da progressione, popolazione generale dello studio |
||
| Numero di eventi, n [%] |
283 (58,5) |
193 (79,8) |
| Mediana PFS [mesi] (95 % CI) |
20,6 (18,6; 24,0) |
12,8 (10,9; 16,3) |
| Rapporto dei rischi (95 % CI) |
0,587 (0,488; 0,705) |
|
| Sottogruppo trattamento di prima lineaa |
Kisqali e fulvestrant n = 237 |
Placebo e fulvestrant n = 128 |
| Numero di eventi, n [%] |
112 (47,3) |
95 (74,2) |
| Mediana PFS [mesi] (95 % CI) |
33,6 (27,1; 41,3) |
19,2 (14,9; 23,6) |
| Rapporto dei rischi (95 % CI) |
0,546 (0,415–0,718) |
|
| Sottogruppo trattamento di seconda linea o recidiva precoceb |
Kisqali e fulvestrant n = 237 |
Placebo e fulvestrant n = 109 |
| Numero di eventi, n [%] |
167 (70,5) |
95 (87,2) |
| Mediana PFS [mesi] (95 % CI) |
14,6 (12,5; 18,6) |
9,1 (5,8; 11,0) |
| Rapporto dei rischi (95 % CI) |
0,571 (0,443; 0,737) |
|
| CI – intervallo di confidenza. a Pazienti con carcinoma mammario metastatico de novo senza precedente terapia endocrina e pazienti con recidiva dopo la fine di un trattamento endocrino (neo)adiuvante di 12 mesi. b Pazienti con recidiva durante la terapia adiuvante o entro 12 mesi dal termine della terapia endocrina (neo)adiuvante e pazienti con progressione dopo una linea di terapia endocrina per malattia metastatica. |
||
| Numero di eventi Ribociclib + fulvestrant: 283, placebo + fulvestrant: 193 Rapporto di rischio = 0,587 IC 95% [0,488, 0,705] Mediana di Kaplan-Meier Ribociclib + fulvestrant: 20,6 mesi Placebo + fulvestrant: 12,8 mesi |
| Moments of censorship Ribociclib + fulvestrant (N = 484) Placebo + fulvestrant (N = 242) |
| 100 |
| 80 |
| 60 |
| 40 |
| 20 |
| 0 |
| 0 |
| 2 |
| 4 |
| 6 |
| 8 |
| 10 |
| 12 |
| 14 |
| 16 |
| 18 |
| 20 |
| 22 |
| 24 |
| 26 |
| 28 |
| 30 |
| 32 |
| 34 |
| 36 |
| 38 |
| 40 |
| 42 |
| 44 |
| 46 |
| Tempi (mesi) |
| Placebo |
| Ribociclib |
| 242 |
| 484 |
| 168 |
| 156 |
| 144 |
| 364 |
| 346 |
| 323 |
| 106 |
| 98 |
| 88 |
| 258 |
| 239 |
| 225 |
| 68 |
| 62 |
| 198 |
| 181 |
| 47 |
| 156 |
| 41 |
| 127 |
| 13 |
| 65 |
| 2 |
| 11 |
| 0 |
| 0 |
| 1 |
| 4 |
| 6 |
| 29 |
| 21 |
| 92 |
| 45 |
| 149 |
| 59 |
| 51 |
| 174 |
| 159 |
| 82 |
| 205 |
| 134 |
| 116 |
| 305 |
| 282 |
| 195 |
| 403 |
Fig. 8. MONALEESA-3 (F2301): curva di Kaplan-Meier per PFS in base alla valutazione dello sperimentatore (BPDV) (data di chiusura del data cut-off – 03.06.2019)
I risultati della valutazione dell'efficacia riguardo alla frequenza di risposta globale (FRG) e alla frequenza di efficacia clinica (FEC) secondo la valutazione dello sperimentatore basata sui criteri RECIST versione 1.1 sono riportati nella Tabella 9.
| Tabella 9. MONALEESA-3: risultati della valutazione dell'efficacia (ORR, CBR) basati sulla valutazione dello sperimentatore (data di chiusura del database – 03.11.2017) |
||
| Analisi |
Kisqali e fulvestrante (%, 95 % CI) |
Placebo e fulvestrante (%, 95 % CI) |
| Popolazione analizzabile |
N = 484 |
N = 242 |
| Tasso di risposta globale (ORR)a |
32,4 (28,3; 36,6) |
21,5 (16,3; 26,7) |
| Tasso di beneficio clinico (CBR)b |
70,2 (66,2; 74,3) |
62,8 (56,7; 68,9) |
| Pazienti con malattia misurabile |
n = 379 |
n = 181 |
| Tasso di risposta globalea |
40,9 (35,9; 45,8) |
28,7 (22,1; 35,3) |
| Tasso di beneficio clinico b |
69,4 (64,8; 74,0) |
59,7 (52,5; 66,8) |
| a ORR: proporzione di pazienti con risposta completa + risposta parziale. b CBR: proporzione di pazienti con risposta completa + risposta parziale + (malattia stabile o risposta incompleta/assenza di progressione della malattia per ≥ 24 settimane). |
||
Il rapporto rischi-benefici basato sull'analisi di sottogruppi predefiniti di pazienti trattati con Kisqali e fulvestrante ha dimostrato un'efficacia costante in diversi sottogruppi, inclusi quelli definiti per età, razza, trattamento precedente (alla stadio precoce o al cancro avanzato), chemioterapia adiuvante/neoadiuvante o terapia ormonale precedente, coinvolgimento epatico e/o polmonare e metastasi solo ossee.
Analisi OS
È stata effettuata una seconda analisi OS, endpoint secondario dello studio, rivelando un miglioramento statisticamente significativo della sopravvivenza globale.
I risultati dell'analisi finale della sopravvivenza globale nell'intera popolazione dello studio e nell'analisi dei sottogruppi sono riportati nella Tabella 10 e nella Figura 9.
| Tabella 10. MONALEESA-3 (F2301): risultati di efficacia (SOP) (data di chiusura del reclutamento dati – 03.06.2019) |
||
| Parametri |
Kisqali e fulvestrante |
Placebo e fulvestrante |
| Popolazione totale dello studio |
N = 484 |
N = 242 |
| Numero di eventi, n [%] |
167 (34,5) |
108 (44,6) |
| Mediana SOP [mesi] (IC 95 %) |
NR, (NR, NR) |
40 (37, NR) |
| Rapporto dei rischi (IC 95 %)a |
0,724 (0,568; 0,924) |
|
| Valore p b |
0,00455 |
|
| Sottogruppo trattamento di prima linea |
n = 237 |
n = 128 |
| Numero di eventi, n [%] |
63 (26,6) |
47 (36,7) |
| Rapporto dei rischi (IC 95 %)c |
0,700 (0,479; 1,021) |
|
| Sottogruppo trattamento di seconda linea o recidiva precoce |
n = 237 |
n = 109 |
| Numero di eventi, n [%] |
102 (43,0) |
60 (55,0) |
| Rapporto dei rischi (IC 95 %)c |
0,730 (0,530; 1,004) |
|
| NR – non raggiunta. a Il rapporto dei rischi è stato ottenuto dal modello di Cox PH stratificato per presenza di metastasi polmonari e/o epatiche e terapia endocrina precedente. b Il valore p unidirezionale è stato ottenuto dal test log-rank stratificato per presenza di metastasi polmonari e/o epatiche e terapia endocrina precedente in base all'IRT. Il valore p è unidirezionale e viene confrontato con il valore limite di 0,01129, determinato secondo la funzione di dipendenza della probabilità di errore di tipo I dalla frazione di informazione richiesta di Lan-DeMets (O'Bryan-Fleming), per un livello di significatività complessivo di 0,025. c Il rapporto dei rischi è stato ottenuto dal modello di Cox PH non stratificato. |
||
| Numero di eventi Ribociclib + fulvestrante: 167; placebo + fulvestrante: 108 Rapporto di rischio = 0,724 IC 95% [0,568; 0,924] Mediana di Kaplan-Meier Ribociclib + fulvestrante: NR Placebo + fulvestrante: 40,0 mesi Valore p del log-rank = 0,00455 |
| Moments of censorship Ribociclib + fulvestrant (N = 484) Placebo + fulvestrant (N = 242) |
| 242 |
| 233 |
| 227 |
| 223 |
| 218 |
| 213 |
| 207 |
| 199 |
| 194 |
| 187 |
| 184 |
| 174 |
| 169 |
| 159 |
| 155 |
| 147 |
| 141 |
| 134 |
| 107 |
| 64 |
| 37 |
| 14 |
| 3 |
| 0 |
| 100 |
| 80 |
| 60 |
| 40 |
| 20 |
| 0 |
| 0 |
| 2 |
| 4 |
| 6 |
| 8 |
| 10 |
| 12 |
| 14 |
| 16 |
| 18 |
| 20 |
| 22 |
| 24 |
| 26 |
| 28 |
| 30 |
| 32 |
| 34 |
| 36 |
| 38 |
| 40 |
| 42 |
| 44 |
| 46 |
| 48 |
| 484 |
| 470 |
| 454 |
| 444 |
| 436 |
| 428 |
| 414 |
| 402 |
| 397 |
| 389 |
| 374 |
| 365 |
| 348 |
| 334 |
| 326 |
| 309 |
| Tempi (mesi) |
| Placebo |
| Ribociclib |
| 300 |
| 287 |
| 237 |
| 159 |
| 92 |
| 41 |
| 14 |
| 2 |
| 0 |
| 0 |
Fig. 9. MONALEESA-3 (F2301): curva di Kaplan-Meier per la SLP (set di dati per l'analisi completa [SDAC]) (data di chiusura del database: 03.06.2019)
Il test log-rank e il modello di Cox sono stati stratificati per la presenza di metastasi polmonari e/o epatiche, chemioterapia precedente per malattia avanzata e componente della combinazione endocrina per IRT.
Il tempo alla progressione durante la terapia di linea successiva o morte (TTP2) nei pazienti del gruppo Kisqali è risultato maggiore rispetto ai pazienti del gruppo placebo (HR [hazard ratio]: 0,670 [IC 95 %: 0,542; 0,830]) nella popolazione complessiva dello studio. La mediana del TTP2 è stata di 39,8 mesi (IC 95 %: 32,5; NR) per il gruppo Kisqali e di 29,4 mesi (IC 95 %: 24,1; 33,1) per il gruppo placebo.
Pazienti di età avanzata
Tra tutti i pazienti trattati con Kisqali negli studi MONALEESA-2 e MONALEESA-3, sono stati inclusi pazienti di età ≥ 65 anni e ≥ 75 anni. Non sono state osservate differenze generali tra questi pazienti e quelli più giovani riguardo alla sicurezza o all'efficacia del trattamento con Kisqali (vedere il paragrafo «Modalità di somministrazione e posologia»).
Pazienti con compromissione renale
Nei tre studi principali (MONALEESA-2, MONALEESA-3 e MONALEESA-7), 510 (53,8 %) pazienti con funzionalità renale normale, 341 (36 %) con compromissione renale lieve e 97 (10,2 %) con compromissione renale moderata hanno ricevuto trattamento con ribociclib. Nessun paziente con compromissione renale grave è stato incluso. I dati relativi alla SLP sono risultati simili nei pazienti con compromissione renale lieve e moderata trattati con ribociclib alla dose iniziale di 600 mg rispetto ai pazienti con normale funzionalità renale. Il profilo di sicurezza nei pazienti con compromissione della funzione renale è stato in generale paragonabile a quello dei pazienti senza compromissione della funzione renale (vedere il paragrafo «Effetti indesiderati»).
Bambini
L’Agenzia europea per i medicinali ha rinunciato all’obbligo di presentare i risultati degli studi con Kisqali in tutte le sottopopolazioni pediatriche nel cancro al seno (vedere le informazioni sull’uso pediatrico nel paragrafo «Modalità di somministrazione e posologia»).
Farmacocinetica
La farmacocinetica del ribociclib è stata studiata in pazienti con cancro avanzato dopo somministrazione orale in dosi giornaliere comprese tra 50 e 1200 mg. Volontari sani hanno ricevuto dosi orali singole nell’intervallo da 400 mg a 600 mg o dosi ripetute di 400 mg al giorno (8 giorni).
Assorbimento
Il valore geometrico medio della biodisponibilità assoluta del ribociclib dopo una singola dose orale di 600 mg è stato del 65,8 % nei volontari sani.
Il tempo per raggiungere la Cmax (Tmax) dopo somministrazione orale di ribociclib è stato di 1–4 ore. È stata osservata una leggera sovraproporzionalità nell’aumento dell’esposizione (Cmax e AUC) del ribociclib nell’intervallo di dosi studiato (da 50 a 1200 mg). Dopo somministrazione ripetuta una volta al giorno, lo stato stazionario è stato generalmente raggiunto entro 8 giorni e la cumulazione del ribociclib si è verificata con un coefficiente geometrico medio di 2,51 (intervallo: da 0,97 a 6,40).
Effetto del cibo
Rispetto alla somministrazione a digiuno, la somministrazione orale di ribociclib in compresse rivestite con film, in dose singola di 600 mg con un pasto ricco di calorie e grassi, non ha influenzato la velocità e l’entità dell’assorbimento del ribociclib.
Distribuzione
Il legame del ribociclib con le proteine plasmatiche umane in vitro è stato di circa il 70 % e non è risultato dipendente dalla concentrazione del farmaco (da 10 a 10 000 ng/ml). Il ribociclib si distribuisce uniformemente tra globuli rossi e plasma con un rapporto medio sangue/plasma in vivo di 1,04. Secondo l’analisi farmacocinetica popolazionale, il volume apparente di distribuzione allo stato stazionario (Vss/F) è stato di 1090 l.
Biotrasformazione
Studi in vitro e in vivo hanno dimostrato che nell’uomo il ribociclib è metabolizzato principalmente nel fegato, principalmente tramite CYP3A4. Dopo somministrazione orale di [14C] ribociclib in dose singola di 600 mg nell’uomo, i principali percorsi metabolici del ribociclib sono stati l’ossidazione (dealchilazione, ossigenazione C e/o N, ossidazione (-2H)) e le loro combinazioni. I metaboliti della Fase I del ribociclib sono stati coniugati nella Fase II tramite acetilazione N, solfatazione, coniugazione con cisteina, glicosilazione e glucuronidazione. Il ribociclib è stato il principale derivato circolante plasmatico del farmaco. I principali metaboliti circolanti sono stati il metabolita M13 (CCI284, idrossilazione N), M4 (LEQ803, demetilazione N) e M1 (glucuronide secondario). L’attività clinica (proprietà farmacologiche e sicurezza) del ribociclib è stata principalmente attribuita al composto originale e solo in minima parte ai metaboliti circolanti.
Il ribociclib è stato ampiamente metabolizzato, con il farmaco immodificato che rappresentava il 17,3 % e il 12,1 % della dose nelle feci e nelle urine rispettivamente. Il metabolita LEQ803 è stato rilevato in quantità significative nelle feci, rappresentando circa il 13,9 % e il 3,74 % della dose somministrata nelle feci e nelle urine rispettivamente. Molti altri metaboliti sono stati identificati nelle feci e nelle urine in quantità minime (≤ 2,78 % della dose somministrata).
Eliminazione
Allo stato stazionario con una dose di 600 mg in pazienti con cancro avanzato, il valore geometrico medio della emivita effettiva nel plasma (basato sul coefficiente di cumulazione) è stato di 32,0 ore (CV 63 %) e il valore geometrico medio del clearance apparente (CL/F) per via orale è stato di 25,5 l/ora (CV 66 %). Sulla base dei dati dell’analisi farmacocinetica popolazionale, si ritiene che l’esposizione al ribociclib nelle pazienti con cancro al seno in stadio precoce sarà leggermente inferiore rispetto a quella delle pazienti con cancro al seno avanzato che ricevono la stessa dose. Il valore geometrico medio dell’emivita terminale apparente nel plasma (T1/2) del ribociclib è stato compreso tra 29,7 e 54,7 ore e il valore geometrico medio del CL/F del ribociclib è stato compreso tra 39,9 e 77,5 l/ora con una dose di 600 mg nei volontari sani in tutti gli studi.
Il ribociclib e i suoi metaboliti sono eliminati principalmente attraverso l’intestino e solo in minima parte attraverso i reni. In 6 volontari sani di sesso maschile, dopo somministrazione orale di una dose singola di [14C] ribociclib, il 91,7 % della dose radioattiva totale somministrata è stato eliminato entro 22 giorni; la principale via di eliminazione è stata attraverso le feci (69,1 %), mentre il 22,6 % della dose è stato eliminato attraverso le urine.
Linearità/non linearità
È stata osservata una leggera sovraproporzionalità nell’aumento dell’esposizione (Cmax e AUC) del ribociclib nell’intervallo di dosi studiato (da 50 a 1200 mg) dopo somministrazione sia singola che ripetuta. Questa analisi è limitata dalla piccola dimensione del campione per la maggior parte delle coorti che ricevono dosi specifiche; il maggior numero di dati è disponibile nella coorte con dose di 600 mg.
Gruppi particolari di pazienti
Compromissione della funzione renale
L’impatto della compromissione renale sulla farmacocinetica del ribociclib è stato valutato in uno studio che ha incluso 14 volontari sani con funzionalità renale normale (velocità di filtrazione glomerulare assoluta [VFGa] ≥ 90 ml/min), 8 soggetti con compromissione renale lieve (VFGa da 60 a < 90 ml/min), 6 con compromissione renale moderata (VFGa da 30 a < 60 ml/min), 7 con compromissione renale grave (VFGa da 15 a < 30 ml/min) e 3 con insufficienza renale allo stadio terminale (VFGa < 15 ml/min), ai quali è stato somministrato ribociclib in dose singola di 400 mg.
L’AUCinf è aumentata rispettivamente di 1,6, 1,9 e 2,7 volte e la Cmax di 1,8, 1,8 e 2,3 volte nei soggetti con compromissione renale lieve, moderata e grave rispetto all’esposizione nei soggetti con normale funzionalità renale. Poiché gli studi di efficacia e sicurezza del ribociclib hanno incluso una grande percentuale di pazienti con compromissione renale lieve (vedere il paragrafo «Farmacodinamica»), i dati relativi ai pazienti con compromissione renale moderata o grave nello studio con compromissione renale sono stati anche confrontati con i dati combinati di soggetti con funzionalità renale normale e compromissione renale lieve. Rispetto ai dati combinati di soggetti con funzionalità renale normale e compromissione renale lieve, l’AUCinf è aumentata di 1,6 e 2,2 volte e la Cmax di 1,5 e 1,9 volte rispettivamente nei soggetti con compromissione renale moderata e grave. Il rapporto molare per i soggetti con insufficienza renale allo stadio terminale non è stato calcolato a causa del numero ridotto di soggetti, ma i risultati indicano un aumento dell’esposizione al ribociclib simile o leggermente maggiore rispetto ai soggetti con compromissione renale grave.
L’impatto della compromissione renale sulla farmacocinetica del ribociclib è stato anche valutato in pazienti con carcinoma mammario localmente avanzato o metastatico inclusi negli studi di efficacia e sicurezza, in cui i pazienti hanno ricevuto una dose iniziale di 600 mg (vedere il paragrafo «Farmacodinamica»). L’analisi dei dati farmacocinetici nei sottogruppi di pazienti con carcinoma mammario localmente avanzato o metastatico dopo assunzione orale di 600 mg di ribociclib come dose singola o dosi ripetute ha mostrato che l’AUCinf e la Cmax del ribociclib nei pazienti con compromissione renale lieve (n = 57) o moderata (n = 14) erano paragonabili a quelli nei pazienti con normale funzionalità renale (n = 86), indicando l’assenza di un impatto clinicamente rilevante della compromissione renale lieve o moderata sull’esposizione al ribociclib.
Compromissione della funzione epatica
Dati da uno studio farmacocinetico in soggetti senza cancro indicano che la compromissione epatica di grado lieve non ha influenzato l’esposizione al ribociclib (vedere il paragrafo «Modalità di somministrazione e posologia»). L’esposizione media al ribociclib nei pazienti con compromissione epatica moderata (rapporto dei valori geometrici medi [RVM]: 1,44 per Cmax; 1,28 per AUCinf) e grave (RVM: 1,32 per Cmax; 1,29 per AUCinf) è risultata aumentata di meno di due volte (vedere il paragrafo «Modalità di somministrazione e posologia»).
I dati dell’analisi farmacocinetica popolazionale, che includevano 160 pazienti con carcinoma mammario localmente avanzato o metastatico e funzionalità epatica normale e 47 pazienti con compromissione epatica di grado lieve, hanno inoltre dimostrato che la compromissione epatica di grado lieve non ha influenzato l’esposizione al ribociclib. L’uso di ribociclib non è stato studiato in pazienti con carcinoma mammario con compromissione epatica moderata o grave.
Effetto dell’età, del peso corporeo, del sesso e dell’etnia del paziente
L’analisi farmacocinetica popolazionale ha mostrato che l’età, il peso corporeo e il sesso del paziente non hanno avuto un impatto clinicamente significativo sull’esposizione sistemica al ribociclib tale da richiedere un aggiustamento della dose. I dati sulle differenze farmacocinetiche legate all’etnia sono troppo limitati per trarre conclusioni.
Dati di interazione in vitro
Effetto del ribociclib sugli enzimi del citocromo P450
In vitro, il ribociclib è un inibitore reversibile di CYP1A2, CYP2E1 e CYP3A4/5 e un inibitore dipendente dal tempo di CYP3A4/5 a concentrazioni clinicamente rilevanti. Studi in vitro hanno mostrato che il ribociclib non inibisce l’attività di CYP2A6, CYP2B6, CYP2C8, CYP2C9, CYP2C19 e CYP2D6 a concentrazioni clinicamente rilevanti. Il ribociclib non ha potenziale di inibizione dipendente dal tempo di CYP1A2, CYP2C9 e CYP2D6.
Dati in vitro indicano che il ribociclib non ha potenziale di indurre enzimi UGT o enzimi CYP come CYP2C9, CYP2C19 e CYP3A4 tramite il recettore pregnano-X (PXR). Pertanto, è improbabile che Kisqali influenzi i substrati di questi enzimi. I dati in vitro non sono sufficienti per escludere il potenziale del ribociclib di indurre CYP2B6 tramite il recettore androstano costitutivo (CAR).
Effetto dei trasportatori sul ribociclib
In vitro, il ribociclib è un substrato di P-gp, ma sulla base dei dati di bilancio di massa, è improbabile che l’inibizione di P-gp o BCRP influenzi l’esposizione al ribociclib alle dosi terapeutiche. Il ribociclib non è un substrato dei trasportatori epatici di uptake OATP1B1, OATP1B3 o OCT-1 in vitro.
Effetto del ribociclib sui trasportatori
Studi in vitro hanno mostrato che il ribociclib ha potenziale di inibire l’attività dei trasportatori di farmaci P-gp, BCRP, OATP1B1/1B3, OCT1, OCT2, MATE1 e BSEP. Il ribociclib non ha inibito OAT1, OAT3 o MRP2 a concentrazioni clinicamente rilevanti in vitro.
Dati preclinici di sicurezza
Sicurezza farmacologica
Studi di sicurezza cardiologica in vivo nei cani hanno dimostrato un prolungamento dose-dipendente e concentrazione-dipendente dell’intervallo QTc a esposizioni raggiungibili nei pazienti dopo la dose raccomandata di 600 mg. Inoltre, a livelli di esposizione più elevati (circa 5 volte superiore alla Cmax clinica prevista) esiste la possibilità di indurre extrasistoli ventricolari.
Tossicità da dosi ripetute
Negli studi di tossicità da dosi ripetute (schema di trattamento: 3 settimane di trattamento / 1 settimana di pausa) della durata fino a 27 settimane nei ratti e fino a 39 settimane nei cani, l’organo principale su cui il ribociclib ha esercitato effetti tossici è risultato essere il sistema epatobiliare (cambiamenti proliferativi, colestasi, calcoli simili alla sabbia nella cistifellea e bile densa). Gli organi bersaglio associati all’azione farmacologica del ribociclib negli studi di dosi ripetute comprendono il midollo osseo (ipocellularità), il sistema linfatico (esaurimento del tessuto linfatico), la mucosa intestinale (atrofia), la pelle (atrofia), le ossa (ridotta formazione ossea), i reni (degenerazione e rigenerazione contemporanea delle cellule epiteliali tubulari) e i testicoli (atrofia). Oltre alle alterazioni atrofiche osservate nei testicoli, che tendevano a essere reversibili, tutti gli altri cambiamenti sono risultati completamente reversibili dopo un periodo di 4 settimane senza trattamento. L’esposizione al ribociclib negli animali negli studi di tossicità è stata generalmente inferiore o paragonabile a quella osservata nei pazienti che ricevono dosi multiple di 600 mg/giorno per carcinoma mammario localmente avanzato o metastatico (basata sull’AUC).
Tossicità riproduttiva / effetto sulla fertilità
Il ribociclib ha mostrato fetotossicità e teratogenicità nei ratti e nei conigli a dosi che non erano tossiche per la femmina gravida. Dopo esposizione prenatale nei ratti, si è osservato un aumento dei casi di perdita fetale post-impianto e riduzione del peso corporeo fetale, mentre nei conigli il ribociclib si è dimostrato teratogeno a esposizioni ≤ 1,5 volte superiori a quelle nell’uomo alla dose raccomandata più alta di 600 mg/giorno (basata sull’AUC).
Nei ratti, si è osservata riduzione del peso corporeo fetale associata a cambiamenti scheletrici considerati temporanei e/o legati al minor peso corporeo fetale. Nei conigli, si è osservato un effetto avverso sullo sviluppo dell’embrione/feto, evidenziato da un aumento della frequenza di anomalie dello sviluppo fetale (malformazioni e anomalie esterne, viscerali e scheletriche) e della crescita fetale (riduzione del peso corporeo fetale). Queste anomalie includevano lobi polmonari ridotti/piccoli, un vaso aggiuntivo sull’arco aortico e ernia diaframmatica, assenza di un lobo aggiuntivo o lobi polmonari parzialmente fusi e lobi polmonari aggiuntivi ridotti/piccoli (30 e 60 mg/kg), tredicesime costole aggiuntive/riduttive, osso ioide deformato e riduzione del numero di falangi della prima dita. Non si sono verificati casi di morte embrionale/fetale.
In uno studio sulla fertilità nelle femmine di ratto, il ribociclib non ha influenzato la funzione riproduttiva, la fertilità o le prime fasi dell’embriogenesi a qualsiasi dose fino a 300 mg/kg/giorno (probabilmente a un’esposizione inferiore o pari all’esposizione clinica nei pazienti alla dose giornaliera raccomandata più alta di 600 mg/giorno basata sull’AUC).
Il ribociclib non è stato valutato in studi sulla fertilità maschile. Tuttavia, negli studi di tossicità nei ratti e nei cani sono stati riportati cambiamenti atrofici nei testicoli a esposizioni inferiori o pari a quelle nell’uomo alla dose giornaliera raccomandata più alta di 600 mg/giorno (basata sull’AUC). Questi effetti potrebbero essere correlati all’effetto antiproliferativo diretto sulle cellule germinali testicolari, portando all’atrofia dei tubuli seminiferi.
Il ribociclib e i suoi metaboliti attraversano facilmente nel latte nei ratti. L’esposizione al ribociclib nel latte è stata superiore rispetto a quella nel plasma.
Genotossicità
Negli studi di genotossicità in sistemi batterici in vitro e in sistemi in vitro e in vivo nei mammiferi con e senza attivazione metabolica, non sono state osservate evidenze di potenziale genotossico del ribociclib.
Carcinogenicità
La carcinogenicità del ribociclib è stata valutata in uno studio di 2 anni nei ratti.
La somministrazione orale di ribociclib per 2 anni ha portato a un aumento della frequenza di tumori epiteliali dell’endometrio, iperplasia ghiandolare e squamosa dell’utero/cervice nelle femmine di ratto a dosi ≥ 300 mg/kg/giorno, nonché a un aumento della frequenza di tumori follicolari della tiroide nei maschi di ratto alla dose di 50 mg/kg/giorno. L’esposizione media allo stato stazionario (AUC0–24 h) nelle femmine e nei maschi di ratto in cui sono state osservate alterazioni neoplastiche è stata rispettivamente 1,2 e 1,4 volte superiore rispetto ai pazienti alla dose raccomandata di 600 mg/giorno. L’esposizione media allo stato stazionario (AUC0–24 h) nelle femmine e nei maschi di ratto in cui sono state osservate alterazioni neoplastiche è stata rispettivamente 2,2 e 2,5 volte superiore rispetto ai pazienti alla dose di 400 mg/giorno.
Altre alterazioni proliferative non neoplastiche includevano un aumento di lesioni focali epatiche (cellule basofile e chiare) e iperplasia delle cellule interstiziali testicolari (cellule di Leydig) nei maschi di ratto a dosi ≥ 5 mg/kg/giorno e 50 mg/kg/giorno rispettivamente.
I potenziali meccanismi delle alterazioni della tiroide nei maschi di ratto includono l’induzione specifica per i roditori di enzimi microsomiali epatici, ritenuta non rilevante per l’uomo. L’effetto sull’utero/cervice e sulle cellule interstiziali testicolari (cellule di Leydig) potrebbe essere correlato a una prolungata ipoprolattinemia dovuta all’inibizione dell’attività CDK4 nelle cellule lattotrofe dell’ipofisi, che altera l’asse ipotalamo-ipofisi-gonadi.
Qualsiasi potenziale aumento del rapporto estrogeni/progesterone nell’uomo dovuto a questo meccanismo è compensato dall’effetto inibitorio della terapia antiestrogenica concomitante sulla sintesi degli estrogeni, poiché Kisqali è indicato per l’uso in combinazione con agenti che riducono i livelli di estrogeni.
Considerando le importanti differenze tra roditori e uomo riguardo alla sintesi e al ruolo della prolattina, si prevede che questo meccanismo non avrà conseguenze nell’uomo.
Caratteristiche cliniche
Indicazioni
Carcinoma mammario in fase precoce
Kisqali è indicato in associazione con un inibitore dell’aromatasi per la terapia adiuvante di donne con carcinoma mammario in fase precoce, ormono-recettore positivo (HR+) ed HER2-negativo (recettore del fattore di crescita epidermico umano di tipo 2), con alto rischio di recidiva (criteri di selezione vedere nel paragrafo «Proprietà farmacologiche»).
Nelle donne in premenopausa o perimenopausa o negli uomini, la terapia con inibitore dell’aromatasi deve essere somministrata in combinazione con un agonista del rilascio dell’ormone luteinizzante (LHRH).
Carcinoma mammario localmente avanzato o metastatico
Kisqali è indicato in associazione con un inibitore dell’aromatasi o con fulvestrant per il trattamento di donne con carcinoma mammario localmente avanzato o metastatico, HR+ e HER2-negativo, come terapia endocrina iniziale oppure per il trattamento di donne precedentemente trattate con terapia endocrina.
Nelle donne in premenopausa o perimenopausa, la terapia endocrina deve essere somministrata in combinazione con un agonista LHRH.
Controindicazioni
Ipersensibilità al principio attivo o all’arachide, alla soia o ad uno qualsiasi degli eccipienti del medicinale.
Interazioni con altri medicinali ed altre forme di interazione
Medicinali che possono aumentare la concentrazione plasmatica di ribociclib
Ribociclib è metabolizzato principalmente tramite CYP3A4. Pertanto, i medicinali che possono influenzare l’attività dell’enzima CYP3A4 possono influenzare anche la farmacocinetica di ribociclib. La somministrazione concomitante di ritonavir (inibitore potente di CYP3A4, 100 mg due volte al giorno per 14 giorni) con ribociclib in dose singola di 400 mg a volontari sani ha determinato un aumento dell’esposizione (AUCinf) e della concentrazione massima (Cmax) di ribociclib rispettivamente di 3,2 e 1,7 volte rispetto alla somministrazione di ribociclib in dose singola di 400 mg da solo. La Cmax e l’AUClast di LEQ803 (metabolita principale di ribociclib, che rappresenta meno del 10% dell’esposizione della sostanza originale) sono diminuite rispettivamente del 96% e del 98%. La modellizzazione farmacocinetica basata su considerazioni fisiologiche (physiologically based pharmacokinetic modelling (PBPK)) con somministrazione concomitante di ritonavir (100 mg due volte al giorno) ha mostrato che la Cmax e l’AUC0-24h di ribociclib allo stato stazionario (400 mg una volta al giorno) sono aumentate rispettivamente di 1,5 e 1,8 volte.
È necessario evitare la somministrazione concomitante di inibitori potenti di CYP3A4, come ad esempio claritromicina, indinavir, itraconazolo, ketoconazolo, lopinavir, ritonavir, nefazodone, nelfinavir, posaconazolo, saquinavir, telaprevir, telitromicina, verapamil e voriconazolo (vedere paragrafo «Avvertenze particolari e precauzioni di impiego»). Si deve considerare l’uso di medicinali alternativi che siano inibitori meno potenti di CYP3A4, e si deve effettuare un monitoraggio degli effetti avversi (EA) associati a ribociclib (vedere paragrafi «Modalità di somministrazione e posologia», «Avvertenze particolari e precauzioni di impiego» e «Farmacocinetica»).
Se non è possibile evitare la somministrazione concomitante di Kisqali con un inibitore potente di CYP3A4, si deve modificare la dose di Kisqali come descritto nel paragrafo «Modalità di somministrazione e posologia». Tuttavia, dati clinici riguardo a tale aggiustamento della dose non sono disponibili. A causa della variabilità interindividuale, l’aggiustamento della dose raccomandato potrebbe non essere ottimale per tutti i pazienti; pertanto, si raccomanda un monitoraggio attento degli EA associati a ribociclib. In caso di comparsa di effetti tossici da ribociclib, si deve aggiustare la dose del medicinale o sospendere temporaneamente il trattamento fino alla risoluzione dei sintomi di tossicità (vedere paragrafi «Modalità di somministrazione e posologia» e «Farmacocinetia»). Dopo l’interruzione dell’inibitore potente di CYP3A4, almeno dopo 5 emivite di eliminazione (vedere il foglio illustrativo dell’inibitore di CYP3A4 in caso di dubbi), il trattamento con Kisqali deve essere ripreso alla stessa dose utilizzata prima dell’inizio dell’inibitore potente di CYP3A4.
La PBPK ha dimostrato che, con ribociclib in dose di 600 mg, un inibitore moderato di CYP3A4 (eritromicina) può aumentare la Cmax e l’AUC di ribociclib allo stato stazionario rispettivamente di 1,1 e 1,1 volte. La PBPK ha dimostrato che, con ribociclib in dose di 400 mg, un inibitore moderato di CYP3A4 può aumentare la Cmax e l’AUC di ribociclib allo stato stazionario rispettivamente di 1,1 e 1,2 volte. Si prevede che, con ribociclib in dose di 200 mg una volta al giorno, si osservi un aumento della Cmax e dell’AUC allo stato stazionario rispettivamente di 1,3 e 1,5 volte. All’inizio del trattamento con inibitori deboli o moderati di CYP3A4, non è necessario aggiustare la dose di ribociclib. Tuttavia, si raccomanda di effettuare un monitoraggio degli EA associati a ribociclib.
I pazienti devono essere informati della necessità di evitare il consumo di pompelmo e succo di pompelmo. È noto che questi prodotti inibiscono gli enzimi del citocromo CYP3A4 e possono aumentare l’esposizione a ribociclib.
Medicinali che possono ridurre la concentrazione plasmatica di ribociclib
La somministrazione concomitante a volontari sani di rifampicina (induttore potente di CYP3A4, 600 mg al giorno per 14 giorni) con ribociclib in dose singola di 600 mg ha ridotto l’AUCinf e la Cmax di ribociclib rispettivamente dell’89% e dell’81% rispetto alla somministrazione di ribociclib in dose singola di 600 mg da solo. La Cmax di LEQ803 è aumentata di 1,7 volte, mentre l’AUCinf è diminuita del 27%. Pertanto, la somministrazione concomitante di induttori potenti di CYP3A4 può determinare una riduzione dell’esposizione e, di conseguenza, un rischio di perdita di efficacia. È necessario evitare la somministrazione concomitante di induttori potenti di CYP3A4, come ad esempio fenitoina, rifampicina, carbamazepina e erba di San Giovanni (Hypericum perforatum). Si deve considerare la possibilità di utilizzare un medicinale alternativo che non induca o abbia una capacità minima di indurre CYP3A4.
L’effetto degli induttori moderati di CYP3A4 sull’esposizione a ribociclib non è stato studiato. La PBPK ha dimostrato che un inibitore moderato di CYP3A4 (efavirenz) può ridurre la Cmax e l’AUC di ribociclib allo stato stazionario rispettivamente del 55% e del 74%, e con ribociclib in dose di 600 mg rispettivamente del 52% e del 71%. Pertanto, la somministrazione concomitante di induttori moderati di CYP3A4 può determinare una riduzione dell’esposizione e, di conseguenza, un rischio di riduzione dell’efficacia, in particolare nei pazienti che ricevono ribociclib in dose di 400 mg o 200 mg una volta al giorno.
Medicinali la cui concentrazione plasmatica può essere influenzata da Kisqali
Ribociclib è un inibitore moderato/potente di CYP3A4 e può interagire con sostanze medicinali metabolizzate tramite CYP3A4, determinando un aumento della concentrazione sierica del medicinale somministrato contemporaneamente.
La somministrazione concomitante a volontari sani di midazolam (substrato di CYP3A4) con dosi ripetute di Kisqali (400 mg) ha aumentato l’esposizione a midazolam del 280% (3,8 volte) rispetto alla somministrazione di midazolam da solo. La modellizzazione con PBPK ha dimostrato che con Kisqali in dose di 600 mg si prevede un aumento dell’AUC di midazolam di 5,2 volte. Pertanto, in generale, quando si somministra ribociclib con altri medicinali, è necessario consultare le raccomandazioni relative alla somministrazione concomitante con inibitori di CYP3A4 riportate nei fogli illustrativi di tali medicinali. Si raccomanda cautela nella somministrazione concomitante con substrati sensibili di CYP3A4 con indice terapeutico stretto (vedere paragrafo «Avvertenze particolari e precauzioni di impiego»). Potrebbe essere necessaria una riduzione della dose di substrati sensibili di CYP3A4 con indice terapeutico stretto, come ad esempio alfentanil, ciclosporina, everolimus, fentanil, sirolimus e tacrolimus, poiché ribociclib può aumentarne l’esposizione.
Si deve evitare la somministrazione concomitante di ribociclib con i seguenti substrati di CYP3A4: alfuzosin, amiodarone, cisapride, pimozide, chinidina, ergotamina, diidroergotamina, quetiapina, lovastatina, simvastatina, sildenafil, midazolam, triazolam.
La somministrazione concomitante a volontari sani di caffeina (substrato di CYP1A2) con dosi ripetute di Kisqali (400 mg) ha aumentato l’esposizione a caffeina del 20% (1,2 volte) rispetto alla somministrazione di caffeina da sola. Con la dose clinicamente rilevante di 600 mg, la modellizzazione farmacocinetica fisiologicamente basata ha previsto solo un debole effetto inibitorio di ribociclib sui substrati di CYP1A2 (aumento dell’AUC < 2 volte).
Medicinali che sono substrati di trasportatori
Studi in vitro hanno dimostrato che ribociclib ha il potenziale di inibire l’attività dei trasportatori farmaci P-gp, BCRP, OATP1B1/1B3, OCT1, OCT2, MATE1 e BSEP. Si raccomanda cautela e monitoraggio per segni di tossicità durante il trattamento concomitante con substrati sensibili di questi trasportatori che hanno un indice terapeutico stretto, come ad esempio digossina, pitavastatina, pravastatina, rosuvastatina e metformina.
Interazione del medicinale con il cibo
Kisqali può essere assunto indipendentemente dall’assunzione di cibo (vedere paragrafi «Modalità di somministrazione e posologia» e «Farmacocinetica»).
Medicinali che aumentano il pH gastrico
Ribociclib è caratterizzato da elevata solubilità a pH 4,5 o inferiore e in ambiente biologico (pH 5,0 e 6,5). La somministrazione concomitante di ribociclib con medicinali che aumentano il pH gastrico non è stata valutata in studi clinici; tuttavia, né nell’analisi farmacocinetica di popolazione né nell’analisi farmacocinetica non compartimentale è stata osservata alterazione dell’assorbimento di ribociclib.
Interazione tra ribociclib e letrozolo
I dati di uno studio clinico su pazienti con carcinoma mammario e di un’analisi farmacocinetica di popolazione hanno dimostrato l’assenza di interazione tra ribociclib e letrozolo dopo somministrazione concomitante di questi medicinali.
Interazione tra ribociclib e anastrozolo
I dati di uno studio clinico su pazienti con carcinoma mammario hanno dimostrato l’assenza di interazione clinicamente significativa tra ribociclib e anastrozolo dopo somministrazione concomitante di questi medicinali.
Interazione tra ribociclib e fulvestrant
I dati di uno studio clinico su pazienti con carcinoma mammario hanno dimostrato l’assenza di effetto clinicamente significativo di fulvestrant sull’esposizione a ribociclib dopo somministrazione concomitante di questi medicinali.
Interazione tra ribociclib e tamoxifene
I dati di uno studio clinico su pazienti con carcinoma mammario hanno dimostrato che, dopo somministrazione concomitante di ribociclib e tamoxifene, l’esposizione a tamoxifene aumenta di circa due volte.
Interazione tra ribociclib e contraccettivi orali
Non sono stati condotti studi di interazione tra ribociclib e contraccettivi orali (vedere paragrafo «Gravidanza e allattamento»).
Interazioni previste
Farmaci antiaritmici e altri medicinali che possono causare prolungamento dell’intervallo QT
Si deve evitare la somministrazione concomitante di Kisqali con medicinali in grado di causare prolungamento dell’intervallo QT, come gli antiaritmici (ad esempio amiodarone, disopiramide, procainamide, chinidina e sotalolo) e altri farmaci (ad esempio clorochina, halofantrina, claritromicina, ciprofloxacina, levofloxacina, azitromicina, aloperidolo, metadone, moxifloxacina, bepridil, pimozide e ondansetron per somministrazione endovenosa) (vedere paragrafo «Avvertenze particolari e precauzioni di impiego»). L’uso concomitante di Kisqali con tamoxifene non è raccomandato (vedere paragrafi «Indicazioni», «Avvertenze particolari e precauzioni di impiego» e «Farmacodinamica»).
Caratteristiche particolari di utilizzo
Malattia viscerale potenzialmente letale
L'efficacia e la sicurezza dell'uso di ribociclib nei pazienti con malattia viscerale potenzialmente letale non sono state studiate.
Neutropenia
A seconda della gravità della neutropenia, il trattamento con Kisqali può richiedere una sospensione temporanea, una riduzione della dose o l'interruzione del trattamento, come descritto nelle sezioni «Modalità di somministrazione e dosi» e «Effetti indesiderati».
Tossicità epatobiliare
Prima di iniziare il trattamento con Kisqali, è necessario effettuare test di funzionalità epatica. Dopo l'inizio del trattamento, la funzionalità epatica deve essere monitorata (vedi sezioni «Modalità di somministrazione e dosi» e «Effetti indesiderati»).
A seconda del grado di aumento delle transaminasi, il trattamento con Kisqali può richiedere una sospensione temporanea, una riduzione della dose o l'interruzione del trattamento, come descritto nelle sezioni «Modalità di somministrazione e dosi» e «Effetti indesiderati». Non sono disponibili raccomandazioni per i pazienti con livelli basali di aspartato aminotransferasi (AST) / alanina aminotransferasi (ALT) ≥ 3.
Allungamento dell'intervallo QT
L'uso di Kisqali deve essere evitato nei pazienti con allungamento dell'intervallo QTc o con un rischio significativo di svilupparlo. Questi includono pazienti con:
- sindrome da QT lungo;
- malattia cardiaca non controllata o significativa, inclusi infarto miocardico recente, insufficienza cardiaca congestizia, angina instabile e bradiaritmie;
- squilibri elettrolitici.
L'uso di Kisqali deve essere evitato con farmaci in grado di prolungare l'intervallo QTc e/o con potenti inibitori del CYP3A4, poiché ciò potrebbe portare a un allungamento clinicamente significativo dell'intervallo QTcF (vedi sezioni «Modalità di somministrazione e dosi», «Interazioni con altri medicinali e altre forme di interazione» e «Farmacodinamica»). Se il trattamento con Kisqali e un potente inibitore del CYP3A4 non può essere evitato, la dose di Kisqali deve essere aggiustata come descritto nella sezione «Modalità di somministrazione e dosi».
In base ai risultati dello studio E2301 (MONALEESA-7), l'uso di Kisqali in combinazione con tamoxifene non è raccomandato (vedi sezioni «Effetti indesiderati» e «Farmacodinamica»).
Carcinoma mammario in stadio precoce
Nello studio O12301C (NATALEE), un aumento dell'intervallo QTcF > 60 ms rispetto al valore basale è stato osservato in 19 (0,8 %) pazienti trattati con Kisqali più un inibitore dell'aromatasi.
Prima dell'inizio del trattamento, deve essere effettuato un ECG. Il trattamento con Kisqali deve essere iniziato solo in pazienti con valori di QTcF inferiori a 450 ms. L'ECG deve essere ripetuto circa al giorno 14 del primo ciclo e successivamente in base alle indicazioni cliniche (vedi sezioni «Modalità di somministrazione e dosi» e «Effetti indesiderati»).
Nei pazienti con carcinoma mammario in stadio precoce, deve essere effettuato un adeguato monitoraggio degli elettroliti sierici (inclusi potassio, calcio, fosforo e magnesio) prima dell'inizio del trattamento, all'inizio dei primi 6 cicli e successivamente in base alle indicazioni cliniche. Eventuali anomalie devono essere corrette prima dell'inizio del trattamento con Kisqali e durante il trattamento stesso.
A seconda dell'allungamento del QT osservato, il trattamento con Kisqali può richiedere una sospensione temporanea, una riduzione della dose o l'interruzione del trattamento, come descritto nella tabella 14 (vedi sezioni «Modalità di somministrazione e dosi», «Effetti indesiderati» e «Farmacocinetica»).
Carcinoma mammario localmente avanzato o metastatico
Nello studio E2301 (MONALEESA-7), un allungamento dell'intervallo QTcF > 60 ms rispetto al valore basale è stato osservato in 14/87 (16,1 %) pazienti trattati con Kisqali in combinazione con tamoxifene e in 18/245 (7,3 %) pazienti trattati con Kisqali in combinazione con un inibitore non steroideo dell'aromatasi (NSAI).
Prima dell'inizio del trattamento, deve essere effettuato un ECG. Il trattamento con Kisqali deve essere iniziato solo in pazienti con valori di QTcF inferiori a 450 ms. L'ECG deve essere ripetuto circa al giorno 14 del primo ciclo e successivamente in base alle indicazioni cliniche (vedi sezioni «Modalità di somministrazione e dosi» e «Effetti indesiderati»).
Nei pazienti con carcinoma mammario localmente avanzato o metastatico, deve essere effettuato un adeguato monitoraggio degli elettroliti sierici (inclusi potassio, calcio, fosforo e magnesio) prima dell'inizio del trattamento, all'inizio dei primi 6 cicli e successivamente in base alle indicazioni cliniche. Eventuali anomalie devono essere corrette prima dell'inizio del trattamento con Kisqali e durante il trattamento stesso.
A seconda dell'allungamento del QT osservato durante il trattamento, il trattamento con Kisqali può richiedere una sospensione temporanea, una riduzione della dose o l'interruzione del trattamento, come descritto nelle sezioni «Modalità di somministrazione e dosi», «Effetti indesiderati» e «Farmacocinetica».
Reazioni cutanee gravi
Durante il trattamento con Kisqali sono stati riportati casi di necrolisi epidermica tossica (TEN). Se si osservano segni e sintomi indicativi di reazioni cutanee gravi (ad esempio eruzioni cutanee progressive e diffuse, spesso con vesciche o coinvolgimento delle mucose), il trattamento con Kisqali deve essere immediatamente interrotto.
Malattia polmonare interstiziale / polmonite
Malattia polmonare interstiziale (ILD) / polmonite sono state osservate con l'uso di Kisqali. I pazienti devono essere monitorati per sintomi polmonari che indicano ILD/polmonite, che possono includere ipossia, tosse e dispnea, e deve essere effettuata una correzione della dose (vedi sezione «Modalità di somministrazione e dosi», tabella 15).
A seconda della gravità dell'ILD / polmonite, che può essere fatale, può essere necessaria la sospensione temporanea, la riduzione della dose o l'interruzione definitiva del trattamento con Kisqali (vedi sezione «Modalità di somministrazione e dosi», tabella 15).
Aumento della creatinina ematica
Ribociclib può causare un aumento della creatinina ematica come inibitore dei trasportatori renali, ovvero il trasportatore cationico organico 2 (OCT2) e la proteina 1 di estrusione multi-farmaco e di tossine (MATE1), coinvolti nella secrezione attiva della creatinina dai tubuli prossimali (vedi sezione «Interazioni con altri medicinali e altre forme di interazione»). In caso di aumento della creatinina ematica durante il trattamento, si raccomanda di effettuare ulteriori valutazioni della funzionalità renale per escludere insufficienza renale.
Substrati del CYP3A4
Ribociclib è un potente inibitore del CYP3A4 alla dose di 600 mg e un inibitore moderato del CYP3A4 alla dose di 400 mg. Pertanto, ribociclib può interagire con farmaci metabolizzati dal CYP3A4, portando a un aumento delle concentrazioni sieriche dei substrati del CYP3A4 (vedi sezione «Interazioni con altri medicinali e altre forme di interazione»). Si raccomanda cautela nell'uso concomitante con substrati sensibili del CYP3A4 con un indice terapeutico stretto e di consultare le istruzioni per l'uso di questi farmaci riguardo all'uso concomitante con inibitori del CYP3A4.
Insufficienza renale
Secondo le stime, la dose iniziale raccomandata di 200 mg nei pazienti con grave insufficienza renale determina un'esposizione approssimativamente del 45 % inferiore rispetto alla dose iniziale standard di 600 mg nei pazienti con carcinoma mammario localmente avanzato o metastatico e funzionalità renale normale. L'efficacia a questa dose iniziale non è stata studiata. Kisqali deve essere usato con cautela nei pazienti con grave insufficienza renale e deve essere effettuato un rigoroso monitoraggio per segni di tossicità (vedi sezioni «Modalità di somministrazione e dosi» e «Farmacocinetica»).
Donne in età fertile
Alle donne in età fertile deve essere raccomandato di utilizzare metodi contraccettivi efficaci durante il trattamento con Kisqali e per almeno 21 giorni dopo l'ultima dose (vedi sezione «Uso in gravidanza o durante l'allattamento»).
Lecitina di soia
Kisqali contiene lecitina di soia. I pazienti con ipersensibilità all'arachide o alla soia non devono assumere Kisqali (vedi sezione «Controindicazioni»).
Uso in gravidanza o durante l'allattamento
Donne in età fertile / contraccezione
Prima di iniziare il trattamento con Kisqali, deve essere effettuato un test di gravidanza.
Alle donne in età fertile che ricevono Kisqali deve essere raccomandato di utilizzare metodi contraccettivi efficaci (ad esempio metodo contraccettivo barriera doppio) durante il trattamento e per almeno 21 giorni dopo l'interruzione del trattamento con Kisqali.
Gravidanza
Non esistono studi adeguati e ben controllati su donne in gravidanza. Sulla base dei dati ottenuti negli animali, ribociclib può avere effetti dannosi sul feto se somministrato a una donna in gravidanza (vedi sezione «Dati di sicurezza preclinici»). L'uso di Kisqali non è raccomandato in donne in gravidanza e in donne in età fertile che non utilizzano metodi contraccettivi.
Allattamento
Non è noto se ribociclib passi nel latte materno umano. Non esistono dati sull'effetto di ribociclib sul neonato allattato al seno o sull'effetto di ribociclib sulla produzione di latte. Ribociclib e i suoi metaboliti passano facilmente nel latte di ratti in allattamento. Le pazienti che ricevono Kisqali non devono allattare per almeno 21 giorni dopo l'ultima dose.
Fertilità
Non esistono dati clinici sull'effetto di ribociclib sulla fertilità. Sulla base degli studi sugli animali, ribociclib può influenzare la fertilità negli uomini con potenziale riproduttivo (vedi sezione «Dati di sicurezza preclinici»).
Capacità di guidare veicoli o di usare macchinari
Kisqali può avere un effetto trascurabile sulla capacità di guidare veicoli o di usare macchinari. Ai pazienti deve essere raccomandato di prestare attenzione durante la guida e l'uso di macchinari in caso di affaticamento, capogiri o vertigini durante il trattamento con Kisqali (vedi sezione «Effetti indesiderati»).
Modalità e dosaggio
Il trattamento con Kisqali deve essere iniziato sotto la supervisione di un medico esperto nell'uso di agenti antineoplastici.
Test per lo stato ormonale positivo HR e stato HER2 negativo
La selezione dei pazienti per il trattamento con Kisqali in base all'espressione di HR e HER2 nel tumore deve essere effettuata mediante un dispositivo diagnostico in vitro (in vitro diagnostic (IVD)) con marcatura CE conforme all'uso previsto. Se un IVD con marcatura CE non è disponibile, deve essere utilizzato un test alternativo validato.
Dosaggio
Carcinoma mammario in fase iniziale
La dose raccomandata è di 400 mg (due compresse rivestite con film da 200 mg) di ribociclib una volta al giorno per 21 giorni consecutivi, seguiti da una pausa di 7 giorni, completando così un ciclo di 28 giorni. Ai pazienti con carcinoma mammario in fase iniziale, Kisqali deve essere somministrato fino al completamento di un trattamento di tre anni, o fino alla ricaduta della malattia o all'insorgenza di tossicità inaccettabile.
Quando Kisqali viene utilizzato in combinazione con un inibitore dell'aromatasi, l'inibitore dell'aromatasi deve essere somministrato per via orale una volta al giorno in modo continuo durante il ciclo di 28 giorni. Per informazioni più dettagliate, si prega di consultare il foglio illustrativo (FI) dell'inibitore dell'aromatasi.
Nel trattamento di donne in premenopausa o perimenopausa o di uomini, l'inibitore dell'aromatasi deve essere associato a un agonista del GnRH.
Carcinoma mammario localmente avanzato o metastatico
La dose raccomandata è di 600 mg (tre compresse rivestite con film da 200 mg) di ribociclib una volta al giorno per 21 giorni consecutivi, seguiti da una pausa di 7 giorni, completando così un ciclo di 28 giorni. Nei pazienti con carcinoma mammario localmente avanzato o metastatico, il trattamento deve proseguire finché persiste l'efficacia clinica della terapia o fino all'insorgenza di tossicità inaccettabile.
Quando Kisqali viene utilizzato in combinazione con un inibitore dell'aromatasi, l'inibitore dell'aromatasi deve essere somministrato per via orale una volta al giorno in modo continuo durante il ciclo di 28 giorni. Per informazioni più dettagliate, si prega di consultare il FI dell'inibitore dell'aromatasi.
Quando Kisqali viene utilizzato in combinazione con fulvestrant, fulvestrant deve essere somministrato per via intramuscolare nel giorno 1, 15 e 29, e successivamente una volta al mese. Per informazioni più dettagliate, si prega di consultare il foglio illustrativo di fulvestrant.
Il trattamento di donne in premenopausa o perimenopausa con le combinazioni approvate di Kisqali deve includere anche agonisti del GnRH, in conformità con le pratiche cliniche locali.
Adattamento del dosaggio
Il trattamento di reazioni avverse gravi o non tollerate può richiedere una sospensione temporanea del trattamento, una riduzione della dose o l'interruzione definitiva di Kisqali. Le indicazioni per la riduzione della dose raccomandata sono riportate nella tabella 11.
Tabella 11. Indicazioni per l'adattamento della dose raccomandata
| Kisqali |
||
| Dose |
Numero di compresse da 200 mg |
|
| Carcinoma mammario in fase iniziale |
||
| Dose iniziale |
400 mg/giorno |
2 |
| Riduzione della dose |
200 mg*/giorno |
1 |
| Carcinoma mammario localmente avanzato o metastatico |
||
| Dose iniziale |
600 mg/giorno |
3 |
| Prima riduzione della dose |
400 mg/giorno |
2 |
| Seconda riduzione della dose |
200 mg*/giorno |
1 |
| * Se fosse necessaria un'ulteriore riduzione della dose al di sotto di 200 mg/giorno, il trattamento deve essere definitivamente interrotto. |
||
Nei tabelle 12-16 sono riassunte le raccomandazioni per l'interruzione del trattamento, la riduzione della dose o la sospensione del farmaco Kisqali nel trattamento di specifiche reazioni avverse. Nella valutazione clinica, il medico deve seguire un piano di gestione personalizzato per ogni paziente, considerando la valutazione del rapporto beneficio/rischio in ciascun singolo caso (vedere la sezione «Proprietà farmacologiche»).
Prima di iniziare il trattamento con Kisqali, deve essere effettuato un emocromo completo (EC). Dopo l'inizio del trattamento, l'EC deve essere eseguito ogni 2 settimane durante i primi 2 cicli, all'inizio di ciascuno dei successivi 4 cicli e successivamente in base alle indicazioni cliniche.
Tabella 12. Adeguamento della dose e trattamento della neutropenia
| Neutropenia |
Grado 1 o 2* (ANC 1000/mm3 – ≤ LNN) |
Grado 3* (ANC 500 – < 1000/mm3) |
Grado 3* neutropenia febbrile** |
Grado 4* (ANC < 500/mm3) |
| Non è necessaria alcuna correzione della dose. |
Sospendere temporaneamente fino al recupero a ≤ grado 2. Riprendere l'assunzione del medicinale Kisqali alla stessa dose. In caso di tossicità di grado 3 ricorrente: sospendere temporaneamente fino al recupero a ≤ grado 2, quindi riprendere l'assunzione del medicinale Kisqali a una dose ridotta di un livello. |
Sospendere temporaneamente fino al recupero a ≤ grado 2. Riprendere l'assunzione del medicinale Kisqali a una dose ridotta di un livello. |
Sospendere temporaneamente fino al recupero a ≤ grado 2. Riprendere l'assunzione del medicinale Kisqali a una dose ridotta di un livello. |
|
| * Grado definito secondo CTCAE, versione 4.03 (CTCAE – Common Terminology Criteria for Adverse Events – criteri comuni di terminologia per la valutazione degli eventi avversi). ** Neutropenia di grado 3 con un singolo episodio di febbre > 38,3 °C (oppure ≥ 38 °C per più di 1 ora e/o infezione concomitante). ANC – conteggio assoluto dei neutrofili; LNN – limite normale inferiore. |
||||
Prima di iniziare il trattamento con Kisqali, è necessario eseguire test di funzionalità epatica (LFT). Dopo l'inizio del trattamento, i LFT devono essere eseguiti ogni 2 settimane durante i primi 2 cicli, all'inizio di ciascuno dei successivi 4 cicli e successivamente in base alle indicazioni cliniche. In caso di alterazioni di grado ≥ 2, si raccomanda un monitoraggio più frequente.
Tabella 13. Aggiustamento della dose e gestione del trattamento in caso di tossicità epatobiliare
| Parametri |
Grado 1* (> LSN – 3 × LSN) |
Grado 2 * (> 3–5 × LSN) |
Grado 3* (> 5–20 × LSN) |
Grado 4* (> 20 × LSN) |
| Aumento di AST e/o ALT rispetto al valore basale** senza aumento della bilirubina totale > 2 × LSN |
Non è necessaria alcuna modifica della dose. |
Valore basale < 2: sospendere temporaneamente fino al ritorno a ≤ valore basale, quindi riprendere Kisqali alla stessa dose. In caso di recidiva di tossicità di grado 2, riprendere Kisqali a una dose ridotta al livello posologico immediatamente inferiore. |
Sospendere temporaneamente Kisqali fino al ritorno a ≤ valore basale, quindi riprendere il trattamento a una dose ridotta al livello posologico immediatamente inferiore. In caso di recidiva di tossicità di grado 3, interrompere definitivamente il trattamento con Kisqali. |
Interrompere definitivamente il trattamento con Kisqali. |
| Valore basale = 2: non è necessaria alcuna interruzione. |
||||
| Aumento contemporaneo di AST e/o ALT insieme ad aumento della bilirubina totale in assenza di colestasi |
Se il paziente presenta un aumento di ALT e/o AST > 3 × LSN insieme ad aumento della bilirubina totale > 2 × LSN, indipendentemente dal valore basale, interrompere definitivamente il trattamento con Kisqali. |
|||
| * Grado definito secondo CTCAE, versione 4.03. ** Valore basale: prima dell'inizio del trattamento. LSN – Limite superiore della norma. |
||||
Prima di iniziare il trattamento con Kisqali, tutti i pazienti devono sottoporsi a un ECG.
Il trattamento con Kisqali deve essere iniziato solo in pazienti con valori di QTcF inferiori a 450 ms.
Dopo l'inizio del trattamento, l'ECG deve essere ripetuto circa il giorno 14 del primo ciclo e successivamente in base alle indicazioni cliniche.
In caso di prolungamento del QTcF durante il trattamento, si raccomanda un monitoraggio più frequente dei pazienti con carcinoma mammario in stadio precoce e con carcinoma mammario localmente avanzato o metastatico.
Tabella 14. Aggiustamento della dose e gestione in caso di prolungamento del QT
| Prolungamento QTcF* |
Cancro al seno in fase precoce |
Cancro al seno localmente avanzato o metastatico |
| > 480 ms – ≤ 500 ms |
Si deve interrompere temporaneamente il trattamento con Kisqali fino al ripristino dell'intervallo QTcF a < 481 ms. |
|
| Riprendere il trattamento con Kisqali alla stessa dose. |
Ridurre la dose al livello immediatamente inferiore. |
|
| Se l'intervallo QTcF aumenta nuovamente a ≥ 481 ms, si deve interrompere temporaneamente il trattamento con Kisqali fino al ripristino dell'intervallo QTcF a < 481 ms, quindi riprendere il trattamento con Kisqali a una dose ridotta al livello immediatamente inferiore. |
||
| > 500 ms |
Si deve interrompere temporaneamente il trattamento con Kisqali fino al ripristino dell'intervallo QTcF a < 481 ms, quindi riprendere il trattamento con Kisqali a una dose ridotta al livello immediatamente inferiore. |
|
| Se l'intervallo QTcF supera 500 ms o se si osserva un aumento superiore a 60 ms rispetto al valore basale in combinazione con tachicardia ventricolare tipo torsione di punta, tachicardia ventricolare polimorfa o segni/sintomi di aritmia grave, si deve interrompere definitivamente il trattamento con Kisqali. |
||
| Nota. Se è necessaria un'ulteriore riduzione della dose al di sotto di 200 mg/giorno, il trattamento con Kisqali deve essere interrotto definitivamente. |
||
Tabella 15. Adeguatezza della dose e trattamento in caso di ICL/pneumonite
| Grado 1* (asintomatico) |
Grado 2* (sintomatico) |
Grado 3 o 4* (grave) |
|
| Malattia polmonare interstiziale/pneumonite |
Non è necessario alcun aggiustamento della dose. Iniziare un’appropriata terapia farmacologica e monitorare clinicamente in base alle indicazioni. |
Sospendere temporaneamente il trattamento con Kisqali fino al recupero fino a ≤ grado 1, quindi riprendere il trattamento alla dose ridotta al livello di dose più vicino**. |
Interrompere definitivamente il trattamento con Kisqali. |
| *La definizione del grado è basata sul CTCAE, versione 4.03. **Nella valutazione della ripresa del trattamento con Kisqali, si deve effettuare una valutazione individuale tra benefici e rischi. Malattia polmonare interstiziale — IHD. |
|||
Tabella 16. Aggiustamento della dose e trattamento in caso di altri effetti tossici*
| Altri effetti tossici |
Grado 1 o 2** |
Grado 3** |
Grado 4** |
| Non è necessaria la modifica della dose. Iniziare la terapia medicamentosa appropriata e monitorare clinicamente in base alle indicazioni. |
Sospendere temporaneamente il trattamento fino al ripristino fino a ≤ grado 1, quindi riprendere il trattamento con Kisqali alla stessa dose. In caso di ricomparsa di tossicità di grado 3, riprendere il trattamento con Kisqali a una dose ridotta al livello posologico immediatamente inferiore. |
Interrompere definitivamente il trattamento con Kisqali. |
|
| * Ad eccezione di neutropenia, epatotossicità, prolungamento dell'intervallo QT e PPE/pneumonite. ** Classificazione del grado secondo CTCAE, versione 4.03. |
|||
Indicazioni per la correzione della dose e altre informazioni sulla sicurezza pertinenti in caso di effetti tossici si trovano nel foglio illustrativo dell'inibitore dell'aromatasi, del fulvestrante o dell'agonista del GnRH utilizzato in associazione.
Correzione della dose in caso di somministrazione concomitante di inibitori potenti del CYP3A4
È consigliabile evitare la somministrazione concomitante di inibitori potenti del CYP3A4 e considerare l'uso di medicinali alternativi che siano inibitori meno potenti del CYP3A4. Se necessario, in caso di somministrazione concomitante di un inibitore potente del CYP3A4 con ribociclib, la dose di Kisqali deve essere ridotta (vedere il paragrafo «Interazioni con altri medicinali ed altre forme di interazione»).
Nei pazienti che assumono 600 mg di ribociclib al giorno e nei quali non è possibile evitare la somministrazione concomitante di un inibitore potente del CYP3A4, la dose deve essere ulteriormente ridotta a 400 mg.
Nei pazienti che assumono 400 mg di ribociclib al giorno e nei quali non è possibile evitare la somministrazione concomitante di un inibitore potente del CYP3A4, la dose deve essere ulteriormente ridotta a 200 mg.
Nei pazienti a cui la dose di ribociclib è già stata ridotta a 200 mg al giorno e nei quali non è possibile evitare la somministrazione concomitante di un inibitore potente del CYP3A4, il trattamento con Kisqali deve essere temporaneamente interrotto.
A causa della variabilità interindividuale, la correzione della dose raccomandata potrebbe non essere ottimale per tutti i pazienti; pertanto, si raccomanda un attento monitoraggio per rilevare segni di tossicità. Dopo l'interruzione dell'inibitore potente del CYP3A4, la dose di Kisqali deve essere riportata alla dose precedente l'inizio dell'inibitore potente del CYP3A4 dopo almeno 5 emivite dell'inibitore potente del CYP3A4 (vedere i paragrafi «Particolari avvertenze e precauzioni per l'uso», «Interazioni con altri medicinali ed altre forme di interazione» e «Farmacocinetica»).
Popolazioni particolari
Alterazioni della funzionalità renale
Non è necessaria alcuna correzione della dose nei pazienti con compromissione renale da lieve a moderata. Nei pazienti con compromissione renale grave si raccomanda una dose iniziale di 200 mg. L'uso di Kisqali nei pazienti con carcinoma mammario e insufficienza renale grave non è stato studiato (vedere i paragrafi «Particolari avvertenze e precauzioni per l'uso», «Farmacodinamica» e «Farmacocinetica»).
Alterazioni della funzionalità epatica
Non è necessaria alcuna correzione della dose nei pazienti con carcinoma mammario in stadio precoce e compromissione della funzionalità epatica (vedere il paragrafo «Farmacocinetica»). Nei pazienti con carcinoma mammario localmente avanzato o metastatico e con compromissione epatica lieve (classe A secondo la classificazione di Child-Pugh) non è necessaria alcuna correzione della dose; nei pazienti con compromissione epatica moderata (classe B secondo la classificazione di Child-Pugh) o grave (classe C secondo la classificazione di Child-Pugh) può verificarsi un'esposizione aumentata (meno del doppio) a ribociclib; pertanto, si raccomanda una dose iniziale di Kisqali di 400 mg una volta al giorno (vedere il paragrafo «Farmacocinetia»).
Pazienti anziani
Nei pazienti di età pari o superiore a 65 anni non è necessaria alcuna correzione della dose (vedere il paragrafo «Farmacocinetica»).
Modalità di somministrazione
Kisqali è destinato all'assunzione orale una volta al giorno indipendentemente dall'assunzione di cibo (vedere il paragrafo «Interazioni con altri medicinali ed altre forme di interazione»). Si raccomanda ai pazienti di assumere la dose giornaliera all'incirca alla stessa ora ogni giorno, preferibilmente al mattino. Se un paziente vomita dopo aver assunto il medicinale o se dimentica di assumere una dose, non deve assumere una dose aggiuntiva nello stesso giorno. La dose successiva deve essere assunta all'ora abituale. Le compresse devono essere inghiottite intere, senza masticarle, frantumarle o dividerle prima di deglutirle. Non assumere una compressa che sia frantumata, crepata o altrimenti compromessa nell'integrità.
Qualsiasi medicinale non utilizzato o i suoi rifiuti devono essere smaltiti in conformità con i requisiti locali.
Uso pediatrico
La sicurezza e l'efficacia di Kisqali nei bambini (sotto i 18 anni) non sono state stabilite. I dati sono mancanti.
Sovradosaggio
I casi di sovradosaggio con Kisqali sono limitati. In caso di sovradosaggio possono manifestarsi sintomi come nausea e vomito. Inoltre, può verificarsi tossicità ematologica (ad esempio neutropenia, trombocitopenia) e allungamento dell'intervallo QT. In tutti i casi di sovradosaggio, se necessario, deve essere istituita una terapia di supporto generale.
Effetti indesiderati
Profilo di sicurezza riassuntivo
Carcinoma mammario in stadio precoce
Nel database combinato, le reazioni avverse (RA) più comuni (≥ 20%), la cui frequenza con l'uso del medicinale Kisqali e di un inibitore dell'aromatasi (IA) era superiore rispetto all'uso dell'IA da solo, sono state neutropenia, infezioni, nausea, cefalea, affaticamento, leucopenia e alterazioni degli indici delle prove funzionali epatiche.
Nel database combinato, le RA più comuni di grado 3/4 (> 2%), la cui frequenza con l'uso del medicinale Kisqali e IA era superiore rispetto all'uso dell'IA da solo, sono state neutropenia, alterazioni degli indici delle prove funzionali epatiche e leucopenia.
Una riduzione della dose a causa di eventi avversi indipendentemente dalla causa è stata osservata nel 22,8% dei pazienti trattati con Kisqali e IA nello studio clinico di Fase III. Il trattamento è stato definitivamente interrotto nell'19,7% dei pazienti trattati con Kisqali e IA nello studio clinico di Fase III.
Carcinoma mammario localmente avanzato o metastatico
Nel database combinato, le RA più comuni (≥ 20%), la cui frequenza con l'uso del medicinale Kisqali in qualsiasi combinazione era superiore rispetto al placebo in qualsiasi combinazione, sono state: neutropenia, infezioni, nausea, affaticamento, diarrea, leucopenia, vomito, cefalea, costipazione, alopecia, tosse, eruzioni cutanee, dolore alla schiena, anemia e alterazioni degli indici delle prove funzionali epatiche.
Nel database combinato, le RA più comuni di grado 3/4 (> 2%), la cui frequenza con l'uso del medicinale Kisqali in qualsiasi combinazione era superiore rispetto al placebo in qualsiasi combinazione, sono state neutropenia, leucopenia, alterazioni degli indici delle prove funzionali epatiche, linfopenia, infezioni, dolore alla schiena, anemia, affaticamento, ipofosfatemia e vomito.
Una riduzione della dose a causa di eventi avversi indipendentemente dalla causa è stata osservata nel 39,5% dei pazienti trattati con Kisqali negli studi clinici di Fase III indipendentemente dalla combinazione. Il trattamento è stato definitivamente interrotto nell'8,7% dei pazienti trattati con Kisqali e qualsiasi combinazione negli studi clinici di Fase III.
Elenco delle reazioni avverse al medicinale
Carcinoma mammario in stadio precoce
Il profilo generale di sicurezza del medicinale Kisqali si basa sui dati ottenuti da 2525 pazienti che hanno assunto Kisqali in combinazione con un IA e sono stati inclusi nello studio clinico randomizzato in aperto di Fase III NATALEE.
La durata media del trattamento con ribociclib nello studio è stata di 33,0 mesi, con il 69,4% dei pazienti che hanno assunto ribociclib per oltre 24 mesi e il 42,8% dei pazienti che hanno completato lo schema terapeutico di 36 mesi con ribociclib.
Carcinoma mammario localmente avanzato o metastatico
Il profilo generale di sicurezza del medicinale Kisqali si basa sui dati combinati ottenuti da 1065 pazienti che hanno ricevuto Kisqali in combinazione con terapia endocrina (582 pazienti in combinazione con un inibitore dell'aromatasi e 483 pazienti in combinazione con fulvestrant) e sono stati inclusi negli studi clinici randomizzati, in doppio cieco, controllati con placebo di Fase III MONALEESA-2, sottogruppo NSAI di MONALEESA-7 e MONALEESA-3.
La durata media del trattamento con il medicinale Kisqali negli studi di Fase III è stata di 19,2 mesi, con il 61,7% dei pazienti che hanno ricevuto il medicinale per ≥ 12 mesi.
Le RA al medicinale osservate negli studi clinici di Fase III (tabella 16) in pazienti con carcinoma mammario in stadio precoce e carcinoma mammario localmente avanzato o metastatico sono elencate per classi di sistemi e organi secondo MedDRA [Medical Dictionary for Regulatory Activities – Dizionario Medico per l'attività regolatoria]. In ciascun gruppo per classi di sistemi e organi, le reazioni avverse al medicinale sono elencate in ordine decrescente di frequenza. In ciascun gruppo per frequenza, le reazioni avverse al medicinale sono elencate in ordine decrescente di gravità. Inoltre, la categoria di frequenza corrispondente per ciascuna reazione avversa al medicinale è definita come segue (CIOMS III): molto comune (≥ 1/10); comune (da ≥ 1/100 a < 1/10); non comune (da ≥ 1/1000 a < 1/100); raro (da ≥ 1/10 000 a < 1/1000); molto raro (< 1/10 000); frequenza non nota (non può essere valutata sulla base dei dati disponibili).
Tabella 17. Reazioni avverse al medicinale osservate negli studi clinici di Fase III e durante la sorveglianza post-marketing
| Frequenza |
Pazienti con carcinoma mammario in stadio precoce che hanno ricevuto ribociclib alla dose iniziale di 400 mg |
Pazienti con carcinoma mammario localmente avanzato o metastatico che hanno ricevuto ribociclib alla dose iniziale di 600 mg |
| Infezioni e infestazioni |
||
| Molto frequente |
Infezioni1 |
Infezioni1 |
| Patologie del sistema emolinfopoietico |
||
| Molto frequente |
Neutropenia, leucopenia |
Neutropenia, leucopenia, anemia, linfopenia |
| Frequente |
Anemia, trombocitopenia, linfopenia |
Trombocitopenia, neutropenia febbrile |
| Non comune |
Neutropenia febbrile |
- |
| Disturbi del metabolismo e della nutrizione |
||
| Molto frequente |
- |
Diminuzione dell'appetito |
| Frequente |
Ipopotassiemia, ipocalcemia, diminuzione dell'appetito |
Ipopotassiemia, ipocalcemia, ipofosfatemia |
| Patologie del sistema nervoso |
||
| Molto frequente |
Cefalea |
Cefalea, capogiro |
| Frequente |
Capogiro |
Vertigini |
| Patologie dell'occhio |
||
| Frequente |
- |
Lacrimazione aumentata, secchezza oculare |
| Patologie cardiache |
||
| Frequente |
- |
Sincopi |
| Patologie respiratorie, toraciche e mediastiniche |
||
| Molto frequente |
Tosse |
Dispnea, tosse |
| Frequente |
Dispnea, malattia polmonare interstiziale (IPD)/polmonite |
Malattia polmonare interstiziale (IPD)/polmonite |
| Patologie gastrointestinali |
||
| Molto frequente |
Nausea, diarrea, costipazione, dolore addominale2 |
Nausea, diarrea, vomito, costipazione, dolore addominale2, stomatite, dispepsia |
| Frequente |
Vomito, stomatite3 |
Disgeusia |
| Patologie epatobiliari |
||
| Frequente |
Epatoressività4 |
Epatoressività4 |
| Patologie della cute e del tessuto sottocutaneo |
||
| Molto frequente |
Alopecia |
Alopecia, eruzione cutanea5, prurito |
| Frequente |
Eruzione cutanea5, prurito |
Secchezza cutanea, eritema, vitiligine |
| Raro |
- |
Eritema multiforme |
| Frequenza non nota |
- |
Necrolisi epidermica tossica (TEN) |
| Patologie del sistema muscoloscheletrico e del tessuto connettivo |
||
| Molto frequente |
- |
Dolore alla schiena |
| Patologie sistemiche e condizioni in relazione al sito di somministrazione |
||
| Molto frequente |
Stanchezza, astenia, aumento della temperatura corporea |
Stanchezza, edema periferico, aumento della temperatura corporea, astenia |
| Frequente |
Edema periferico, dolore orofaringeo |
Dolore orofaringeo, secchezza orale |
| Indagini diagnostiche |
||
| Molto frequente |
Anomalie degli esami di funzionalità epatica6 |
Anomalie degli esami di funzionalità epatica6 |
| Frequente |
Aumento della creatinina nel sangue, allungamento dell'intervallo QT nell'elettrocardiogramma |
Aumento della creatinina nel sangue, allungamento dell'intervallo QT nell'elettrocardiogramma |
| 1 Infezioni: infezioni del tratto urinario, infezioni delle vie respiratorie, gastroenterite (solo nei pazienti con carcinoma mammario localmente avanzato o metastatico), sepsi (solo in < 1 % dei pazienti con carcinoma mammario localmente avanzato o metastatico). 2 Dolore addominale: dolore addominale, dolore nell'area superiore dell'addome. 3 Stomatite nel carcinoma mammario in stadio precoce: stomatite, mucosite. 4 Epatoressività: colisi epatica, danno epatocellulare (solo nei pazienti con carcinoma mammario localmente avanzato o metastatico), danno epatico da farmaco (solo in < 1 % dei pazienti con carcinoma mammario in stadio precoce e nei pazienti con carcinoma mammario localmente avanzato o metastatico), epatoressività, insufficienza epatica (solo nei pazienti con carcinoma mammario localmente avanzato o metastatico), epatite autoimmune (un caso in una paziente con carcinoma mammario in stadio precoce e un caso in una paziente con carcinoma mammario localmente avanzato o metastatico). 5 Eruzione cutanea: eruzione cutanea, eruzione maculopapulare, eruzione cutanea con prurito. 6 Anomalie degli esami di funzionalità epatica: aumento dei livelli di ALT, aumento dei livelli di AST, aumento dei livelli di bilirubina nel sangue. |
||
Descrizione di reazioni avverse specifiche
Neutropenia
Nello studio di Fase III coinvolgente pazienti con carcinoma mammario in stadio precoce, la reazione avversa più comune era la neutropenia (62,5%), mentre una riduzione dei neutrofili di grado 3 o 4 (basata su dati di laboratorio) è stata osservata nel 45,1% dei pazienti trattati con Kisqali e IA.
Nei pazienti con carcinoma mammario in stadio precoce nei quali si è verificata neutropenia di grado 2, 3 o 4, il tempo medio all'insorgenza dell'evento è stato di 0,6 mesi. Il tempo medio per la risoluzione della neutropenia di grado ≥ 3 (fino alla normalizzazione o a un grado < 3) è stato di 0,3 mesi nel gruppo trattato con Kisqali e IA dopo interruzione del trattamento e/o riduzione della dose e/o sospensione del trattamento. La neutropenia febbrile è stata osservata nello 0,3% dei pazienti trattati con Kisqali e IA. La frequenza di interruzione del trattamento a causa della neutropenia è stata bassa (1,1%) nei pazienti che ricevevano Kisqali e IA (vedi sezioni «Modalità di somministrazione e dosaggio» e «Avvertenze particolari e precauzioni di impiego»).
Negli studi di Fase III condotti su pazienti con carcinoma mammario localmente avanzato o metastatico, la reazione avversa più comune era la neutropenia (75,4%), mentre una riduzione dei neutrofili di grado 3 o 4 (basata su dati di laboratorio) è stata osservata nel 62,0% dei pazienti trattati con Kisqali e qualsiasi combinazione.
Nei pazienti con carcinoma mammario localmente avanzato o metastatico nei quali si è verificata neutropenia di grado 2, 3 o 4, il tempo medio all'insorgenza dell'evento è stato di 17 giorni. Il tempo medio per la risoluzione della neutropenia di grado ≥ 3 (fino alla normalizzazione o a un grado < 3) è stato di 12 giorni nei gruppi trattati con Kisqali e qualsiasi combinazione dopo interruzione del trattamento e/o riduzione della dose e/o sospensione del trattamento. La neutropenia febbrile è stata osservata in circa l'1,7% dei pazienti trattati con Kisqali negli studi di Fase III.
La frequenza di interruzione del trattamento a causa della neutropenia è stata bassa (0,8%) (vedi sezioni «Modalità di somministrazione e dosaggio» e «Avvertenze particolari e precauzioni di impiego»).
Tutti i pazienti devono essere istruiti sulla necessità di segnalare immediatamente qualsiasi aumento della temperatura corporea.
Tossicità epatobiliare
Negli studi clinici di Fase III condotti su pazienti con carcinoma mammario in stadio precoce e con carcinoma mammario localmente avanzato o metastatico, è stato osservato un aumento dei livelli delle transaminasi.
Nello studio di Fase III su pazienti con carcinoma mammario in stadio precoce, i casi di tossicità epatobiliare si sono verificati più frequentemente nel gruppo trattato con Kisqali e IA rispetto al gruppo trattato solo con IA (26,4% contro 11,2%), con effetti avversi di grado 3/4 più comuni nei pazienti trattati con Kisqali e IA (8,6% contro 1,7%). Un aumento contemporaneo dei livelli di ALT o AST superiore a tre volte il limite superiore della norma e dei livelli di bilirubina totale superiore a due volte il limite superiore della norma, con fosfatasi alcalina normale, è stato osservato in 8 pazienti trattati con Kisqali e IA (in 6 pazienti i livelli di ALT o AST si sono normalizzati entro 65 e 303 giorni dalla sospensione del trattamento con Kisqali).
L'interruzione temporanea del trattamento a causa di tossicità epatobiliare si è verificata nel 12,4% delle pazienti con carcinoma mammario in stadio precoce trattate con Kisqali e IA, principalmente a causa di aumento dei livelli di ALT (10,1%) e/o aumento dei livelli di AST (6,8%). L'adeguamento della dose a causa di tossicità epatobiliare è stato osservato nel 2,6% dei pazienti trattati con Kisqali e IA, principalmente a causa di aumento dei livelli di ALT (1,9%) e/o aumento dei livelli di AST (0,6%). La sospensione definitiva del trattamento con Kisqali a causa di alterazioni dei test di funzionalità epatica o epatotossicità è stata osservata rispettivamente nell'8,9% e nello 0,1% dei pazienti (vedi sezioni «Modalità di somministrazione e dosaggio» e «Avvertenze particolari e precauzioni di impiego»).
Negli studi clinici di Fase III su pazienti con carcinoma mammario in stadio precoce, l'80,9% (165/204) dei casi di aumento di ALT o AST di grado 3 o 4 si è verificato entro i primi 6 mesi di trattamento. Nei pazienti con aumento di ALT/AST di grado 3 o 4, il tempo medio all'insorgenza di tali eventi è stato di 2,8 mesi nel gruppo trattato con Kisqali e IA. Il tempo medio per la normalizzazione dei valori (fino alla normalizzazione o a un grado ≤ 2) è stato di 0,7 mesi nel gruppo trattato con Kisqali e IA.
Negli studi clinici di Fase III su pazienti con carcinoma mammario localmente avanzato o metastatico, i casi di tossicità epatobiliare si sono verificati più frequentemente nei gruppi trattati con Kisqali e qualsiasi combinazione rispetto ai gruppi trattati con placebo e qualsiasi combinazione (27,3% contro 19,6%), con effetti avversi di grado 3/4 più comuni nei pazienti trattati con Kisqali e qualsiasi combinazione (13,2% contro 6,1%). Aumenti di ALT (11,2% contro 1,7%) e AST (7,8% contro 2,1%) di grado 3 o 4 sono stati osservati rispettivamente nei gruppi trattati con Kisqali e placebo. Un aumento contemporaneo di ALT o AST superiore a tre volte il limite superiore della norma e di bilirubina totale superiore a due volte il limite superiore della norma, con fosfatasi alcalina normale e in assenza di colestasi, è stato osservato in 6 pazienti (4 pazienti nello studio A2301 [MONALEESA-2], nei quali i valori si sono normalizzati entro 154 giorni, e 2 pazienti nello studio F2301 [MONALEESA-3], nei quali i valori si sono normalizzati entro 121 e 532 giorni rispettivamente dopo la sospensione del trattamento con Kisqali). Nessun caso è stato riportato nello studio E2301 (MONALEESA-7).
L'interruzione temporanea del trattamento e/o l'adeguamento della dose a causa di tossicità epatobiliare sono stati necessari nel 12,3% dei pazienti con carcinoma mammario localmente avanzato o metastatico trattati con Kisqali e qualsiasi combinazione, principalmente a causa di aumento dei livelli di ALT (7,9%) e/o aumento dei livelli di AST (7,3%). La sospensione definitiva del trattamento con Kisqali e qualsiasi combinazione a causa di alterazioni dei test di funzionalità epatica o epatotossicità è stata osservata rispettivamente nel 2,4% e nello 0,3% dei pazienti (vedi sezioni «Modalità di somministrazione e dosaggio» e «Avvertenze particolari e precauzioni di impiego»).
Negli studi clinici di Fase III su pazienti con carcinoma mammario localmente avanzato o metastatico, il 70,9% (90/127) dei casi di aumento di ALT o AST di grado 3 o 4 si è verificato entro i primi 6 mesi di trattamento. Nei pazienti con aumento di ALT/AST di grado 3 o 4, il tempo medio all'insorgenza di tali eventi è stato di 92 giorni nei gruppi trattati con Kisqali e qualsiasi combinazione. Il tempo medio per la normalizzazione dei valori (fino alla normalizzazione o a un grado ≤ 2) è stato di 21 giorni nei gruppi trattati con Kisqali e qualsiasi combinazione.
Prolungamento dell'intervallo QT
Nello studio di Fase III su pazienti con carcinoma mammario in stadio precoce, un prolungamento dell'intervallo QT è stato osservato nel 5,3% dei pazienti nel gruppo trattato con Kisqali e IA e nell'1,4% dei pazienti nel gruppo trattato solo con IA. Nel gruppo trattato con Kisqali e IA, i casi di prolungamento dell'intervallo QT erano principalmente rappresentati da prolungamento dell'intervallo QT all'ECG (4,3%), che è stata l'unica reazione avversa confermata associata all'uso di Kisqali. L'interruzione del trattamento a causa di prolungamento dell'intervallo QT all'ECG e síncope è stata necessaria nell'1,1% dei pazienti trattati con Kisqali. L'adeguamento della dose a causa di prolungamento dell'intervallo QT all'ECG è stato necessario nello 0,1% dei pazienti trattati con Kisqali.
L'analisi centrale dei dati ECG ha mostrato che almeno un episodio di prolungamento di QTcF > 480 ms dopo l'inizio del trattamento si è verificato in 10 pazienti (0,4%) nel gruppo trattato con Kisqali e IA e in 4 pazienti (0,2%) nel gruppo trattato solo con IA. Nei pazienti con prolungamento di QTcF > 480 ms nel gruppo trattato con Kisqali e IA, il tempo medio all'insorgenza di tali eventi è stato di 15 giorni e tali modifiche sono state reversibili con interruzione temporanea del trattamento e/o adeguamento della dose. Un cambiamento di QTcF > 60 ms rispetto al valore basale è stato osservato in 19 pazienti (0,8%) nel gruppo trattato con Kisqali e IA e un intervallo QTcF > 500 ms dopo l'inizio del trattamento è stato osservato in 3 pazienti (0,1%) nel gruppo trattato con Kisqali e IA.
Nello studio E2301 (MONALEESA-7) su pazienti con carcinoma mammario localmente avanzato o metastatico, il prolungamento medio osservato dell'intervallo QTcF rispetto al valore basale è stato di circa 10 ms maggiore nel sottogruppo trattato con tamoxifene e placebo rispetto al sottogruppo trattato con inibitori della aromatasi (IA) e placebo, indicando che il tamoxifene da solo causa un prolungamento di QTcF e può contribuire ai valori di QTcF osservati nel gruppo trattato con Kisqali e tamoxifene. Nel gruppo placebo, un prolungamento di QTcF > 60 ms rispetto al valore basale è stato osservato in 6/90 (6,7%) pazienti trattati con tamoxifene e in nessun paziente trattato con IA (vedi sezione «Farmacocinetica»). Un prolungamento di QTcF > 60 ms rispetto al valore basale è stato osservato in 14/87 (16,1%) pazienti trattati con Kisqali e tamoxifene e in 18/245 (7,3%) pazienti trattati con Kisqali e IA. L'uso di Kisqali in combinazione con tamoxifene non è raccomandato (vedi sezione «Farmacodinamica»).
Negli studi clinici di Fase III, almeno un episodio di prolungamento dell'intervallo QT (incluso prolungamento dell'intervallo QT all'ECG e síncope) è stato osservato nel 9,3% dei pazienti con carcinoma mammario localmente avanzato o metastatico nei gruppi trattati con Kisqali e IA o fulvestrant e nel 3,5% dei pazienti nei gruppi trattati con placebo e IA o fulvestrant. L'analisi dei dati ECG ha mostrato che in 15 pazienti (1,4%) il valore di QTcF dopo l'inizio del trattamento è stato > 500 ms e in 61 pazienti (5,8%) è stato osservato un aumento di QTcF > 60 ms rispetto al valore basale. Nessun caso di tachicardia ventricolare tipo torsione di punta è stato riportato. L'interruzione del trattamento / adeguamento della dose a causa di prolungamento dell'intervallo QT all'ECG e síncope è stato necessario nel 2,9% dei pazienti trattati con Kisqali e IA o fulvestrant.
L'analisi dei dati ECG ha mostrato che almeno un episodio di prolungamento di QTcF > 480 ms dopo l'inizio del trattamento si è verificato in 55 pazienti (5,2%) nei gruppi trattati con Kisqali e inibitore della aromatasi o fulvestrant e in 12 pazienti (1,5%) nei gruppi trattati con placebo e inibitore della aromatasi o fulvestrant. Nei pazienti con prolungamento di QTcF > 480 ms, il tempo medio all'insorgenza di tale evento è stato di 15 giorni indipendentemente dalla combinazione, e tali modifiche sono state reversibili con interruzione temporanea del trattamento e/o riduzione della dose (vedi sezioni «Modalità di somministrazione e dosaggio», «Avvertenze particolari e precauzioni di impiego» e «Farmacocinetica»).
Pazienti con insufficienza renale
Negli studi clinici di Fase III su pazienti con carcinoma mammario in stadio precoce, 983 pazienti con compromissione renale lieve e 71 pazienti con compromissione renale moderata hanno ricevuto ribociclib. Nessun paziente con compromissione renale grave è stato incluso nello studio (vedi sezione «Farmacodinamica»).
Nei tre studi principali, 341 pazienti con carcinoma mammario localmente avanzato o metastatico e insufficienza renale lieve e 97 pazienti con insufficienza renale moderata hanno ricevuto trattamento con ribociclib. Nessun paziente con insufficienza renale grave è stato incluso nello studio (vedi sezione «Farmacodinamica»). È stata osservata una correlazione tra il grado di insufficienza renale al basale e i livelli di creatinina durante il trattamento. Nei pazienti con insufficienza renale lieve o moderata, è stata osservata una leggera aumento della frequenza di prolungamento dell'intervallo QT e trombocitopenia. Per le raccomandazioni sul monitoraggio e l'adeguamento della dose in caso di tali effetti tossici, vedere le sezioni «Modalità di somministrazione e dosaggio» e «Avvertenze particolari e precauzioni di impiego».
Segnalazione di reazioni avverse sospette
La segnalazione delle reazioni avverse dopo la commercializzazione del medicinale è di grande importanza. Permette un monitoraggio continuo del rapporto beneficio/rischio del medicinale. Il personale medico e farmaceutico, nonché i pazienti o i loro rappresentanti legali, devono segnalare tutti i casi sospetti di reazioni avverse e di mancata efficacia del medicinale attraverso il Sistema Informatizzato Automatizzato di Farmacovigilanza al seguente indirizzo: https://aisf.dec.gov.ua
Periodo di validità. 1 anno.
Condizioni di conservazione
Conservare in frigorifero (da 2 a 8 °C) per 10 mesi nella zona di magazzino.
Conservare a una temperatura non superiore a 25 °C per 2 mesi durante l'uso del medicinale da parte del paziente. Conservare nella confezione originale. Conservare fuori dalla portata dei bambini.
Confezionamento
- 21 compresse in un blister, 3 blister in una scatola di cartone.
- 21 compresse in un blister, 3 blister in una scatola di cartone, 3 scatole in una scatola di cartone.
Categoria di prescrizione. Sotto prescrizione medica.
Produttore
Novartis Pharmaceuticals Manufacturing LLC.
Sede del produttore e indirizzo del luogo di esercizio dell'attività
Via Verovškova 57, Lubiana, 1000, Slovenia.