Ilaris
UkrainaSpis treści
INSTRUKCJA DOTYCZĄCA STOSOWANIA LEKU ILARIS (ILARIS®)
Skład:
substancja czynna: canakinumab;
1 fiolka zawiera 150 mg canakinumabu;
substancje pomocnicze: sacharoza, L-histydyna, L-histydyny chlorowodorek monohydrat, polisorbat 80.
Postać leku. Proszek do sporządzenia roztworu do wstrzykiwań.
Główne właściwości fizykochemiczne: proszek liofilizowany białego koloru.
Grupa farmakoterapeutyczna. Leki przeciwnowotworowe i immunomodulujące. Immunosupresanty. Inhibitory interleukiny. Kod ATC L04A C08.
Właściwości farmakologiczne.
Farmakodynamika.
Mechanizm działania
Kanakinumab to monoklonalne przeciwciało o całkowicie ludzkim pochodzeniu, izotypu IgG1/κ, skierowane przeciwko interleukinie-1 beta (IL-1 beta). Kanakinumab wiąże się specyficznie z wysokim powinowactwem do ludzkiej IL-1 beta i neutralizuje jej aktywność biologiczną, blokując jej interakcję z receptorami IL-1, zapobiegając w ten sposób aktywacji indukowanego przez IL-1 beta genu oraz produkcji mediatorów zapalnych.
Skutki farmakodynamiczne
Zespoły okresowe związane z krioptyriną (CAP-zespoły), okresowy zespół związany z receptorem czynnika martwicy nowotworów (TRAP-zespół), zespół hiperimmunoglobulinemii D (HID-zespół) / niedobór mewalonianokinazy (MKD), rodzinna gorączka śródziemnomorska (FMF)
W badaniach klinicznych u pacjentów z CAP-zespołami, TRAP-zespołem, HID-zespołem / niedoborem mewalonianokinazy (MKD) oraz rodzinną gorączką śródziemnomorską (FMF), u których występuje niekontrolowane wydzielanie IL-1 beta, obserwowano szybką reakcję na terapię kanakinumabem, tj. szybkie normalizowanie się wskaźników laboratoryjnych, takich jak wysoki poziom C-reaktywnej białka (CRP) i amyloidu surowicy A (SAA), wysoki poziom neutrofili i płytek krwi oraz leukocytoza.
Choroba Stilla (w tym choroba Stilla u dorosłych (AOSD) i systematyczny młodzieńczy idiopatyczny zapalenie stawów (SJIA))
Choroba Stilla u dorosłych oraz systematyczny młodzieńczy idiopatyczny zapalenie stawów to ciężkie wrodzone choroby autozapalne, wywołane przez prozapalne cytokiny, z których kluczową jest IL-1 beta.
Główne objawy systematycznego młodzieńczego idiopatycznego zapalenia stawów obejmują gorączkę, wysypkę, hepatosplenomegalię, limfadenopatię, poliserozit i zapalenie stawów.
Leczenie kanakinumabem powodowało szybkie i trwałe zmniejszenie zarówno objawów stawowych, jak i ogólnoustrojowych systematycznego młodzieńczego idiopatycznego zapalenia stawów, z istotnym zmniejszeniem liczby zapalonych stawów, ustępowaniem gorączki oraz obniżeniem oznak reakcji ostrej fazy u większości pacjentów.
Zapalenie stawów podagrycznych
Przyczyną napadu zapalenia stawów podagrycznych są kryształy uranów (monohydraty jednonatrowe uranów) w stawach i otaczających tkankach, które aktywują makrofagi tkankowe do wytwarzania IL-1 beta poprzez kompleks inflazomowy NALP3. Aktywacja makrofagów i towarzyszący temu zwiększony wydzielanie IL-1 beta prowadzi do ostrych, bolesnych reakcji zapalnych. Inne aktywatory wrodzonego układu odpornościowego, takie jak endogenne agonisty receptorów Toll-podobnych, mogą przyczyniać się do transkrypcyjnej aktywacji genu IL-1 beta, inicjując napad zapalenia stawów podagrycznych. Po leczeniu kanakinumabem markery zapalenia, tj. białko C-reaktywne (CRP) i amyloid surowicy A (SAA), oraz objawy ostrego zapalenia (np. ból, obrzęk, zaczerwienienie) w dotkniętym stawie szybko ustępują.
Skuteczność kliniczna i bezpieczeństwo
Zespoły okresowe związane z krioptyriną (CAP-zespoły)
Skuteczność i bezpieczeństwo leku Ilaris zostały zademonstrowane u pacjentów z różnym stopniem nasilenia choroby i różnymi fenotypami (w tym ciężkie formy rodinnego zimowego autozapalnego zespołu / rodzinnej zimowej pokrzywki, zespołu Muckle’a-Wella i noworodkowego wieloukładowego zapalnego schorzenia / przewlekłego dziecięcego neurologicznego, skórno-stawowego zespołu). Do kluczowego badania włączone zostały wyłącznie pacjenci z potwierdzoną mutacją NLRP3.
W fazie badania I/II lek Ilaris wykazywał szybki początek działania: objawy ustępowały lub znacznie słabły w ciągu jednego dnia. Wskaźniki laboratoryjne, takie jak poziom CRP i SAA, poziom neutrofili i płytek krwi, szybko normalizowały się w ciągu kilku dni po podaniu Ilaris.
Kluczowe 48-tygodniowe, wieloośrodkowe badanie składało się z trzech części: 8-tygodniowego otwartego okresu (część I), 24-tygodniowego randomizowanego, podwójnie ślepego, placebo-kontrolowanego okresu odstawiania (część II) oraz 16-tygodniowego okresu otwartego (część III). Celem badania było ocenienie skuteczności, bezpieczeństwa i tolerancji Ilaris (150 mg lub 2 mg/kg co 8 tygodni) u pacjentów z CAP-zespołami.
- Część I: pełna kliniczna odpowiedź i odpowiedź biomarkerów na Ilaris (określona jako ocena lekarza dotycząca autozapalnej choroby i choroby skóry ≤ minimum oraz wartość CRP lub SAA < 10 mg/l) obserwowano u 97% pacjentów i pojawiały się one w ciągu 7 dni od rozpoczęcia leczenia. Znacząca poprawa została odnotowana w klinicznej ocenie lekarza dotyczącej aktywności autozapalenia: ogólna ocena aktywności autozapalnej choroby, ocena choroby skóry (pokrzywka, wysypka skórna), ból stawów, mięśniówka, ból głowy/migrena, zapalenie spojówek, zmęczenie/niedoból, ocena innych objawów związanych, ocena objawów przez pacjenta.
- Część II: w okresie odstawiania głównym punktem końcowym była frakcja pacjentów z nawrotem choroby/powtórnym zaostrzeniem: 0% pacjentów z nawrotem przy stosowaniu Ilaris w porównaniu do 81% pacjentów randomizowanych do grupy placebo.
- Część III: pacjenci, którzy otrzymywali placebo w części II, u których wystąpił nawrót i którzy zachowali kliniczną i serologiczną odpowiedź po wejściu do otwartej części przedłużonego badania Ilaris.
Tabela 1. Skuteczność w fazie III kluczowego badania placebo-kontrolowanego w okresie odstawiania (część II)
| Wskaźnik |
Ilaris N=15 n(%) |
Placebo N=16 n(%) |
p -poziom |
| Pierwotny punkt końcowy (nasilenie) Liczba pacjentów z nawrotem choroby w części II |
0 (0 %) |
13 (81 %) |
<0,001 |
| Markery stanu zapalnego* Białko C-reaktywne, mg/l Amyloid A osocza, mg/l |
1,10 (0,40) 2,27 (-0,20) |
19,93 (10,50) 71,09 (14,35) |
<0,001 0,002 |
| *Średnia zmiana od początku części II |
|||
Przeprowadzono dwa otwarte, niekontrolowane, długoterminowe badania fazy III. W jednym z nich oceniano bezpieczeństwo, skuteczność i skuteczność kanakynumabu u pacjentów z chorobami okresowymi związanymi z kriopiryną. Całkowity czas leczenia wynosił od 6 miesięcy do 2 lat. Drugie badanie było otwartym badaniem kanakynumabu mającym na celu ocenę skuteczności i bezpieczeństwa u japońskich pacjentów z chorobami okresowymi związanymi z kriopiryną, trwającym 24 tygodnie z fazą rozszerzoną do 48 tygodni. Głównym celem była ocena frakcji pacjentów bez nawrotu w 24. tygodniu, w tym tych, u których dawkę zwiększono.
W połączonym analizie skuteczności tych dwóch badań, 65,6% pacjentów wcześniej nie leczonych kanakynumabem osiągnęło pełną odpowiedź przy dawce 150 mg lub 2 mg/kg, podczas gdy 85,2% pacjentów osiągnęło pełną remisję przy dowolnej dawce. Wśród pacjentów, którzy otrzymywali 600 mg lub 8 mg/kg (lub nawet wyższą dawkę), pełną odpowiedź osiągnęło 43,8%. Liczba pacjentów w wieku od 2 do 4 lat, którzy osiągnęli pełną odpowiedź (57,1%), była mniejsza w porównaniu z starszymi dziećmi i dorosłymi pacjentami. Wśród pacjentów, którzy osiągnęli pełną odpowiedź, 89,3% utrzymywało odpowiedź bez nawrotu.
Doświadczenie z zastosowania u poszczególnych pacjentów, którzy osiągnęli pełną odpowiedź po zwiększeniu dawki do 600 mg (8 mg/kg) co 8 tygodni, wskazuje, że wyższa dawka może być korzystna dla pacjentów, którzy nie osiągnęli pełnej odpowiedzi lub nie utrzymali pełnej odpowiedzi przy zalecanych dawkach (150 mg lub 2 mg/kg dla pacjentów o masie ciała ≥ 15 kg do ≤ 40 kg). Zwiększona dawka była częściej stosowana u pacjentów w wieku od 2 do 4 lat oraz u pacjentów z objawami NOMID/CINCA w porównaniu z FCAS lub MWS.
Przeprowadzono 6-letnie badanie rejestracyjne mające na celu uzyskanie danych na temat długoterminowego bezpieczeństwa i skuteczności leczenia kanakynumabem u pediatrycznych i dorosłych pacjentów z chorobami okresowymi związanymi z kriopiryną w warunkach rzeczywistej praktyki klinicznej. Badanie obejmowało 243 pacjentów z chorobami okresowymi związanymi z kriopiryną (w tym 85 pacjentów w wieku do 18 lat). Aktywność choroby oceniano jako brak lub lekką/umiarkowaną u ponad 90% pacjentów we wszystkich okresach po wstępnym okresie badania, a średnie serologiczne markery zapalenia (CRP i SAA) były w normie (<10 mg/litr) we wszystkich punktach czasowych po okresie podstawowym. Choć około 22% pacjentów leczonych kanakynumabem wymagało dostosowania dawki, jedynie niewielki odsetek pacjentów (1,2%) przerwał leczenie kanakynumabem z powodu braku efektu terapeutycznego.
Populacja pediatryczna
W badaniach chorób okresowych związanych z kriopiryną wzięło udział ogółem 80 pacjentów pediatrycznych w wieku od 2 do 17 lat (ok. połowie z nich dawkę dostosowano do masy ciała). Ogólnie rzecz biorąc, u pacjentów pediatrycznych nie stwierdzono żadnych klinicznie istotnych różnic w skuteczności, bezpieczeństwie i profilu tolerancji Ilaris w porównaniu z populacją ogólną. Większość pacjentów pediatrycznych osiągnęła poprawę objawów klinicznych i obiektywnych markerów zapalenia (np. SAA i CRP).
Skuteczność, bezpieczeństwo i tolerancję leku Ilaris oceniano w otwartym, 56-tygodniowym badaniu z udziałem dzieci w wieku do 4 lat z chorobami okresowymi związanymi z kriopiryną. Oceny dokonano u 17 pacjentów (w tym 6 pacjentów w wieku do 2 lat), stosując dawki początkowe obliczone na podstawie masy ciała – 2–8 mg/kg. W badaniu oceniano również wpływ kanakynumabu na powstawanie przeciwciał przeciwko standardowym szczepionkom dziecięcym. Nie zaobserwowano różnic w bezpieczeństwie ani skuteczności u pacjentów w wieku do 2 lat w porównaniu z pacjentami w wieku od 2 lat. Wszyscy pacjenci, którzy otrzymali nieżywe standardowe szczepionki dziecięce (N = 7), osiągnęli poziomy przeciwciał zapewniające ochronę.
Choroba okresowa związana z receptorem czynnika martwicy nowotworów (TRAPS), zespół hiperimmunoglobulinemii D (HIDS) / niedobór mewalonianokinazy (MKD), rodzinna gorączka śródziemnomorska (FMF)
Skuteczność i bezpieczeństwo kanakynumabu w leczeniu choroby okresowej związanej z receptorem czynnika martwicy nowotworów (TRAPS), zespołu hiperimmunoglobulinemii D (HIDS) / niedoboru mewalonianokinazy (MKD) oraz rodzinnej gorączki śródziemnomorskiej (FMF) wykazano w badaniu fazy III jednego kluczowego badania (N2301) składającego się z czterech części i obejmującego trzy oddzielne kohorty chorób.
- Część I: Pacjenci każdej grupy w wieku od 2 lat zostali zakwalifikowani do 12-tygodniowego okresu skriningowego, podczas którego oceniano ich pod kątem wystąpienia zaostrzenia choroby.
- Część II: Pacjenci z zaostrzeniem choroby zostali losowo przydzieleni do 16-tygodniowego podwójnie ślepego, placebo-kontrolowanego okresu leczenia, podczas którego otrzymywali 150 mg kanakynumabu (2 mg/kg dla pacjentów o masie ciała ≤ 40 kg) podskórnie lub placebo co 4 tygodnie. Pacjentom w wieku od 28 dni, ale < 2 lat, umożliwiono udział w otwartej części II części badania jako pacjentów nierandomizowanych (i zostali wykluczeni z pierwotnej analizy skuteczności).
- Część III: Pacjenci, którzy ukończyli 16-tygodniowy okres leczenia i zostali sklasyfikowani jako responderzy (odpowiadający na leczenie), zostali ponownie zrandomizowani do 24-tygodniowego podwójnie ślepego okresu odstawiania, podczas którego otrzymywali 150 mg kanakynumabu (2 mg/kg dla pacjentów o masie ciała ≤ 40 kg) podskórnie lub placebo co 8 tygodni.
- Część IV: Wszyscy pacjenci z części III, którzy otrzymywali kanakynumab, mieli prawo wziąć udział w 72-tygodniowym otwartym okresie leczenia kontynuacyjnego.
Łącznie zakwalifikowano 185 pacjentów w wieku od 28 dni, a ogólnie 181 pacjentów w wieku od 2 lat zostało zrandomizowanych w części II badania.
Pierwotnym punktem końcowym skuteczności w okresie leczenia randomizowanym (część II) była frakcja responderów w każdej grupie, którzy do 15. dnia odnotowali zmniejszenie się indeksu zaostrzenia choroby i nie doświadczyli nowego zaostrzenia w pozostałym okresie 16-tygodniowego leczenia (określone jako pełna odpowiedź). Zmniejszenie indeksu zaostrzenia choroby definiowano jako osiągnięcie ogólnej oceny lekarza (PGA) w skali aktywności choroby < 2 („choroba minimalna lub brak”) oraz poziomu CRP w normie (≤ 10 mg/l) lub zmniejszenia się o ≥ 70% od wartości wyjściowej. Nowe zaostrzenie definiowano jako PGA ≥ 2 („łagodna, umiarkowana lub ciężka choroba”) oraz poziom CRP ≥ 30 mg/l. Punkty końcowe wtórne, oparte na wynikach z 16. tygodnia (koniec części II), obejmowały frakcję pacjentów, którzy osiągnęli PGA < 2, frakcję pacjentów z remisją serologiczną (określoną jako CRP ≤ 10 mg/l) oraz frakcję pacjentów z znormalizowanym poziomem SAA (określonym jako SAA ≤ 10 mg/l).
W odniesieniu do pierwotnego punktu końcowego skuteczności kanakynumab okazał się lepszy niż placebo we wszystkich trzech grupach chorób. Kanakynumab wykazał również lepszą skuteczność niż placebo we wtórnych punktach końcowych PGA < 2 oraz CRP ≤ 10 mg/l we wszystkich trzech grupach. Większa frakcja pacjentów znormalizowała poziom SAA (≤ 10 mg/l) w 16. tygodniu podczas leczenia kanakynumabem w porównaniu z placebo we wszystkich trzech grupach, przy czym istotna statystycznie różnica została zaobserwowana u pacjentów z TRAPS.
Tabela 2. Skuteczność w fazie III kluczowego randomizowanego, placebo-kontrolowanego badania w okresie leczenia (część II)
| Wskaźnik |
Kanakimumab, n/N (%) |
Placebo, n/N (%) |
p -poziom |
| Pierwotny punkt końcowy (zaostrzenie) Liczba pacjentów, u których w 15. dniu stwierdzono wskaźnik zaostrzenia choroby i którzy nie doświadczyli nowego zaostrzenia w pozostałym 16-tygodniowym okresie leczenia |
|||
| FMF Zespół HID/MKD Zespół TRAP |
19/31 (61,29) 13/37 (35,14) 10/22 (45,45) |
2/32 (6,25) 2/35 (5,71) 2/24 (8,33) |
<0,0001* 0,0020* 0,0050* |
| Punkt końcowy wtórny (choroba i markery zapalenia) |
|||
| Ogólna ocena lekarza < 2 FMF Zespół HID/MKD Zespół TRAP |
20/31 (64,52) 17/37 (45,95) 10/22 (45,45) |
3/32 (9,38) 2/35 (5,71) 1/24 (4,17) |
<0,0001** 0,0006** 0,0028** |
| Białko C-reaktywne ≤ 10 mg/l FMF Zespół HID/MKD Zespół TRAP |
21/31 (67,74) 15,37 (40,54) 8/22 (36,36) |
2/32 (6,25) 2/35 (5,71) 2/24 (8,33) |
<0,0001** 0,0010** 0,0149** |
| Surowiczy białek amyloidu A ≤ 10 mg/l FMF Zespół HID/MKD Zespół TRAP |
8/31 (25,81) 5/37 (13,51) 6/22 (27,27) |
0,32 (0,00) 1/35 (2,86) 0,24 (0,00) |
0,0286 0,0778 0,0235* |
| n – liczba reagujących; N – liczba ocenianych pacjentów. *Wskazuje na istotność statystyczną (jednostronną) na poziomie 0,025 na podstawie dokładnego testu Fishera. **Wskazuje na istotność statystyczną (jednostronną) na poziomie 0,025 na podstawie modelu regresji logistycznej z grupą leczenia oraz wyjściowym PGA, CRP lub SAA odpowiednio jako zmiennymi wyjaśniającymi dla każdej grupy. |
|||
Tytrowanie dawki
W części II badania pacjenci otrzymujący kanakimumab, u których nadal występowała aktywność choroby, otrzymywali dawkę uzupełniającą 150 mg (lub 2 mg/kg u pacjentów o masie ciała ≤ 40 kg) w ciągu pierwszego miesiąca. Dawkę uzupełniającą można podać już po 7 dniach od pierwszej dawki leczenia. Wszyscy pacjenci z tytrowaniem pozostawali w grupie zwiększonej dawki 300 mg (lub 4 mg/kg u pacjentów o masie ciała ≤ 40 kg) co 4 tygodnie.
W trakcie badawczego analizy punktu końcowego pierwotnego zauważono, że u pacjentów z nieadekwatną odpowiedzią po pierwszej dawce zwiększenie dawki w ciągu pierwszego miesiąca do dawki 300 mg (lub 4 mg/kg) co 4 tygodnie dodatkowo poprawiało kontrolę nasilenia, zmniejszenie aktywności choroby oraz normalizację poziomów CRP i SAA.
Pacjenci pediatryczni
Dwóch niezrandomizowanych pacjentów z zespołem HID/MKD w wieku >28 dni – <2 lat zostało włączonych do badania i otrzymywało kanakimumab. U jednego pacjenta stwierdzono ustąpienie wskaźnika nasilenia do 15. dnia po podaniu pojedynczej dawki kanakimumabu 2 mg/kg, jednak leczenie zostało przerwane po tej pierwszej dawce z powodu poważnych działań niepożądanych (pancytopenia i niewydolność wątroby). Przed włączeniem do badania pacjent ten zgłosił wywiadem zespół immunologicznej małopłytkowości oraz stan medyczny z zaburzeniami funkcji wątroby. Drugi pacjent otrzymał początkową dawkę kanakimumabu 2 mg/kg oraz dawkę uzupełniającą 2 mg/kg w 3. tygodniu, a w 5. tygodniu otrzymał tytrowanie do dawki 4 mg/kg, którą stosowano co 4 tygodnie do końca części II badania. Ustąpienie nasilenia choroby osiągnięto do 5. tygodnia, a pacjent nie doświadczył nowych nasileń do końca części II badania (16. tydzień).
Choroba Still’a (choroba Still’a u dorosłych (AOSD) oraz systematyczny młodzieńczy zapalenie stawów idiopatyczne (SJIA))
Systematyczne młodzieńcze zapalenie stawów idiopatyczne
Skuteczność leku Ilaris w leczeniu aktywnego systematycznego młodzieńczego zapalenia stawów idiopatycznego oceniano w dwóch badaniach podstawowych (G2305 i G2301). Do badań włączono pacjentów w wieku od 2 do 20 lat (średni wiek 8,5 roku i średnia długość trwania choroby 3,5 roku na początek badania), którzy mieli aktywną chorobę, określoną obecnością 2 lub więcej stawów z aktywnym zapaleniem stawów, gorączką i podwyższonym CRP.
Badanie G2305
G2305 – randomizowane, podwójnie ślepe, kontrolowane placebo 4-tygodniowe badanie. W badaniu tym oceniano krótkoterminową skuteczność leku Ilaris u 84 pacjentów, którzy zostali zrandomizowani do otrzymania jednej dawki 4 mg/kg (do 300 mg) leku Ilaris lub placebo. Głównym celem było ustalenie frakcji pacjentów, którzy do 15. dnia osiągnęli minimalne poprawienie o 30% zgodnie z kryteriami pediatrycznymi Amerykańskiego Kolegium Reumatologów (ACR). Kryteria te zostały dostosowane, aby umożliwić włączenie pacjentów bez gorączki. Leczenie lekiem Ilaris poprawiło wszystkie wskaźniki ACR w porównaniu z placebo w 15. i 29. dniu (tabela 3).
Tabela 3. Pediatryczne wskaźniki ACR i stan choroby w 15. i 29. dniu
| Wskaźnik |
15. dzień |
29. dzień |
||
| Ilaris N=43 |
Placebo N=41 |
Ilaris N=43 |
Placebo N=41 |
|
| ACR30 |
84 % |
10 % |
81 % |
10 % |
| ACR50 |
67 % |
5 % |
79 % |
5 % |
| ACR70 |
61 % |
2 % |
67 % |
2 % |
| ACR90 |
42 % |
0 % |
47 % |
2 % |
| ACR100 |
33 % |
0 % |
33 % |
2 % |
| Bezobjawowe przebieg choroby |
33 % |
0 % |
30 % |
0 % |
| Różnica w leczeniu dla wszystkich wskaźników ACR była istotna (p ≤ 0,0001) |
||||
Wyniki dostosowanych pediatrycznych wskaźników ACR, które obejmowały składniki systemowe i artretyczne, były zgodne z ogólnymi wynikami wskaźników ACR. W 15. dniu średnia zmiana od wartości wyjściowej w liczbie stawów z aktywnym zapaleniem stawów i ograniczonym zakresem ruchu wynosiła odpowiednio –67 % i –73 % dla Ilaris (N = 43) w porównaniu z medianą zmiany 0 % i 0 % w grupie placebo (N = 41). Średnia zmiana w skali bólu u pacjenta (0–100 mm wizualnej skali analogowej) w 15. dniu wyniosła 50,0 mm dla Ilaris (N = 43) w porównaniu z +4,5 mm dla placebo (N = 25). Średnia zmiana wskaźnika bólu u pacjentów była zgodna w 29. dniu.
Dane badania G2301
G2301 – randomizowane, podwójnie ślepe, kontrolowane placebo badanie dotyczące zapobiegania nasileniom leczenia lekiem Ilaris. Badanie składało się z dwóch części z dwoma niezależnymi punktami końcowymi pierwotnymi (udane zmniejszenie dawki steroidów i czas do nasilenia). W części I (otwartej) włączono 177 pacjentów, którzy otrzymywali 4 mg/kg (do maksymalnie 300 mg) leku Ilaris co 4 tygodnie przez okres do 32 tygodni. Pacjenci w części II (podwójnie ślepej) otrzymywali Ilaris 4 mg/kg lub placebo co 4 tygodnie do momentu zaobserwowania 37 nasileń.
Zmniejszenie dawki kortykosteroidów
Spośród 128 pacjentów przyjmujących kortykosteroidy i uczestniczących w części I badania, 92 pacjentów próbowało zmniejszyć dawkę kortykosteroidów. Pięćdziesiąt siedem (62 %) z nich zdołało znacząco zmniejszyć dawkę kortykosteroidów, a 42 (46 %) całkowicie odstawiło kortykosteroidy.
Czas do pierwszego nasilenia
U pacjentów przyjmujących lek Ilaris w części II badania zaobserwowano 64 % zmniejszenie ryzyka nasilenia choroby w porównaniu z grupą placebo (stosunek ryzyka 0,36; 95 % CI: 0,17–0,75; p = 0,0032). U sześćdziesięciu trzech z 100 pacjentów uczestniczących w części II badania w grupie placebo lub kanakimumabu nie zaobserwowano nasilenia choroby w okresie obserwacji (maksymalnie do 80 tygodni).
Wyniki badań G2305 i G2301 dotyczące stanu zdrowia i jakości życia
W trakcie leczenia lekiem Ilaris zaobserwowano klinicznie istotne poprawy funkcji fizycznej i jakości życia pacjentów. W badaniu G2305, według danych z ankiety oceny stanu zdrowia dzieci, poprawa wyniosła 0,69 w grupie leku Ilaris w porównaniu z grupą placebo, co przekraczałoby minimalną klinicznie istotną różnicę 0,19 aż 3,6 razy (p = 0,0002). Średnia poprawa w porównaniu z wartością wyjściową na końcu części II badania G2301 wyniosła 0,88 (79 %). Statystycznie istotne poprawy w wynikach ankiety oceny stanu zdrowia dzieci PF50 zaobserwowano w grupie leku Ilaris w porównaniu z grupą placebo w badaniu G2305 (stan fizyczny p = 0,0012; dobrostan psychospołeczny p = 0,0017).
Zespolony analiza skuteczności
Dane z pierwszych 12 tygodni leczenia lekiem Ilaris z badań G2305, G2301 oraz rozszerzonego badania zostały połączone w celu oceny skuteczności. Dane te wskazują na jednolite poprawy w porównaniu z wartością wyjściową w dostosowanych pediatrycznych wskaźnikach ACR i ich składnikach już w 12. tygodniu w porównaniu z grupą kontrolną placebo (G2305). W 12. tygodniu odsetek pacjentów osiągających dostosowane pediatryczne wskaźniki ACR30, 50, 70, 90 i 100 wynosił odpowiednio 70 %, 69 %, 61 %, 49 % i 30 %, podczas gdy 28 % pacjentów (N = 178) osiągnęło stan nieaktywny choroby.
Skuteczność obserwowana w trakcie badań G2305 i G2301 utrzymywała się w długoterminowym otwartym rozszerzeniu badania, które trwa obecnie (dostępne dane z okresu obserwacji o medianie 49 tygodni). W tym badaniu 25 pacjentów, którzy mieli silną odpowiedź według ACR przez co najmniej 5 miesięcy, zmniejszyło dawkę leku Ilaris do 2 mg/kg co 4 tygodnie i utrzymywało odpowiedź pediatryczną ACR100 podczas stosowania zmniejszonej dawki (mediana 32 tygodnie, zakres 8–124 tygodnie).
Mimo ograniczonej liczby danych, wyniki badań klinicznych wskazują, że pacjenci, którzy nie odpowiadają na leczenie tocylizumabem lub anakinrą, mogą odpowiadać na leczenie kanakimumabem.
Badanie G2306
Badanie G2306 było otwartym badaniem mającym na celu ocenę utrzymania odpowiedzi na leczenie przy zmniejszonej dawce kanakimumabu (2 mg/kg co 4 tygodnie) lub wydłużonym interwale dawkowania (4 mg/kg co 8 tygodni) u pacjentów z młodzieńczym idiopatycznym zapaleniem stawów typu systemowego, którzy otrzymywali kanakimumab 4 mg/kg co 4 tygodnie. Siedemdziesięciu pięciu pacjentów w wieku od 2 do 22 lat, którzy utrzymywali stan nieaktywny choroby przez co najmniej 6 miesięcy z rzędu (remisja kliniczna) na monoterapii kanakimumabem, w tym pacjentów, którzy utrzymywali stan nieaktywny choroby po odstawieniu współistniejącego leczenia kortykosteroidami i/lub metotreksatem przez co najmniej 4 tygodnie, zostało zrandomizowanych do otrzymywania kanakimumabu 2 mg/kg co 4 tygodnie (N = 38) lub kanakimumabu 4 mg/kg co 8 tygodni (N = 37). Po 24 tygodniach 71 % (27/38) pacjentów otrzymujących zmniejszoną dawkę (2 mg/kg co 4 tygodnie) i 84 % (31/37) pacjentów z wydłużonym interwałem dawkowania (4 mg/kg co 8 tygodni) mogło utrzymać stan nieaktywny choroby przez 6 miesięcy. Spośród pacjentów, którzy pozostawali w remisji klinicznej i kontynuowali dalsze zmniejszanie dawki (1 mg/kg co 4 tygodnie) lub wydłużenie interwału dawkowania (4 mg/kg co 12 tygodni), odpowiednio 93 % (26/28) i 91 % (30/33) pacjentów mogło utrzymać stan nieaktywny choroby przez 6 miesięcy. Pacjentom, którzy utrzymywali stan nieaktywny choroby przez dodatkowe 6 miesięcy przy tej minimalnej dawce, umożliwiono odstawienie kanakimumabu. Ogółem 33 % (25/75) pacjentów zrandomizowanych do zmniejszenia dawki lub wydłużenia interwału dawkowania mogło odstawić leczenie kanakimumabem i utrzymać stan nieaktywny choroby przez 6 miesięcy. Częstość występowania działań niepożądanych w obu grupach leczenia była podobna do tej obserwowanej u pacjentów otrzymujących kanakimumab 4 mg/kg co 4 tygodnie.
Choroba Stilla u dorosłych (AOSD)
Skuteczność kanakimumabu w dawce 4 mg/kg (maksymalnie do 300 mg) podawanego co 4 tygodnie pacjentom z AOSD została potwierdzona w randomizowanym, podwójnie ślepym, kontrolowanym placebo badaniu z udziałem 36 pacjentów (w wieku od 22 do 70 lat) i była podobna do tej obserwowanej u pacjentów z SJIA. W badaniu GDE01T większy odsetek pacjentów (12/18, 66,7 %) w grupie kanakimumabu niż w grupie placebo (7/17, 41,2 %) wykazał poprawę w porównaniu z wartością wyjściową w Skali Aktywności Choroby 28 (DAS28-ESR) > 1,2 w 12. tygodniu, co nie osiągnęło istotności statystycznej (stosunek szans 2,86, różnica w leczeniu [%] 25,49 [95 % CI: 9,43, 55,80]). Do 4. tygodnia 7 z 18 pacjentów (38,9 %) przyjmujących kanakimumab osiągnęło remisję DAS28-ESR w porównaniu z 2 z 17 pacjentów (11,8 %) otrzymujących placebo.
Te dane są zgodne z wynikami połączonej analizy skuteczności 418 pacjentów z SJIA, które wykazały, że skuteczność kanakimumabu w podgrupie pacjentów z SJIA w wieku od 16 do < 20 lat (n = 34) odpowiadała skuteczności obserwowanej u pacjentów poniżej 16 roku życia (n = 384).
Zapalenie stawów podagrycznych
Skuteczność Ilaris w leczeniu napadów ostrych zapalenia stawów podagrycznych została wykazana w dwóch wieloośrodkowych, randomizowanych, podwójnie ślepych, kontrolowanych aktywnie badaniach u pacjentów z częstymi napadami zapalenia stawów podagrycznych (3 lub więcej napadów w ciągu ostatnich 12 miesięcy) i niemożnością stosowania niesteroidowych leków przeciwzapalnych (NLPZ) lub kolkochiny (z powodu przeciwwskazań, nietolerancji lub niewystarczającej skuteczności). Badania trwały 12 tygodni z 12-tygodniowym podwójnie ślepym rozszerzeniem. Ogółem 225 pacjentom podano Ilaris podskórnie w dawce 150 mg, a 229 pacjentom podano wewnątrzmięśniowo triamcinolonu acetonid (TA) w dawce 40 mg w momencie rozpoczęcia badania i po nawrocie napadu. Średnia liczba napadów zapalenia stawów podagrycznych w ciągu ostatnich 12 miesięcy wynosiła 6,5. Ponad 85 % pacjentów miało choroby współistniejące, w tym nadciśnienie tętnicze (60 %), cukrzycę (15 %), chorobę wieńcową (12 %) i przewlekłe choroby nerek stadium ≥ 3 (25 %). Około jedna trzecia pacjentów włączonych do badania (76 [33,8 %] w grupie Ilaris i 84 [36,7 %] w grupie triamcinolonu) nie mogła stosować NLPZ i kolkochiny (nietolerancja, przeciwwskazania lub brak odpowiedzi). Leczenie towarzyszące obniżające poziom kwasu moczowego (ULT) stosowano u 42 % pacjentów przy włączeniu do badania.
Pierwotnymi punktami końcowymi były: (I) nasilenie bólu w zapaleniu stawów podagrycznych (według wizualnej skali analogowej, VAS) po 72 godzinach od podania dawki oraz (II) czas do pierwszego nowego napadu zapalenia stawów podagrycznych.
W ogólnej populacji badania nasilenie bólu było statystycznie istotnie niższe przy stosowaniu Ilaris 150 mg w porównaniu z triamcinolonu acetonidem po 72 godzinach. Ilaris zmniejsza również ryzyko kolejnych napadów (patrz tabela 4).
Wyniki skuteczności w podgrupie pacjentów, którzy nie mogą stosować NLPZ i kolkochiny, oraz tych, którzy stosowali ULT, nie odpowiadali na ULT lub mieli przeciwwskazania do ULT (N = 101), były zgodne z wynikami badania w ogólnej populacji, z istotnymi statystycznie różnicami w porównaniu z triamcinolonu acetonidem w zakresie nasilenia bólu po 72 godzinach (–10,2 mm, p = 0,0208) i zmniejszenia ryzyka kolejnych napadów (stosunek ryzyka 0,39, p = 0,0047 w 24. tygodniu).
Wyniki skuteczności dla skróconej podgrupy ograniczonej do pacjentów stosujących ULT (N = 62) przedstawiono w tabeli 3. Leczenie Ilaris przyczyniło się do zmniejszenia bólu i zmniejszenia ryzyka kolejnych napadów u pacjentów stosujących ULT, którzy nie mogą stosować NLPZ i kolkochiny, choć różnica w leczeniu w porównaniu z triamcinolonu acetonidem była mniej wyraźna niż w ogólnej populacji badania.
Tabela 4. Skuteczność w ogólnej populacji badania i podgrupie pacjentów stosujących ULT, którzy nie mogą stosować NLPZ lub kolkochiny
| Punkt końcowy skuteczności |
Całkowita populacja badawcza N=454 |
Pacjenci, którzy nie mogą stosować NSAID i kolkochiny, otrzymujący ULT N=62 |
| Leczenie napadów artretyzmu podagrycznego (intensywność bólu (VAS) po 72 godzinach) |
||
| Ocena średniej różnicy metodą najmniejszych kwadratów dla triamcinolonu acetonidu CI p – poziom, jednostronny |
−10,7 (−15,4, −6,0) p < 0,0001* |
−3,8 (−16,7, 9,1) p = 0,2798 |
| Ryzyko zmniejszenia kolejnych napadów artretyzmu podagrycznego oceniane jako czas do pierwszego nasilenia (24 tygodnie) |
||
| Stosunek ryzyka dla triamcinolonu acetonidu CI p – poziom, jednostronny |
0,44 (0,32, 0,60) p < 0,0001* |
0,71 (0,29, 1,77) p = 0,2337 |
| *Oznacza istotny poziom p ≤0,025. |
||
Wyniki badań bezpieczeństwa wykazały większą liczbę przypadków niepożądanych zdarzeń po podaniu kanakinumabu w porównaniu z triamcinolonem acetonidem: 66 % wobec 53 % pacjentów, u których wystąpiły jakiekolwiek niepożądane zdarzenia, oraz 20 % wobec 10 % pacjentów, u których wystąpiły przypadki infekcji, w ciągu 24 tygodni.
Pacjenci w wieku podeszłym
Ogólnie skuteczność, bezpieczeństwo i profil tolerancji Ilaris u pacjentów w wieku podeszłym (≥ 65 lat) były porównywalne z takimi u pacjentów poniżej 65 roku życia.
Pacjenci leczeni terapią obniżającą poziom kwasu moczowego (ULT)
W badaniach klinicznych Ilaris można bezpiecznie stosować razem z ULT. W ogólnej populacji badanej pacjenci leczeni ULT wykazywali mniejsze zmniejszenie bólu i niższy spadek ryzyka kolejnych napadów artretyzmu podagrycznego w porównaniu z pacjentami nieleczonymi ULT.
Immunogenność
Nie zaobserwowano żadnych reakcji anafilaktycznych u pacjentów otrzymujących Ilaris.
Przeciwciała przeciwko lekowi Ilaris obserwowano u około 1,5 %, 3 % i 2 % pacjentów otrzymujących Ilaris w leczeniu periodycznych zespołów związanych z kriopiryną, uogólnionego młodzieńczego idiopatycznego zapalenia stawów (SJIA) i artretyzmu podagrycznego, odpowiednio.
Przeciwciał przeciwkanakinumabowi nie obserwowano u pacjentów z zespołem TRAP, zespołem HID/MKD oraz FMF, którzy otrzymywali dawki 150 mg i 300 mg przez 16 tygodni leczenia.
Populacja pediatryczna
Właściciel zakończył cztery plany badań pediatrycznych dotyczących kanakinumabu (dla zespołu CAP, SJIA, FMF – zespół HID/MKD oraz zespół TRAP, odpowiednio). Informacje o leku zostały zaktualizowane, aby uwzględnić wyniki badań zastosowania kanakinumabu w populacji pediatrycznej.
Europejska Agencja Leków odmówiła obowiązku przedstawienia wyników badań Ilaris we wszystkich podkategorii badań populacji pediatrycznej w przypadku artretyzmu podagrycznego.
Farmakokinetyka.
Periodyczne zespoły związane z kriopiryną (CAPS)
Wchłanianie
Szczotkowa stężenie surowicy kanakinumabu (Cmax) obserwowano około 7 dni po jednorazowym podaniu podskórnie dawki 150 mg dorosłym pacjentom z CAPS. Średni okres półtrwania wynosił 26 dni. Średnie wartości Cmax i AUCinf po jednorazowym podaniu podskórnie dawki 150 mg typowemu dorosłemu pacjentowi z CAPS (70 kg) wynosiły 15,9 µg/ml i 708 µg*d/ml. Szacowana biodostępność po podaniu podskórnym kanakinumabu wynosi 66 %. Parametry ekspozycji (np. AUC i Cmax) zwiększały się proporcjonalnie do dawki w zakresie dawek od 0,30 do 10,0 mg/kg podanych w formie wlewów dożylnych lub od 150 do 600 mg w formie iniekcji podskórnej. Prognozowane wartości stężenia w stanie stacjonarnym (Cmin,ss, Cmax,ss, AUC,ss,8w) po podaniu podskórnym dawki 150 mg (lub 2 mg/kg) co 8 tygodni były nieco wyższe u pacjentów o masie ciała 40–70 kg (6,6 µg/ml, 24,3 µg/ml, 767 µg*d/ml) w porównaniu z pacjentami o masie ciała < 40 kg (4,0 µg/ml, 19,9 µg/ml, 566 µg*d/ml) i > 70 kg (4,6 µg/ml, 17,8 µg/ml, 545 µg*d/ml). Oczekiwany współczynnik kumulacji wynosił 1,3 po 6 miesiącach podawania podskórnego dawki 150 mg kanakinumabu co 8 tygodni.
Rozkład
Kanakinumab wiąże się z IL-1 beta w osoczu krwi. Objętość rozkładu (Vss) kanakinumabu zależy od masy ciała. Szacuje się, że wynosi ona 6,2 litra u pacjentów z periodycznymi zespołami związanymi z kriopiryną o masie ciała 70 kg.
Eliminacja
Widoczny klirens (CL/F) kanakinumabu zwiększa się wraz z masą ciała. Szacuje się, że wynosi on 0,17 l/dzień u pacjentów z periodycznymi zespołami związanymi z kriopiryną o masie ciała 70 kg oraz 0,11 l/dzień u pacjentów z uogólnionym młodzieńczym idiopatycznym zapaleniem stawów o masie ciała 33 kg.
Nie zaobserwowano żadnych oznak przyspieszonego klirensu ani zależnych od czasu zmian właściwości farmakokinetycznych kanakinumabu po wielokrotnym podaniu. Po skorygowaniu masy ciała nie zaobserwowano różnic farmakokinetycznych w zależności od płci ani wieku pacjenta.
Zespół TRAP, zespół HID/MKD i FMF
Biodostępność u chorych na zespół TRAP, zespół HID/MKD oraz FMF nie była odrębnie określana. Widoczny klirens (CL/F) w populacji z zespołem TRAP, zespołem HID/MKD oraz FMF przy masie ciała 55 kg (0,14 l/d) był porównywalny z takim w populacji z zespołem CAP przy masie ciała 70 kg (0,17 l/dzień). Widoczna objętość rozkładu (V/F) wynosiła 4,96 l przy masie ciała 55 kg.
Po wielokrotnym podaniu podskórnemu dawki 150 mg co 4 tygodnie minimalne stężenie kanakinumabu w 16. tygodniu (Cmin) szacowano na 15,4±6,6 µg/ml. Szacowana stacjonarna AUCtau wynosiła 636,7±260,2 µg*d/ml.
Choroba Stilla (uogólnione młodzieńcze idiopatyczne zapalenie stawów (SJIA) i choroba Stilla u dorosłych (AOSD))
Biodostępność u pacjentów z uogólnionym młodzieńczym idiopatycznym zapaleniem stawów nie była oddzielnie określana. Widoczny klirens na kilogram masy ciała (CL/F na 1 kg) porównywano w grupach pacjentów z uogólnionym młodzieńczym idiopatycznym zapaleniem stawów oraz periodycznymi zespołami związanymi z kriopiryną (0,004 l/dzień/kg). Widoczna objętość rozkładu na kilogram masy ciała (V/F na kg) wynosiła 0,14 l/kg.
Po podaniu powtarzanych dawek 4 mg/kg co 4 tygodnie współczynnik kumulacji kanakinumabu był 1,6 razy wyższy u pacjentów z uogólnionym młodzieńczym idiopatycznym zapaleniem stawów. Stan stacjonarny osiągany był po 110 dniach. Ogólne prognozowane średnie wartości (±SD) Cmin,ss, Cmax,ss oraz AUC,ss4w wynosiły odpowiednio 14,7±8,8 µg/ml, 36,5±14,9 µg/ml oraz 696,1±326,5 µg*dzień/ml.
W podziale na grupy wiekowe AUCss4w wynosił 692, 615, 707 oraz 742 µg*dzień/ml u pacjentów w wieku 2–3, 4–5, 6–11 oraz 12–19 lat, odpowiednio. Po podziale według masy ciała zaobserwowano niższą (30–40 %) medianę ekspozycji Cmin,ss (11,4 w porównaniu z 19 µg/ml) oraz AUCss (594 w porównaniu z 880 µg*dzień/ml) w kategorii pacjentów o niższej masie ciała (≤ 40 kg) w porównaniu z pacjentami o wyższej masie ciała (> 40 kg).
Na podstawie analizy modelowania farmakokinetycznego populacyjnego farmakokinetyka kanakinumabu u młodych dorosłych pacjentów z SJIA w wieku od 16 do 20 lat była podobna do takiej u pacjentów poniżej 16 roku życia. Prognozowana ekspozycja kanakinumabu w stanie stacjonarnym przy dawce 4 mg/kg (maksymalnie 300 mg) u pacjentów powyżej 20 roku życia była porównywalna z taką u pacjentów z SJIA poniżej 20 roku życia.
Pacjenci z artretyzmem podagrycznym
Biodostępność u pacjentów z artretyzmem podagrycznym nie była określana. Widoczny klirens na kilogram masy ciała (CL/F na 1 kg) porównywano w grupach pacjentów z artretyzmem podagrycznym i CAPS (0,004 l/dzień/kg). Średnia ekspozycja u typowego pacjenta z artretyzmem podagrycznym (93 kg) po jednorazowej dawce podskórnej 150 mg (Cmax: 10,8 µg/ml i AUCinf: 495 µg*d/ml) była niższa niż u typowych pacjentów z CAPS o masie ciała 70 kg (15,9 µg/ml i 708 µg*d/ml). Jest to zgodne z obserwowanym wzrostem CL/F w zależności od masy ciała.
Oczekiwany współczynnik kumulacji był 1,1 razy wyższy po podaniu podskórnemu kanakinumabu w dawce 150 mg co 12 tygodni.
Dzieci
Stężenie szczytowe kanakinumabu osiągano po 2–7 dniach po jednorazowym podaniu podskórnemu kanakinumabu w dawce 150 mg lub 2 mg/kg u chorych pediatrycznych powyżej 4 roku życia. Okres półtrwania wahał się od 22,9 do 25,7 dnia, podobnie jak u dorosłych. Na podstawie analizy modelowania farmakokinetycznego farmakokinetyka kanakinumabu u dzieci w wieku od 2 do 4 lat była analogiczna do tej u chorych powyżej 4 roku życia.
Stwierdzono, że po podaniu podskórnemu stopień absorpcji zmniejsza się z wiekiem i przyspiesza u młodszych pacjentów. W związku z tym Tmax był krótszy (3,6 dnia) u młodszych pacjentów z uogólnionym młodzieńczym idiopatycznym zapaleniem stawów (2–3 lata) w porównaniu z starszymi pacjentami z uogólnionym młodzieńczym idiopatycznym zapaleniem stawów (12–19 lat; Tmax: 6 dni). Nie wykazano negatywnego wpływu na biodostępność (AUCss).
Dodatkowa analiza farmakokinetyczna wykazała, że farmakokinetyka kanakinumabu u 6 pacjentów poniżej 2 roku życia z periodycznymi zespołami związanymi z kriopiryną była podobna do farmakokinetyki u dzieci w wieku 2–4 lat. Modelowanie populacyjne farmakokinetyki wskazuje, że prognozowane poziomy ekspozycji po dawce 2 mg/kg były porównywalne u dzieci z periodycznymi zespołami związanymi z kriopiryną, ale o 40 % niższe u pacjentów o bardzo niskiej masie ciała, np. 10 kg, w porównaniu z dorosłymi pacjentami (dawka 150 mg/kg). Jest to zgodne z wyższymi poziomami ekspozycji w grupach pacjentów z periodycznymi zespołami związanymi z kriopiryną o wyższej masie ciała.
Farmakokinetyka jest taka sama u dzieci z periodycznymi zespołami związanymi z kriopiryną, zespołem TRAP, zespołem HID/MKD i FMF oraz uogólnionym młodzieńczym idiopatycznym zapaleniem stawów.
Pacjenci w wieku podeszłym
Nie zaobserwowano żadnych zmian parametrów farmakokinetycznych opartych na klirensie lub objętości rozkładu u pacjentów w wieku podeszłym i dorosłych poniżej 65 roku życia.
Dane przedkliniczne dotyczące bezpieczeństwa
Dane przedkliniczne nie wykazały specyficznych zagrożeń dla człowieka na podstawie badań reaktywności krzyżowej, wielokrotnego podawania dawek, toksyczności immunologicznej, toksyczności rozrodczej i toksyczności u dzieci, przeprowadzonych z kanakinumabem lub przeciwciałami mysimi przeciwko IL-1 beta u myszy.
Ponieważ kanakinumab wiąże się z IL-1 beta u zwierząt (C. jacchus) i człowieka z podobnym powinowactwem, bezpieczeństwo kanakinumabu badano na zwierzętach. Nie zaobserwowano żadnych skutków ubocznych kanakinumabu po podaniu zwierzętom dwa razy w tygodniu przez 26 tygodni ani w badaniu toksyczności rozwoju embrionalno-płodowego u ciężarnych zwierząt. Stężenia w osoczu, które były dobrze tolerowane u zwierząt, przekraczały co najmniej 42 razy (Cmax) i 78 razy (CAVG) stężenia w osoczu u pacjentów pediatrycznych z CAPS (masa ciała 10 kg), którym podawano dawki kliniczne kanakinumabu do 8 mg/kg podskórnie co 8 tygodni. Ponadto przeciwciała przeciwkanakinumabowi nie zostały wykryte w tych badaniach. Nie wykazano żadnej niespecyficznej tkankowej reaktywności krzyżowej po nałożeniu kanakinumabu na zdrowe tkanki człowieka.
Formalnych badań kanakinumabu pod kątem rakotwórczości nie przeprowadzono.
W badaniu rozwoju embrionalno-płodowego u zwierząt kanakinumab nie wykazał toksycznego wpływu na organizm matki, embriotoksyczności ani działania teratogennego po podaniu w trakcie organogenezy.
Nie zaobserwowano żadnych skutków ubocznych z przeciwciałami mysimi przeciw IL-1 beta w szeregu badań rozrodczych i badań na młodych myszach. Przeciwciała mysie przeciw IL-1 beta nie wykazały negatywnego wpływu na płód ani na wzrost noworodka po podaniu matce w późnym okresie ciąży, podczas porodu i karmienia piersią. Wysokie dawki stosowane w tych badaniach były maksymalnie skuteczne pod względem hamowania aktywności IL-1 beta.
Badania immunotoksykologiczne na myszach z przeciwciałami mysimi przeciw IL-1 beta wykazały, że neutralizacja IL-1 beta nie ma żadnego wpływu na parametry immunologiczne i nie powoduje zaburzeń funkcji immunologicznej u myszy.
Charakterystyki kliniczne.
Wskazania.
Zespoły okresowej gorączki
Ilaris wskazany jest w leczeniu następujących autozakładczych zespółów okresowej gorączki u dorosłych, nastolatków i dzieci od 2. roku życia:
Okresowe zespoły związane z kriopiryną
Leczenie okresowych zespołów związanych z kriopiryną u dorosłych, nastolatków i dzieci od 2. roku życia o masie ciała 7,5 kg lub wyższej, w tym:
- zespół Muckle’a–Wellsa;
- noworodkowe wieloukładowe choroby zapalne / przewlekły dziecięcy neurologiczny, skórno-stawowy zespół;
- ciężkie postacie dziedzicznego zimowego autozakładcowego zespołu / dziedzicznej zimnej pokrzywki z objawami niecharakterystycznymi dla zimnej pokrzywki.
Okresowy zespół związany z receptorem czynnika martwicy nowotworów (TRAPS).
Zespół hiperimmunoglobuliny D (HID) / niedobór mewalonokinazy (MKD).
Rodzinna gorączka śródziemnomorska (FMF).
Należy stosować w połączeniu z kolkichiną, jeśli to konieczne.
Ilaris jest również wskazany w leczeniu następujących chorób:
Choroba Stilla
Ilaris wskazany jest w leczeniu aktywnej choroby Stilla, w tym dorosłej choroby Stilla (AOSD) i uogólnionego młodzieńczego zapalenia stawów idiopatycznego (SJIA) u pacjentów od 2. roku życia, u których obserwowano niewystarczającą odpowiedź na wcześniejszą terapię niesteroidowymi lekami przeciwzapalnymi (NSAID) i kortykosteroidami doustnymi. Ilaris może być stosowany jako monoterapia lub w połączeniu z metotreksatem.
Choroba podagra
Leczenie objawowe dorosłych pacjentów z częstymi napadami podagrycznego zapalenia stawów (co najmniej 3 napady w ciągu ostatnich 12 miesięcy), gdy niesteroidowe leki przeciwzapalne (NSAID) i kolkichina są przeciwwskazane, nie są tolerowane lub nie zapewniają wystarczającego efektu, a powtarzające się cykle leczenia kortykosteroidami nie są uznawane za odpowiednie.
Przeciwwskazania.
Podwyższona wrażliwość na substancję czynną lub którąkolwiek z substancji pomocniczych. Aktywne, ciężkie infekcje.
Interakcje z innymi lekami i inne rodzaje interakcji.
Interakcje kanakimumabu z innymi lekami nie były oceniane w oficjalnych badaniach.
Zwiększenie liczby przypadków ciężkich infekcji było związane z podawaniem innego inhibitorem IL-1 w połączeniu z inhibitorami czynnika martwicy nowotworów (TNF). Stosowanie kanakimumabu w połączeniu z inhibitorami TNF nie jest zalecane, ponieważ zwiększa ryzyko ciężkich infekcji.
Aktywność enzymów wątrobowych CYP450 może być hamowana przez cytokiny stymulujące przewlekłe zapalenie, takie jak interleukina-1 beta (IL-1 beta). W związku z tym aktywność CYP450 może ulec zmianie podczas stosowania silnej terapii hamującej cytokiny, np. podczas podawania kanakimumabu. Ma to znaczenie kliniczne dla substratów CYP450 o wąskim indeksie terapeutycznym, gdy dawkowanie jest dostosowywane indywidualnie. Na początku stosowania kanakimumabu w połączeniu z tym typem leku należy przeprowadzić terapeutyczny monitoring efektu lub stężenia substancji czynnej i w razie potrzeby dostosować dawkę.
Brak danych dotyczących wpływu szczepionek żywych lub wtórnego przeniesienia infekcji szczepionkowych żywych u pacjentów stosujących kanakimumab. W związku z tym szczepionek żywych nie należy podawać jednocześnie z Ilarisem, z wyjątkiem przypadków, gdy korzyści wyraźnie przewyższają ryzyko. Jeśli szczepienie żywymi szczepionkami jest planowane po rozpoczęciu leczenia kanakimumabem, zaleca się przerwę co najmniej 3 miesięcy po ostatniej iniekcji kanakimumabu i przed kolejną iniekcją.
Wyniki badania przeprowadzonego u zdrowych dorosłych ochotników wykazały, że jedna dawka 300 mg leku Ilaris nie wpływa na indukcję i utrzymanie odpowiedzi przeciwciał po szczepieniu przeciw grypie lub szczepionką przeciwko meningokokom opartą na białku glikozylowanym.
Wyniki 56-tygodniowego badania otwartego u pacjentów z okresowymi zespołami związanymi z kriopiryną w wieku do 4 lat wykazały, że u wszystkich pacjentów, którzy otrzymali szczepionki nieżywe, które są standardem szczepień dziecięcych, wytworzyły się poziomy przeciwciał ochronnych.
Szczególne środki ostrożności.
Śledzenie
W celu poprawy śledzenia produktów biologicznych należy dokładnie dokumentować nazwę i numer serii podanego leku.
Infekcje
Zastosowanie kanakinumabu wiązało się ze zwiększoną liczbą przypadków ciężkich infekcji. Dlatego pacjentów należy dokładnie monitorować pod kątem objawów infekcji zarówno podczas, jak i po leczeniu kanakinumabem. Lekarze powinni zachować ostrożność przy stosowaniu kanakinumabu u pacjentów z infekcjami, nawracającymi infekcjami w wywiadzie lub z obecnością stanów, które mogą prowadzić do infekcji.
Leczenie okresowych zespołów związanych z kriopiryną (CAP), zespołu TRAP, zespołu HID/MKD, choroby FMF oraz choroby Still’a (SJIA i AOSD)
Nie należy stosować kanakinumabu w trakcie aktywnej infekcji wymagającej interwencji medycznej.
Leczenie artretyzmu podagrycznego
Nie należy stosować kanakinumabu w trakcie aktywnej infekcji.
Jednoczesne stosowanie leku Ilaris z inhibitorami czynnika martwicy nowotworu (TNF) nie jest zalecane, ponieważ zwiększa ryzyko ciężkich infekcji.
Zarejestrowano pojedyncze przypadki nietypowych lub oportunisticznych infekcji (w tym aspergilozy, nietypowych infekcji mikobakteriami, opryszczu pospolitego) podczas leczenia kanakinumabem. Jednak związek przyczynowy kanakinumabu z tymi zdarzeniami nie może być wykluczony.
Badanie przesiewowe na gruźlicę
W badaniach klinicznych około 12% pacjentów z okresowymi zespołami związanymi z kriopiryną, którym podawano kanakinumab bez klinicznych objawów ukrytej lub aktywnej infekcji gruźliczej, miało pozytywny wynik testu skórnego tuberkulinowego (PDD).
Nie wiadomo, czy stosowanie inhibitorów interleukiny-1 (IL-1), takich jak kanakinumab, zwiększa ryzyko reaktywacji gruźlicy. Przed rozpoczęciem terapii należy przebadać wszystkich pacjentów pod kątem obecności aktywnej i utajonej gruźlicy. Lekarz powinien dokładnie przeanalizować wywiad medyczny. U wszystkich pacjentów (możliwe są lokalne zalecenia) należy przeprowadzić odpowiednie badania przesiewowe (np. test skórny tuberkulinowy, test wydzielania interferonu gamma lub rentgen klatki piersiowej). Należy dokładnie monitorować pacjentów pod kątem rozwoju objawów gruźlicy zarówno podczas, jak i po leczeniu kanakinumabem. Pacjent powinien wiedzieć, że jeśli podczas terapii kanakinumabem pojawią się objawy wskazujące na gruźlicę (np. trwały kaszel, utrata masy ciała, subfebrilia), musi skontaktować się z lekarzem. Jeśli próba Mantoux jest pozytywna, szczególnie u pacjentów z wysokim ryzykiem, należy rozważyć alternatywne metody badania przesiewowego na infekcję gruźliczą.
Neutropenia i leukopenia
Neutropenia (bezwzględna liczba neutrofili [ANC] < 1,5 × 10⁹/l) oraz leukopenia występowały przy stosowaniu leków hamujących IL-1, w tym kanakinumabu. Nie należy rozpoczynać leczenia kanakinumabem u pacjentów z neutropenią lub leukopenią. Zaleca się ocenę poziomu białych krwinek, w tym liczby neutrofili, przed rozpoczęciem leczenia oraz po 1 i 2 miesiącach od jego rozpoczęcia. U pacjentów z przewlekłym stanem lub poddawanych ponownemu leczeniu zaleca się również okresowe oceny poziomu białych krwinek podczas terapii. Jeśli pacjent wejdzie w stan neutropenii lub leukopenii, należy dokładnie monitorować poziom białych krwinek i rozważyć konieczność przerwania leczenia.
Nowotwory złośliwe
Zarejestrowano przypadki nowotworów złośliwych u pacjentów stosujących kanakinumab. Ryzyko rozwoju nowotworów złośliwych przy stosowaniu inhibitorów IL-1 nie jest znane.
Reakcje nadwrażliwości
Zarejestrowano przypadki wskazujące na reakcje nadwrażliwości podczas stosowania kanakinumabu. Większość z tych przypadków miała łagodny stopień ciężkości. W trakcie klinicznego opracowywania Ilarisu u ponad 2600 pacjentów nie odnotowano reakcji anafilaktycznych ani anafilaktydowych. Jednak ryzyko ciężkich reakcji nadwrażliwości, które nie są rzadkością przy białkowych zastrzykach, nie może być wykluczone.
Funkcja wątroby
W badaniach klinicznych zarejestrowano krótkotrwałe i bezobjawowe przypadki podwyższenia poziomu transaminaz surowicy lub bilirubiny.
Szczepienia
Brak danych dotyczących ryzyka wtórnego przeniesienia infekcji z żywymi (osłabionymi) szczepionkami na pacjentów stosujących kanakinumab. W związku z tym szczepionek żywych nie należy podawać jednocześnie z kanakinumabem, z wyjątkiem przypadków, gdy korzyści znacznie przewyższają ryzyko.
Przed rozpoczęciem terapii kanakinumabem dorosłym pacjentom i dzieciom zaleca się, w razie potrzeby, podanie wszystkich szczepień, w tym szczepionki przeciwko pneumokokom oraz inaktywowanej szczepionki przeciwko grypie.
Mutacja genu NLRP3 u pacjentów z okresowymi zespołami związanymi z kriopiryną
Doświadczenie kliniczne u pacjentów z okresowymi zespołami związanymi z kriopiryną bez potwierdzonej mutacji w genie NLRP3 jest ograniczone.
Zespół aktywacji makrofagów u pacjentów z chorobą Still’a
Zespół aktywacji makrofagów to znany stan zagrażający życiu, który może się rozwijać u pacjentów z chorobami reumatycznymi, w szczególności u pacjentów z chorobą Still’a. W przypadku rozwoju zespołu aktywacji makrofagów lub podejrzenia takiego stanu ocena i leczenie powinny zostać rozpoczęte jak najszybciej. Lekarze powinni uważnie obserwować objawy infekcji lub nasilenia przebiegu choroby Still’a, znane jako mechanizm wyzwalający zespołu aktywacji makrofagów. Dane z badań klinicznych wskazują, że kanakinumab najprawdopodobniej nie zwiększa ryzyka rozwoju zespołu aktywacji makrofagów u pacjentów z chorobą Still’a, jednak nie pozwalają one na ostateczne wnioski.
Reakcja leku z eozynofilią i objawami systemowymi (DRESS)
Rzadko u pacjentów otrzymujących Ilaris, głównie z młodzieńczym idiopatycznym zapaleniem stawów systemowym (SJIA), zgłaszano reakcje leku z eozynofilią i objawami systemowymi (DRESS). Pacjenci z DRESS mogą wymagać hospitalizacji, ponieważ ten stan może prowadzić do śmiertelnego wyniku. Jeśli występują objawy i znaki DRESS i nie można ustalić alternatywnej etiologii, Ilaris nie powinien być ponownie przepisywany; należy rozważyć inne leczenie.
Stosowanie w okresie ciąży lub karmienia piersią.
Kobiety w wieku rozrodczym
Kobiety powinny stosować skuteczne środki antykoncepcyjne podczas leczenia kanakinumabem oraz przez 3 miesiące po podaniu ostatniej dawki.
Ciąża
Dane dotyczące stosowania leku Ilaris u kobiet w ciąży są ograniczone. Badania na zwierzętach nie wskazują na bezpośredni lub pośredni niekorzystny wpływ leku na funkcję rozrodczą. Ryzyko dla płodu/maty jest nieznane. Dlatego kobiety w ciąży lub kobiety planujące zajść w ciążę powinny stosować lek tylko po dokładnej ocenie korzyści i możliwego ryzyka.
Badania na zwierzętach wykazały, że kanakinumab przenika przez łożysko i występuje w organizmie płodu. Brak danych u ludzi, jednak ponieważ kanakinumab jest immunoglobuliną klasy G (IgG1), oczekuje się jego przenikania przez łożysko. Kliniczne znaczenie tego zjawiska jest nieznane. Jednak nie zaleca się podawania żywych szczepionek noworodkom, które były narażone na działanie kanakinumabu in utero, w ciągu 16 tygodni po ostatniej dawce kanakinumabu podanej matce przed porodem. Kobiety, które otrzymywały kanakinumab w trakcie ciąży, powinny zostać poinstruowane o konieczności poinformowania lekarza noworodka przed podaniem szczepionki.
Karmienie piersią
Nie wiadomo, czy kanakinumab wydzielany jest w mleku matki. Decyzję o stosowaniu leku Ilaris u kobiet karmiących piersią należy rozważyć tylko wtedy, gdy oczekiwana korzyść dla kobiety przewyższa potencjalne ryzyko dla dziecka.
Plodność
Nie przeprowadzono badań potencjalnego wpływu Ilarisu na płodność człowieka.
Kanakinumab nie wpływał na parametry płodności u samców zwierząt (C. jacchus). Przeciwciała anty-myszacze IL-1 beta nie wywierały niepożądanych efektów na płodność samców ani samic myszy.
Wpływ na zdolność prowadzenia pojazdów lub obsługi mechanizmów.
Ilaris ma nieznaczny wpływ na zdolność prowadzenia pojazdów lub obsługi mechanizmów. Leczenie lekiem Ilaris może prowadzić do zawrotów głowy/vertigo lub osłabienia. Pacjenci doświadczający takich objawów podczas leczenia lekiem Ilaris powinni odczekać, aż objawy ustąpią, zanim będą prowadzić samochód lub obsługiwać inne mechanizmy.
Sposób stosowania i dawki
Zespoły okresowe związane z kriopiryną (zespoły CAP), zespół okresowy związany z receptorem czynnika martwicy nowotworów (zespoł TRAP), zespół hiperimmunoglobulinemii D (zespoł HID) / niedobór mewalonianokinazy (MKD), rodzinna gorączka śródziemnomorska (FMF) oraz choroba Still’a
Leczenie należy rozpoczynać na pisemne zlecenie i pod nadzorem lekarza posiadającego doświadczenie w diagnostyce i leczeniu odpowiednich stanów.
Po odpowiednim przeszkoleniu w zakresie techniki wstrzykiwania pacjenci lub ich opiekunowie mogą samodzielnie podawać Ilaris, jeśli lekarz uzna to za stosowne i uzasadnione pod względem medycznym.
Zalecane dawki początkowe leku Ilaris u dorosłych, młodzieży i dzieci od 2. roku życia z zespołem okresowym związanym z kriopiryną (zespoł CAP):
Dorośli i dzieci od 4. roku życia:
- 150 mg u pacjentów o masie ciała > 40 kg;
- 2 mg/kg u pacjentów o masie ciała ≥ 15 kg i ≤ 40 kg;
- 4 mg/kg u pacjentów o masie ciała ≥ 7,5 kg i < 15 kg.
Dzieci w wieku od 2 do 4 lat:
- 4 mg/kg u pacjentów o masie ciała ≥ 7,5 kg.
Te dawki podaje się co osiem tygodni jako pojedynczą dawkę w formie iniekcji podskórnej.
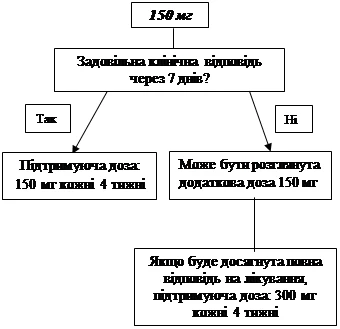
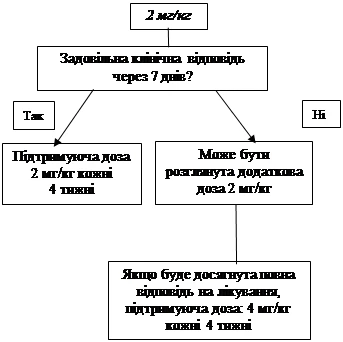
Jeśli po 7 dniach od rozpoczęcia leczenia nie osiągnięto zadowalającego efektu klinicznego (zniknięcie wysypek i innych objawów ogólnych) przy dawce początkowej 150 mg lub 2 mg/kg, możliwe jest podanie drugiej dawki leku Ilaris 150 mg lub 2 mg/kg. Po osiągnięciu efektu klinicznego należy kontynuować zintensyfikowany schemat dawkowania: 300 mg lub 2 mg/kg co 8 tygodni. Jeśli po tej zwiększonej dawce nie osiągnięto zadowalającego efektu klinicznego w ciągu 7 dni, możliwe jest podanie trzeciej dawki leku Ilaris 300 mg lub 4 mg/kg. Po osiągnięciu pełnego efektu klinicznego należy kontynuować zintensyfikowany schemat dawkowania: 600 mg lub 8 mg/kg co 8 tygodni, na podstawie indywidualnej oceny klinicznej.
Jeśli po 7 dniach od rozpoczęcia leczenia nie osiągnięto zadowalającego efektu klinicznego przy dawce początkowej 4 mg/kg, możliwe jest podanie drugiej dawki leku Ilaris 4 mg/kg. Po osiągnięciu pełnego efektu klinicznego należy kontynuować zintensyfikowany schemat dawkowania: 8 mg/kg co 8 tygodni, na podstawie indywidualnej oceny klinicznej.
Doświadczenie kliniczne w zakresie stosowania dawek w odstępach krótszych niż 4 tygodnie oraz dawek wyższych niż 600 mg lub 8 mg/kg jest ograniczone.

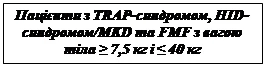
 |
|||
 |
|||
Choroba Stilla (SJIA i AOSD)
Zalecana dawka leku Ilaris u pacjentów z chorobą Still (choroba Still dorosłych (AOSD) i młodzieńcze idiopatyczne zapalenie stawów typu systemowego, o masie ciała ≥ 7,5 kg), wynosi 4 mg/kg (maksymalnie 300 mg) co cztery tygodnie w formie podskórnej iniekcji. Decyzję o kontynuacji leczenia lekiem Ilaris u pacjentów bez poprawy klinicznej podejmuje lekarz.
Choroba podagra
Leczenie powinno być prowadzone pod nadzorem lekarzy posiadających doświadczenie w rozpoznawaniu i leczeniu choroby podagry oraz stosowaniu leków biologicznych. Lek Ilaris powinien być podawany przez personel medyczny.
Należy rozpocząć leczenie hiperurykemii za pomocą odpowiedniej terapii obniżającej lub optymalizującej stężenie moczanów (ULT). Kanakinumab powinien być stosowany jako terapia na żądanie w leczeniu napadów podagrycznego zapalenia stawów.
Wymagana jest kontrola hiperurykemii za pomocą odpowiedniej terapii obniżającej poziom moczanów. Ilaris należy stosować jako terapię na żądanie w leczeniu zapaleń stawów spowodowanych podagrą.
Zalecana dawka leku Ilaris u dorosłych pacjentów z podagrycznym zapaleniem stawów wynosi 150 mg podskórnie jako dawka pojedyncza podczas napadu. Aby osiągnąć maksymalny efekt, Ilaris należy podawać jak najszybciej po wystąpieniu napadu podagrycznego zapalenia stawów.
Pacjentom, u których nie stwierdzono efektu w wczesnym okresie leczenia, nie należy ponownie podawać Ilaris. U pacjentów, u których zaobserwowano efekt i którzy wymagają ponownego leczenia, odstęp między dawkami powinien wynosić co najmniej 12 tygodni.
Grupy specjalne
Pacjenci w podeszłym wieku
Dostosowanie dawki nie jest wymagane.
Nie stwierdzono istotnych różnic w profilu bezpieczeństwa u pacjentów w wieku powyżej 65 lat.
Niewydolność wątroby
Brak danych dotyczących stosowania leku Ilaris u pacjentów z zaburzeniami funkcji wątroby.
Niewydolność nerek
Dostosowanie dawki nie jest wymagane u pacjentów z niewydolnością nerek. Jednakże doświadczenie kliniczne w stosowaniu leku u tych pacjentów jest ograniczone.
Sposób podania
Ilaris, 150 mg, proszek do sporządzenia roztworu do wstrzykiwania, dostarczany jest w fiolce jednorazowej do indywidualnego użytku. Nieużywany lek lub odpady należy zutylizować zgodnie z lokalnymi przepisami.
Instrukcje rozpuszczania
Z zachowaniem techniki jałowej rozpuścić zawartość fiolki w temperaturze pokojowej: powoli wstrzyknąć 1,0 ml wody do wstrzykiwania za pomocą strzykawki 1 ml i igły 18 G × 50 mm. Delikatnie obracać fiolkę pod kątem około 45° przez około 1 minutę, a następnie pozostawić na 5 minut. Następnie ostrożnie odwrócić fiolkę do góry nogami i z powrotem dziesięć razy. Jeśli to możliwe, nie dotykać gumowego zatyczki palcami. Pozostawić na 15 minut w temperaturze pokojowej, aż do uzyskania przezroczystego lub opalescencyjnego roztworu. Nie wstrząsać. Nie należy stosować leku, jeśli w roztworze występują cząstki.
Płukać ścianki fiolki, aby usunąć resztki cieczy z zatyczki. Roztwór powinien być wolny od widocznych cząstek, przejrzysty lub opalescencyjny. Roztwór powinien być bezbarwny, ale może mieć lekko brązowozłoty odcień. Jeśli roztwór ma intensywnie brązowy kolor, nie należy go stosować. Jeśli roztwór nie został użyty natychmiast po rozpuszczeniu, należy go przechowywać w temperaturze od 2 do 8 °C i zużyć w ciągu 24 godzin.
Instrukcje dotyczące podania
Delikatnie napełnić strzykawkę niezbędną ilością roztworu w zależności od dawki (od 0,2 ml do 1,0 ml) i podać podskórnie za pomocą igły 27 G × 13 mm.
Miejsca iniekcji: górna część uda, brzuch, ramię lub pośladki. Należy unikać obszarów z uszkodzoną skórą, siniakami lub wysypką. Należy unikać wstrzykiwania w tkankę bliznową, ponieważ może to zmniejszyć działanie leku Ilaris.
Utylizacja
Pacjentów lub ich opiekunów należy poinstruować w zakresie utylizacji fiolki, strzykawek i igieł zgodnie z lokalnymi przepisami.
Dzieci.
Okresowe zespoły związane z kriopiryną (CAP), zespół TRAP, zespół HID/MKD oraz FMF
Nie ustalono bezpieczeństwa i skuteczności stosowania leku Ilaris u dzieci poniżej 2 roku życia z okresowymi zespołami związanymi z kriopiryną, zespołem TRAP, zespołem HID/MKD oraz FMF. Dane dostępne obecnie opisano w punktach „Farmakokinetyka”, „Farmakodynamika”, „Działania niepożądane”, jednak nie można sformułować żadnych zaleceń dotyczących dawkowania.
Młodzieńcze idiopatyczne zapalenie stawów typu systemowego (SJIA)
Nie ustalono bezpieczeństwa i skuteczności stosowania leku Ilaris u dzieci poniżej 2 roku życia z młodzieńczym idiopatycznym zapaleniem stawów typu systemowego.
Choroba podagra
Brak doświadczenia w stosowaniu leku Ilaris u dzieci wskazanych do leczenia chorobą podagry.
Przedawkowanie.
Informacje dotyczące przedawkowania są ograniczone. W badaniach wczesnej fazy pacjenci i zdrowi ochotnicy otrzymywali dawki do 10 mg/kg dożylnie lub podskórnie bez żadnych objawów ostrej toksyczności leku.
W przypadku przedawkowania zaleca się monitorowanie stanu pacjenta i, w razie potrzeby, natychmiastowe rozpoczęcie odpowiedniej terapii objawowej.
Efekty uboczne.
Do ślepych otwartych badań klinicznych włączono około 2300 pacjentów, w tym około 250 dzieci (w wieku od 2 do 17 lat) z rozpoznanymi syndromami okresowymi związanymi z kryopiryną, młodzieńczym idiopatycznym zapaleniem stawów typu systemowego, artretyzmem podagrycznym lub innymi chorobami pośrednio związanymi z IL-1β, a także zdrowych ochotników. Najczęstszymi reakcjami ubocznymi były infekcje (np. infekcje dróg oddechowych górnych). Większość reakcji miała charakter łagodny lub umiarkowany. Długotrwałe leczenie nie wpływało na rodzaj ani częstość występowania działań niepożądanych.
U pacjentów stosujących Ilaris obserwowano przypadki reakcji nadwrażliwości.
Podczas leczenia lekiem Ilaris odnotowano infekcje oportunistyczne.
Syndromy okresowe związane z kryopiryną
Do badań klinicznych włączono 211 dorosłych pacjentów oraz dzieci i młodzież (z rozpoznaniami: rodzinny zimowy autozapalny syndrom/rodzinna zimowa pokrzywka, zespół Muckle’a–Wellsa oraz noworodkowe wieloukładowe zapalenie/chroniczny dziecięcy neurologiczny, skórno-stawowy syndrom). Ocena bezpieczeństwa Ilarisu była porównywana z placebo w badaniu podstawowym fazy III, które składało się z 8-tygodniowego okresu otwartego (część 1), 24-tygodniowego okresu randomizowanego, podwójnie ślepego i kontrolowanego placebo (część 2) oraz 16-tygodniowego okresu otwartego z podawaniem Ilarisu (część 3). Wszyscy pacjenci otrzymywali 150 mg leku Ilaris podskórnie lub 2 mg/kg masy ciała w przypadku masy ciała ≥ 15 kg i ≤ 40 kg.
Młodzieńcze idiopatyczne zapalenie stawów typu systemowego
Do badań klinicznych Ilarisu włączono 201 pacjentów w wieku od 2 do 20 lat z rozpoznanym młodzieńczym idiopatycznym zapaleniem stawów typu systemowego. Bezpieczeństwo leku Ilaris porównywano z placebo w dwóch badaniach pilotażowych fazy III.
Artretyzm podagryczny
Do randomizowanych, podwójnie ślepych, aktywnie kontrolowanych badań klinicznych trwających do 24 tygodni włączono ponad 700 pacjentów z artretyzmem podagrycznym, którym podawano dawki od 10 mg do 300 mg. Ponad 250 pacjentów otrzymało leczenie w zalecanej dawce 150 mg w badaniach fazy II i III.
Reakcje niepożądane wymieniono zgodnie z klasami układów narządów MedDRA oraz częstością występowania. W ramach każdej klasy układów narządów działania niepożądane sklasyfikowano według kategorii częstości, przy czym najczęściej występujące wymieniono jako pierwsze. Kategorie częstości określono następująco: bardzo często (≥ 1/10); często (≥ 1/100 do < 1/10); rzadko (≥ 1/1000 do < 1/100); niezwykle rzadko (≥ 1/10000 do < 1/1000); bardzo rzadko (< 1/10000); nieznana (częstość nie może być oszacowana na podstawie dostępnych danych). W każdej grupie według częstości działania niepożądane wymieniono w kolejności malejącej pod względem nasilenia.
Tabela 5. Reakcje niepożądane
| Klasy układów narządów |
Zespół CAP, zespół TRAP, zespół HID/MKD, FMF, młodzieńczy idiopatyczny zapalenie stawów, artretyzm podagrny |
| Infekcje i inwazje |
|
| Bardzo często |
Infekcje dróg oddechowych (w tym zapalenie płuc, zapalenie oskrzeli, grypa, infekcje wirusowe, zapalenie zatok, zapalenie nosa, zapalenie gardła, zapalenie migdałków, zapalenie nosogardła, infekcje górnych dróg oddechowych) Infekcje ucha Łożyskowica Przeziębienie żołądka i jelit Infekcje dróg moczowych |
| Często |
Kandydoza pochwy i sromu |
| Zaburzenia ze strony układu nerwowego |
|
| Często |
Zawroty głowy/obrzydzenie |
| Zaburzenia ze strony układu pokarmowego |
|
| Bardzo często |
Ból brzucha (górnego)1 |
| Niekonie |
Choroba refluksowa przełyku2 |
| Zaburzenia ze strony skóry i tkanek podskórnych |
|
| Bardzo często |
Reakcje w miejscu wstrzyknięcia |
| Zaburzenia ze strony układu mięśniowo-szkieletowego i tkanki łącznej |
|
| Bardzo często |
Ból stawów1 |
| Często |
Ból mięśniowo-szkieletowy1 Ból pleców2 |
| Zaburzenia ogólne |
|
| Często |
Zmęczenie/astenia2 |
| Badania |
|
| Bardzo często |
Obniżenie klirensu kreatyniny nerkowej1,3 Proteinuria1,4 Leukopenia1,5 |
| Często |
Neutropenia |
| Niekonie |
Obniżenie liczby płytek krwi |
| 1Przy młodzieńczym idiopatycznym zapaleniu stawów. 2Przy artretyzmie podagrycznym. 3Zgodnie z oszacowanym klirens kreatyniny, większość przypadków była przejściowa. 4Większość przypadków była reprezentowana przez przejściowe ślady lub reakcję na poziomie 1+ białka w moczu metodą pasków testowych. 5Zobacz inne informacje poniżej. |
|
W podgrupie młodych dorosłych pacjentów z młodzieńczym idiopatycznym zapaleniem stawów systemicznym w wieku od 16 do 20 lat (n = 31) profil bezpieczeństwa kanakinumabu był zgodny z obserwowanym u pacjentów z młodzieńczym idiopatycznym zapaleniem stawów systemicznym w wieku do 16 lat. Na podstawie opublikowanych doniesień oczekuje się, że profil bezpieczeństwa u pacjentów z dorosłopoczynkową chorobą Still’a będzie podobny do takiego u pacjentów z młodzieńczym idiopatycznym zapaleniem stawów systemicznym.
Dane długoterminowych badań i odchylenia laboratoryjne u pacjentów z okresowymi zespołami związanymi z kriopiryną
W trakcie badań klinicznych Ilaris u pacjentów z okresowymi zespołami związanymi z kriopiryną obserwowano wzrost średnich wartości hemoglobiny oraz spadek stężenia białych krwinek, neutrofili i płytek krwi.
Podwyższenie poziomu transaminaz obserwowano rzadko.
Beobjawowe i umiarkowane wzrosty stężenia bilirubiny osocza bez jednoczesnego wzrostu poziomu transaminaz odnotowano u pacjentów z okresowymi zespołami związanymi z kriopiryną leczonych kanakinumabem.
W długoterminowych, otwartych badaniach z zwiększaniem dawki częstotliwość występowania infekcji (gastroenteropatia, infekcje dróg oddechowych, infekcje górnych dróg oddechowych), wymioty i zawroty głowy były wyższe w grupie otrzymującej dawkę 600 mg lub 8 mg/kg niż w innych grupach dawkowych.
Odchylenia laboratoryjne u pacjentów z zespołem TRAP, zespołem HID/MKD i FMF
Neutrofile
Chociaż zmniejszenie neutrofili ≥ stopnia 2 wystąpiło u 6,5 % pacjentów (często), a zmniejszenie stopnia 1 wystąpiło u 9,5 % pacjentów, zmiany te były zazwyczaj przemijające i nie stwierdzono infekcji związanej z neutropenią jako działania niepożąganego.
Płytki krwi
Chociaż zmniejszenie liczby płytek krwi (≥ stopnia 2) wystąpiło u 0,6 % pacjentów, nie stwierdzono krwawienia jako działania niepożąganego. Lekkie i przemijające zmniejszenie płytek krwi stopnia 1 wystąpiło u 15,9 % pacjentów bez żadnych działań niepożądanych związanych z krwawieniem.
Odchylenia laboratoryjne u pacjentów z młodzieńczym idiopatycznym zapaleniem stawów systemicznym
Hematologia
W ramach ogólnego programu leczenia młodzieńczego idiopatycznego zapalenia stawów systemicznego przemijające zmniejszenia liczby białych krwinek ≤ 0,8 × NDS odnotowano u 33 pacjentów (16,5 %). Przemijające zmniejszenia bezwzględnej liczby neutrofili (BLN) poniżej 1 × 109/l obserwowano u 12 pacjentów (6,0 %). Przemijające zmniejszenia liczby płytek krwi (< NDS) odnotowano u 19 pacjentów (9,5 %).
ALT/AST
W ramach ogólnego programu leczenia młodzieńczego idiopatycznego zapalenia stawów systemicznego wysokie stężenia ALT i/lub AST (powyżej 3-krotnej górnej granicy normy (GGN)) obserwowano u 19 pacjentów (9,5 %).
Odchylenia laboratoryjne u pacjentów z zapaleniem stawów podagrycznych
Hematologia
Zmniejszenia liczby białych krwinek ≤ 0,8 × NDS (dolna granica normy) odnotowano u 6,7 % pacjentów leczonych kanakinumabem w porównaniu do 1,4 % pacjentów leczonych triamcinolonem acetonidem. Zmniejszenia bezwzględnej liczby neutrofili (BLN) poniżej 1 × 109/l odnotowano u 2 % pacjentów w badaniach porównawczych. Obserwowano również pojedyncze przypadki stwierdzenia BLN < 0,5 × 109/l.
Umiarkowane (< NDS i > 75 × 109/l) i przemijające zmniejszenia liczby płytek krwi występowały częściej (12,7 %) po podaniu kanakinumabu w aktywnie kontrolowanych badaniach klinicznych w porównaniu z lekiem porównawczym (7,7 %) u pacjentów z zapaleniem stawów podagrycznych.
Kwas moczowy
Wzrost stężenia kwasu moczowego (0,7 mg/dl po 12 tygodniach i 0,5 mg/dl po 24 tygodniach) obserwowano po leczeniu kanakinumabem w badaniach porównawczych u pacjentów z zapaleniem stawów podagrycznych. W innym badaniu u pacjentów poddawanych ULT nie obserwowano wzrostu stężenia kwasu moczowego. Wzrostu stężenia kwasu moczowego nie obserwowano w badaniach klinicznych w grupie pacjentów bez zapalenia stawów podagrycznych.
ALT/AST
Średnie i medianowe wzrosty alaninotransaminazy (ALT) odpowiednio do 3,0 j/l i 2,0 j/l oraz asparginianotransaminazy (AST) odpowiednio do 2,7 j/l i 2,0 j/l w porównaniu z poziomem wyjściowym do końca badania obserwowano w grupach kanakinumabu i triamcinolonu acetonidu, jednak częstość zmian klinicznie istotnych (≥ 3 × GGN) była wyższa u pacjentów otrzymujących triamcinolon acetonid (2,5 % dla AST i ALT) w porównaniu z grupą leczoną kanakinumabem (1,6 % dla ALT i 0,8 % dla AST).
Triglicerydy
W aktywnie kontrolowanych badaniach klinicznych z udziałem pacjentów z zapaleniem stawów podagrycznych średnie zwiększenie stężenia triglicerydów wyniosło 33,5 mg/dl w grupie leczonej kanakinumabem w porównaniu z umiarkowanym spadkiem −3,1 mg/dl w grupie triamcinolonu acetonidu. Częstość przypadków podwyższenia stężenia triglicerydów > 5 × GGN wyniosła 2,4 % dla kanakinumabu i 0,7 % dla triamcinolonu acetonidu. Kliniczne znaczenie tego obserwowania jest nieznane.
Dane długoterminowego badania obserwacyjnego
W trakcie długotrwałego badania (średni okres ekspozycji na kanakinumab – 3,8 roku) 85 dziecięcych pacjentów w wieku ≥ 2 do ≤ 17 lat oraz 158 dorosłych pacjentów w wieku ≥ 18 lat z młodzieńczym idiopatycznym zapaleniem stawów systemicznym otrzymywało kanakinumab w warunkach standardowej praktyki klinicznej. Profil bezpieczeństwa kanakinumabu obserwowany po długotrwałym leczeniu w tych warunkach był zgodny z profilem obserwowanym w badaniach interwencyjnych u pacjentów z młodzieńczym idiopatycznym zapaleniem stawów systemicznym.
Populacja pediatryczna
Do badania zakwalifikowano 80 dziecięcych pacjentów w wieku 2–17 lat z okresowymi zespołami związanymi z kriopiryną. Ogólnie nie stwierdzono żadnych klinicznie istotnych różnic dotyczących bezpieczeństwa stosowania i profilu tolerancji Ilaris u pacjentów pediatrycznych w porównaniu z ogólną populacją pacjentów z okresowymi zespołami związanymi z kriopiryną (składającą się z dorosłych i dziecięcych pacjentów, N = 211), w tym co do ogólnej częstości i nasilenia epizodów infekcyjnych. Infekcje górnych dróg oddechowych były najczęściej występującymi chorobami zakaźnymi.
Dodatkowo oceniono 6 pacjentów pediatrycznych w wieku poniżej 2 lat w małym, otwartym badaniu klinicznym. Profil bezpieczeństwa leku Ilaris był podobny do profilu u pacjentów w wieku od 2 lat.
W trakcie 16-tygodniowego badania 102 pacjentów z zespołem TRAP, zespołem HID/MKD oraz FMF (w wieku 2–17 lat) otrzymywało kanakinumab. Ogólnie nie stwierdzono klinicznie istotnych różnic w profilu bezpieczeństwa i tolerancji kanakinumabu u pacjentów pediatrycznych w porównaniu z ogólną populacją.
Okres ważności. 3 lata.
Warunki przechowywania.
Przechowywać w temperaturze 2–8 °C w oryginalnym opakowaniu w celu ochrony przed światłem. Nie zamarzać. Przechowywać w miejscu niedostępnym dla dzieci.
Niezgodność.
Z uwagi na brak badań dotyczących zgodności, niniejszego leku nie należy mieszać z innymi lekami.
Opakowanie.
150 mg proszku do sporządzenia roztworu do wstrzykiwań w fiolce 6 ml z szkła bezbarwnego; 1 fiolka w opakowaniu kartonowym. Albo 4 opakowania kartonowe, z których każde zawiera 150 mg proszku do sporządzenia roztworu do wstrzykiwań w fiolce 6 ml z szkła bezbarwnego, w pudełku.
Kategoria recepturowa. Na receptę.
Producent.
Novartis Pharma Stein AG.
Miejsce produkcji oraz adres siedziby producenta. Schaffhauserstrasse, 4332 Stein, Szwajcaria.