Ilaris
UcraniaContenido
INSTRUCCIONES para uso médico del medicamento ILARIS (ILARIS®)
Composición:
Principio activo: canakinumab;
1 vial contiene 150 mg de canakinumab;
Excipientes: sacarosa, L-histidina, clorhidrato de L-histidina monohidrato, polisorbato 80.
Forma farmacéutica. Polvo para solución inyectable.
Características físicas y químicas principales: polvo liofilizado de color blanco.
Grupo farmacoterapéutico. Agentes antineoplásicos e inmunomoduladores. Inmunosupresores. Inhibidores de interleucina. Código ATC L04A C08.
Propiedades farmacológicas.
Farmacodinámica.
Mecanismo de acción
Canacínumab es un anticuerpo monoclonal totalmente humano del isotipo IgG1/κ frente a la interleucina-1 beta (IL-1 beta). Canacínumab se une específicamente con alta afinidad a la IL-1 beta humana y neutraliza su actividad biológica, bloqueando su interacción con los receptores de IL-1, previniendo así la activación del gen inducido por IL-1 beta y la producción de mediadores inflamatorios.
Efectos farmacodinámicos
Síndromes periódicos asociados a criopirina (CAP-síndrome), síndrome periódico asociado al receptor del factor de necrosis tumoral (TRAP-síndrome), síndrome de hiperinmunoglobulinemia D (HID-síndrome)/deficiencia de mevalonato quinasa (MKD), fiebre familiar del Mediterráneo (FMF)
En estudios clínicos realizados en pacientes con síndromes periódicos asociados a criopirina (CAP-síndrome), síndrome periódico asociado al receptor del factor de necrosis tumoral (TRAP-síndrome), síndrome de hiperinmunoglobulinemia D (HID-síndrome)/deficiencia de mevalonato quinasa (MKD) y fiebre familiar del Mediterráneo (FMF), enfermedades en las que se produce una liberación descontrolada de IL-1 beta, se observó una rápida respuesta al tratamiento con canacínumab, es decir, parámetros de laboratorio como niveles elevados de proteína C reactiva (PCR) y amiloide sérico A (ASA), leucocitosis, elevación del recuento de neutrófilos y plaquetas, se normalizaron rápidamente.
Enfermedad de Still (incluyendo la enfermedad de Still del adulto (AOSD) y la artritis idiopática juvenil sistémica (SJIA))
La enfermedad de Still del adulto y la artritis idiopática juvenil sistémica son enfermedades autoinflamatorias congénitas graves causadas por citocinas proinflamatorias, siendo la IL-1 beta una clave.
Las manifestaciones principales de la artritis idiopática juvenil sistémica incluyen fiebre, erupción cutánea, hepatosplenomegalia, linfadenopatía, poliserositis y artritis.
El tratamiento con canacínumab provocó una reducción rápida y sostenida tanto de las manifestaciones articulares como sistémicas de la artritis idiopática juvenil sistémica, con una reducción significativa del número de articulaciones inflamadas, mejoría de la fiebre y disminución de los marcadores de fase aguda en la mayoría de los pacientes.
Artritis gotosa
La causa de un ataque de artritis gotosa son los cristales de urato (urato monosódico monohidratado) en las articulaciones y tejidos circundantes, que activan macrófagos residentes para producir IL-1 beta a través del complejo inflamasoma NALP3. La activación de macrófagos y la consiguiente liberación elevada de IL-1 beta provocan una reacción inflamatoria aguda y dolorosa. Otros activadores del sistema inmunitario innato, como agonistas endógenos de los receptores tipo Toll, pueden contribuir a la activación transcripcional del gen de IL-1 beta, iniciando así un ataque de artritis gotosa. Tras el tratamiento con canacínumab, los marcadores de inflamación como la proteína C reactiva (PCR) y el amiloide sérico A (ASA), junto con signos de inflamación aguda (por ejemplo, dolor, hinchazón, enrojecimiento) en la articulación afectada, desaparecen rápidamente.
Eficacia clínica y seguridad
Síndromes periódicos asociados a criopirina (CAP-síndrome)
La eficacia y seguridad del medicamento Ilaris se han demostrado en pacientes con diferentes grados de gravedad de la enfermedad y distintos fenotipos (incluyendo formas graves del síndrome autoinflamatorio familiar por frío/urticaria familiar por frío, síndrome de Muckle-Wells y enfermedad inflamatoria multisistémica neonatal/síndrome neurológico cutáneo-articular infantil crónico). Solo se incluyeron pacientes con mutación NLRP3 confirmada en el estudio clave.
En la fase I/II del estudio, Ilaris mostró un inicio rápido de acción: los síntomas desaparecieron o disminuyeron significativamente en un día. Los parámetros de laboratorio, como los niveles de PCR y ASA, así como el recuento de neutrófilos y plaquetas, se normalizaron rápidamente en cuestión de días tras la administración de Ilaris.
El estudio clave multicéntrico de 48 semanas constó de tres partes: un período abierto de 8 semanas (parte I), un período aleatorizado, doble ciego y controlado con placebo de 24 semanas de retirada (parte II) y un período abierto de 16 semanas (parte III). El objetivo del estudio fue evaluar la eficacia, seguridad y tolerabilidad de Ilaris (150 mg o 2 mg/kg cada 8 semanas) en pacientes con síndromes periódicos asociados a criopirina.
- Parte I: respuesta clínica completa y respuesta de biomarcadores a Ilaris (definida como evaluación global del médico sobre la enfermedad autoinflamatoria y la enfermedad cutánea ≤ mínima, y valores de PCR o ASA < 10 mg/l) se observó en el 97 % de los pacientes, alcanzándose en 7 días desde el inicio del tratamiento. Se observó una mejora significativa en la evaluación clínica del médico sobre la actividad autoinflamatoria: evaluación global de la actividad de la enfermedad autoinflamatoria, evaluación de la enfermedad cutánea (urticaria, erupción cutánea), dolor articular, mialgia, cefalea/migraña, conjuntivitis, fatiga/malestar, evaluación de otros síntomas relacionados, evaluación de los síntomas por el paciente.
- Parte II: durante el período de retirada del estudio principal, el criterio principal de valoración fue la proporción de pacientes con recidiva/recaída de la enfermedad: 0 % de pacientes con recidiva en el grupo Ilaris frente al 81 % de pacientes asignados al azar al grupo placebo.
- Parte III: los pacientes que recibieron placebo en la parte II y que experimentaron recidiva, mantuvieron respuesta clínica y serológica tras ingresar en la parte abierta del estudio de extensión con Ilaris.
Tabla 1. Eficacia en la fase III del estudio clave controlado con placebo durante el período de retirada (parte II)
| Indicador |
Ilaris N=15 n(%) |
Placebo N=16 n(%) |
p-valor |
| Punto final primario (exacerbación) Número de pacientes con recidiva de la enfermedad en la parte II |
0 (0 %) |
13 (81 %) |
<0,001 |
| Marcadores de inflamación* Proteína C reactiva, mg/l Amiloide sérico A, mg/l |
1,10 (0,40) 2,27 (-0,20) |
19,93 (10,50) 71,09 (14,35) |
<0,001 0,002 |
| *Cambio medio desde el inicio de la parte II |
|||
Se realizaron dos estudios abiertos no controlados de fase III a largo plazo. En uno de ellos se evaluó la seguridad, tolerabilidad y eficacia de canakinumab en pacientes con síndromes periódicos asociados a criopirina. La duración total del tratamiento fue de entre 6 meses y 2 años. El otro estudio fue un ensayo abierto con canakinumab para evaluar la eficacia y seguridad en pacientes japoneses con síndromes periódicos asociados a criopirina, con una duración de 24 semanas y una fase de extensión hasta las 48 semanas. El objetivo principal fue evaluar la proporción de pacientes sin recidiva en la semana 24, incluidos aquellos a quienes se aumentó la dosis.
Según el análisis combinado de eficacia de estos dos estudios, el 65,6 % de los pacientes que previamente no habían sido tratados con canakinumab alcanzaron una respuesta completa con una dosis de 150 mg o 2 mg/kg, mientras que el 85,2 % de los pacientes alcanzaron remisión completa con cualquier dosis. Entre los pacientes que recibieron 600 mg o 8 mg/kg (o incluso dosis superiores), el 43,8 % lograron una respuesta completa. El número de pacientes entre 2 y 4 años que alcanzaron respuesta completa (57,1 %) fue menor en comparación con niños mayores y pacientes adultos. Entre los pacientes que alcanzaron respuesta completa, el 89,3 % mantuvieron la respuesta sin recidiva.
La experiencia clínica en pacientes individuales que alcanzaron respuesta completa tras aumentar la dosis a 600 mg (8 mg/kg) cada 8 semanas indica que una dosis más alta puede ser útil en pacientes que no alcanzaron respuesta completa o que no mantienen una respuesta completa con las dosis recomendadas (150 mg o 2 mg/kg para pacientes con peso corporal ≥ 15 kg y ≤ 40 kg). La dosis aumentada se administró más frecuentemente a pacientes de entre 2 y 4 años y a pacientes con síntomas de NOMID/CINCA en comparación con FCAS o MWS.
Se realizó un estudio de registro observacional de 6 años con el fin de obtener datos sobre la seguridad y eficacia a largo plazo del tratamiento con canakinumab en pacientes pediátricos y adultos con síndromes periódicos asociados a criopirina en la práctica clínica habitual. El estudio incluyó a 243 pacientes con síndromes periódicos asociados a criopirina (incluyendo 85 pacientes menores de 18 años). La actividad de la enfermedad se evaluó como ausente o leve/moderada en más del 90 % de los pacientes en todos los periodos posteriores al inicio del estudio, y los marcadores serológicos promedio de inflamación (PCR y SAP) se mantuvieron dentro de lo normal (<10 mg/litro) en todos los momentos posteriores al período basal. Aunque aproximadamente el 22 % de los pacientes que recibieron canakinumab requirieron ajuste de dosis, solo un pequeño porcentaje de pacientes (1,2 %) interrumpió el tratamiento con canakinumab debido a la falta de efecto terapéutico.
Población pediátrica
En los estudios sobre síndromes periódicos asociados a criopirina participaron un total de 80 pacientes pediátricos de entre 2 y 17 años de edad (a aproximadamente la mitad de ellos se les administró una dosis ajustada según el peso corporal). En general, no se observaron diferencias clínicamente relevantes en la eficacia, seguridad y perfil de tolerabilidad de Ilaris en pacientes pediátricos en comparación con la población general. La mayoría de los pacientes pediátricos mostraron mejoría en los síntomas clínicos y en los marcadores objetivos de inflamación (por ejemplo, SAP y PCR).
La eficacia, seguridad y tolerabilidad del medicamento Ilaris se evaluaron en un estudio abierto de 56 semanas que incluyó pacientes pediátricos (≤ 4 años) con síndromes periódicos asociados a criopirina. Se evaluaron diecisiete pacientes (incluyendo 6 pacientes menores de 2 años), utilizando dosis iniciales basadas en el peso corporal de 2–8 mg/kg. En el estudio también se evaluó el impacto de canakinumab sobre la formación de anticuerpos frente a vacunas infantiles estándar. No se observaron diferencias en seguridad ni eficacia entre pacientes menores de 2 años y pacientes de 2 años o más. Todos los pacientes que recibieron vacunas infantiles estándar inactivadas (N = 7) alcanzaron niveles protectores de anticuerpos.
Síndrome periódico asociado al receptor del factor de necrosis tumoral (TRAPS), síndrome de hiperinmunoglobulinemia D (HIDS)/deficiencia de mevalonato quinasa (MKD), fiebre mediterránea familiar (FMF)
La eficacia y seguridad de canakinumab para el tratamiento del síndrome periódico asociado al receptor del factor de necrosis tumoral (TRAPS), el síndrome de hiperinmunoglobulinemia D (HIDS)/deficiencia de mevalonato quinasa (MKD) y la fiebre mediterránea familiar (FMF) fueron demostradas en un estudio clave de fase III que constó de cuatro partes (N2301) e incluyó tres cohortes independientes de enfermedades.
- Parte I: Los pacientes de cada grupo, a partir de los 2 años de edad, fueron incluidos en un período de cribado de 12 semanas, durante el cual se evaluó la presencia de brotes de la enfermedad.
- Parte II: Los pacientes con brotes de la enfermedad fueron aleatorizados a un período de tratamiento doble ciego controlado con placebo de 16 semanas, durante el cual recibieron 150 mg de canakinumab (2 mg/kg para pacientes con peso corporal ≤ 40 kg) subcutáneamente o placebo cada 4 semanas. A los pacientes de entre 28 días y menos de 2 años se les permitió participar directamente en la parte abierta de la Parte II del estudio como pacientes no aleatorizados (y fueron excluidos del análisis primario de eficacia).
- Parte III: Los pacientes que completaron el tratamiento de 16 semanas y fueron clasificados como respondedores (aquellos que respondieron al tratamiento) fueron realeatorizados a un período doble ciego de retirada de 24 semanas, durante el cual recibieron 150 mg de canakinumab (2 mg/kg para pacientes con peso corporal ≤ 40 kg) subcutáneamente o placebo cada 8 semanas.
- Parte IV: Todos los pacientes de la Parte III que recibieron canakinumab tuvieron derecho a participar en un período abierto de tratamiento continuado de 72 semanas.
En total, se incluyeron 185 pacientes desde los 28 días de edad, y 181 pacientes a partir de los 2 años fueron aleatorizados en la Parte II del estudio.
El punto final primario de eficacia del período aleatorizado de tratamiento (Parte II) fue la proporción de respondedores en cada grupo que mostraron una reducción del índice de brote de la enfermedad al día 15 y no experimentaron un nuevo brote durante el resto del período de tratamiento de 16 semanas (definido como respuesta completa). La reducción del índice de brote de la enfermedad se definió como una evaluación global del médico (PGA) en la escala de actividad de la enfermedad < 2 («enfermedad mínima o ausente») y niveles de PCR dentro de lo normal (≤ 10 mg/l) o una reducción ≥ 70 % respecto al valor basal. Un nuevo brote se definió como una puntuación PGA ≥ 2 («enfermedad leve, moderada o grave») y niveles de PCR ≥ 30 mg/l. Los puntos finales secundarios basados en los resultados de la semana 16 (final de la Parte II) incluyeron la proporción de pacientes con PGA < 2, la proporción de pacientes con remisión serológica (definida como PCR ≤ 10 mg/l) y la proporción de pacientes con niveles normalizados de SAP (definida como SAP ≤ 10 mg/l).
Respecto al punto final primario de eficacia, canakinumab fue superior al placebo en los tres grupos de enfermedades. Canakinumab también demostró mayor eficacia en comparación con placebo en los puntos finales secundarios de PGA < 2 y PCR ≤ 10 mg/l en los tres grupos. Una mayor proporción de pacientes normalizó los niveles de SAP (≤ 10 mg/l) en la semana 16 con tratamiento con canakinumab en comparación con placebo en los tres grupos, con una diferencia estadísticamente significativa observada en los pacientes con TRAPS.
Tabla 2. Eficacia en el estudio clave de fase III aleatorizado y controlado con placebo durante el período de tratamiento (Parte II)
| Indicador |
Canakinumab, n/N (%) |
Placebo, n/N (%) |
p-valor |
| Punto final primario (exacerbación) Número de pacientes con un índice de exacerbación de la enfermedad detectado en el día 15 y que no experimentaron una nueva exacerbación durante el resto del período de tratamiento de 16 semanas |
|||
| FMF Síndrome HID/MKD Síndrome TRAP |
19/31 (61,29) 13/37 (35,14) 10/22 (45,45) |
2/32 (6,25) 2/35 (5,71) 2/24 (8,33) |
<0,0001* 0,0020* 0,0050* |
| Punto final secundario (enfermedad y marcadores de inflamación) |
|||
| Evaluación global del médico < 2 FMF Síndrome HID/MKD Síndrome TRAP |
20/31 (64,52) 17/37 (45,95) 10/22 (45,45) |
3/32 (9,38) 2/35 (5,71) 1/24 (4,17) |
<0,0001** 0,0006** 0,0028** |
| Proteína C reactiva ≤ 10 mg/l FMF Síndrome HID/MKD Síndrome TRAP |
21/31 (67,74) 15/37 (40,54) 8/22 (36,36) |
2/32 (6,25) 2/35 (5,71) 2/24 (8,33) |
<0,0001** 0,0010** 0,0149** |
| Amiloide A sérico ≤ 10 mg/l FMF Síndrome HID/MKD Síndrome TRAP |
8/31 (25,81) 5/37 (13,51) 6/22 (27,27) |
0/32 (0,00) 1/35 (2,86) 0/24 (0,00) |
0,0286 0,0778 0,0235* |
| n – número de pacientes respondedores; N – número de pacientes evaluados. *Indica significación estadística (unilateral) a un nivel de 0,025 basado en la prueba exacta de Fisher. **Indica significación estadística (unilateral) a un nivel de 0,025 basado en un modelo de regresión logística con el grupo de tratamiento y la PGA, PCR o AA basal, según corresponda, como variables explicativas para cada grupo. |
|||
Titulación de la dosis
En la parte II del estudio, los pacientes que recibían canakinumab y que tenían actividad persistente de la enfermedad recibieron una dosis adicional de 150 mg (o 2 mg/kg para pacientes con peso corporal ≤ 40 kg) durante el primer mes. Esta dosis adicional puede administrarse ya a los 7 días después de la primera dosis del tratamiento. Todos los pacientes con ajuste de dosis permanecieron en el grupo de dosis aumentada de 300 mg (o 4 mg/kg para pacientes con peso corporal ≤ 40 kg) cada 4 semanas.
Durante el análisis investigador del punto final primario, se observó que en los pacientes con respuesta inadecuada tras la primera dosis, el aumento de la dosis durante el primer mes hasta una dosis de 300 mg (o 4 mg/kg) cada 4 semanas mejoró adicionalmente el control de la exacerbación, la reducción de la actividad de la enfermedad y la normalización de los niveles de PCR y SAP.
Pacientes pediátricos
Dos pacientes no aleatorizados con síndrome HID/MKD de entre >28 días y <2 años de edad fueron incluidos en los estudios y recibieron canakinumab. Un paciente logró la supresión del índice de exacerbación al día 15 tras recibir una dosis única de canakinumab de 2 mg/kg, pero interrumpió el tratamiento tras esta primera dosis debido a reacciones adversas graves (pancitopenia e insuficiencia hepática). Antes de su inclusión en el estudio, este paciente había informado de antecedentes de púrpura trombocitopénica inmune y un estado médico activo con alteraciones de la función hepática. El segundo paciente recibió una dosis inicial de canakinumab de 2 mg/kg y una dosis adicional de 2 mg/kg en la semana 3, y en la semana 5 recibió un ajuste de dosis hasta 4 mg/kg, que se administró cada 4 semanas hasta el final de la parte II del estudio. La supresión de la exacerbación de la enfermedad se logró antes de la semana 5, y el paciente no experimentó nuevas exacerbaciones hasta el final de la parte II del estudio (semana 16).
Enfermedad de Still (enfermedad de Still del adulto (AOSD) y artritis idiopática juvenil sistémica (AIJS))
Artritis idiopática juvenil sistémica
La eficacia de Ilaris para el tratamiento de la artritis idiopática juvenil sistémica activa se evaluó en dos estudios principales (G2305 y G2301). Los pacientes incluidos en los estudios tenían entre 2 y 20 años de edad (edad media de 8,5 años y duración media de la enfermedad de 3,5 años al inicio del estudio) y presentaban enfermedad activa definida por la presencia de 2 o más articulaciones con artritis activa, fiebre y niveles elevados de PCR.
Estudio G2305
G2305 fue un estudio aleatorizado, doble ciego, controlado con placebo, de 4 semanas de duración. En este estudio se evaluó la eficacia a corto plazo de Ilaris en 84 pacientes, asignados aleatoriamente para recibir una dosis única de 4 mg/kg (hasta un máximo de 300 mg) de Ilaris o placebo. El objetivo principal fue determinar la proporción de pacientes que alcanzaron una mejora mínima del 30 % según los criterios pediátricos de la American College of Rheumatology (ACR). Estos criterios fueron adaptados para permitir la inclusión de pacientes sin fiebre. El tratamiento con Ilaris mejoró todos los parámetros ACR en comparación con placebo en los días 15 y 29 (tabla 3).
Tabla 3. Parámetros pediátricos ACR y estado de la enfermedad en los días 15 y 29
| Indicador |
Día 15 |
Día 29 |
||
| Ilaris N=43 |
Placebo N=41 |
Ilaris N=43 |
Placebo N=41 |
|
| ACR30 |
84 % |
10 % |
81 % |
10 % |
| ACR50 |
67 % |
5 % |
79 % |
5 % |
| ACR70 |
61 % |
2 % |
67 % |
2 % |
| ACR90 |
42 % |
0 % |
47 % |
2 % |
| ACR100 |
33 % |
0 % |
33 % |
2 % |
| Enfermedad inactiva |
33 % |
0 % |
30 % |
0 % |
| La diferencia en el tratamiento para todos los parámetros ACR fue significativa (p ≤ 0,0001) |
||||
Los resultados de los criterios adaptados pediátricos ACR, que incluían componentes sistémicos y artríticos, fueron coherentes con los resultados generales de los criterios ACR. Al día 15, el cambio medio desde el valor basal en el número de articulaciones con artritis activa y con rango de movimiento limitado fue de −67 % y −73 %, respectivamente, para Ilaris (N = 43), en comparación con un cambio mediano del 0 % y 0 % en el grupo de placebo (N = 41). El cambio medio en la escala de dolor del paciente (escala visual analógica de 0–100 mm) al día 15 fue de 50,0 mm para Ilaris (N = 43) frente a +4,5 mm para placebo (N = 25). El cambio medio en el indicador de dolor del paciente fue coherente al día 29.
Estudio G2301
G2301 fue un estudio aleatorizado, doble ciego, controlado con placebo, sobre la prevención de brotes con tratamiento con Ilaris. El estudio constó de dos partes con dos puntos finales primarios independientes (reducción exitosa de corticosteroides y tiempo hasta la inflamación). En la parte I (abierta), 177 pacientes fueron incluidos y recibieron 4 mg/kg (hasta 300 mg) de Ilaris administrado cada 4 semanas durante un periodo de hasta 32 semanas. Los pacientes en la parte II (doble ciego) recibieron Ilaris 4 mg/kg o placebo cada 4 semanas hasta la observación de 37 brotes.
Reducción de la dosis de corticosteroides
De los 128 pacientes que tomaban corticosteroides y participaron en la parte I del estudio, 92 intentaron reducir la dosis de corticosteroides. De ellos, 57 (62 %) lograron reducir significativamente la dosis de corticosteroides y 42 (46 %) suspendieron completamente el uso de corticosteroides.
Tiempo hasta el primer brote
En los pacientes que recibieron Ilaris en la parte II del estudio, se observó una reducción del 64 % en el riesgo de brote de la enfermedad en comparación con el grupo placebo (razón de riesgos 0,36; IC del 95 %: 0,17–0,75; p = 0,0032). En 63 de los 100 pacientes que participaron en la parte II del estudio en el grupo de placebo o canakinumab, no se observó ningún brote durante el periodo de observación (hasta un máximo de 80 semanas).
Resultados de los estudios G2305 y G2301 relacionados con la salud y la calidad de vida
Durante el tratamiento con Ilaris se observó una mejora clínicamente significativa en la función física y la calidad de vida de los pacientes. En el estudio G2305, según la evaluación mediante cuestionario de salud infantil, la mejora fue de 0,69 en el grupo Ilaris en comparación con el grupo placebo, lo que supera en 3,6 veces la diferencia mínima clínicamente significativa de 0,19 (p = 0,0002). La mejora media respecto al valor basal al final de la segunda parte del estudio G2301 fue de 0,88 (79 %). Se observaron mejoras estadísticamente significativas en los indicadores del cuestionario de evaluación de salud infantil PF50 en el grupo Ilaris en comparación con el grupo placebo en el estudio G2305 (estado físico p = 0,0012; bienestar psicosocial p = 0,0017).
Análisis combinado de eficacia
Los datos de las primeras 12 semanas de tratamiento con Ilaris procedentes de los estudios G2305, G2301 y un estudio ampliado se combinaron para evaluar la eficacia. Estos datos indican mejoras similares respecto al valor basal en los criterios pediátricos adaptados ACR y sus componentes ya a la semana 12, en comparación con los observados en el estudio controlado con placebo (G2305). A la semana 12, los porcentajes de respuesta pediátrica adaptada ACR30, 50, 70, 90 y 100 fueron del 70 %, 69 %, 61 %, 49 % y 30 %, respectivamente, mientras que el 28 % de los pacientes presentaban enfermedad inactiva (N = 178).
La eficacia observada durante los estudios G2305 y G2301 se mantuvo en un estudio ampliado abierto a largo plazo, que continúa actualmente (datos disponibles con una mediana de seguimiento de 49 semanas). En este estudio, 25 pacientes que tuvieron una respuesta fuerte según los criterios ACR durante al menos 5 meses redujeron la dosis de Ilaris a 2 mg/kg cada 4 semanas y mantuvieron la respuesta pediátrica ACR100 durante el tratamiento con la dosis reducida (mediana de 32 semanas, rango 8–124 semanas).
Aunque los datos son limitados, los estudios clínicos indican que los pacientes que no responden al tratamiento con tocilizumab o anakinra pueden responder al tratamiento con canakinumab.
Estudio G2306
El estudio G2306 fue un estudio abierto para evaluar el mantenimiento de la respuesta al tratamiento con la reducción de la dosis de canakinumab (2 mg/kg cada 4 semanas) o el alargamiento del intervalo (4 mg/kg cada 8 semanas) en pacientes con artritis idiopática juvenil sistémica que recibían canakinumab 4 mg/kg cada 4 semanas. Setenta y cinco pacientes de 2 a 22 años que mantenían un estado inactivo de la enfermedad durante al menos 6 meses consecutivos (remisión clínica) con monoterapia de canakinumab, incluyendo pacientes que lograron mantener el estado inactivo tras la suspensión del uso concomitante de corticosteroides y/o metotrexato durante al menos 4 semanas, fueron aleatorizados para recibir canakinumab 2 mg/kg cada 4 semanas (N = 38) o canakinumab 4 mg/kg cada 8 semanas (N = 37). Tras 24 semanas, el 71 % (27/38) de los pacientes que recibieron la dosis reducida (2 mg/kg cada 4 semanas) y el 84 % (31/37) de los pacientes con intervalo alargado (4 mg/kg cada 8 semanas) lograron mantener el estado inactivo de la enfermedad durante 6 meses. De los pacientes que permanecieron en remisión clínica y continuaron con una reducción adicional de la dosis (1 mg/kg cada 4 semanas) o alargamiento del intervalo (4 mg/kg cada 12 semanas), el 93 % (26/28) y el 91 % (30/33), respectivamente, lograron mantener el estado inactivo durante 6 meses. A los pacientes que mantuvieron un estado inactivo durante 6 meses adicionales con esta dosis mínima se les permitió suspender el tratamiento con canakinumab. En total, el 33 % (25/75) de los pacientes aleatorizados para reducir la dosis o alargar el intervalo lograron suspender el tratamiento con canakinumab y mantener el estado inactivo durante 6 meses. La tasa de eventos adversos en ambos grupos de tratamiento fue similar a la observada en pacientes que recibieron canakinumab 4 mg/kg cada 4 semanas.
Enfermedad de Still del adulto (AOSD)
La eficacia del canakinumab en una dosis de 4 mg/kg (hasta un máximo de 300 mg), administrado cada 4 semanas a pacientes con AOSD, en un estudio aleatorizado, doble ciego, controlado con placebo con 36 pacientes (de 22 a 70 años) fue similar a la observada en pacientes con SJIA. En el estudio GDE01T, una mayor proporción de pacientes (12/18, 66,7 %) en el grupo de canakinumab, en comparación con el grupo placebo (7/17, 41,2 %), mostró una mejora respecto al valor basal en el Índice de Actividad de la Enfermedad 28 con Velocidad de Sedimentación de Eritrocitos (DAS28-ESR) > 1,2 a la semana 12, aunque no alcanzó significación estadística (razón de odds 2,86, diferencia en el tratamiento [%] 25,49 [IC del 95 %: 9,43, 55,80]). A la semana 4, 7 de 18 pacientes (38,9 %) que recibieron canakinumab ya habían alcanzado remisión DAS28-ESR, frente a 2 de 17 pacientes (11,8 %) que recibieron placebo.
Estos datos son coherentes con los resultados del análisis combinado de eficacia en 418 pacientes con SJIA, que mostró que la eficacia del canakinumab en el subgrupo de pacientes con SJIA de 16 a < 20 años (n = 34) fue comparable con la observada en pacientes menores de 16 años (n = 384).
Artritis gotosa
La eficacia de Ilaris para el tratamiento de los brotes agudos de artritis gotosa se demostró en dos estudios multicéntricos, aleatorizados, doble ciego, controlados activamente, en pacientes con artritis gotosa frecuente (3 o más brotes en los últimos 12 meses) y con imposibilidad de usar AINE o colchicina (debido a contraindicaciones, intolerancia o falta de eficacia). Los estudios duraron 12 semanas con una extensión doble ciego de 12 semanas adicional. En total, 225 pacientes recibieron Ilaris subcutáneo a una dosis de 150 mg y 229 pacientes recibieron acetónido de triamcinolona (TA) intramuscular a una dosis de 40 mg en el inicio del estudio y tras la recurrencia de un brote. El número medio de brotes de artritis gotosa en los últimos 12 meses fue de 6,5. Más del 85 % de los pacientes tenían enfermedades concomitantes, incluyendo hipertensión arterial (60 %), diabetes mellitus (15 %), enfermedad coronaria (12 %) y enfermedad renal crónica estadio ≥ 3 (25 %). Aproximadamente un tercio de los pacientes incluidos en el estudio (76 [33,8 %] en el grupo Ilaris y 84 [36,7 %] en el grupo triamcinolona) no podían usar AINE ni colchicina (intolerancia, contraindicaciones o falta de respuesta). El 42 % de los pacientes recibía tratamiento concomitante para la reducción de uratos (ULT) al momento del ingreso al estudio.
Los puntos finales primarios combinados fueron: (I) intensidad del dolor en la artritis gotosa (mediante escala visual analógica, VAS) a las 72 horas tras la administración de la dosis y (II) tiempo hasta el primer nuevo brote de artritis gotosa.
En la población general del estudio, la intensidad del dolor fue estadísticamente significativamente menor con Ilaris 150 mg en comparación con triamcinolona acetonida a las 72 horas. Ilaris también reduce el riesgo de brotes posteriores (ver tabla 4).
Los resultados de eficacia en el subgrupo de pacientes que no pueden usar AINE ni colchicina, y aquellos que recibieron ULT, no obtuvieron respuesta a ULT o tenían contraindicaciones para ULT (N = 101), fueron coherentes con el estudio en la población general, mostrando diferencias estadísticamente significativas en comparación con triamcinolona acetonida respecto a la intensidad del dolor a las 72 horas (−10,2 mm, p = 0,0208) y reducción del riesgo de brotes posteriores (razón de riesgos 0,39, p = 0,0047 a la semana 24).
Los resultados de eficacia en un subgrupo reducido, limitado a pacientes que recibieron ULT (N = 62), se presentan en la tabla 3. El tratamiento con Ilaris favoreció la reducción del dolor y el riesgo de brotes posteriores en pacientes que reciben ULT y que no pueden usar AINE ni colchicina, aunque la diferencia respecto al tratamiento con triamcinolona acetonida fue menos pronunciada que en la población general del estudio.
Tabla 4. Eficacia en la población general del estudio y en el subgrupo de pacientes que reciben ULT y que no pueden usar AINE o colchicina
| Punto final de eficacia |
Población total del estudio N=454 |
Pacientes que no pueden usar AINE ni colchicina y que reciben ULT N=62 |
| Tratamiento de los ataques de artritis gotosa (intensidad del dolor [EVA] a las 72 horas) |
||
| Estimación de la diferencia media mediante el método de mínimos cuadrados para el acetonido de triamcinolona IC p-valor, unilateral |
−10,7 (−15,4, −6,0) p < 0,0001* |
−3,8 (−16,7, 9,1) p = 0,2798 |
| Riesgo de reducción de los siguientes ataques de artritis gotosa, evaluado según el tiempo hasta la primera exacerbación (24 semanas) |
||
| Relación de riesgos para el acetonido de triamcinolona IC p-valor, unilateral |
0,44 (0,32, 0,60) p < 0,0001* |
0,71 (0,29, 1,77) p = 0,2337 |
| *Indica un nivel de significación p ≤ 0,025. |
||
Los resultados del estudio de seguridad mostraron un mayor número de eventos adversos tras la administración de canakinumab en comparación con el acetónido de triamcinolona: el 66 % frente al 53 % de los pacientes con cualquier evento adverso, y el 20 % frente al 10 % de los pacientes con casos de infección, durante 24 semanas.
Pacientes de edad avanzada
En general, la eficacia, seguridad y perfil de tolerabilidad de Ilaris en pacientes de edad avanzada (≥ 65 años) fueron comparables a los de pacientes menores de 65 años.
Pacientes en tratamiento con terapia uricosurica (ULT)
En estudios clínicos, Ilaris se administró de forma segura junto con la ULT. En la población general del estudio, los pacientes en tratamiento con ULT mostraron una reducción menos pronunciada del dolor y una disminución del riesgo de futuros episodios de artritis gotosa en comparación con los pacientes que no recibieron ULT.
Inmunogenicidad
No se observaron reacciones anafilácticas en pacientes que recibieron Ilaris.
Los anticuerpos contra el medicamento Ilaris se observaron en aproximadamente el 1,5 %, 3 % y 2 % de los pacientes que recibieron Ilaris para el tratamiento de los síndromes periódicos asociados a criopirina, artritis idiopática juvenil sistémica (SJIA) y artritis gotosa, respectivamente.
No se observaron anticuerpos contra canakinumab en pacientes con síndrome TRAP, síndrome HID/MKD y FMF que recibieron dosis de 150 mg y 300 mg durante 16 semanas de tratamiento.
Población pediátrica
El solicitante ha completado cuatro planes pediátricos de investigación sobre canakinumab (para el síndrome CAP, SJIA, FMF – síndrome HID/MKD y síndrome TRAP, respectivamente). Esta información del medicamento se ha actualizado para incluir los resultados de los estudios sobre el uso de canakinumab en la población pediátrica.
La Agencia Europea de Medicamentos ha eximido la obligación de presentar los resultados de los estudios de Ilaris en todos los subgrupos de la población pediátrica con artritis gotosa.
Farmacocinética.
Síndromes periódicos asociados a criopirina (CAPS)
Absorción
La concentración máxima en suero de canakinumab (Cmax) se observó aproximadamente a los 7 días tras una administración subcutánea única de 150 mg en adultos con CAPS. El periodo medio de semivida fue de 26 días. Los valores medios de Cmax y AUCinf tras la administración de una dosis subcutánea única de 150 mg en un paciente adulto típico con CAPS (70 kg) fueron de 15,9 µg/ml y 708 µg*d/ml, respectivamente. La biodisponibilidad absoluta tras la administración subcutánea de canakinumab se estima en un 66 %. Los parámetros de exposición (por ejemplo, AUC y Cmax) aumentaron de forma proporcional a la dosis en el rango de dosis de 0,30 a 10,0 mg/kg administradas como infusión intravenosa, o de 150 a 600 mg como inyección subcutánea. Los valores predichos de exposición en estado de equilibrio (Cmin,ss, Cmax,ss, AUC,ss,8w) tras la administración subcutánea de 150 mg (o 2 mg/kg) cada 8 semanas fueron ligeramente más altos en pacientes con un peso corporal de 40–70 kg (6,6 µg/ml, 24,3 µg/ml, 767 µg*d/ml) en comparación con los de pacientes con un peso corporal < 40 kg (4,0 µg/ml, 19,9 µg/ml, 566 µg*d/ml) y > 70 kg (4,6 µg/ml, 17,8 µg/ml, 545 µg*d/ml). El coeficiente de acumulación esperado fue de 1,3 tras 6 meses de administración subcutánea de 150 mg de canakinumab cada 8 semanas.
Disposición
Canakinumab se une a la IL-1 beta en suero. El volumen de distribución (Vss) de canakinumab varía según el peso corporal. Se estima en 6,2 litros en pacientes con síndromes periódicos asociados a criopirina con un peso corporal de 70 kg.
Eliminación
La depuración aparente (CL/F) de canakinumab aumenta con el peso corporal. Se estima en 0,17 l/día en pacientes con síndromes periódicos asociados a criopirina con un peso corporal de 70 kg y en 0,11 l/día en pacientes con artritis idiopática juvenil sistémica con un peso corporal de 33 kg.
No se observaron signos de aclaramiento acelerado ni cambios dependientes del tiempo en las propiedades farmacocinéticas de canakinumab tras la administración repetida. Tras la corrección por peso corporal, no se observaron diferencias farmacocinéticas según el sexo o la edad del paciente.
Síndrome TRAP, síndrome HID/MKD y FMF
La biodisponibilidad en pacientes con síndrome TRAP, síndrome HID/MKD y FMF no se determinó por separado. La depuración aparente (CL/F) en la población con síndrome TRAP, síndrome HID/MKD y FMF con un peso corporal de 55 kg (0,14 l/día) fue comparable a la de la población con síndrome CAP con un peso corporal de 70 kg (0,17 l/día). El volumen aparente de distribución (V/F) fue de 4,96 l con un peso corporal de 55 kg.
Tras la administración subcutánea repetida de 150 mg cada 4 semanas, la concentración mínima de canakinumab en la semana 16 (Cmin) se estimó en 15,4 ± 6,6 µg/ml. La AUCtau en estado estable se estimó en 636,7 ± 260,2 µg*d/ml.
Enfermedad de Still (artritis idiopática juvenil sistémica (SJIA) y enfermedad de Still del adulto (AOSD))
La biodisponibilidad en pacientes con artritis idiopática juvenil sistémica no se determinó por separado. La depuración aparente por kilogramo de peso corporal (CL/F por 1 kg) se comparó entre grupos de pacientes con artritis idiopática juvenil sistémica y síndromes periódicos asociados a criopirina (0,004 l/día/kg). El volumen aparente de distribución por kilogramo de peso corporal (V/F por kg) fue de 0,14 l/kg.
Tras la administración de dosis repetidas de 4 mg/kg cada 4 semanas, el coeficiente de acumulación de canakinumab fue 1,6 veces mayor en pacientes con artritis idiopática juvenil sistémica. Se alcanzó el estado estable a los 110 días. Los valores medios predichos generales (± DE) de Cmin,ss, Cmax,ss y AUC,ss4w fueron de 14,7 ± 8,8 µg/ml, 36,5 ± 14,9 µg/ml y 696,1 ± 326,5 µg*día/ml, respectivamente.
Según grupos de edad, la AUCss4w fue de 692, 615, 707 y 742 µg*día/ml en pacientes de 2–3, 4–5, 6–11 y 12–19 años, respectivamente. Tras la estratificación por peso corporal, se observó una mediana de exposición más baja (30–40 %) de Cmin,ss (11,4 frente a 19 µg/ml) y AUCss (594 frente a 880 µg*día/ml) en la categoría de pacientes con menor peso corporal (≤ 40 kg) en comparación con pacientes con mayor peso corporal (> 40 kg).
Basándose en el análisis de modelado farmacocinético poblacional, la farmacocinética de canakinumab en pacientes jóvenes adultos con SJIA de 16 a 20 años fue similar a la de pacientes menores de 16 años. La exposición predicha en estado estable con una dosis de 4 mg/kg (máximo 300 mg) en pacientes mayores de 20 años fue comparable a la de pacientes con SJIA menores de 20 años.
Pacientes con artritis gotosa
La biodisponibilidad en pacientes con artritis gotosa no se determinó. La depuración aparente por kilogramo de peso corporal (CL/F por 1 kg) se comparó entre grupos de pacientes con artritis gotosa y CAPS (0,004 l/día/kg). La exposición media en un paciente típico con artritis gotosa (93 kg) tras una dosis subcutánea única de 150 mg (Cmax: 10,8 µg/ml y AUCinf: 495 µg*d/ml) fue menor que en pacientes típicos con CAPS con un peso corporal de 70 kg (15,9 µg/ml y 708 µg*d/ml). Esto concuerda con el aumento observado en CL/F en función del peso corporal.
El coeficiente esperado de acumulación fue 1,1 veces mayor tras la administración subcutánea de canakinumab a una dosis de 150 mg cada 12 semanas.
Niños
La concentración máxima de canakinumab se alcanzó entre 2 y 7 días tras la administración subcutánea única de canakinumab a una dosis de 150 mg o 2 mg/kg en pacientes pediátricos a partir de 4 años de edad. El periodo de semivida osciló entre 22,9 y 25,7 días, similar al de los adultos. Según el análisis de modelado farmacocinético, la farmacocinética de canakinumab en niños de 2 a 4 años fue análoga a la de pacientes a partir de 4 años.
Se determinó que, tras la administración subcutánea, el grado de absorción disminuye con la edad y es más rápido en pacientes jóvenes. Por consiguiente, el Tmax fue más corto (3,6 días) en pacientes jóvenes con artritis idiopática juvenil sistémica (2–3 años) en comparación con pacientes mayores con artritis idiopática juvenil sistémica (12–19 años; Tmax: 6 días). No se detectó ningún efecto negativo sobre la biodisponibilidad (AUCss).
Un análisis farmacocinético adicional mostró que la farmacocinética de canakinumab en 6 pacientes menores de 2 años con síndromes periódicos asociados a criopirina fue similar a la de niños de 2 a 4 años. El modelado farmacocinético poblacional indica que los niveles predichos de exposición tras una dosis de 2 mg/kg fueron comparables en pacientes pediátricos con síndromes periódicos asociados a criopirina, pero un 40 % menores en pacientes con peso corporal muy bajo, por ejemplo 10 kg, en comparación con pacientes adultos (dosis de 150 mg/kg). Esto concuerda con los niveles más altos de exposición en grupos de pacientes con síndromes periódicos asociados a criopirina con mayor peso corporal.
La farmacocinética es la misma en niños con síndromes periódicos asociados a criopirina, síndrome TRAP, síndrome HID/MKD y FMF y artritis idiopática juvenil sistémica.
Pacientes de edad avanzada
No se detectaron cambios en los parámetros farmacocinéticos basados en el aclaramiento o el volumen de distribución en pacientes de edad avanzada en comparación con adultos menores de 65 años.
Datos preclínicos de seguridad
Los datos preclínicos no mostraron peligros específicos para el ser humano basados en estudios de reactividad cruzada, administración de dosis repetidas, inmunotoxicidad, toxicidad reproductiva y juvenil, realizados con canakinumab o con anticuerpos murinos anti-IL-1 beta murina.
Dado que canakinumab se une a la IL-1 beta de origen animal (C. jacchus) y humano con afinidad similar, la seguridad de canakinumab se estudió en animales. No se observaron efectos adversos tras la administración del fármaco dos veces por semana durante 26 semanas ni en estudios de toxicidad embriofetal en animales gestantes. Las concentraciones en plasma bien toleradas en animales excedieron al menos 42 veces (Cmax) y 78 veces (CAVG) las concentraciones en plasma en pacientes pediátricos con CAPS (peso corporal 10 kg) que recibieron dosis clínicas de canakinumab hasta 8 mg/kg subcutáneamente cada 8 semanas. Además, no se detectaron anticuerpos contra canakinumab en estos estudios. No se demostró reactividad cruzada tisular no específica tras la aplicación de canakinumab en tejidos humanos sanos.
No se realizaron estudios formales de carcinogenicidad con canakinumab.
En estudios de desarrollo embriofetal en animales, canakinumab no mostró efectos tóxicos en la madre, embriotóxicos ni teratogénicos tras la administración durante la organogénesis.
No se observaron efectos adversos con anticuerpos murinos anti-IL-1 beta murina en una serie de estudios reproductivos y en estudios con ratones juveniles. Los anticuerpos anti-IL-1 beta murina no mostraron efectos adversos sobre el feto ni sobre el crecimiento del recién nacido cuando se administraron a la madre en etapas tardías del embarazo, durante el parto y la lactancia. Las altas dosis utilizadas en estos estudios fueron las máximamente eficaces en términos de supresión de la actividad de IL-1 beta.
Estudios inmunotoxicológicos en ratones con anticuerpos murinos anti-IL-1 beta murina mostraron que la neutralización de IL-1 beta no tiene ningún efecto sobre los parámetros inmunológicos ni causa alteraciones en la función inmune en ratones.
Características clínicas.
Indicaciones.
Síndromes febriles periódicos
Ilaris está indicado para el tratamiento de los siguientes síndromes febriles periódicos autoinflamatorios en adultos, adolescentes y niños a partir de 2 años de edad:
Síndromes periódicos asociados a criopirina
Tratamiento de los síndromes periódicos asociados a criopirina en adultos, adolescentes y niños a partir de 2 años de edad con un peso corporal de 7,5 kg o superior, incluyendo:
- Síndrome de Muckle-Wells;
- Enfermedad inflamatoria multisistémica neonatal/síndrome neurológico cutáneo articular crónico infantil;
- Formas graves del síndrome autoinflamatorio familiar inducido por frío/sarpullido familiar por frío con síntomas no característicos del urticaria inducida por frío.
Síndrome periódico asociado al receptor del factor de necrosis tumoral (TRAPS).
Síndrome de hiperinmunoglobulinemia D (HID) / deficiencia de mevalonato quinasa (MKD).
Fiebre familiar del Mediterráneo (FMF).
Debe utilizarse en combinación con colchicina si es necesario.
Ilaris también está indicado para el tratamiento de las siguientes enfermedades:
Enfermedad de Still
Ilaris está indicado para el tratamiento de la enfermedad de Still activa, incluyendo la enfermedad de Still del adulto (AOSD) y la artritis idiopática juvenil sistémica (SJIA) en pacientes a partir de 2 años de edad que hayan tenido una respuesta inadecuada al tratamiento previo con antiinflamatorios no esteroideos (AINE) y corticosteroides sistémicos. Ilaris puede utilizarse como monoterapia o en combinación con metotrexato.
Artritis gotosa
Tratamiento sintomático de pacientes adultos con episodios frecuentes de artritis gotosa (al menos 3 episodios durante los últimos 12 meses) cuando los antiinflamatorios no esteroideos (AINE) y la colchicina están contraindicados, no son tolerados o no proporcionan un efecto adecuado, y cuando la administración repetida de corticosteroides no es aceptable.
Contraindicaciones.
Hipersensibilidad al principio activo o a cualquiera de los excipientes. Infecciones activas y graves.
Interacción con otros medicamentos y otras formas de interacción.
Las interacciones de canakinumab con otros medicamentos no han sido evaluadas en estudios oficiales.
Se ha relacionado el aumento en el número de infecciones graves con la administración de otro inhibidor de IL-1 en combinación con inhibidores del factor de necrosis tumoral (FNT). No se recomienda el uso de canakinumab junto con inhibidores del FNT, ya que esto incrementa el riesgo de infecciones graves.
La actividad de los enzimas hepáticos CYP450 puede estar suprimida por citoquinas que estimulan la inflamación crónica, como la interleucina-1 beta (IL-1 beta). Por lo tanto, la actividad del CYP450 puede modificarse durante una terapia potente inhibidora de citoquinas, como con la administración de canakinumab. Esto tiene relevancia clínica para los sustratos del CYP450 con un índice terapéutico estrecho, cuando la dosis se ajusta individualmente. Al iniciar el tratamiento con canakinumab junto con este tipo de medicamento, se debe realizar un monitoreo terapéutico del efecto o de la concentración del principio activo y ajustar la dosis si es necesario.
No existen datos sobre el efecto de las vacunas vivas o la transmisión secundaria de infección con vacunas vivas en pacientes que toman canakinumab. Por lo tanto, no se deben administrar vacunas vivas simultáneamente con Ilaris, excepto cuando los beneficios superen claramente los riesgos. Si se planea la vacunación con vacunas vivas después del inicio del tratamiento con canakinumab, se recomienda hacer una pausa de al menos 3 meses tras la última inyección de canakinumab y antes de la siguiente inyección.
Los resultados de un estudio realizado con voluntarios adultos sanos mostraron que una dosis única de 300 mg de Ilaris no afecta la inducción ni el mantenimiento de la respuesta de anticuerpos tras la vacunación contra la gripe o con vacuna contra meningococo basada en proteína glicosilada.
Los resultados de un estudio abierto de 56 semanas en pacientes con síndromes periódicos asociados a criopirina menores de 4 años mostraron que todos los pacientes que recibieron vacunas inactivadas, que son parte del calendario estándar de vacunación infantil, desarrollaron niveles protectores de anticuerpos.
Características de uso.
Rastreabilidad
Para mejorar el rastreo de medicamentos biológicos, se debe registrar claramente el nombre y el número de lote del producto administrado.
Infecciones
El uso de canakinumab se ha asociado con un aumento en la frecuencia de infecciones graves. Por lo tanto, los pacientes deben ser vigilados cuidadosamente en busca de signos y síntomas de infección durante y después del tratamiento con canakinumab. Los médicos deben tener precaución al administrar canakinumab a pacientes con infecciones activas, antecedentes de infecciones recurrentes o condiciones que puedan predisponer a infecciones.
Tratamiento de los síndromes periódicos asociados a criopirina (CAP), síndrome TRAP, síndrome HID/MKD, FMF y enfermedad de Still (SJIA y AOSD)
No se debe administrar canakinumab durante una infección activa que requiera tratamiento médico.
Tratamiento de la artritis gotosa
No se debe administrar canakinumab durante una infección activa.
No se recomienda la administración concomitante del medicamento Ilaris con inhibidores del factor de necrosis tumoral (FNT), ya que esto aumenta el riesgo de infecciones graves.
Se han notificado casos aislados de infecciones inusuales u oportunistas (incluyendo aspergilosis, infecciones micobacterianas atípicas y herpes zóster) durante el tratamiento con canakinumab. Sin embargo, no puede descartarse una relación causal entre canakinumab y estos eventos.
Detección de tuberculosis
Aproximadamente el 12 % de los pacientes con síndromes periódicos asociados a criopirina que se sometieron a la prueba cutánea de tuberculina (PPD) en estudios clínicos dieron resultados positivos, aunque se les administró canakinumab sin signos clínicos de infección tuberculosa latente o activa.
No se sabe si el uso de inhibidores de la interleucina-1 (IL-1), como canakinumab, aumenta el riesgo de reactivación de la tuberculosis. Antes de iniciar la terapia, todos los pacientes deben ser evaluados para descartar tuberculosis activa o latente. El médico debe realizar una historia clínica detallada. A todos los pacientes (pueden aplicarse recomendaciones locales) se les deben realizar pruebas de cribado adecuadas (por ejemplo, prueba cutánea de tuberculina, prueba de liberación de interferón gamma o radiografía de tórax). Se debe observar cuidadosamente a los pacientes en busca de síntomas de tuberculosis durante y después del tratamiento con canakinumab. El paciente debe saber que, si durante la terapia con canakinumab aparecen síntomas sugestivos de tuberculosis (por ejemplo, tos persistente, pérdida de peso, fiebre subfebril), debe consultar inmediatamente a su médico. Si la prueba de Mantoux es positiva, especialmente en pacientes con alto riesgo, se debe considerar la posibilidad de utilizar métodos alternativos de cribado para la infección tuberculosa.
Neutropenia y leucopenia
Se han observado neutropenia (recuento absoluto de neutrófilos [RAN] < 1,5 × 10⁹/l) y leucopenia con el uso de medicamentos que inhiben la IL-1, incluyendo canakinumab. No se debe iniciar el tratamiento con canakinumab en pacientes con neutropenia o leucopenia. Se recomienda evaluar el recuento de glóbulos blancos, incluyendo el número de neutrófilos, antes de iniciar el tratamiento y a los 1 y 2 meses después del inicio. En pacientes con tratamiento crónico o en aquellos que requieran tratamiento repetido, también se recomienda evaluar periódicamente el recuento de glóbulos blancos durante el tratamiento. Si un paciente entra en un estado neutropénico o leucopénico, se debe vigilar estrechamente el recuento de glóbulos blancos y considerar la necesidad de suspender el tratamiento.
Neoplasias malignas
Se han notificado casos de neoplasias malignas en pacientes que recibieron canakinumab. El riesgo de desarrollar tumores malignos con el uso de anti-IL-1 no se conoce.
Reacciones de hipersensibilidad
Se han notificado casos que sugieren reacciones de hipersensibilidad con el uso de canakinumab. La mayoría de estos casos fueron de gravedad leve. Durante el desarrollo clínico de Ilaris en más de 2600 pacientes, no se observaron reacciones anafilactoides ni anafilácticas. Sin embargo, el riesgo de reacciones graves de hipersensibilidad, que no es infrecuente con proteínas inyectables, no puede descartarse.
Función hepática
En estudios clínicos se han registrado casos breves y asintomáticos de aumento de las transaminasas séricas o de la bilirrubina.
Vacunación
No existen datos sobre el riesgo de transmisión secundaria de infección con vacunas vivas (atenuadas) en pacientes que reciben canakinumab. Por lo tanto, no se deben administrar vacunas vivas simultáneamente con canakinumab, excepto cuando los beneficios superen claramente los riesgos.
Antes de iniciar el tratamiento con canakinumab, se recomienda que adultos y niños reciban todas las vacunas necesarias, incluyendo la vacuna antineumocócica y la vacuna contra la gripe inactivada.
Mutación en el gen NLRP3 en pacientes con síndromes periódicos asociados a criopirina
La experiencia clínica en pacientes con síndromes periódicos asociados a criopirina sin una mutación confirmada en el gen NLRP3 es limitada.
Síndrome de activación de macrófagos en pacientes con enfermedad de Still
El síndrome de activación de macrófagos es una condición grave, potencialmente mortal, que puede desarrollarse en pacientes con enfermedades reumáticas, particularmente en aquellos con enfermedad de Still. En caso de desarrollo o sospecha de este síndrome, se debe iniciar la evaluación y el tratamiento lo antes posible. Los médicos deben estar atentos a los signos de infección o empeoramiento de la enfermedad de Still, conocidos como desencadenantes del síndrome de activación de macrófagos. Los datos de estudios clínicos indican que canakinumab probablemente no aumenta el riesgo de este síndrome en pacientes con enfermedad de Still, aunque no permiten conclusiones definitivas.
Reacción al fármaco con eosinofilia y síntomas sistémicos (DRESS)
Rara vez, en pacientes que recibieron Ilaris, principalmente con artritis idiopática juvenil sistémica (SJIA), se han notificado reacciones al fármaco con eosinofilia y síntomas sistémicos (DRESS). Los pacientes con DRESS pueden requerir hospitalización, ya que esta condición puede ser fatal. Si están presentes signos y síntomas de DRESS y no puede identificarse una etiología alternativa, no se debe volver a administrar Ilaris; debe considerarse otro tratamiento.
Uso durante el embarazo o la lactancia.
Mujeres en edad fértil
Las mujeres deben utilizar métodos anticonceptivos eficaces durante el tratamiento con canakinumab y durante 3 meses después de la última dosis.
Embarazo
Los datos sobre el uso de Ilaris en mujeres embarazadas son limitados. Los estudios en animales no indican efectos desfavorables directos ni indirectos sobre la función reproductiva. El riesgo para el feto/la madre es desconocido. Por lo tanto, el medicamento solo debe usarse durante el embarazo o en mujeres que deseen quedar embarazadas tras una evaluación cuidadosa del beneficio y el riesgo potencial.
Los estudios en animales muestran que canakinumab atraviesa la placenta y se detecta en el organismo fetal. No existen datos en humanos, pero dado que canakinumab es una inmunoglobulina de clase G (IgG1), se espera su paso transplacentario. El significado clínico de esto es desconocido. Sin embargo, no se recomienda la administración de vacunas vivas a recién nacidos expuestos a canakinumab in utero durante las 16 semanas posteriores a la última dosis administrada a la madre antes del parto. Las mujeres que recibieron canakinumab durante el embarazo deben ser instruidas para informar al médico del recién nacido antes de que se administren vacunas al bebé.
Lactancia
No se sabe si canakinumab se excreta en la leche materna humana. La administración de Ilaris a mujeres que amamantan solo debe considerarse si el beneficio esperado para la madre supera cualquier riesgo potencial para el lactante.
Fertilidad
No se han realizado estudios sobre el posible efecto de Ilaris sobre la fertilidad humana.
Canakinumab no afectó los parámetros de fertilidad en machos de animales (C. jacchus). Los anticuerpos anti-IL-1 beta murinos no tuvieron efectos adversos sobre la fertilidad en machos ni hembras de ratones.
Efecto sobre la capacidad para conducir y usar máquinas.
Ilaris tiene un efecto insignificante sobre la capacidad para conducir vehículos o usar maquinaria. El tratamiento con Ilaris puede provocar mareo/vertigo o astenia. Los pacientes que experimenten estos síntomas durante el tratamiento con Ilaris deben esperar a que desaparezcan antes de conducir o manipular maquinaria.
Vía de administración y dosis.
Síndromes periódicos asociados a la criopirina (síndrome CAP), síndrome periódico asociado al receptor del factor de necrosis tumoral (síndrome TRAP), síndrome de hiperinmunoglobulinemia D (síndrome HID)/deficiencia de mevalonato quinasa (DMK), fiebre mediterránea familiar (FMF) y enfermedad de Still
El tratamiento debe iniciarse bajo prescripción y supervisión de un médico con experiencia en el diagnóstico y tratamiento de estas afecciones.
Tras la debida formación en la técnica de administración por inyección, los pacientes o sus cuidadores podrán autoinyectarse Ilaris, siempre que el médico considere adecuado y médicamente necesario.
Dosis iniciales recomendadas de Ilaris para adultos, adolescentes y niños a partir de los 2 años con síndromes periódicos asociados a la criopirina (síndrome CAP).
Adultos y niños a partir de 4 años de edad:
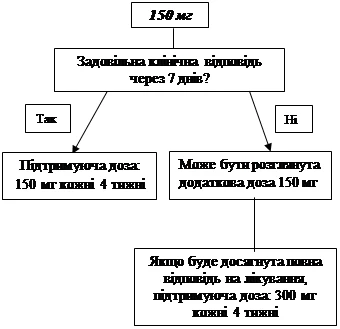
- 150 mg para pacientes con peso corporal > 40 kg;
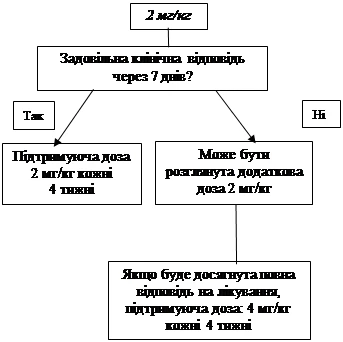
- 2 mg/kg para pacientes con peso corporal ≥ 15 kg y ≤ 40 kg;
- 4 mg/kg para pacientes con peso corporal ≥ 7,5 kg y < 15 kg.
Niños de 2 a 4 años de edad:
- 4 mg/kg para pacientes con peso corporal ≥ 7,5 kg.
Estas dosis deben administrarse cada ocho semanas como dosis única mediante inyección subcutánea.
Si no se logra un efecto clínico satisfactorio (desaparición del rash cutáneo y otros síntomas sistémicos) con la dosis inicial de 150 mg o 2 mg/kg en los 7 días posteriores al inicio del tratamiento, puede administrarse una segunda dosis de Ilaris de 150 mg o 2 mg/kg. Tras alcanzar el efecto clínico deseado, debe mantenerse un régimen de dosificación intensificada de 300 mg o 4 mg/kg cada 8 semanas. Si no se logra un efecto clínico satisfactorio a los 7 días tras este aumento de dosis, puede administrarse una tercera dosis de Ilaris de 300 mg o 4 mg/kg. Tras alcanzar el efecto clínico completo, debe mantenerse un régimen de dosificación intensificada de 600 mg o 8 mg/kg cada 8 semanas, según evaluación clínica individual.
Si no se logra un efecto clínico satisfactorio con la dosis inicial de 4 mg/kg en los 7 días posteriores al inicio del tratamiento, puede administrarse una segunda dosis de Ilaris de 4 mg/kg. Tras alcanzar el efecto clínico completo, debe mantenerse un régimen de dosificación intensificada de 8 mg/kg cada 8 semanas, según evaluación clínica individual.
La experiencia clínica con dosis administradas con intervalos inferiores a 4 semanas, o con dosis superiores a 600 mg o 8 mg/kg, es limitada.

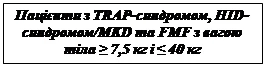
 |
|||
 |
|||
Enfermedad de Still (SJIA y AOSD)
La dosis recomendada de Ilaris para pacientes con enfermedad de Still (enfermedad de Still del adulto [AOSD] y artritis idiopática juvenil sistémica [SJIA]) con un peso corporal ≥ 7,5 kg es de 4 mg/kg (máximo 300 mg) cada cuatro semanas mediante inyección subcutánea. La decisión sobre continuar el tratamiento con Ilaris en pacientes que no presenten mejorías clínicas la tomará el médico.
Artritis gotosa
El tratamiento debe realizarse bajo la supervisión de médicos con experiencia en el diagnóstico y tratamiento de la artritis gotosa y en el uso de medicamentos biológicos. El medicamento Ilaris debe ser administrado por personal médico.
Debe iniciarse el tratamiento de la hiperuricemia con una terapia adecuada que reduzca u optimice la concentración de uratos (ULT). Canakinumab debe utilizarse como terapia a demanda para el tratamiento de los ataques de artritis gotosa.
Es necesario controlar la hiperuricemia con una terapia adecuada para reducir los niveles de uratos. Ilaris debe utilizarse como terapia según necesidad para el tratamiento de la artritis gotosa.
La dosis recomendada de Ilaris para adultos con artritis gotosa es de 150 mg administrados subcutáneamente como dosis única durante un ataque. Para lograr el máximo efecto, Ilaris debe administrarse lo más rápidamente posible tras el inicio del ataque de artritis gotosa.
No se debe volver a administrar Ilaris a pacientes que no hayan respondido al tratamiento inicial. Para pacientes que hayan respondido y que requieran tratamiento repetido, el intervalo entre dosis debe ser de al menos 12 semanas.
Poblaciones especiales
Pacientes de edad avanzada
No se requiere ajuste de dosis.
No existen diferencias significativas en el perfil de seguridad observado en pacientes mayores de 65 años.
Insuficiencia hepática
No hay datos disponibles sobre el uso de Ilaris en pacientes con disfunción hepática.
Insuficiencia renal
No se requiere ajuste de dosis en pacientes con insuficiencia renal. Sin embargo, la experiencia clínica con el uso del medicamento en estos pacientes es limitada.
Vía de administración
Ilaris, 150 mg, polvo para solución inyectable, se suministra en un frasco de un solo uso para uso individual. Cualquier medicamento no utilizado o residuos deben eliminarse de acuerdo con los requisitos locales.
Instrucciones para la reconstitución
Utilizando técnica aséptica, reconstituir el contenido del frasco a temperatura ambiente: inyectar lentamente 1,0 ml de agua para inyección mediante una jeringa de 1 ml y una aguja de 18 G × 50 mm. Agitar suavemente el frasco inclinándolo aproximadamente 45° durante aproximadamente 1 minuto y dejar reposar durante 5 minutos. Luego, girar cuidadosamente el frasco boca abajo y de vuelta diez veces. Si es posible, evitar tocar el tapón de goma con los dedos. Dejar reposar durante 15 minutos a temperatura ambiente hasta obtener una solución transparente u opalescente. No agitar. No utilizar el medicamento si están presentes partículas en la solución.
Golpear suavemente la pared del frasco para eliminar los restos de líquido del tapón. La solución debe estar libre de partículas visibles, ser transparente u opalescente. Debe ser incolora, aunque puede tener un ligero tono amarillento. No debe utilizarse si la solución tiene un color marrón intenso. Si la solución no se utiliza inmediatamente tras la reconstitución, debe conservarse a una temperatura de entre 2 y 8 °C y utilizarse dentro de las 24 horas.
Instrucciones para la administración
Llenar cuidadosamente la jeringa con la cantidad necesaria de solución según la dosis (entre 0,2 ml y 1,0 ml) y administrar subcutáneamente mediante una aguja de 27 G × 13 mm.
Lugares para la inyección: parte superior del muslo, abdomen, hombro o glúteos. Deben evitarse las áreas con piel dañada, hematomas o erupciones. Debe evitarse la administración en tejido cicatricial, ya que podría reducir el efecto del medicamento Ilaris.
Eliminación
Los pacientes o sus cuidadores deben recibir instrucciones sobre la eliminación adecuada de frascos, jeringas y agujas según los requisitos locales.
Pediatría
Síndromes periódicos asociados a criopirina (CAP), síndrome TRAP, síndrome HID/MKD y FMF
No se ha establecido la seguridad ni la eficacia del uso de Ilaris en pacientes menores de 2 años con síndromes periódicos asociados a criopirina, síndrome TRAP, síndrome HID/MKD y FMF. Los datos actualmente disponibles se describen en las secciones «Farmacocinética», «Farmacodinamia» y «Reacciones adversas», pero no pueden hacerse recomendaciones sobre la dosificación.
Artritis idiopática juvenil sistémica (SJIA)
No se ha establecido la seguridad ni la eficacia del uso de Ilaris en pacientes menores de 2 años con artritis idiopática juvenil sistémica (SJIA).
Artritis gotosa
No existe experiencia en el uso de Ilaris en niños para el tratamiento de la artritis gotosa.
Sobredosis
La información sobre sobredosis es limitada. Durante estudios iniciales, pacientes y voluntarios sanos recibieron dosis de hasta 10 mg/kg por vía intravenosa o subcutánea sin signos de toxicidad aguda del medicamento.
En caso de sobredosis, se recomienda vigilar al paciente y, si es necesario, iniciar inmediatamente un tratamiento sintomático adecuado.
Reacciones adversas
Aproximadamente 2300 pacientes, incluyendo cerca de 250 niños (de 2 a 17 años de edad) con síndromes periódicos asociados con criopirina, artritis idiopática juvenil sistémica, artritis gotosa u otras enfermedades mediadas por IL-1 beta, así como voluntarios sanos, participaron en estudios clínicos abiertos y ciegos. Las reacciones adversas más frecuentes fueron infecciones (por ejemplo, infecciones de las vías respiratorias superiores). La mayoría de las reacciones fueron de intensidad leve o moderada. El tratamiento a largo plazo no influyó en el tipo ni en la frecuencia de las reacciones adversas.
En pacientes que recibieron Ilaris se han observado casos de reacciones de hipersensibilidad.
Durante el tratamiento con Ilaris se han notificado infecciones oportunistas.
Síndromes periódicos asociados con criopirina
En los estudios clínicos participaron 211 pacientes adultos y pediátricos (con diagnósticos de síndrome autoinflamatorio familiar inducido por frío/urticaria familiar fría, síndrome de Muckle-Wells y enfermedad inflamatoria multisistémica neonatal/síndrome neurológico cutáneo articular crónico infantil). La seguridad de Ilaris se comparó con placebo en un estudio de fase III que incluyó un período abierto de 8 semanas (parte 1), un período aleatorizado, doble ciego y controlado con placebo de 24 semanas de exclusión (parte 2) y un período abierto de 16 semanas con tratamiento con Ilaris (parte 3). Todos los pacientes recibieron Ilaris 150 mg por vía subcutánea o 2 mg/kg de peso corporal si el peso corporal era ≥ 15 kg y ≤ 40 kg.
Artritis idiopática juvenil sistémica
En los estudios clínicos con Ilaris participaron 201 pacientes de 2 a 20 años de edad con diagnóstico de artritis idiopática juvenil sistémica. La seguridad de Ilaris se comparó con placebo en dos estudios piloto de fase III.
Artritis gotosa
Más de 700 pacientes con artritis gotosa fueron incluidos en estudios clínicos aleatorizados, doble ciego y controlados activamente, con una duración de hasta 24 semanas, en los que se utilizaron dosis de 10 mg a 300 mg. Más de 250 pacientes recibieron tratamiento con la dosis recomendada de 150 mg en estudios de fase II y III.
Las reacciones adversas se enumeran por clasificación de órganos y sistemas MedDRA y por frecuencia. Dentro de cada clasificación de órganos y sistemas, las reacciones adversas se presentan por categorías de frecuencia, comenzando por las más frecuentes. Las categorías de frecuencia se definen de la siguiente manera: muy frecuentes (≥ 1/10); frecuentes (≥ 1/100 a < 1/10); poco frecuentes (≥ 1/1000 a < 1/100); raras (≥ 1/10000 a < 1/1000); muy raras (< 1/10000); no conocidas (la frecuencia no puede estimarse a partir de los datos disponibles). Dentro de cada grupo por frecuencia, las reacciones adversas se presentan en orden decreciente de gravedad.
Tabla 5. Reacciones adversas
| Clases de sistemas orgánicos |
Síndrome CAP, síndrome TRAP, síndrome HID/MKD, FMF, artritis juvenil idiopática sistémica, artritis gotosa |
| Infecciones e infestaciones |
|
| Muy frecuentes |
Infecciones de las vías respiratorias (incluyendo neumonía, bronquitis, gripe, infecciones virales, sinusitis, rinitis, faringitis, amigdalitis, rinofaringitis, infecciones de las vías respiratorias superiores) Infecciones del oído Celulitis Gastroenteritis Infecciones del tracto urinario |
| Frecuentes |
Candidiasis vulvovaginal |
| Alteraciones del sistema nervioso |
|
| Frecuentes |
Mareo/vertigo |
| Alteraciones del aparato gastrointestinal |
|
| Muy frecuentes |
Dolor abdominal (parte superior)1 |
| No frecuentes |
Enfermedad por reflujo gastroesofágico2 |
| Alteraciones de la piel y tejidos subcutáneos |
|
| Muy frecuentes |
Reacciones en el lugar de inyección |
| Alteraciones del sistema músculo-esquelético y del tejido conjuntivo |
|
| Muy frecuentes |
Artalgia1 |
| Frecuentes |
Dolor músculo-esquelético1 Dolor de espalda2 |
| Alteraciones generales |
|
| Frecuentes |
Cansancio/astenia2 |
| Pruebas |
|
| Muy frecuentes |
Disminución del aclaramiento renal de la creatinina1,3 Proteinuria1,4 Leucopenia1,5 |
| Frecuentes |
Neutropenia |
| No frecuentes |
Disminución del número de plaquetas |
| 1En artritis juvenil idiopática sistémica. 2En artritis gotosa. 3Según la evaluación del aclaramiento de creatinina, la mayoría de los casos fueron transitorios. 4La mayoría de los casos se presentaron como trazas transitorias o como reacción de nivel 1+ de proteína en orina mediante tira reactiva. 5Véase otra información más abajo. |
|
En un subgrupo de adultos jóvenes con artritis idiopática juvenil sistémica de 16 a 20 años de edad (n = 31), el perfil de seguridad de canaquinumab fue coherente con el observado en pacientes con artritis idiopática juvenil sistémica menores de 16 años. Según informes publicados, se espera que el perfil de seguridad en pacientes con enfermedad de Still del adulto sea similar al de pacientes con artritis idiopática juvenil sistémica.
Datos de estudios a largo plazo y alteraciones de laboratorio en pacientes con síndromes periódicos asociados a criopirina
Durante los ensayos clínicos con Ilaris en pacientes con síndromes periódicos asociados a criopirina, los valores medios de hemoglobina aumentaron, mientras que los niveles de leucocitos, neutrófilos y plaquetas disminuyeron.
Raramente se observó un aumento de las transaminasas.
Se han notificado aumentos asintomáticos y moderados de la bilirrubina sérica sin aumento concomitante de transaminasas en pacientes con síndromes periódicos asociados a criopirina que recibieron canaquinumab.
En estudios abiertos a largo plazo con aumento de dosis, las infecciones (gastroenteritis, infecciones respiratorias, infecciones de las vías respiratorias superiores), vómitos y mareos fueron más frecuentes en el grupo que recibió 600 mg o 8 mg/kg que en otros grupos de dosis.
Alteraciones de laboratorio en pacientes con síndrome TRAP, síndrome HID/MKD y FMF
Neutrófilos
Aunque la disminución de grado ≥ 2 en neutrófilos ocurrió en el 6,5 % de los pacientes (frecuente), y la disminución de grado 1 ocurrió en el 9,5 % de los pacientes, dicha disminución fue generalmente transitoria y no se identificó infección asociada a neutropenia como reacción adversa.
Plaquetas
Aunque la disminución en el recuento de plaquetas (grado ≥ 2) ocurrió en el 0,6 % de los pacientes, no se identificó hemorragia como reacción adversa. Una disminución leve y transitoria de grado 1 en plaquetas ocurrió en el 15,9 % de los pacientes sin reacciones adversas asociadas a hemorragia.
Alteraciones de laboratorio en pacientes con artritis idiopática juvenil sistémica
Hematología
Dentro del programa general de tratamiento de la artritis idiopática juvenil sistémica, se observaron disminuciones transitorias en el recuento de leucocitos ≤ 0,8 × LMR en 33 pacientes (16,5 %). Disminuciones transitorias en el recuento absoluto de neutrófilos (CAN) hasta niveles inferiores a 1 × 109/l se observaron en 12 pacientes (6,0 %). Disminuciones transitorias en el recuento de plaquetas (< LMR) se observaron en 19 pacientes (9,5 %).
ALT/AST
Dentro del programa general de tratamiento de la artritis idiopática juvenil sistémica, se observaron niveles elevados de ALT y/o AST (más de 3 veces por encima del límite superior normal (LSN)) en 19 pacientes (9,5 %).
Alteraciones de laboratorio en pacientes con artritis gotosa
Hematología
Se registraron disminuciones en el recuento de leucocitos ≤ 0,8 × LMR (límite inferior normal) en el 6,7 % de los pacientes que recibieron canaquinumab, en comparación con el 1,4 % de los pacientes tratados con acetónido de triamcinolona. Disminuciones en el recuento absoluto de neutrófilos (CAN) hasta niveles inferiores a 1 × 109/l se observaron en el 2 % de los pacientes en ensayos comparativos. También se observaron casos aislados con CAN < 0,5 × 109/l.
Una disminución moderada (< LMR y > 75 × 109/l) y transitoria en el recuento de plaquetas se observó con mayor frecuencia (12,7 %) tras la administración de canaquinumab en estudios clínicos controlados activamente, en comparación con el fármaco de comparación (7,7 %) en pacientes con artritis gotosa.
Ácido úrico
Se observó un aumento en los niveles de ácido úrico (0,7 mg/dl a las 12 semanas y 0,5 mg/dl a las 24 semanas) tras el tratamiento con canaquinumab en ensayos comparativos en pacientes con artritis gotosa. En otro estudio en pacientes que recibían tratamiento uricosúrico (ULT), no se observó aumento del ácido úrico. No se observó aumento del ácido úrico en ensayos clínicos en grupos de pacientes sin artritis gotosa.
ALT/AST
Se observaron aumentos medios y medianos de alanina aminotransferasa (ALT) de 3,0 U/l y 2,0 U/l, respectivamente, y de aspartato aminotransferasa (AST) de 2,7 U/l y 2,0 U/l, respectivamente, en comparación con los niveles basales al final del estudio en los grupos de canaquinumab y acetónido de triamcinolona. Sin embargo, la frecuencia de cambios clínicamente significativos (≥ 3 × LSN) fue mayor en pacientes que recibieron acetónido de triamcinolona (2,5 % para AST y ALT) en comparación con el grupo tratado con canaquinumab (1,6 % para ALT y 0,8 % para AST).
Triglicéridos
En ensayos clínicos controlados activamente con pacientes con artritis gotosa, el aumento medio en los niveles de triglicéridos fue de 33,5 mg/dl en el grupo tratado con canaquinumab, en comparación con una leve disminución de −3,1 mg/dl en el grupo tratado con acetónido de triamcinolona. La frecuencia de casos con niveles de triglicéridos > 5 × LSN fue del 2,4 % con canaquinumab y del 0,7 % con acetónido de triamcinolona. La relevancia clínica de esta observación es desconocida.
Datos a largo plazo de estudios de vigilancia
Durante un estudio prolongado (exposición media a canaquinumab de 3,8 años), se administró canaquinumab según la práctica clínica habitual a pacientes con artritis idiopática juvenil sistémica (85 pacientes pediátricos de ≥ 2 a ≤ 17 años y 158 adultos de ≥ 18 años). El perfil de seguridad de canaquinumab observado tras el tratamiento prolongado en estas condiciones fue coherente con el observado en estudios de intervención en pacientes con artritis idiopática juvenil sistémica.
Población pediátrica
El estudio incluyó 80 pacientes pediátricos de 2 a 17 años con síndromes periódicos asociados a criopirina. En general, no hubo diferencias clínicamente relevantes en cuanto a la seguridad y el perfil de tolerabilidad de Ilaris en pacientes pediátricos en comparación con la población general de pacientes con síndromes periódicos asociados a criopirina (compuesta por adultos y pediátricos, N = 211), incluyendo la frecuencia general y gravedad de episodios infecciosos. Las infecciones de las vías respiratorias superiores fueron las infecciones más frecuentes.
Además, se evaluaron 6 pacientes pediátricos menores de 2 años en un pequeño estudio clínico abierto. El perfil de seguridad de Ilaris es similar al observado en pacientes de 2 años o más.
Durante un estudio de 16 semanas, 102 pacientes con síndrome TRAP, síndrome HID/MKD y FMF (de 2 a 17 años) recibieron canaquinumab. En general, no hubo diferencias clínicamente relevantes en el perfil de seguridad y tolerabilidad de canaquinumab en pacientes pediátricos en comparación con la población general.
Duración del medicamento. 3 años.
Condiciones de almacenamiento.
Conservar a 2–8 °C en el envase original para protegerlo de la luz. No congelar. Mantener fuera del alcance de los niños.
Incompatibilidad.
Debido a la falta de estudios de compatibilidad, este medicamento no debe mezclarse con otros medicamentos.
Envase.
150 mg de polvo para solución inyectable en un frasco de vidrio incoloro de 6 ml; 1 frasco por caja. O bien, 4 cajas, cada una conteniendo 150 mg de polvo para solución inyectable en un frasco de vidrio incoloro de 6 ml, en un estuche.
Categoría de dispensación. Bajo receta médica.
Fabricante.
Novartis Pharma Stein AG.
Dirección del fabricante y lugar de actividad. Schaffhauserstrasse, 4332 Stein, Suiza.