Ilaris
UkraineTable of Contents
INSTRUCTION FOR MEDICAL USE OF THE MEDICINAL PRODUCT ILARIS (ILARIS®)
Composition:
Active substance: canakinumab;
1 vial contains 150 mg of canakinumab;
Excipients: sucrose, L-histidine, L-histidine hydrochloride monohydrate, polysorbate 80.
Pharmaceutical form. Powder for solution for injection.
Main physicochemical characteristics: lyophilized white powder.
Pharmacotherapeutic group. Antineoplastic and immunomodulating agents. Immunosuppressants. Interleukin inhibitors. ATC code L04A C08.
Pharmacological Properties
Pharmacodynamics
Mechanism of action
Canakinumab is a fully human monoclonal antibody of IgG1/κ isotype directed against interleukin-1 beta (IL-1 beta). Canakinumab specifically binds with high affinity to human IL-1 beta and neutralizes the biological activity of human IL-1 beta by blocking its interaction with IL-1 receptors, thereby preventing IL-1 beta-induced gene activation and production of inflammatory mediators.
Pharmacodynamic effects
Periodic syndromes associated with cryopyrin (CAP syndromes), periodic syndrome associated with tumor necrosis factor receptor (TRAPS), hyperimmunoglobulinemia D syndrome (HIDS)/mevalonate kinase deficiency (MKD), familial Mediterranean fever (FMF)
In clinical studies of patients with periodic syndromes associated with cryopyrin (CAP syndromes), tumor necrosis factor receptor-associated periodic syndrome (TRAPS), hyperimmunoglobulinemia D syndrome (HIDS)/mevalonate kinase deficiency (MKD), and familial Mediterranean fever (FMF), in whom uncontrolled release of IL-1 beta occurs, a rapid response to canakinumab therapy was observed. Laboratory markers such as elevated levels of C-reactive protein (CRP) and serum amyloid A (SAA), increased neutrophil and platelet counts, and leukocytosis rapidly normalized.
Systemic autoinflammatory diseases (including adult-onset Still’s disease (AOSD) and systemic juvenile idiopathic arthritis (SJIA))
Adult-onset Still’s disease and systemic juvenile idiopathic arthritis are severe autoinflammatory inherited diseases driven by pro-inflammatory cytokines, with IL-1 beta playing a key role.
Key features of systemic juvenile idiopathic arthritis include fever, rash, hepatosplenomegaly, lymphadenopathy, polyserositis, and arthritis.
Treatment with canakinumab led to rapid and sustained reduction of both joint-related and systemic manifestations of systemic juvenile idiopathic arthritis, with significant reduction in the number of inflamed joints, resolution of fever, and decreased acute-phase reactants in the majority of patients.
Gouty arthritis
Gouty arthritis flare is caused by urate crystals (monosodium urate monohydrate) in joints and surrounding tissues, which activate resident macrophages to produce IL-1 beta via the NALP3 inflammasome complex. Activation of macrophages and the consequent increased release of IL-1 beta lead to an acute painful inflammatory reaction. Other activators of the innate immune system, such as endogenous toll-like receptor agonists, may contribute to transcriptional activation of the IL-1 beta gene, initiating a gouty arthritis flare. After treatment with canakinumab, inflammatory markers such as C-reactive protein (CRP) and serum amyloid A (SAA), along with signs of acute inflammation (e.g., pain, swelling, redness) in the affected joint, rapidly resolve.
Clinical efficacy and safety
Periodic syndromes associated with cryopyrin (CAP syndromes)
The efficacy and safety of Ilaris have been demonstrated in patients with varying degrees of disease severity and different phenotypes (including severe forms of familial cold autoinflammatory syndrome/familial cold urticaria, Muckle-Wells syndrome, and neonatal-onset multisystem inflammatory disease/chronic infantile neurological cutaneous and articular syndrome). Only patients with a confirmed NLRP3 mutation were included in the pivotal study.
In the phase I/II study, Ilaris demonstrated a rapid onset of action: symptoms resolved or markedly diminished within one day. Laboratory markers such as CRP and SAA levels, neutrophil and platelet counts, rapidly normalized within several days after administration of Ilaris.
The pivotal 48-week multicenter study consisted of three parts: an 8-week open-label period (Part I), a 24-week randomized, double-blind, placebo-controlled withdrawal period (Part II), and a 16-week open-label period (Part III). The objective of the study was to evaluate the efficacy, safety, and tolerability of Ilaris (150 mg or 2 mg/kg every 8 weeks) in patients with cryopyrin-associated periodic syndromes.
- Part I: Complete clinical and biomarker response to Ilaris (defined as physician’s global assessment of autoinflammatory disease and skin disease ≤ minimum, and CRP or SAA level < 10 mg/L) was observed in 97% of patients, with responses appearing within 7 days of treatment initiation. Significant improvement was observed in physician-assessed clinical measures of autoinflammation: global assessment of autoinflammatory disease activity, assessment of skin disease (urticaria, skin rash), joint pain, myalgia, headache/migraine, conjunctivitis, fatigue/malaise, assessment of other related symptoms, and patient-reported symptom assessment.
- Part II: During the withdrawal phase of the main study, the primary endpoint was defined as the proportion of patients experiencing disease relapse/recurrent flare: 0% of patients relapsed while receiving Ilaris compared to 81% of patients randomized to placebo.
- Part III: Patients who received placebo in Part II and experienced relapse were eligible to enter the open-label extension phase and resume Ilaris treatment, maintaining clinical and serological response.
Table 1. Efficacy in the phase III pivotal placebo-controlled study during the withdrawal period (Part II)
| Parameter |
Ilaris N=15 n(%) |
Placebo N=16 n(%) |
p-value |
| Primary endpoint (flare) Number of patients with disease flare in part II |
0 (0 %) |
13 (81 %) |
<0,001 |
| Inflammatory markers* C-reactive protein, mg/L Serum amyloid A, mg/L |
1.10 (0.40) 2.27 (-0.20) |
19.93 (10.50) 71.09 (14.35) |
<0.001 0.002 |
| *Mean change from start of part II |
|||
Two open-label, uncontrolled, long-term phase III studies were conducted. In one study, the safety, tolerability, and efficacy of canakinumab were investigated in patients with cryopyrin-associated periodic syndromes (CAPS). The total treatment duration ranged from 6 months to 2 years. The other was an open-label study of canakinumab to evaluate efficacy and safety in Japanese patients with CAPS, lasting 24 weeks with an extension phase up to 48 weeks. The primary objective was to assess the proportion of patients without relapse at week 24, including those whose dose was increased.
In the combined efficacy analysis of these two studies, 65.6% of patients who had not previously been treated with canakinumab achieved complete response at a dose of 150 mg or 2 mg/kg, while 85.2% of patients achieved complete remission at any dose. Among patients who received 600 mg or 8 mg/kg (or even higher), 43.8% achieved complete response. The proportion of patients aged 2 to 4 years achieving complete response (57.1%) was lower compared to older children and adult patients. Among patients who achieved complete response, 89.3% maintained response without relapse.
Experience in individual patients who achieved complete response after dose escalation to 600 mg (8 mg/kg) every 8 weeks indicates that a higher dose may be beneficial for patients who did not achieve complete response or who do not maintain complete response at the recommended doses (150 mg or 2 mg/kg for patients with body weight ≥15 kg to ≤40 kg). The higher dose was more frequently administered to patients aged 2 to 4 years and to patients with NOMID/CINCA symptoms compared to those with FCAS or MWS.
A 6-year registry observational study was conducted to obtain data on the long-term safety and efficacy of canakinumab treatment in pediatric and adult patients with cryopyrin-associated periodic syndromes (CAPS) under routine clinical practice. The study included 243 patients with CAPS (including 85 patients under 18 years of age). Disease activity was assessed as absent or mild/moderate in more than 90% of patients at all time points after the initial study period, and mean serological inflammatory markers (CRP and SAA) were within normal limits (<10 mg/liter) at all time points after baseline. Although approximately 22% of patients receiving canakinumab required dose adjustments, only a small percentage of patients (1.2%) discontinued canakinumab due to lack of therapeutic effect.
Pediatric population
A total of 80 pediatric patients aged 2 to 17 years participated in the cryopyrin-associated periodic syndromes (CAPS) studies (approximately half of them received weight-based dosing). Overall, no clinically significant differences in efficacy, safety, or tolerability profile of Ilaris were observed in pediatric patients compared to the overall population. The majority of pediatric patients achieved improvement in clinical symptoms and objective markers of inflammation (e.g., SAA and CRP).
The efficacy, safety, and tolerability of Ilaris were evaluated in an open-label 56-week study involving young pediatric patients (≤4 years of age) with cryopyrin-associated periodic syndromes. Seventeen patients (including 6 patients under 2 years of age) were evaluated using initial weight-based dosing of 2–8 mg/kg. The study also assessed the impact of canakinumab on the immune response to standard childhood vaccines. No differences in safety or efficacy were observed in patients under 2 years of age compared to those aged 2 years and older. All patients who received inactivated standard childhood vaccines (N = 7) developed protective antibody levels.
TNF receptor-associated periodic syndrome (TRAPS), hyperimmunoglobulinemia D syndrome (HIDS)/mevalonate kinase deficiency (MKD), familial Mediterranean fever (FMF)
The efficacy and safety of canakinumab for the treatment of TNF receptor-associated periodic syndrome (TRAPS), hyperimmunoglobulinemia D syndrome (HIDS)/mevalonate kinase deficiency (MKD), and familial Mediterranean fever (FMF) were demonstrated in a phase III pivotal study consisting of four parts (N2301), which included three separate disease cohorts.
- Part I: Patients in each cohort aged ≥2 years were included in a 12-week screening period during which they were evaluated for disease flare.
- Part II: Patients with active disease flare were randomized into a 16-week double-blind, placebo-controlled treatment period, during which they received subcutaneous canakinumab 150 mg (2 mg/kg for patients with body weight ≤40 kg) or placebo every 4 weeks. Patients aged 28 days but <2 years were permitted to enter the open-label Part II directly as non-randomized patients (and were excluded from the primary efficacy analysis).
- Part III: Patients who completed the 16-week treatment period and were classified as responders (those who responded to treatment) were re-randomized into a 24-week double-blind withdrawal period, during which they received subcutaneous canakinumab 150 mg (2 mg/kg for patients with body weight ≤40 kg) or placebo every 8 weeks.
- Part IV: All patients from Part III who received canakinumab were eligible to participate in a 72-week open-label extension treatment period.
A total of 185 patients aged 28 days and older were enrolled, and overall, 181 patients aged ≥2 years were randomized in Part II of the study.
The primary efficacy endpoint of the randomized treatment period (Part II) was the proportion of responders in each group who achieved a reduction in disease flare index by Day 15 and did not experience a new flare during the remainder of the 16-week treatment period (defined as complete response). Reduction in disease flare index was defined as a physician’s global assessment (PGA) score for disease activity <2 ("minimal or no disease") and either CRP within normal limits (≤10 mg/L) or a reduction ≥70% from baseline. A new flare was defined as a PGA score ≥2 ("mild, moderate, or severe disease") and CRP ≥30 mg/L. Secondary endpoints based on Week 16 results (end of Part II) included the proportion of patients achieving PGA <2, the proportion with serological remission (defined as CRP ≤10 mg/L), and the proportion with normalized SAA levels (defined as SAA ≤10 mg/L).
For the primary efficacy endpoint, canakinumab was superior to placebo in all three disease groups. Canakinumab also demonstrated superior efficacy compared to placebo in the secondary endpoints of PGA <2 and CRP ≤10 mg/L in all three groups. A greater proportion of patients normalized SAA (≤10 mg/L) at Week 16 with canakinumab treatment compared to placebo in all three groups, with a statistically significant difference observed in patients with TRAPS.
Table 2. Efficacy in the phase III pivotal randomized placebo-controlled study during the treatment period (Part II)
| Parameter |
Canakinumab, n/N (%) |
Placebo, n/N (%) |
p-value |
| Primary endpoint (flare) Number of patients who had a disease flare index on day 15 and did not experience a new flare during the remaining 16-week treatment period |
|||
| FMF HID syndrome/MKD TRAPS |
19/31 (61.29) 13/37 (35.14) 10/22 (45.45) |
2/32 (6.25) 2/35 (5.71) 2/24 (8.33) |
<0.0001* 0.0020* 0.0050* |
| Secondary endpoint (disease and inflammatory markers) |
|||
| Physician’s Global Assessment < 2 FMF HID syndrome/MKD TRAPS |
20/31 (64.52) 17/37 (45.95) 10/22 (45.45) |
3/32 (9.38) 2/35 (5.71) 1/24 (4.17) |
<0.0001** 0.0006** 0.0028** |
| C-reactive protein ≤ 10 mg/L FMF HID syndrome/MKD TRAPS |
21/31 (67.74) 15/37 (40.54) 8/22 (36.36) |
2/32 (6.25) 2/35 (5.71) 2/24 (8.33) |
<0.0001** 0.0010** 0.0149** |
| Serum amyloid A ≤ 10 mg/L FMF HID syndrome/MKD TRAPS |
8/31 (25.81) 5/37 (13.51) 6/22 (27.27) |
0/32 (0.00) 1/35 (2.86) 0/24 (0.00) |
0.0286 0.0778 0.0235* |
| n – number of responders; N – number of evaluated patients. *Indicates statistical significance (one-sided) at the 0.025 level based on Fisher’s exact test. **Indicates statistical significance (one-sided) at the 0.025 level based on logistic regression model with treatment group and baseline PGA, CRP, or SAA as explanatory variables accordingly for each group. |
|||
Dose Titration
In Part II of the study, patients receiving canakinumab who had persistent disease activity received an additional dose of 150 mg (or 2 mg/kg for patients with body weight ≤ 40 kg) within the first month. This additional dose could be administered as early as 7 days after the first treatment dose. All patients requiring dose titration remained on the higher dose of 300 mg (or 4 mg/kg for patients with body weight ≤ 40 kg) every 4 weeks.
During the investigator analysis of the primary endpoint, it was observed that in patients with an inadequate response after the first dose, increasing the dose within the first month to 300 mg (or 4 mg/kg) every 4 weeks further improved flare control, reduction in disease activity, and normalization of CRP and SAA levels.
Paediatric Patients
Two non-randomized patients with HIDS/MKD aged >28 days to <2 years were included in the study and received canakinumab. One patient achieved resolution of the flare index by Day 15 after receiving a single dose of canakinumab 2 mg/kg but discontinued treatment after this first dose due to serious adverse events (pancytopenia and hepatic failure). Prior to study entry, this patient had a history of immune thrombocytopenic purpura and an ongoing medical condition with impaired liver function. The second patient received an initial dose of canakinumab 2 mg/kg, an additional dose of 2 mg/kg at Week 3, and dose titration to 4 mg/kg at Week 5, which was administered every 4 weeks until the end of Part II of the study. Resolution of the disease flare was achieved by Week 5, and the patient experienced no further flares until the end of Part II of the study (Week 16).
Still’s Disease (Adult-Onset Still’s Disease (AOSD) and Systemic Juvenile Idiopathic Arthritis (SJIA))
Systemic Juvenile Idiopathic Arthritis
The efficacy of Ilaris for the treatment of active systemic juvenile idiopathic arthritis was evaluated in two pivotal studies (G2305 and G2301). Patients included in the studies were aged 2 to 20 years (mean age 8.5 years, mean disease duration 3.5 years at study start) and had active disease defined by the presence of ≥2 joints with active arthritis, fever, and elevated CRP.
Study G2305
G2305 was a randomized, double-blind, placebo-controlled, 4-week study evaluating the short-term efficacy of Ilaris in 84 patients randomized to receive a single dose of 4 mg/kg (up to 300 mg) of Ilaris or placebo. The primary objective was to determine the proportion of patients achieving a 30% improvement according to the pediatric American College of Rheumatology (ACR) criteria. These criteria were adapted to allow inclusion of patients without fever. Treatment with Ilaris improved all ACR criteria compared to placebo on Day 15 and Day 29 (Table 3).
Table 3. Pediatric ACR criteria and disease status on Day 15 and Day 29
| Parameter |
Day 15 |
Day 29 |
||
| Ilaris N=43 |
Placebo N=41 |
Ilaris N=43 |
Placebo N=41 |
|
| ACR30 |
84 % |
10 % |
81 % |
10 % |
| ACR50 |
67 % |
5 % |
79 % |
5 % |
| ACR70 |
61 % |
2 % |
67 % |
2 % |
| ACR90 |
42 % |
0 % |
47 % |
2 % |
| ACR100 |
33 % |
0 % |
33 % |
2 % |
| Inactive disease |
33 % |
0 % |
30 % |
0 % |
| Treatment difference for all ACR outcomes was significant (p ≤ 0.0001) |
||||
Results of adapted pediatric ACR criteria, which included systemic and arthritic components, were consistent with overall ACR criteria outcomes. On Day 15, the median change from baseline in the number of joints with active arthritis and limited range of motion was −67% and −73%, respectively, for Ilaris (N = 43) compared to a median change of 0% and 0% in the placebo group (N = 41). The mean change in patient pain score (0–100 mm visual analog scale) on Day 15 was −50.0 mm for Ilaris (N = 43) compared to +4.5 mm for placebo (N = 25). Mean change in patient pain score was consistent on Day 29.
Study G2301
G2301 was a randomized, double-blind, placebo-controlled study evaluating Ilaris for the prevention of flares. The study consisted of two parts with two independent primary endpoints (successful steroid reduction and time to flare). In Part I (open-label), 177 patients were enrolled and received 4 mg/kg (up to 300 mg) of Ilaris administered every 4 weeks for up to 32 weeks. Patients in Part II (double-blind) received Ilaris 4 mg/kg or placebo every 4 weeks until observation of 37 flares.
Reduction of corticosteroid dose
Of the 128 patients receiving corticosteroids who participated in Part I of the study, 92 attempted corticosteroid dose reduction. Fifty-seven (62%) of these patients were able to significantly reduce their corticosteroid dose, and 42 (46%) discontinued corticosteroids.
Time to first flare
In patients receiving Ilaris in Part II of the study, a 64% reduction in the risk of disease flare was observed compared to the placebo group (hazard ratio 0.36; 95% CI: 0.17–0.75; p = 0.0032). Sixty-three of the 100 patients enrolled in Part II of the study in the placebo or canakinumab group did not experience a disease flare during the observation period (up to 80 weeks).
Health-related outcomes and quality of life from G2305 and G2301 studies
Treatment with Ilaris was associated with clinically meaningful improvements in physical function and quality of life. In the G2305 study, based on the Pediatric Quality of Life Inventory, least squares mean improvement was 0.69 in the Ilaris group compared to placebo, exceeding the minimal clinically important difference of 0.19 by 3.6-fold (p = 0.0002). Mean improvement from baseline at the end of Part II of the G2301 study was 0.88 (79%). Statistically significant improvements in Pediatric Quality of Life Inventory PF50 scores were observed in the Ilaris group compared to placebo in the G2305 study (physical functioning p = 0.0012; psychosocial well-being p = 0.0017).
Pooled efficacy analysis
Data from the first 12 weeks of Ilaris treatment from studies G2305, G2301, and an extension study were pooled to assess efficacy. These data show consistent improvements from baseline in adapted pediatric ACR criteria and their components by Week 12 compared to those observed in the placebo-controlled study (G2305). At Week 12, adapted pediatric ACR30, 50, 70, 90, and 100 responses were 70%, 69%, 61%, 49%, and 30%, respectively, while 28% of patients had inactive disease (N = 178).
Efficacy observed in studies G2305 and G2301 was maintained in the ongoing long-term open-label extension study (with available data from a median observation period of 49 weeks). In this study, 25 patients who maintained a strong ACR response for at least 5 months had their Ilaris dose reduced to 2 mg/kg every 4 weeks and maintained pediatric ACR100 response during reduced-dose treatment (median 32 weeks, range 8–124 weeks).
Although data are limited, clinical trial results suggest that patients who do not respond to tocilizumab or anakinra treatment may respond to canakinumab therapy.
Study G2306
Study G2306 was an open-label study evaluating maintenance of treatment response with dose reduction (2 mg/kg every 4 weeks) or extended dosing interval (4 mg/kg every 8 weeks) in patients with systemic juvenile idiopathic arthritis who were receiving canakinumab 4 mg/kg every 4 weeks. Seventy-five patients aged 2 to 22 years who maintained inactive disease status for at least 6 consecutive months (clinical remission) on canakinumab monotherapy, including those who discontinued concomitant corticosteroids and/or methotrexate for at least 4 weeks, were randomized to receive canakinumab 2 mg/kg every 4 weeks (N = 38) or canakinumab 4 mg/kg every 8 weeks (N = 37). After 24 weeks, 71% (27/38) of patients receiving the reduced dose (2 mg/kg every 4 weeks) and 84% (31/37) of patients with extended dosing interval (4 mg/kg every 8 weeks) maintained inactive disease status over 6 months. Among patients who remained in clinical remission and continued further dose reduction (1 mg/kg every 4 weeks) or extended dosing interval (4 mg/kg every 12 weeks), 93% (26/28) and 91% (30/33), respectively, maintained inactive disease status for 6 months. Patients who maintained inactive disease status for an additional 6 months at this minimal dose were allowed to discontinue canakinumab. Overall, 33% (25/75) of patients randomized to dose reduction or extended dosing interval were able to discontinue canakinumab and maintain inactive disease status for 6 months. The adverse event rate in both treatment groups was similar to that observed in patients receiving canakinumab 4 mg/kg every 4 weeks.
Adult-Onset Still’s Disease (AOSD)
The efficacy of canakinumab 4 mg/kg (up to a maximum of 300 mg) administered every 4 weeks in patients with AOSD was evaluated in a randomized, double-blind, placebo-controlled study involving 36 patients (aged 22 to 70 years) and was similar to that observed in SJIA patients. In study GDE01T, a greater proportion of patients (12/18, 66.7%) in the canakinumab group compared to placebo (7/17, 41.2%) achieved an improvement from baseline in Disease Activity Score 28 Erythrocyte Sedimentation Rate (DAS28-ESR) > 1.2 at Week 12, which did not reach statistical significance (odds ratio 2.86, treatment difference [%] 25.49 [95% CI: 9.43, 55.80]). By Week 4, 7 of 18 patients (38.9%) receiving canakinumab achieved DAS28-ESR remission compared to 2 of 17 patients (11.8%) receiving placebo.
These data are consistent with results from a pooled efficacy analysis of 418 SJIA patients, which showed that the efficacy of canakinumab in the subgroup of SJIA patients aged 16 to <20 years (n = 34) was comparable to that observed in patients under 16 years of age (n = 384).
Gouty arthritis
The efficacy of Ilaris for the treatment of acute gouty arthritis flares was demonstrated in two multicenter, randomized, double-blind, active-controlled studies in patients with frequent gout flares (3 or more flares in the previous 12 months) who were unable to take NSAIDs or colchicine (due to contraindications, intolerance, or inadequate efficacy). The studies lasted 12 weeks with a subsequent 12-week double-blind extension. A total of 225 patients received subcutaneous Ilaris 150 mg and 229 patients received intramuscular triamcinolone acetonide (TA) 40 mg at study initiation and after flare recurrence. The mean number of gout flares in the previous 12 months was 6.5. More than 85% of patients had comorbid conditions, including arterial hypertension (60%), diabetes mellitus (15%), ischemic heart disease (12%), and chronic kidney disease stage ≥3 (25%). Approximately one-third of patients enrolled in the study (76 [33.8%] in the Ilaris group and 84 [36.7%] in the triamcinolone group) could not use NSAIDs and colchicine (intolerance, contraindications, or lack of response). Concomitant urate-lowering therapy (ULT) was used by 42% of patients at study entry.
Primary efficacy endpoints were: (I) intensity of pain due to gouty arthritis (on a visual analog scale, VAS) at 72 hours after dose administration and (II) time to first new gouty arthritis flare.
In the overall study population, pain intensity was statistically significantly lower with Ilaris 150 mg compared to triamcinolone acetonide at 72 hours. Ilaris also reduced the risk of subsequent flares (see Table 4).
Efficacy results in the subgroup of patients unable to use NSAIDs and colchicine, and those receiving ULT, not benefiting from ULT, or contraindicated to ULT (N = 101), were consistent with the overall study population, showing statistically significant differences compared to triamcinolone acetonide in pain intensity at 72 hours (−10.2 mm, p = 0.0208) and reduced risk of subsequent flares (hazard ratio 0.39, p = 0.0047 at Week 24).
Efficacy results for the smaller subgroup limited to patients receiving ULT (N = 62) are presented in Table 3. Treatment with Ilaris led to reduced pain and lower risk of subsequent flares in patients receiving ULT who cannot use NSAIDs and colchicine, although the treatment difference compared to triamcinolone acetonide was less pronounced than in the overall study population.
Table 4. Efficacy in the overall study population and the subgroup of patients receiving ULT who cannot use NSAIDs or colchicine
| Primary efficacy endpoint |
Overall study population N=454 |
Patients unable to take NSAIDs and colchicine receiving ULT N=62 |
| Treatment of acute gouty arthritis (pain intensity [VAS] at 72 hours) |
||
| Least squares mean difference estimate for triamcinolone acetonide CI p-value, 1-sided |
−10.7 (−15.4, −6.0) p < 0.0001* |
−3.8 (−16.7, 9.1) p = 0.2798 |
| Risk reduction of subsequent gout flares assessed by time to first flare (24 weeks) |
||
| Hazard ratio for triamcinolone acetonide CI p-value, 1-sided |
0.44 (0.32, 0.60) p < 0.0001* |
0.71 (0.29, 1.77) p = 0.2337 |
| *Indicates significant p-value ≤0.025. |
||
The safety study results showed an increased number of adverse events following canakinumab compared to triamcinolone acetonide: 66% versus 53% of patients experiencing any adverse events, and 20% versus 10% of patients experiencing infections, over 24 weeks.
Elderly patients
Overall, the efficacy, safety, and tolerability profile of Ilaris in elderly patients (≥65 years) was comparable to that in patients under 65 years of age.
Patients on urate-lowering therapy (ULT)
Ilaris was safely used concomitantly with ULT in clinical studies. In the overall study population, patients receiving ULT had less pronounced pain reduction and a lower reduction in the risk of subsequent gouty arthritis flares compared to patients not receiving ULT.
Immunogenicity
No anaphylactic reactions were observed in patients receiving Ilaris.
Antibodies against Ilaris were observed in approximately 1.5%, 3%, and 2% of patients receiving Ilaris for the treatment of cryopyrin-associated periodic syndromes (CAPS), systemic juvenile idiopathic arthritis (SJIA), and gouty arthritis, respectively.
Antibodies against canakinumab were not observed in patients with TRAPS, HID/MKD, and FMF who received doses of 150 mg and 300 mg over 16 weeks of treatment.
Paediatric population
The applicant has completed four paediatric study plans for canakinumab (for CAPS, SJIA, FMF – HID/MKD, and TRAPS, respectively). This medicinal product information has been updated to include the results of canakinumab studies in the paediatric population.
The European Medicines Agency has waived the obligation to submit results of studies of Ilaris in all subcategories of the paediatric population with gouty arthritis.
Pharmacokinetics
Cryopyrin-Associated Periodic Syndromes (CAPS)
Absorption
The peak serum concentration of canakinumab (Cmax) was observed approximately 7 days after a single subcutaneous dose of 150 mg in adult CAPS patients. The mean elimination half-life was 26 days. The mean Cmax and AUCinf values after a single subcutaneous dose of 150 mg in a typical adult CAPS patient (70 kg) were 15.9 µg/mL and 708 µg*d/mL, respectively. The estimated absolute bioavailability following subcutaneous administration of canakinumab is 66%. Exposure parameters (e.g., AUC and Cmax) increased proportionally with dose in the range of 0.30 to 10.0 mg/kg administered as intravenous infusion, or 150 to 600 mg as subcutaneous injection. Predicted steady-state exposure values (Cmin,ss, Cmax,ss, AUC,ss,8w) after subcutaneous administration of 150 mg (or 2 mg/kg) every 8 weeks were slightly higher in patients with body weight 40–70 kg (6.6 µg/mL, 24.3 µg/mL, 767 µg*d/mL) compared to those with body weight < 40 kg (4.0 µg/mL, 19.9 µg/mL, 566 µg*d/mL) and > 70 kg (4.6 µg/mL, 17.8 µg/mL, 545 µg*d/mL). The expected accumulation ratio was 1.3-fold after 6 months of subcutaneous administration of 150 mg canakinumab every 8 weeks.
Distribution
Canakinumab binds to IL-1 beta in blood serum. The volume of distribution (Vss) of canakinumab varies with body weight. It is estimated to be 6.2 liters in CAPS patients with a body weight of 70 kg.
Elimination
The apparent clearance (CL/F) of canakinumab increases with body weight. It is estimated to be 0.17 L/day in CAPS patients with a body weight of 70 kg and 0.11 L/day in patients with systemic juvenile idiopathic arthritis with a body weight of 33 kg.
There were no signs of accelerated clearance or time-dependent changes in the pharmacokinetic properties of canakinumab after repeated administration. After body weight adjustment, no pharmacokinetic differences based on patient sex or age were observed.
TRAPS, HID/MKD, and FMF
Bioavailability in patients with TRAPS, HID/MKD, and FMF was not determined separately. The apparent clearance (CL/F) in the population with TRAPS, HID/MKD, and FMF at a body weight of 55 kg (0.14 L/day) was comparable to that in the CAPS population at a body weight of 70 kg (0.17 L/day). The apparent volume of distribution (V/F) was 4.96 L at a body weight of 55 kg.
After repeated subcutaneous administration of 150 mg every 4 weeks, the minimum concentration of canakinumab at week 16 (Cmin) was estimated at 15.4±6.6 µg/mL. The estimated steady-state AUCtau was 636.7±260.2 µg*d/mL.
Still’s disease (systemic juvenile idiopathic arthritis (SJIA) and adult-onset Still’s disease (AOSD))
Bioavailability in patients with systemic juvenile idiopathic arthritis was not determined separately. The apparent clearance per kilogram of body weight (CL/F per 1 kg) was compared between patients with SJIA and CAPS (0.004 L/day/kg). The apparent volume of distribution per kilogram of body weight (V/F per kg) was 0.14 L/kg.
After repeated dosing of 4 mg/kg every 4 weeks, the accumulation ratio of canakinumab was 1.6 times higher in patients with SJIA. Steady state was achieved by day 110. The overall predicted mean values (±SD) for Cmin,ss, Cmax,ss, and AUC,ss4w were 14.7±8.8 µg/mL, 36.5±14.9 µg/mL, and 696.1±326.5 µg*day/mL, respectively.
By age groups, AUCss4w was 692, 615, 707, and 742 µg*day/mL in patients aged 2–3, 4–5, 6–11, and 12–19 years, respectively. When stratified by body weight, lower (30–40%) median exposure for Cmin,ss (11.4 vs. 19 µg/mL) and AUCss (594 vs. 880 µg*day/mL) was observed in the lower body weight category (≤40 kg) compared to patients with higher body weight (>40 kg).
Based on population pharmacokinetic modeling analysis, the pharmacokinetics of canakinumab in young adult SJIA patients aged 16 to 20 years were similar to those in patients under 16 years. Predicted steady-state exposure to canakinumab at a dose level of 4 mg/kg (maximum 300 mg) in patients aged 20 years and above was comparable to that in SJIA patients under 20 years.
Patients with gouty arthritis
Bioavailability in patients with gouty arthritis was not determined. The apparent clearance per kilogram of body weight (CL/F per 1 kg) was compared between gouty arthritis and CAPS patient groups (0.004 L/day/kg). The mean exposure in a typical gouty arthritis patient (93 kg) after a single subcutaneous dose of 150 mg (Cmax: 10.8 µg/mL and AUCinf: 495 µg*d/mL) was lower than in typical CAPS patients with a body weight of 70 kg (15.9 µg/mL and 708 µg*d/mL). This is consistent with the observed increase in CL/F with body weight.
The expected accumulation ratio was 1.1-fold after subcutaneous administration of canakinumab 150 mg every 12 weeks.
Children
Peak concentration of canakinumab was reached within 2–7 days after a single subcutaneous dose of 150 mg or 2 mg/kg in paediatric patients aged 4 years and older. The elimination half-life ranged from 22.9 to 25.7 days, similar to that in adults. Based on pharmacokinetic modeling analysis, the pharmacokinetics of canakinumab in children aged 2 to 4 years were similar to those in patients aged 4 years and older.
It was determined that the extent of absorption after subcutaneous administration decreases with age and is faster in younger patients. Accordingly, Tmax was shorter (3.6 days) in younger patients with systemic juvenile idiopathic arthritis (2–3 years) compared to older patients (12–19 years; Tmax: 6 days). No negative impact on bioavailability (AUCss) was observed.
Additional pharmacokinetic analysis showed that the pharmacokinetics of canakinumab in 6 patients under 2 years of age with CAPS were similar to those in children aged 2–4 years. Population pharmacokinetic modeling indicates that predicted exposure levels after a 2 mg/kg dose were comparable in paediatric patients with CAPS, but 40% lower in patients with very low body weight (e.g., 10 kg) compared to adult patients (150 mg dose). This is consistent with higher exposure levels in CAPS patient groups with higher body weight.
Pharmacokinetics are similar in children with CAPS, TRAPS, HID/MKD, FMF, and systemic juvenile idiopathic arthritis.
Elderly patients
No changes in pharmacokinetic parameters based on clearance or volume of distribution were observed in elderly patients compared to adult patients under 65 years of age.
Preclinical safety data
Preclinical data did not reveal specific hazards for humans based on cross-reactivity, repeat-dose, immunotoxicity, reproductive, and juvenile toxicity studies conducted with canakinumab or murine anti-murine IL-1 beta antibodies.
Since canakinumab binds to both animal (C. jacchus) and human IL-1 beta with similar affinity, the safety of canakinumab was studied in animals. No adverse effects of canakinumab were observed after administration to animals twice weekly for 26 weeks or in embryofetal development toxicity studies in pregnant animals. Plasma concentrations well tolerated in animals exceeded at least 42 times (Cmax) and 78 times (CAVG) the plasma concentrations in paediatric CAPS patients (body weight 10 kg) receiving clinical doses of canakinumab up to 8 mg/kg subcutaneously every 8 weeks. Furthermore, antibodies to canakinumab were not detected in these studies. No non-specific tissue cross-reactivity was demonstrated after application of canakinumab to healthy human tissues.
Formal carcinogenicity studies with canakinumab were not conducted.
In animal embryofetal development studies, canakinumab showed no toxic effects on the maternal organism, embryotoxic, or teratogenic effects when administered during organogenesis.
No adverse effects were observed with murine anti-murine IL-1 beta antibodies in a range of reproductive and juvenile animal studies. Anti-murine IL-1 beta did not show adverse effects on the fetus or newborn growth when administered to the mother during late pregnancy, delivery, and breastfeeding. The high doses used in these studies were maximally effective in terms of suppressing IL-1 beta activity.
Immunotoxicological studies in mice with murine anti-murine IL-1 beta antibodies showed that neutralization of IL-1 beta had no effect on immunological parameters and did not cause immune dysfunction in mice.
Clinical Characteristics
Indications
Periodic fever syndromes
Ilaris is indicated for the treatment of the following periodic autoinflammatory fever syndromes in adults, adolescents, and children aged 2 years and older:
Cryopyrin-associated periodic syndromes (CAPS)
Treatment of cryopyrin-associated periodic syndromes in adults, adolescents, and children aged 2 years and older with a body weight of 7.5 kg or higher, including:
- Muckle–Wells syndrome;
- Neonatal-onset multisystem inflammatory disease / chronic infantile neurological, cutaneous, articular syndrome (NOMID/CINCA);
- Severe forms of familial cold autoinflammatory syndrome / familial cold urticaria with systemic symptoms not typical of cold-induced urticaria.
Tumor necrosis factor receptor-associated periodic syndrome (TRAPS)
Hyperimmunoglobulinemia D syndrome (HIDS) / mevalonate kinase deficiency (MKD)
Familial Mediterranean fever (FMF)
Should be used in combination with colchicine when necessary.
Ilaris is also indicated for the treatment of the following conditions:
Still’s disease
Ilaris is indicated for the treatment of active Still’s disease, including adult-onset Still’s disease (AOSD) and systemic juvenile idiopathic arthritis (SJIA) in patients aged 2 years and older who have had an inadequate response to prior therapy with nonsteroidal anti-inflammatory drugs (NSAIDs) and systemic corticosteroids. Ilaris may be used as monotherapy or in combination with methotrexate.
Gouty arthritis
Symptomatic treatment of adult patients with frequent gouty arthritis flares (at least 3 flares during the previous 12 months) when nonsteroidal anti-inflammatory drugs (NSAIDs) and colchicine are contraindicated, not tolerated, or do not provide adequate control, and when repeated courses of corticosteroid therapy are not considered appropriate.
Contraindications
Hypersensitivity to the active substance or to any of the excipients. Active, severe infections.
Interaction with other medicinal products and other forms of interaction
Interactions of canakinumab with other medicinal products have not been evaluated in formal studies.
An increased incidence of serious infections has been associated with the use of another IL-1 blocker in combination with tumor necrosis factor (TNF) inhibitors. Concomitant use of canakinumab with TNF inhibitors is not recommended, as it may increase the risk of serious infections.
The activity of hepatic CYP450 enzymes may be suppressed by cytokines involved in chronic inflammation, such as interleukin-1 beta (IL-1 beta). Thus, the activity of CYP450 may be altered during potent cytokine-inhibiting therapy, such as with canakinumab. This is clinically relevant for CYP450 substrates with a narrow therapeutic index, when dose adjustments are individually required. When initiating canakinumab with such medicinal products, therapeutic monitoring of effect or active substance concentration is recommended, and dose adjustments should be made if necessary.
There are no data on the effect of live vaccines or secondary transmission of infection from live vaccines in patients receiving canakinumab. Therefore, live vaccines should not be administered concurrently with Ilaris, except when the benefits clearly outweigh the risks. If live vaccination is planned after initiation of canakinumab treatment, a waiting period of at least 3 months after the last canakinumab injection is recommended before the next vaccination.
Results from a study conducted in healthy adult volunteers showed that a single 300 mg dose of Ilaris did not affect the induction or maintenance of antibody response following influenza or meningococcal glycoconjugate vaccination.
Results from a 56-week open-label study in patients with cryopyrin-associated periodic syndromes (CAPS) aged up to 4 years showed that protective antibody levels were achieved in all patients who received inactivated vaccines included in routine childhood immunization schedules.
Special precautions for use
Traceability
To improve traceability of biological medicinal products, the name and batch number of the administered product should be clearly recorded.
Infections
The use of canakinumab has been associated with an increased incidence of serious infections. Therefore, patients should be closely monitored for signs and symptoms of infection during and after treatment with canakinumab. Physicians should exercise caution when administering canakinumab to patients with current infections, recurrent infections in their history, or underlying conditions that may predispose to infections.
Treatment of cryopyrin-associated periodic syndromes (CAPS), TRAPS, HID/MKD, FMF, and Still’s disease (SJIA and AOSD)
Canakinumab should not be administered during active infection requiring medical intervention.
Treatment of gouty arthritis
Canakinumab should not be administered during active infection.
Concomitant use of Ilaris with tumour necrosis factor (TNF) inhibitors is not recommended, as this increases the risk of serious infections.
Isolated cases of unusual or opportunistic infections (including aspergillosis, atypical mycobacterial infections, and herpes zoster) have been reported during treatment with canakinumab. However, a causal relationship between canakinumab and these events cannot be excluded.
Tuberculosis screening
Approximately 12% of patients with cryopyrin-associated periodic syndromes tested positive in the tuberculin skin test (TST) in clinical trials while receiving canakinumab, despite having no clinical evidence of latent or active tuberculosis infection.
It is unknown whether the use of interleukin-1 (IL-1) inhibitors such as canakinumab increases the risk of tuberculosis reactivation. Prior to initiating therapy, all patients should be evaluated for both active and latent tuberculosis. Physicians should carefully review the patient’s medical history. All patients (local guidelines may apply) should undergo appropriate screening tests (e.g., tuberculin skin test, interferon-gamma release assay, or chest X-ray). Patients should be closely monitored for signs and symptoms of tuberculosis during and after treatment with canakinumab. Patients should be informed that if symptoms suggestive of tuberculosis (e.g., persistent cough, weight loss, low-grade fever) develop during treatment with canakinumab, they must seek medical attention. If the Mantoux test is positive, particularly in patients at high risk, alternative methods for screening tuberculosis infection should be considered.
Neutropenia and leukopenia
Neutropenia (absolute neutrophil count [ANC] < 1.5 × 10⁹/L) and leukopenia have been observed with IL-1 inhibitors, including canakinumab. Treatment with canakinumab should not be initiated in patients with neutropenia or leukopenia. It is recommended to assess white blood cell counts, including neutrophil counts, prior to starting treatment and at 1 and 2 months after initiation. For patients undergoing chronic treatment or repeat therapy, periodic monitoring of white blood cell counts during treatment is also recommended. If a patient develops neutropenia or leukopenia, white blood cell counts should be closely monitored and discontinuation of treatment should be considered.
Malignant neoplasms
Malignant neoplasms have been reported in patients treated with canakinumab. The risk of developing malignancies with anti-interleukin-1 (IL-1) therapy is unknown.
Hypersensitivity reactions
Cases suggesting hypersensitivity reactions have been reported with the use of canakinumab. Most of these cases were mild in severity. During clinical development of Ilaris in over 2600 patients, anaphylactoid or anaphylactic reactions were not observed. However, the risk of severe hypersensitivity reactions, which are not uncommon with protein-based injectables, cannot be excluded.
Liver function
Transient, asymptomatic elevations in serum transaminases or bilirubin levels have been reported in clinical trials.
Vaccination
There are no data on the risk of secondary transmission of infection from live (attenuated) vaccines to patients receiving canakinumab. Therefore, live vaccines should not be administered concurrently with canakinumab, except when the benefits clearly outweigh the risks.
Prior to initiating canakinumab therapy, adults and children should receive all recommended vaccinations, including pneumococcal and inactivated influenza vaccines, if needed.
NLRP3 gene mutation in patients with cryopyrin-associated periodic syndromes
Clinical experience in patients with cryopyrin-associated periodic syndromes without a confirmed NLRP3 gene mutation is limited.
Macrophage activation syndrome in patients with Still’s disease
Macrophage activation syndrome (MAS) is a known life-threatening condition that may develop in patients with rheumatic diseases, particularly in those with Still’s disease. If MAS develops or is suspected, prompt evaluation and treatment should be initiated. Physicians should be vigilant for signs of infection or worsening of Still’s disease, which are known triggers for MAS. Clinical trial data suggest that canakinumab is unlikely to increase the risk of MAS in patients with Still’s disease, but definitive conclusions cannot be drawn.
Drug reaction with eosinophilia and systemic symptoms (DRESS)
Rarely, patients receiving Ilaris, primarily those with systemic juvenile idiopathic arthritis (SJIA), have reported drug reactions with eosinophilia and systemic symptoms (DRESS). Patients with DRESS may require hospitalization, as this condition can be fatal. If signs and symptoms of DRESS are present and no alternative etiology can be identified, Ilaris should not be re-administered; alternative treatments should be considered.
Use during pregnancy or breastfeeding
Women of childbearing potential
Women should use effective contraception during treatment with canakinumab and for 3 months after the last dose.
Pregnancy
Data on the use of Ilaris in pregnant women are limited. Animal studies do not indicate a direct or indirect adverse effect on reproductive toxicity. The risk to the fetus/mother is unknown. Therefore, Ilaris should be used in pregnant women or women planning pregnancy only after careful assessment of benefit versus potential risk.
Animal studies show that canakinumab crosses the placenta and is detectable in the fetus. There are no human data, but since canakinumab is an immunoglobulin G1 (IgG1), transplacental passage is expected. The clinical significance of this is unknown. However, live vaccines should not be administered to newborns exposed to canakinumab in utero within 16 weeks after the last dose of canakinumab received by the mother before delivery. Women who received canakinumab during pregnancy should be instructed to inform the newborn’s physician before any vaccination is administered.
Breastfeeding
It is unknown whether canakinumab is excreted in human breast milk. The use of Ilaris in breastfeeding women should only be considered if the expected benefit to the mother outweighs any potential risk to the infant.
Fertility
Studies on the potential effect of Ilaris on human fertility have not been conducted.
Canakinumab did not affect fertility parameters in male animals (C. jacchus). Mouse anti-mouse IL-1 beta antibodies showed no adverse effects on fertility in male or female mice.
Ability to influence the speed of reactions while driving or operating machinery
Ilaris has negligible influence on the ability to drive or operate machinery. However, treatment with Ilaris may cause dizziness/vertigo or asthenia. Patients experiencing these symptoms during treatment with Ilaris should wait until symptoms resolve before driving or operating machinery.
Method of administration and dosage.
Cryopyrin-associated periodic syndromes (CAPS), tumor necrosis factor receptor-associated periodic syndrome (TRAPS), hyperimmunoglobulinemia D syndrome (HIDS)/mevalonate kinase deficiency (MKD), familial Mediterranean fever (FMF), and Still's disease
Treatment should be initiated and supervised by a physician experienced in the diagnosis and treatment of the respective conditions.
After appropriate training in injection techniques, patients or their caregivers may self-administer Ilaris, if the physician determines that it is appropriate and medically necessary.
Recommended initial doses of Ilaris for adults, adolescents, and children aged 2 years and older with cryopyrin-associated periodic syndromes (CAPS):
Adults and children aged 4 years and older:
- 150 mg for patients with body weight > 40 kg;
- 2 mg/kg for patients with body weight ≥ 15 kg and ≤ 40 kg;
- 4 mg/kg for patients with body weight ≥ 7.5 kg and < 15 kg.
Children aged 2 to 4 years:
- 4 mg/kg for patients with body weight ≥ 7.5 kg.
These doses are administered every eight weeks as a single dose by subcutaneous injection.
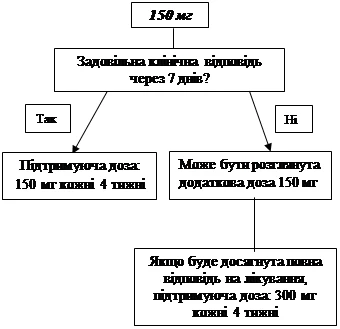
If an adequate clinical response (resolution of rash and other systemic symptoms) has not been achieved within 7 days after the initial dose of 150 mg or 2 mg/kg, administration of a second dose of Ilaris 150 mg or 2 mg/kg may be considered. Upon achieving clinical response, an intensified dosing regimen of 300 mg or 4 mg/kg every 8 weeks should be maintained. If an adequate clinical response is not achieved within 7 days after this dose increase, administration of a third dose of Ilaris 300 mg or 4 mg/kg may be considered. Upon achieving full clinical response, an intensified dosing regimen of 600 mg or 8 mg/kg every 8 weeks should be maintained, based on individual clinical assessment.
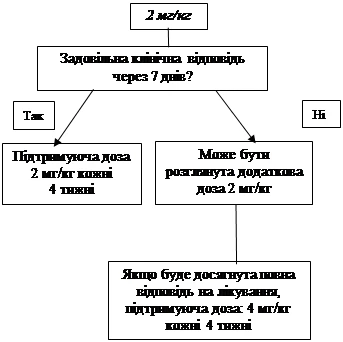
If an adequate clinical response has not been achieved within 7 days after the initial dose of 4 mg/kg, administration of a second dose of Ilaris 4 mg/kg may be considered. Upon achieving full clinical response, an intensified dosing regimen of 8 mg/kg every 8 weeks should be maintained, based on individual clinical assessment.
Clinical experience with dosing intervals shorter than 4 weeks and with doses exceeding 600 mg or 8 mg/kg is limited.

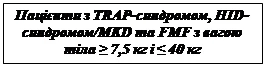
 |
|||
 |
|||
Still's Disease (SJIA and AOSD)
The recommended dose of Ilaris for patients with Still's disease (adult-onset Still's disease (AOSD) and systemic juvenile idiopathic arthritis) with body weight ≥ 7.5 kg is 4 mg/kg (maximum 300 mg) every four weeks by subcutaneous injection. The decision on continuing Ilaris treatment in patients without clinical improvement is made by the physician.
Gouty Arthritis
Treatment should be administered under the supervision of physicians experienced in diagnosing and treating gouty arthritis and using biopharmaceuticals. The medicinal product Ilaris must be administered by a healthcare professional.
Treatment of hyperuricemia should be initiated with appropriate therapy to lower or optimize urate concentrations (ULT). Canakinumab should be used as on-demand therapy for the treatment of gouty arthritis flares.
Control of hyperuricemia with appropriate urate-lowering therapy is required. Ilaris should be used as on-demand therapy for the treatment of gouty arthritis flares.
The recommended dose of Ilaris for adult patients with gouty arthritis is 150 mg subcutaneously as a single dose during a flare. To achieve maximum effect, Ilaris should be administered as soon as possible after the onset of a gouty arthritis flare.
Patients who did not respond to initial treatment should not receive repeated doses of Ilaris. For patients who responded and require repeat treatment, the interval between doses should be at least 12 weeks.
Special Populations
Geriatric Patients
Dose adjustment is not required.
There are no significant differences in the safety profile observed in patients over 65 years of age.
Hepatic Impairment
Data on the use of Ilaris in patients with hepatic impairment are not available.
Renal Impairment
Dose adjustment is not required for patients with renal impairment. However, clinical experience with the use of the drug in such patients is limited.
Method of Administration
Ilaris 150 mg, powder for solution for injection, is supplied in a single-use vial for individual administration. Unused medicinal product or waste material must be disposed of in accordance with local requirements.
Reconstitution Instructions
Using aseptic technique, reconstitute the vial contents at room temperature: slowly inject 1.0 mL of water for injection using a 1 mL syringe and an 18 G × 50 mm needle. Gently rotate the vial at an angle of approximately 45° for about 1 minute and leave it to stand for 5 minutes. Then gently invert the vial upside down and back ten times. Avoid touching the rubber stopper with fingers if possible. Allow to stand for 15 minutes at room temperature to obtain a clear or opalescent solution. Do not shake. Do not use the medicinal product if particles are present in the solution.
Tap the side of the vial to dislodge any residual liquid from the stopper. The solution should be free of visible particles, clear or opalescent. The solution should be colorless, but may have a slight brownish-yellow tint. If the solution is dark brown, it must not be used. If the solution is not used immediately after reconstitution, it should be stored at 2 to 8 °C and used within 24 hours.
Administration Instructions
Carefully fill the syringe with the required volume of solution depending on the dose (from 0.2 mL to 1.0 mL) and administer subcutaneously using a 27 G × 13 mm needle.
Injection sites: upper thigh, abdomen, shoulder, or buttocks. Avoid areas with damaged skin, bruises, or rashes. Administration into scar tissue should be avoided, as this may reduce the effect of Ilaris.
Disposal
Patients or their caregivers should be instructed on the proper disposal of vials, syringes, and needles in accordance with local requirements.
Children
Periodic syndromes associated with cryopyrin (CAP syndromes), TRAPS, HIDS/MKD, and FMF
The safety and efficacy of Ilaris in patients under 2 years of age with periodic syndromes associated with cryopyrin, TRAPS, HIDS/MKD, and FMF have not been established. Available data are described in the sections "Pharmacokinetics", "Pharmacodynamics", and "Adverse Reactions", but no dosing recommendations can be made.
Systemic Juvenile Idiopathic Arthritis (SJIA)
The safety and efficacy of Ilaris in patients under 2 years of age with systemic juvenile idiopathic arthritis have not been established.
Gouty Arthritis
There is no experience with the use of Ilaris in children for the indication of gouty arthritis.
Overdose
Information regarding overdose is limited. In early studies, patients and healthy volunteers received doses up to 10 mg/kg intravenously or subcutaneously without any signs of acute toxic effects of the drug.
In case of overdose, patients should be monitored and appropriate symptomatic treatment should be initiated immediately if necessary.
Adverse Reactions
Approximately 2300 patients were enrolled in blinded, open-label clinical trials, including about 250 pediatric patients (aged 2 to 17 years) diagnosed with cryopyrin-associated periodic syndromes, systemic juvenile idiopathic arthritis, gouty arthritis, or other IL-1 beta-mediated diseases, as well as healthy volunteers. The most common adverse reactions were infections (e.g., upper respiratory tract infections). Most reactions were mild or moderate in severity. Long-term treatment did not affect the type or frequency of adverse reactions.
Cases of hypersensitivity reactions have been observed in patients treated with Ilaris.
Opportunistic infections have been reported during treatment with Ilaris.
Cryopyrin-Associated Periodic Syndromes
A total of 211 adult and pediatric patients (with diagnoses including familial cold autoinflammatory syndrome/familial cold urticaria, Muckle-Wells syndrome, and neonatal-onset multisystem inflammatory disease/chronic infantile neurologic cutaneous and articular syndrome) were enrolled in clinical trials. The safety of Ilaris was compared to placebo in a phase III study consisting of an 8-week open-label period (Part 1), a 24-week randomized, double-blind, placebo-controlled withdrawal period (Part 2), and a 16-week open-label period with Ilaris treatment (Part 3). All patients received 150 mg of Ilaris subcutaneously or 2 mg/kg body weight for patients with body weight ≥ 15 kg and ≤ 40 kg.
Systemic Juvenile Idiopathic Arthritis
A total of 201 patients aged 2 to 20 years with systemic juvenile idiopathic arthritis were enrolled in clinical trials of Ilaris. The safety of Ilaris was compared to placebo in two phase III pilot studies.
Gouty Arthritis
More than 700 patients with gouty arthritis were enrolled in randomized, double-blind, active-controlled clinical trials lasting up to 24 weeks and received doses ranging from 10 mg to 300 mg. Over 250 patients were treated at the recommended dose of 150 mg in phase II and III trials.
Adverse reactions are listed below by MedDRA system organ class and frequency. Within each system organ class, adverse reactions are categorized by frequency, with the most frequent listed first. Frequency categories are defined as follows: very common (≥ 1/10); common (≥ 1/100 to < 1/10); uncommon (≥ 1/1,000 to < 1/100); rare (≥ 1/10,000 to < 1/1,000); very rare (< 1/10,000); not known (frequency cannot be estimated from available data). Within each frequency group, adverse reactions are listed in order of decreasing severity.
Table 5. Adverse Reactions
| System organ classes |
CAP syndrome, TRAP syndrome, HID syndrome/MKD, FMF, systemic juvenile idiopathic arthritis, gouty arthritis |
| Infections and infestations |
|
| Very common |
Respiratory tract infections (including pneumonia, bronchitis, influenza, viral infections, sinusitis, rhinitis, pharyngitis, tonsillitis, nasopharyngitis, upper respiratory tract infections) Ear infections Cellulitis Gastroenteritis Urinary tract infections |
| Common |
Vulvovaginal candidiasis |
| Nervous system disorders |
|
| Common |
Dizziness/vertigo |
| Gastrointestinal disorders |
|
| Very common |
Abdominal pain (upper)1 |
| Uncommon |
Gastroesophageal reflux disease2 |
| Skin and subcutaneous tissue disorders |
|
| Very common |
Injection site reactions |
| Musculoskeletal and connective tissue disorders |
|
| Very common |
Arthralgia1 |
| Common |
Musculoskeletal pain1 Back pain2 |
| General disorders |
|
| Common |
Fatigue/asthenia2 |
| Investigations |
|
| Very common |
Decreased creatinine renal clearance1,3 Proteinuria1,4 Leukopenia1,5 |
| Common |
Neutropenia |
| Uncommon |
Decreased platelet count |
| 1In systemic juvenile idiopathic arthritis. 2In gouty arthritis. 3According to estimated creatinine clearance, most cases were transient. 4Most cases were either transient traces or test strip proteinuria at 1+ level. 5See additional information below. |
|
In a subset of young adult patients with systemic juvenile idiopathic arthritis aged 16 to 20 years (n = 31), the safety profile of canakinumab was consistent with that observed in patients with systemic juvenile idiopathic arthritis under 16 years of age. Based on published reports, the safety profile in adult-onset Still’s disease patients is expected to be similar to that in patients with systemic juvenile idiopathic arthritis.
Long-term study data and laboratory abnormalities in patients with cryopyrin-associated periodic syndromes
During clinical trials of Ilaris in patients with cryopyrin-associated periodic syndromes, mean hemoglobin levels increased, while levels of white blood cells, neutrophils, and platelets decreased.
Elevations in transaminases were observed infrequently.
Asymptomatic and mild elevations in serum bilirubin levels without concomitant increases in transaminases were reported in patients with cryopyrin-associated periodic syndromes treated with canakinumab.
In long-term open-label dose-escalation studies, the incidence of infections (gastroenteritis, respiratory tract infections, upper respiratory tract infections), vomiting, and dizziness was higher in the group receiving 600 mg or 8 mg/kg compared to other dose groups.
Laboratory abnormalities in patients with TRAPS, HIDS/MKD, and FMF
Neutrophils
Although grade ≥ 2 decreases in neutrophils occurred in 6.5% of patients (common), and grade 1 decreases occurred in 9.5% of patients, such reductions were generally transient, and infection associated with neutropenia was not identified as an adverse reaction.
Platelets
Although grade ≥ 2 decreases in platelet counts occurred in 0.6% of patients, bleeding was not reported as an adverse reaction. Mild and transient grade 1 decreases in platelets occurred in 15.9% of patients without any bleeding-related adverse reactions.
Laboratory abnormalities in patients with systemic juvenile idiopathic arthritis
Hematology
Within the overall systemic juvenile idiopathic arthritis treatment program, transient decreases in white blood cell counts ≤ 0.8 × ULN were observed in 33 patients (16.5%). Transient decreases in absolute neutrophil count (ANC) to less than 1 × 10⁹/L were observed in 12 patients (6.0%). Transient decreases in platelet counts (< ULN) were observed in 19 patients (9.5%).
ALT/AST
Within the overall systemic juvenile idiopathic arthritis treatment program, elevated ALT and/or AST levels (> 3 times upper limit of normal (ULN)) were observed in 19 patients (9.5%).
Laboratory abnormalities in patients with gouty arthritis
Hematology
Decreases in white blood cell counts ≤ 0.8 × ULN (lower limit of normal) were reported in 6.7% of patients treated with canakinumab, compared to 1.4% of patients treated with triamcinolone acetonide. Decreases in absolute neutrophil count (ANC) to levels below 1 × 10⁹/L were observed in 2% of patients in comparative trials. Isolated cases of ANC levels < 0.5 × 10⁹/L were also observed.
Mild (< ULN and > 75 × 10⁹/L) and transient decreases in platelet counts were observed more frequently (12.7%) following canakinumab administration in active-controlled clinical trials compared to the comparator (7.7%) in patients with gouty arthritis.
Uric acid
Elevations in uric acid levels (0.7 mg/dL at 12 weeks and 0.5 mg/dL at 24 weeks) were observed after canakinumab treatment in comparative trials in patients with gouty arthritis. In another study in patients receiving ULT, no increase in uric acid levels was observed. No increase in uric acid levels was observed in clinical trials in patient groups without gouty arthritis.
ALT/AST
Mean and median increases in alanine aminotransferase (ALT) of 3.0 U/L and 2.0 U/L, respectively, and aspartate aminotransferase (AST) of 2.7 U/L and 2.0 U/L, respectively, from baseline to end of study were observed in both the canakinumab and triamcinolone acetonide groups; however, the frequency of clinically significant changes (≥ 3 × ULN) was higher in patients receiving triamcinolone acetonide (2.5% for AST and ALT) compared to the canakinumab treatment group (1.6% for ALT and 0.8% for AST).
Triglycerides
In active-controlled clinical trials involving patients with gouty arthritis, mean increase in triglyceride levels was 33.5 mg/dL in the canakinumab treatment group compared to a modest decrease of −3.1 mg/dL in the triamcinolone acetonide group. The frequency of triglyceride elevations > 5 × ULN was 2.4% for canakinumab and 0.7% for triamcinolone acetonide. The clinical significance of this observation is unknown.
Long-term observational study data
In a long-term study (mean exposure to canakinumab – 3.8 years), canakinumab was administered in routine clinical practice to patients with systemic juvenile idiopathic arthritis (85 pediatric patients aged ≥ 2 to ≤ 17 years and 158 adult patients aged ≥ 18 years). The safety profile of canakinumab observed after long-term treatment under these conditions was consistent with that observed in interventional studies in patients with systemic juvenile idiopathic arthritis.
Pediatric population
The study included 80 pediatric patients aged 2–17 years with cryopyrin-associated periodic syndromes. Overall, there were no clinically significant differences in safety and tolerability profile of Ilaris in pediatric patients compared to the general population of patients with cryopyrin-associated periodic syndromes (including both adult and pediatric patients, N = 211), including with respect to the overall frequency and severity of infectious episodes. Upper respiratory tract infections were the most common infectious events.
Additionally, 6 pediatric patients under 2 years of age were evaluated in a small open-label clinical study. The safety profile of Ilaris was similar to that in patients aged 2 years and older.
During a 16-week study, 102 patients with TRAPS, HIDS/MKD, and FMF (aged 2–17 years) received canakinumab. Overall, there were no clinically significant differences in the safety and tolerability profile of canakinumab in pediatric patients compared to the general population.
Shelf life. 3 years.
Storage conditions.
Store at 2–8°C in the original packaging to protect from light. Do not freeze. Keep out of reach of children.
Incompatibilities.
Due to lack of compatibility studies, this medicinal product should not be mixed with other medicinal products.
Packaging.
150 mg of powder for solution for injection in a 6 mL vial made of clear glass; 1 vial per carton. Or 4 cartons, each containing 150 mg of powder for solution for injection in a 6 mL vial made of clear glass, in an outer package.
Prescription category. Prescription only.
Manufacturer.
Novartis Pharma Stein AG.
Manufacturer’s location and address of place of business. Schaffhauserstrasse, 4332 Stein, Switzerland.