Heloplazma
Ukraina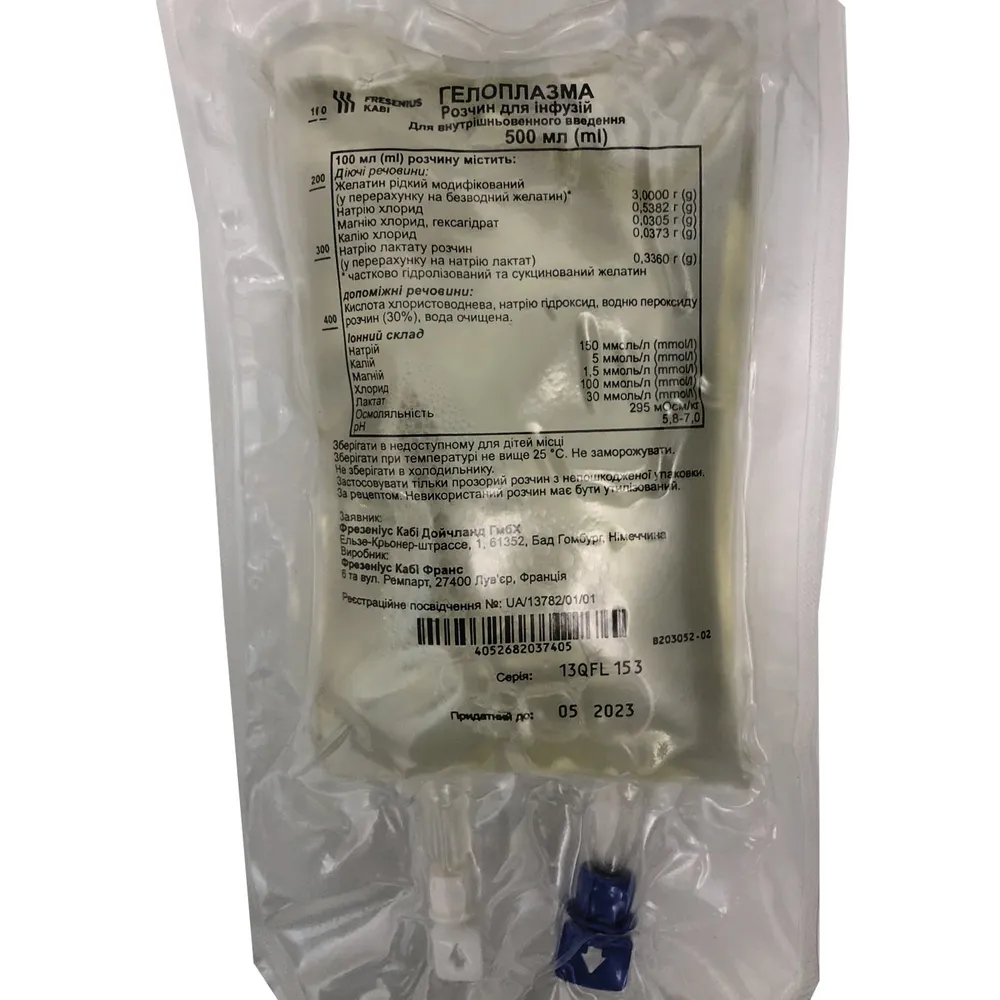
Spis treści
ULOTKA DOTYCZĄCA STOSOWANIA LEKU GELOPLAZMA (GELOPLAZMA)
Skład:
Substancje czynne:
100 ml roztworu zawiera: żelatyny ciekłej modyfikowanej, przeliczonej na żelatynę bezwodną – 3,0000 g, chlorku sodu – 0,5382 g, chlorku magnezu, heksahydrat – 0,0305 g, chlorku potasu – 0,0373 g, roztworu lakatanu sodu, przeliczonego na lakatan sodu – 0,3360 g;
Substancje pomocnicze: kwas solny, wodorotlenek sodu, roztwór nadtlenku wodoru (30%), woda do wstrzykiwania.
Skład jonowy:
sód 150 mmol/l
potas 5 mmol/l
magnez 1,5 mmol/l
chlorek 100 mmol/l
lakatan 30 mmol/l
Osmolalność 295 mOsm/kg
pH 5,8–7,0
Postać leku. Roztwór do wlewania.
Główne właściwości fizykochemiczne: przezroczysty, bezbarwny lub lekko żółty roztwór.
Grupa farmakoterapeutyczna. Zamienniki osocza i roztwory do perfuzji. Pochodne żelatyny.
Kod ATX B05A A06.
Właściwości farmakodynamiczne.
Farmakodynamika.
Heloplazma to roztwór modyfikowanego żelu w roztworze fizjologicznym o średniej masie cząsteczkowej (Mw) 45 000 Daltonów. W wyniku sukcynilacji cząsteczki żelu stają się bardziej ujemnie naładowane, a tym samym bardziej wydłużone. Heloplazma jest substytutem osocza, ale nie wywiera działania plazmoplastycznego.
Roztwór leku wpływa na przywrócenie równowagi jonowej i korygencję acidозy oraz nieznacznie zwiększa diurezę.
Roztwór leku stosuje się samodzielnie, bez przetaczania krwi, w przypadkach utraty krwi w ilości od 0 do 20% całkowitej objętości krwi oraz przy wlewowym podawaniu ograniczonej objętości (około 500 ml). Takie podania nie wpływają na zmianę grupy krwi i wywierają obojętne działanie na mechanizmy krzepnięcia krwi.
W przypadku silnego krwawienia naprzemienne podawanie krwi oraz leków na bazie płynnego żelu zapewnia odpowiednią hemodylucję (przywrócenie objętości krwi i utrzymanie ciśnienia onkotycznego).
Farmakokinetyka.
Po wlewie lek szybko rozprzestrzenia się w przestrzeni wewnątrznaczyniowej i częściowo – z uwagi na niską frakcję mas cząsteczkowych – w przestrzeni międzykomórkowej. Obecność substancji o niskiej masie cząsteczkowej wpływa na funkcję nerek i zwiększenie diurezy. Efekt zastępczy objętościowy leku utrzymuje się przez 4–5 godzin po wlewie. Lek jest szybko wydalany (75% w ciągu 24 godzin), głównie z moczem przez nerki.
Dane kliniczne.
Wskazania.
Leczenie nagłych stanów wstrząsu:
- wstrząs hipowolemiczny spowodowany krwawieniem, odwodnieniem, utratą krwi podczas operacji, oparzeniami;
- wstrząs wazoplegiczny o pochodzeniu traumatycznym, chirurgicznym, septycznym lub toksycznym.
Leczenie współistniejącej hipowolemii związanej z hipotensją w kontekście wazoplegii spowodowanej lekami obniżającymi ciśnienie, w szczególności podczas znieczulenia.
Przeciwwskazania.
- Nadwrażliwość na roztwory zawierające żelatynę lub na którykolwiek z substancji czynnych lub pomocniczych;
- podwyższona wrażliwość na galaktozę-α-1,3-galaktozę (alfa-Gal) lub znana alergia na czerwone mięso (mięso ssaków) i przetwory mięsne;
- hiperkaliemia;
- przede wszystkim pozakomórkowe przeciążenie płynami;
- alkaloza metaboliczna;
- koniec ciąży (poród).
Interakcje z innymi lekami i inne rodzaje interakcji.
Jednoczesne dożylne podawanie innych substancji nie jest zalecane, ponieważ farmakokinetyka składników tych mieszanek nie została zbadana. Ze względu na zawartość potasu w tym roztworze, należy unikać podawania potasu oraz leków, które mogą powodować hiperkaliemię (np. moczopędne oszczędzające potas, inhibitory ACE).
Właściwości stosowania.
Nie wolno podawać roztworu w sposób dożylny.
Przed przeprowadzeniem infuzji lekarz powinien dokonać wizualnej kontroli worków z lekiem. Lek jest nadany do użycia pod warunkiem zachowania szczelności opakowania.
Preparat powinien być klarowny. Zakazane jest stosowanie mętnych roztworów.
Lek może powodować alkaloza metaboliczną ze względu na obecność jonów laktycznych.
Niewydolność wątroby
Preparat może nie wykazywać działania alkalizującego u pacjentów z niewydolnością wątroby z powodu zaburzonego metabolizmu laktyku.
Nie można jednoczesnie (w jednym systemie) podawać roztworu z krwią lub jej pochodnymi (koncentrat erytrocytów, osocze i frakcje osocza). Podawanie możliwe tylko przez oddzielne systemy infuzyjne.
Określenie grupy krwi, testy na obecność przeciwciał oraz inne badania laboratoryjne krwi są możliwe u pacjentów, którzy otrzymali do 2 litrów preparatu, choć interpretacja wyników jest utrudniona z powodu rozcieńczenia krwi. Dlatego próbki krwi do wykonania tych badań należy zbierać przed infuzją preparatu żelatyny.
Należy prowadzić monitorowanie stanu pacjenta ze względu na możliwość wystąpienia reakcji alergicznych (reakcje anafilaktyczne/anafilaktoidealne).
Ze względu na możliwe reakcje krzyżowe, u pacjentów z podwyższoną wrażliwością na galaktozę-α-1,3-galaktozę (alfa-Gal) ryzyko podwyższonej wrażliwości i odpowiedniej reakcji anafilaktycznej na roztwory zawierające żelatynę może być znacznie zwiększone u pacjentów z znaną alergią na czerwone mięso (mięso ssaków) i przetwory mięsne oraz/lub z dodatnim wynikiem testu na przeciwciała anty-alfa-Gal IgE. Takim pacjentom nie należy przepisywać roztworów koloidalnych zawierających żelatynę.
W przypadku wystąpienia reakcji alergicznych należy natychmiast przerwać infuzję leku i rozpocząć odpowiednie leczenie.
Roztwór zawiera 5 mmol/l potasu i 150 mmol/l sodu. Należy wziąć pod uwagę te informacje przy przepisywaniu roztworu pacjentom z niewydolnością nerek oraz tym, którzy przestrzegają diety kontrolowanej pod względem zawartości potasu i sodu.
Zasady bezpieczeństwa
Stosowanie tego leku wymaga monitorowania stanu klinicznego pacjenta oraz wskaźników laboratoryjnych:
- pomiar ciśnienia tętniczego i w razie potrzeby – ciśnienia w centralnej żyłach;
- kontrola diurezy;
- oznaczenie poziomu hematokrytu i elektrolitów.
Szczególnie ważne w przypadku:
- niewydolności serca z zastojem;
- zaburzeń funkcji płuc;
- ciężkich zaburzeń funkcji nerek;
- obrzęków/zatrzymania soli;
- przeciążenia krążenia;
- leczenia kortykosteroidami i ich pochodnymi;
- ciężkich zaburzeń krzepnięcia krwi.
Hematokryt nie powinien opaść poniżej 25%; u pacjentów w podeszłym wieku nie powinien być niższy niż 30%. Należy unikać zaburzeń krzepnięcia krwi spowodowanych rozcieńczeniem czynników krzepnięcia. W przypadku przewlekłej niewydolności serca infuzję należy przeprowadzać powoli ze względu na możliwość przeciążenia krążenia. Jeśli przed i podczas operacji podano więcej niż 2000–3000 ml Heloplazmy, zaleca się po operacji sprawdzenie stężenia białka w surowicy krwi, szczególnie w obecności objawów obrzęku tkanek.
Stosowanie w czasie ciąży lub karmienia piersią.
Brakuje wystarczających danych dotyczących stosowania zamienników osocza w czasie ciąży lub karmienia piersią. Nie odnotowano przypadków działania embriotoksycznego leku w tej grupie pacjentów, jednak istnieje ryzyko wystąpienia reakcji anafilaktycznych/anafilaktoidealnych, co może prowadzić do stanu zagrożenia płodu lub noworodka w wyniku hipotensji tętniczej u matki. W związku z tym mogą wystąpić reakcje alergiczne, dlatego lek nie jest zalecany w trzecim trymestrze ciąży. W czasie ciąży preparat należy stosować tylko wtedy, gdy oczekiwany pozytywny efekt dla kobiety ciężarnej przewyższa potencjalne ryzyko dla płodu, ponieważ nie można całkowicie wykluczyć ryzyka reakcji anafilaktoidealnych/anafilaktycznych.
Nie można stosować leku do zapobiegania hipowolemii podczas znieczulenia porodu lub znieczulenia podpajęczynówkowego. Można go jednak stosować do leczenia hipowolemii, gdy istnieje potrzeba uzupełnienia objętości osocza w czasie ciąży.
Brak danych dotyczących przenikania leku do mleka matki. Brakuje wystarczającego doświadczenia w stosowaniu w czasie karmienia piersią. Ryzyko dla noworodków/maluchów nie może być wykluczone.
Niepłodność
Brak kontrolowanych badań wpływu leku na niepłodność u zwierząt lub ludzi.
Wpływ na zdolność kierowania pojazdami lub obsługiwania maszyn.
Brak informacji dotyczących wpływu leku na zdolność kierowania pojazdami lub pracy z złożonymi mechanizmami.
Sposób stosowania i dawki.
Tylko do wstrzykiwania dożylnego.
Objętość i szybkość wlewu zależą od indywidualnego stanu pacjenta, warunków stosowania oraz reakcji na wypełnienie naczyń.
Lek podaje się w postaci wlewu dożylnego (kroplowego). Stosowanie pompy wlewniczej może zwiększyć szybkość wlewu.
Dawkowanie i szybkość wlewu zależą od potrzeb pacjenta, objętości zastępowanej krwi oraz stanu hemodynamicznego pacjenta.
Zwykle podawana dawka wynosi od 500 do 1000 ml (1–2 worki), a czasem więcej.
Dorośli i dzieci o masie ciała powyżej 25 kg otrzymują 500 ml (1 worek) z odpowiednią szybkością zależną od stanu pacjenta. Szybkość wlewu można zwiększyć w przypadku ciężkiej utraty krwi.
Jeśli u dorosłego pacjenta utrata krwi przekracza 1,5 l (czyli więcej niż 20 % objętości krwi), zazwyczaj należy dodatkowo podać krew oprócz Heloplazmy. Należy kontrolować wskaźniki hemodynamiczne, hematologiczne oraz czynniki krzepnięcia krwi.
Dzieci.
Lek stosuje się w praktyce pediatrycznej (patrz sekcja „Sposób stosowania i dawki”).
Przedawkowanie.
Wyższe dawki leku mogą spowodować przeciążenie krążenia z istotnym obniżeniem hematokrytu i stężenia białek osocza. Zwiększenie ciśnienia w krążeniu płucnym może prowadzić do wycieku płynu do przestrzeni pozakomórkowej i spowodować obrzęk płuc. W razie pojawienia się objawów przeciążenia krążenia w wyniku przedawkowania należy natychmiast przerwać wlew.
W przypadku przedawkowania należy przerwać wlew i podać moczopędne o szybkim działaniu.
Wymagane jest leczenie objawowe pacjenta oraz kontrola poziomu elektrolitów.
Niepożądane działania.
Do oceny częstości występowania działań niepożądanych stosowana jest następująca klasyfikacja: bardzo często (≥ 1/10), często (≥ 1/100, < 1/10), rzadko (≥ 1/1 000, < 1/100), niezbyt często (≥ 1/10 000, < 1/1 000), bardzo rzadko (< 1/10 000), częstość nieznana (nie można ustalić częstości na podstawie dostępnych danych).
Niepożądane efekty obserwowane podczas podawania leku:
| Rzadko (≥ 1/10 000, < 1/1 000) |
Bardzo rzadko (< 1/10 000) |
|
| Zaburzenia ze strony układu immunologicznego |
Szok anafilaktyczny, szczególnie nadwrażliwość na galaktozę-α-1,3-galaktozę (alfa-Gal) oraz alergie na czerwone mięso i przetwory |
|
| Zaburzenia ze strony skóry i tkanki podskórnej |
Reakcje alergiczne skóry |
|
| Zaburzenia ze strony układu naczyniowego |
Obniżenie ciśnienia |
|
| Zaburzenia ze strony serca |
Spowolnienie rytmu serca |
|
| Zaburzenia oddechowe, klatki piersiowej i jamy śródpiersia |
Trudności w oddychaniu |
|
| Zaburzenia ogólne i w miejscu podania |
Podgorączkowość, dreszcze |
Zgłaszanie podejrzewanych działań niepożądanych
Zgłaszanie działań niepożądanych po rejestracji leku ma istotne znaczenie. Pozwala to na monitorowanie stosunku korzyści do ryzyka w trakcie stosowania tego leku. Osoby pracujące zawodowo w sektorze medycznym i farmaceutycznym, jak również pacjenci lub ich uprawnieni przedstawiciele, powinni zgłaszać wszystkie przypadki podejrzewanych działań niepożądanych oraz braku skuteczności leku poprzez Automatyczny System Informacyjny do Nadzoru Farmakologicznego pod adresem: https://aisf.dec.gov.ua
Okres ważności.
2 lata.
Warunki przechowywania.
Przechowywać w miejscu niedostępnym dla dzieci w temperaturze nie wyższej niż 25°C. Nie zamrażać. Nie przechowywać w lodówce.
Niezgodność.
Niekompatybilność fizyko-chemiczna z niektórymi antybiotykami (chlortetracyklina, amfoterycyna B (IV), oksytetracyklina, wancomycyna).
Z powodu braku badań dotyczących zgodności, niniejszego leku nie wolno mieszać z innymi lekami.
Opakowanie.
500 ml w worku Freeflex.
500 ml w worku Freeflex; 20 worków Freeflex w kartonowym pudełku.
Kategoria wydawania.
Na receptę.
Producent.
Fresenius Kabi France.
Siedziba producenta oraz adres miejsca prowadzenia działalności.
6 rue Rempart, 27400 Louviers, Francja.
Wnioskodawca.
Fresenius Kabi Deutschland GmbH.
Siedziba wnioskodawcy i/lub przedstawiciela wnioskodawcy.
Else-Kröner-Straße 1, 61352 Bad Homburg, Niemcy.