Fibryga
UkrainaSpis treści
INSTRUKCJA dot. stosowania leku Fibryga (FIBRYGA®)
Skład:
substancja czynna: ludzki fibrynogen;
1 fiolka proszku do sporządzenia roztworu do wstrzykiwań/wlewu zawiera 1 g ludzkiego fibrynogenu;
substancje pomocnicze: sodu chloridum; sodu cytrynian, dihydret; glicyna; L-argininy hydrochlorid.
rozpuszczalnik: woda do wstrzykiwań.
Postać farmaceutyczna. Proszek i rozpuszczalnik do sporządzenia roztworu do wstrzykiwań/wlewu.
Główne właściwości fizykochemiczne:
proszek: proszek biały lub bladożółty, higroskopijny, lub krucha masa;
rozpuszczalnik: przezroczysta, bezbarwna ciecz, bez zawiesiny.
Grupa farmakoterapeutyczna. Środki przeciwkrwotoczne. Ludzki fibrynogen.
Kod ATC B02B B01.
Właściwości farmakologiczne.
Farmakodynamika.
Ludzki fibrynogen (czynnik krzepnięcia krwi I) w obecności trombiny, aktywowanego czynnika krzepnięcia krwi XIII (FXIIIa) oraz jonów wapnia przekształca się w stabilny i elastyczny trójwymiarowy (przestrzenny) skrzep fibrynowy o działaniu hemostatycznym.
Podanie ludzkiego fibrynogenu prowadzi do zwiększenia stężenia fibrynogenu w osoczu krwi i może tymczasowo korygować zaburzenia krzepnięcia krwi u pacjentów z niedoborem fibrynogenu.
W otwartym, prospektywnym, randomizowanym, kontrolowanym, krzyżowym badaniu farmakokinetycznym fazy II z udziałem 22 pacjentów z wrodzonym niedoborem fibrynogenu (afibrynogenemią), w którym oceniano jednokrotne podanie leku, oceniano również maksymalną stabilność skrzepu (МСЗ) jako zmienną zastępczą skuteczności hemostatycznej (FORMA-01). МСЗ była oznaczana metodą tromboelastometrii (ROTEM). У кожnym pacjencie МСЗ była oznaczana przed jednokrotnym podaniem (wartość wyjściowa) oraz po upływie jednej godziny po jednokrotnym podaniu leku. Wartości МСЗ były istotnie wyższe po podaniu leku Fibryga niż w okresie wyjściowym (patrz tabela 1).
Tabela 1
Maksymalna stabilność skrzepu МСЗ [mm] (populacja ITT), n = 22
| Moment czasu |
Średnia wartość + SD |
Wartość środkowa (zakres) |
| Przed infuzją |
0 + 0 |
0 (0–0) |
| 1 godzina po zakończeniu infuzji |
9,7 + 3,0 |
10,0 (4,0–16,0) |
| Zmiana średniej wartości (analiza pierwotna)* |
9,7 + 3,0 |
10,0 (4,0–16,0) |
MCZ – maksymalna stabilność skrzepu; ITT – wszyscy pacjenci, którzy zostali włączeni do badania i rozpoczęli leczenie; SD – odchylenie standardowe.
*p ˂ 0,0001 (95 % przedział ufności 8,37; 10,99).
Przeprowadzono analizę pośrednią trwającego prospektywnego, otwartego, niekontrolowanego badania wieloośrodkowego III fazy (FORMA-02) z udziałem 13 pacjentów z wrodzonym deficytem fibrynogenu (afibrynogenemia i hipofibrynogenemia) w wieku od 13 do 53 lat (2 nastolatków, 11 dorosłych). Badanie obejmowało leczenie 23 krwawień oraz 4 interwencje chirurgiczne. Obserwowano istotne zmiany w porównaniu z wartościami wyjściowymi w MCZ, określonej metodą ROTEM, oraz w stężeniach fibrynogenu w osoczu krwi. Wszystkie leczone krwawienia i interwencje chirurgiczne u badanych oceniono jako skuteczne (częstość dobrej lub doskonałej skuteczności) przez badacza i niezależny komitet ekspertów za pomocą obiektywnej skali oceny.
W prospektywnym, randomizowanym, kontrolowanym badaniu FORMA-05 oceniano skuteczność hemostatyczną i bezpieczeństwo leku Fibryga w porównaniu z krioprecypitatem jako dodatkowym źródłem fibrynogenu u pacjentów, u których rozwinił się nabyte niedobory fibrynogenu podczas operacji cytoradykalnej z powodu rozległego, złośliwego pseudomiększa otrzewnowego. Badanie obejmowało 43 dorosłych pacjentów w populacji analizowanej według zasady per-protokół (PP): 21 pacjentów leczonych lekiem Fibryga oraz 22 pacjentów otrzymujących terapię krioprecypitatem. Intraoperacyjne uzupełnienie fibrynogenu podawano wcześnie (tj. 60–90 minut po rozpoczęciu operacji, gdy stwierdzono znaczną utratę krwi, ale nie więcej niż 2 litry) w dawce 4 g leku Fibryga lub 2 worki po 5 jednostek krioprecypitatu, powtarzane w razie potrzeby. Podczas 7,8 + 1,7 godziny trwania operacji zastosowano 6,5 + 3 g leku Fibryga (89 + 39 mg/kg masy ciała) oraz 4,1 + 2,2 worki po 5 jednostek krioprecypitatu. Średnio 1 jednostkę i 0,5 jednostki krwinki czerwonych (RBC) podano pacjentom podczas operacji odpowiednio w grupie leczonej lekiem Fibryga i krioprecypitatem; średnio zastosowano 0 jednostek krwinek czerwonych w ciągu pierwszych 24 godzin po operacji w obu grupach (patrz tabela 2). Podczas badania nie przeprowadzono transfuzji masy krwinek zastyżalnych świeżo mrożonych ani płytek krwi. Terapię hemostatyczną opartą na uzupełnieniu fibrynogenu oceniono jako skuteczną w 100 % interwencji chirurgicznych w obu grupach przez niezależny komitet ekspertów za pomocą obiektywnej skali oceny.
Tabela 2
Transfuzja RBC* [jednostki] podczas operacji i w ciągu pierwszych 24 godzin po operacji (populacja PP)
| Okres czasu |
Grupa leczona lekiem Fibryga (n = 21) Wartość środkowa (zakres) |
Grupa leczona krzepną krupią mrożoną (n = 22) Wartość środkowa (zakres) |
| W trakcie operacji |
1 (0–4) |
0,5 (0–5) |
| W pierwszych 24 godzinach po operacji |
0 (0–2) |
0 (0–2) |
RBC – koncentraty erytrocytów; PP – grupa pacjentów spełniających kryteria protokołu.
*Nie przeprowadzono transfuzji innych allogennych leków krwiowych, takich jak koncentraty świeżo mrożonej osocza lub trombocytów.
Pacjenci w wieku dziecięcym
Lek Fibryga został podany 8 pacjentom w wieku od 12 do 18 lat w ramach dwóch badań klinicznych. Europejska Agencja Leków odroczyła obowiązek przedstawienia wyników badań stosowania leku Fibryga w leczeniu wrodzonego niedoboru fibrynogenu u pacjentów poniżej 12. roku życia (informacje dotyczące stosowania u dzieci znajdują się w sekcji „Sposób stosowania i dawki”).
Farmakokinetyka.
Ludzki fibrynogen jest naturalnym składnikiem osocza krwi człowieka i działa jako endogenny fibrynogen. W osoczu krwi okres półtrwania fibrynogenu wynosi 3–4 dni. Lek Fibryga podaje się dożylnie i natychmiast pojawia się we krwi w stężeniu odpowiadającym podanej dawce. W otwartym, prospektywnym, randomizowanym, kontrolowanym badaniu krzyżowym fazy II z udziałem 22 pacjentów w wieku od 12 do 53 lat (6 nastolatków, 16 dorosłych) z wrodzonym niedoborem fibrynogenu (afibrynogenemią), w dwóch równoległych grupach, porównywano właściwości farmakokinetyczne leku Fibryga po pojedynczym podaniu z właściwościami farmakokinetycznymi innego dostępnego na rynku koncentratu fibrynogenu u tych samych pacjentów (FORMA-01). Każdy pacjent otrzymał pojedynczą dawkę dożylną 70 mg/kg leku Fibryga oraz leku porównawczego. Pobierano próbki krwi w celu oznaczenia aktywności fibrynogenu w stanach wyjściowych oraz w ciągu 14 dni po infuzji. Parametry farmakokinetyczne leku Fibryga w grupie pacjentów analizowanych zgodnie z protokołem (PP) (n = 21) przedstawiono w tabeli 3.
Tabela 3
Parametry farmakokinetyczne (n = 21) aktywności fibrynogenu (populacja PP*)
| Parametr |
Średnia wartość + SD |
Zakres |
| Okres półwyladowania [godz.] |
75,9 + 23,8 |
40,0–157,0 |
| Cmax [mg/dl] |
139,0 + 36,9 |
83,0–216,0 |
| AUCnorm dla dawki 70 mg/kg [mg*godz./ml] |
113,7 + 31,5 |
59,7–175,5 |
| Klirens [ml/godz./kg] |
0,67 + 0,2 |
0,4–1,2 |
| Średni czas utrzymywania [godz.] |
106,3 + 30,9 |
58,7–205,5 |
| Objętość rozkładu w stanie równowagi [ml/kg] |
70,2 + 29,9 |
36,9–149,1 |
*Jeden pacjent został wykluczony z populacji per-protokół (PP), ponieważ otrzymał < 90 % zaplanowanej dawki leku Fibryga oraz leku porównawczego.
Cmax – maksymalne stężenie we krwi; AUCnorm – pole pod krzywą stężenia, znormalizowane w stosunku do dawki; SD – odchylenie standardowe.
Wzrost in vivo odtwarzany stopniowo, mierzony w ciągu 4 godzin po zakończeniu infuzji. Mediana odtworzenia in vivo wynosiła
1,8 mg/dl (zakres 1,08–2,62 mg/dl) wzrostu na mg/kg. Mediana odtworzenia in vivo wskazuje, że dawka 70 mg/kg zwiększy stężenie fibrynogenu we krwi pacjenta o około 125 mg/dl.
Farmakokinetyka u szczególnych grup pacjentów
Nie zaobserwowano istotnej statystycznie różnicy w aktywności fibrynogenu u mężczyzn i kobiet uczestniczących w badaniu. W analizie zgodnej z protokołem stwierdzono niewielką różnicę w okresie półtrwania u pacjentów w wieku poniżej 18 lat (n = 5), który wynosił 72,8 + 16,5 godziny, w porównaniu do 76,9 + 26,1 godziny w grupie dorosłych (n = 16). Klirens był niemal identyczny w obu grupach wiekowych i wynosił odpowiednio 0,68 + 0,18 ml/godz/kg oraz 0,66 + 0,21 ml/godz/kg.
Pacjenci w wieku pediatrycznym
Brak danych farmakokinetycznych dotyczących pacjentów pediatrycznych w wieku < 12 lat.
Charakterystyka kliniczna.
Wskazania.
Leczenie krwawień oraz profilaktyka w okresie okołochirurgicznym u pacjentów z wrodzoną hipofibrynogenemią lub afibrynogenemią z predyspozycją do krwawień.
Jako terapia wspomagająca w leczeniu niekontrolowanych silnych krwawień u pacjentów z nabytą hipofibrynogenemią podczas zabiegu chirurgicznego.
Przeciwwskazania.
Podwyższona wrażliwość na substancję czynną lub na którąkolwiek z substancji pomocniczych.
Interakcje z innymi lekami i inne rodzaje interakcji.
Nie znano interakcji leków ludzkiego fibrynogenu z innymi lekami.
Szczególne środki ostrożności dotyczące stosowania.
Jeden fiolka leku Fibryga zawiera 1 g ludzkiego fibrynogenu. Po rozcieńczeniu rozpuszczalnikiem, tj. 50 ml wody do wstrzykiwań, Fibryga zawiera około 20 mg/ml ludzkiego fibrynogenu.
Zawartość białka zdolnego do tworzenia skrzeplin określa się zgodnie z Europejską Farmakopeą dla ludzkiego fibrynogenu. Lek ten wytwarza się z osocza dawców ludzkich.
Tromboembolia
Istnieje ryzyko wystąpienia zakrzepicy podczas leczenia pacjentów z wrodzonym lub nabytym niedoborem ludzkiego fibrynogenu, w szczególności w przypadku stosowania wysokich dawek lub dawek powtarzanych. U pacjentów otrzymujących ludzki fibrynogen należy dokładnie obserwować występowanie objawów lub symptomów zakrzepicy.
U pacjentów z chorobą niedokrwienną serca lub z przebytym zawałem mięśnia sercowym, u pacjentów z chorobami wątroby, u pacjentów w okresie okołochirurgicznym i pozachirurgicznym, u noworodków oraz u pacjentów z ryzykiem powikłań tromboembolicznych lub ogólnego zespołu trombohemoragicznego należy ocenić przewidywaną korzyść z leczenia ludzkim fibrynogenem w porównaniu z ryzykiem wystąpienia powikłań tromboembolicznych. Należy zachować ostrożność i dokładnie monitorować stan takich pacjentów.
Nabyty hipofibrynogenemia wiąże się z niskimi stężeniami wszystkich czynników krzepnięcia krwi (a nie tylko fibrynogenu) we krwi oraz obecnością inhibitorów, dlatego należy rozważyć leczenie lekami krwi zawierającymi czynniki krzepnięcia. Wymagany jest dokładny monitoring układu krzepnięcia krwi.
Reakcje alergiczne lub anafilaktyczne
W przypadku wystąpienia reakcji alergicznych lub anafilaktycznych należy natychmiast przerwać wstrzykiwanie/infuzję. W przypadku wstrząsu anafilaktycznego należy zastosować standardowe leczenie wstrząsu.
Zawartość sodu
Lek Fibryga zawiera do 132 mg (5,8 mmol) sodu na fiolkę, co odpowiada 6,6% rekomendowanego przez WHO maksymalnego dziennego spożycia 2 g sodu dla dorosłego. Należy to uwzględnić u pacjentów przestrzegających bezsodowej diety.
Bezpieczeństwo wirusowe
Standardowe środki zapobiegające zakażeniom w wyniku stosowania leków wytwarzanych z krwi lub osocza ludzkiego obejmują selekcję dawców, badanie krwi poszczególnych dawców i partii osocza dawcy pod kątem obecności specyficznych markerów zakażeń oraz włączenie skutecznych etapów procesu produkcyjnego mających na celu inaktywację/usunięcie wirusów. Pomimo tego, przy podawaniu leków wytwarzanych z krwi lub osocza ludzkiego, nie można całkowicie wykluczyć możliwości przeniesienia czynników zakaźnych. Dotyczy to również nieznanych lub nowych wirusów oraz innych patogennych mikroorganizmów.
Środki, które są stosowane, uważane są za skuteczne wobec wirusów otoczkowych, takich jak wirus HIV (ludzkiego wirusa niedoboru odporności), HBV (wirus zapalenia wątroby typu B) i HCV (wirus zapalenia wątroby typu C), a także wobec wirusa zapalenia wątroby typu A (HAV) bez otoczki. Środki te mogą mieć ograniczoną skuteczność wobec wirusów bez otoczki, takich jak parwowirus B19. Zakażenie parwowirusem B19 może być niebezpieczne dla kobiet w ciąży (zakażenie płodu) oraz dla osób z niedoborem odporności lub zwiększonym erytropoezą (np. anemia hemolityczna).
Należy rozważyć odpowiednie szczepienia (zapalenie wątroby typu A i B) u pacjentów, którzy otrzymują powtarzalnie leki wytwarzane z osocza ludzkiego.
Zaleca się zapisywanie nazwy i numeru serii leku Fibryga przy każdym jego podaniu pacjentowi, aby możliwe było śledzenie związku między stanem pacjenta a podaniem konkretnego leku z danej serii.
Imunogenność
W przypadku terapii zastępczej czynnikami krzepnięcia krwi przy innych wrodzonych niedoborach obserwowano reakcje z udziałem przeciwciał, jednak obecnie nie ma danych dotyczących stężenia fibrynogenu.
Szczególne środki ostrożności dotyczące usuwania i ponownego przetwarzania
Ogólne wskazówki
Rozcieńczony roztwór powinien być prawie bezbarwny i lekko opalizujący.
Nie należy stosować roztworów, które są mętne lub zawierają osad.
Rozcieńczanie
- Proszek (lek Fibryga) i rozpuszczalnik (woda do wstrzykiwań) w zamkniętych fiolkach należy ogrzać do temperatury pokojowej. Temperaturę pokojową należy również utrzymywać podczas przygotowywania roztworu. Jeśli do ogrzewania stosuje się łaźnię wodną, należy zadbać, aby woda nie dotykała gumowych pokrywek ani kapturków fiol. Temperatura wody w łaźni nie powinna przekraczać +37°C.
- Zdjąć kapturki z fiol zawierających proszek (lek Fibryga) i rozpuszczalnik, aby uwolnić środkową część przekładki przeznaczonej do infuzji. Przetrzeć gumowe przekładki watą nasączoną alkoholem i pozostawić je do wyschnięcia.
- Zdjąć pokrywę z zewnętrznej opakowania urządzenia do przenoszenia Octajet. Aby zachować sterylność, urządzenie Octajet należy trzymać lub układać w czystym zewnętrznym opakowaniu.
|
Rysunek 1 |
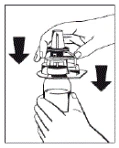
|
Rysunek 2 |
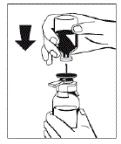
|
Rysunek 3 Rysunek 4 |
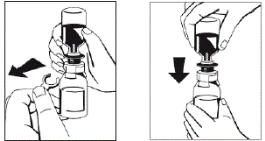
- Gdy rozpuszczalnik całkowicie spłynie do fiolki, należy ostrożnie obracać fiolkę zawierającą lek, aż proszek całkowicie się rozpuści. Nie należy wstrząsać fiolką, aby uniknąć powstawania piany. Rozpuszczenie proszku powinno zakończyć się w ciągu około 5 minut. Zazwyczaj rozpuszczanie proszku trwa nie dłużej niż 20 minut. Jeśli proszek nie rozpuści się w ciągu 20 minut, lek należy zutylizować.
W rzadkich przypadkach, gdy nierozpuszczony proszek w postaci kruchej masy unosi się podczas przemieszczania rozpuszczalnika lub czas rozpuszczania nieoczekiwanie się wydłuża, należy unikać pionowego wstrząsania w celu zapobieżenia wyciekowi cieczy przez konektor fiolki. Procesowi rozpuszczania można sprzyjać intensywniejszym poziomym mieszaniem zawartości fiolki. Gdy krucha masa proszku całkowicie się rozpuści, odbudowa przebiegnie w oczekiwanym czasie.
|
Rysunek 5 |
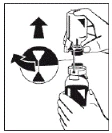
Wciągnąć roztwór do strzykawki przez filtr (rysunek 8). |
|
Rysunek 6 Rysunek 7 Rysunek 8 |
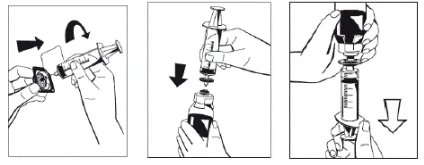
- Odłączyć wypełniony strzykawka od filtra i wyrzucić pusty fiolka.
Zaleca się standardowy zestaw do infuzji dożylnych do stosowania rozcieńczonego roztworu w temperaturze pokojowej.
Wszelkie niewykorzystane leki lub odpady należy utylizować zgodnie z wymaganiami lokalnych władz.
Stosowanie w okresie ciąży lub karmienia piersią.
Ciąża
Badania funkcji rozrodczych na zwierzętach nie były prowadzone z lekiem Fibryga. Ponieważ substancja czynna pochodzi od człowieka, ulega ona katabolizmowi w sposób podobny do własnego białka pacjenta. Nie przewiduje się, że te fizjologiczne składniki krwi człowieka mogą wywierać działanie niepożądane na funkcję rozrodczą lub na płód.
Bezpieczeństwo stosowania leku Fibryga w okresie ciąży nie zostało ustalone w kontrolowanych badaniach klinicznych.
Doświadczenie kliniczne z lekami fibrynogenu w leczeniu powikłań położniczych wskazuje, że nie należy oczekiwać szkodliwego wpływu na przebieg ciąży ani na zdrowie płodu lub noworodka.
Laktacja
Nie wiadomo, czy lek Fibryga wydostaje się do mleka matki. Stosowania leku Fibryga u kobiet karmiących piersią nie badano w badaniach klinicznych.
Plodność
Brak danych dotyczących wpływu na płodność.
Wpływ na zdolność prowadzenia pojazdów lub obsługiwania maszyn.
Lek Fibryga nie wpływa na zdolność prowadzenia pojazdów i pracy z innymi mechanizmami.
Sposób stosowania i dawki
Leczenie należy prowadzić pod nadzorem lekarza posiadającego doświadczenie w leczeniu zaburzeń krzepnięcia krwi.
Dawki
Dawkowanie i długość trwania terapii zastępczej zależą od ciężkości zaburzenia, lokalizacji i objętości krwawienia oraz stanu klinicznego pacjenta.
Należy oznaczać (funkcjonalny) poziom fibrynogenu, aby obliczyć indywidualną dawkę, a liczbę i częstotliwość podawania należy dobierać dla każdego pacjenta indywidualnie, na podstawie ciągłego monitorowania stężenia fibrynogenu we krwi oraz stanu klinicznego pacjenta i innej stosowanej terapii zastępczej.
W przypadku dużych zabiegów chirurgicznych istotne jest ścisłe monitorowanie terapii zastępczej poprzez analizę krzepnięcia krwi (badanie aktywności krzepnącej).
Profilaktyka u pacjentów z wrodzoną hipofibrynogenemią lub afibrynogenemią i znaną skłonnością do krwawień.
W celu zapobiegania nadmiernym krwawieniom podczas operacji chirurgicznych zaleca się leczenie profilaktyczne w celu podniesienia poziomu fibrynogenu do 1 g/l oraz utrzymania fibrynogenu na tym poziomie aż do osiągnięcia hemostazy i powyżej 0,5 g/l do pełnego gojenia się rany.
W przypadku operacji chirurgicznej lub leczenia krwawienia dawkę należy obliczać następująco:
Dawka (mg/kg masy ciała) = [poziom docelowy (g/l) – zmierzony poziom (g/l)]
0,018 (g/l na mg/kg masy ciała)
Dalsze dawkowanie (dawki i częstotliwość wstrzyknięć) należy dostosowywać w zależności od stanu klinicznego i wyników badań laboratoryjnych pacjenta.
Okres półtrwania fibrynogenu w organizmie wynosi 3–4 dni. W związku z tym, w przypadku braku zużycia fibrynogenu podczas operacji, ponowne leczenie ludzkim fibrynogenem zazwyczaj nie jest wymagane. W przypadku akumulacji, która może wystąpić przy powtarzanym podawaniu w celu profilaktyki, dawkę i częstotliwość należy dobierać zgodnie z celami terapeutycznymi lekarza dla danego pacjenta.
Dawki dla określonych grup pacjentów
Pacjenci w wieku dziecięcym
Dostępne obecnie dane opisano w sekcjach „Efekty uboczne” i „Farmakodynamika”, jednak nie podano rekomendacji dotyczących dawkowania u dzieci.
Pacjenci w wieku podeszłym
Badania kliniczne z udziałem leku Fibryga nie były prowadzone wśród pacjentów w wieku 65 lat i więcej, aby dostarczyć wiarygodnych danych dotyczących ich odpowiedzi na leczenie w porównaniu z młodszych pacjentami.
Leczenie krwawień
Krwawienia u pacjentów z wrodzoną hipofibrynogenemią lub afibrynogenemią
Krwawienia należy leczyć w celu osiągnięcia zalecanego docelowego poziomu fibrynogenu we krwi wynoszącego 1 g/l. Ten poziom należy utrzymywać aż do osiągnięcia hemostazy.
Krwawienia u pacjentów z nabytym niedoborem fibrynogenu
Dorośli
Zazwyczaj podaje się początkowo 1–2 g leku, a następnie przeprowadza dalsze infuzje w razie potrzeby.
W przypadku silnych krwawień, np. podczas dużych zabiegów chirurgicznych, może być wymagana większa ilość fibrynogenu (4–8 g).
Sposób stosowania
Wlewanie lub wstrzyknięcie dożylnie.
Lek Fibryga należy podawać powoli dożylnie z zalecaną maksymalną prędkością 5 ml na minutę u pacjentów z wrodzoną hipofibrynogenemią lub afibrynogenemią oraz z zalecaną maksymalną prędkością 10 ml na minutę u pacjentów z nabytym niedoborem fibrynogenu.
Informacje dotyczące rozcieńczania leku przed podaniem znajdują się w sekcji „Szczególne wskazania dotyczące stosowania”.
Dzieci.
Dawkę należy dobierać zgodnie z masą ciała i potrzebami klinicznymi. Zazwyczaj wynosi ona 20–30 mg/kg.
Przedawkowanie.
W celu uniknięcia przedawkowania podczas leczenia należy prowadzić stały monitoring stężenia fibrynogenu we krwi.
W przypadku przedawkowania zwiększa się ryzyko wystąpienia powikłań zakrzepowo-embolicznych.
Niepożądane działania
Streszczenie profilu bezpieczeństwa
Nie ma wiarygodnych danych dotyczących częstości występowania niepożądanych działań uzyskanych w badaniach klinicznych tego leku.
W badaniach klinicznych zgłaszano następujące niepożądane działania: umiarkowane podwyższenie temperatury ciała zaobserwowane u jednego pacjenta oraz lekowy zapalenie skóry w postaci łagodnej reakcji skórnej – świądu i zaczerwienienia – po podaniu leku, zaobserwowane również u jednego pacjenta.
Podczas stosowania leku Fibryga oraz innych stężeń fibrynogenu zgłaszano niepożądane działania wymienione w tabeli 4.
Tabela 4
| Klasy układów narządów |
Reakcja niepożądana |
Częstotliwość |
| Poruszenia ze strony układu odpornościowego: |
Reakcje alergiczne lub anafilaktyczne Reakcje skórne |
Nieznane |
| Poruszenia ze strony naczyń |
Powikłania zakrzepowo-zatorowe (w tym zawał mięśnia sercowego i zatorowość płucna) Tromboflebita |
Nieznane |
| Ogólne poruszenia i reakcje w miejscu podania |
Podwyższenie temperatury ciała (gorączka) |
Nieznane |
Informację o ryzyku przeniesienia czynnika zakaźnego patrz w punkcie „Szczególne ostrzeżenia i środki ostrożności stosowania”.
Pacjenci w wieku pediatrycznym
Ośmiu pacjentów w wieku od 12 do 18 lat z wrodzonym niedoborem fibrynogenu zostało włączonych do badania dotyczącego bezpieczeństwa.
Ogólne profile bezpieczeństwa nie różnią się u dorosłych i u nastolatków.
Brak danych dotyczących stosowania leku Fibryga u pacjentów w wieku pediatrycznym z nabytym niedoborem fibrynogenu.
Zgłaszanie podejrzewanych działań niepożądanych
Zgłaszanie podejrzewanych działań niepożądanych po rejestracji produktu leczniczego jest ważne. Pozwala to na ciągłe monitorowanie stosunku korzyści do ryzyka stosowania produktu leczniczego. Lekarzy prosi się o zgłaszanie wszelkich podejrzewanych działań niepożądanych.
Okres ważności.
2 lata.
Stabilność chemiczna i fizyczna rozcieńczonego roztworu została wykazana przez 24 godziny w temperaturze pokojowej (nie wyższej niż 25 °C). Z mikrobiologicznego punktu widzenia, lek należy stosować natychmiast po rozcieńczeniu. Jeśli tego nie uczyni się, odpowiedzialność za czas i warunki przechowywania po otwarciu opakowania ponosi użytkownik. Rozcieńczonego roztworu nie wolno zamrażać ani przechowywać w lodówce. Niecałkowicie wykorzystane fiolki z lekiem należy zutylizować.
Warunki przechowywania.
Przechowywać w temperaturze poniżej 25 °C. Nie zamrażać.
Przechowywać fiolkę w zewnętrznej tekturowej opakowaniu w celu ochrony przed światłem.
Przechowywać w miejscu niedostępnym dla dzieci.
Informacje o warunkach przechowywania po rozcieńczeniu leku patrz w punkcie „Okres ważności”.
Niezgodność.
Ten produkt leczniczy nie powinien być mieszany z innymi lekami.
Opakowanie.
1 g proszku do sporządzenia roztworu do wstrzykiwań/infuzji w fiolce szklanej zatopionej korkiem i aluminiową odkręcaną nakrętką; 50 ml rozpuszczalnika (woda do wstrzykiwań) w fiolce szklanej zatopionej korkiem i aluminiową odkręcaną nakrętką. 1 fiolka z proszkiem, 1 fiolka z rozpuszczalnikiem, 1 urządzenie Octajet do przenoszenia, 1 filtr w pudełku tekturowym.
Kategoria wydawania.
Na receptę.
Producent.
- Octapharma AB.
- Octapharma Pharmazeutika Produktionsges. m.b.H.
Miejsce produkcji i adres miejsca prowadzenia działalności.
- Lars Forssells gata 23, Sztokholm, 11275, Szwecja.
- Oberlaaer Straße 235, 1100 Wiedeń, Austria.