Fibriga
UkraineTable of Contents
INSTRUCTIONS FOR MEDICAL USE OF THE MEDICINAL PRODUCT FIBRYGA (FIBRYGA®)
Composition:
Active substance: human fibrinogen;
1 vial of powder for solution for injection/infusion contains 1 g of human fibrinogen;
Excipients: sodium chloride; sodium citrate, dihydrate; glycine; L-arginine hydrochloride.
Solvent: water for injections.
Pharmaceutical form. Powder and solvent for solution for injection/infusion.
Main physicochemical properties:
Powder: white or pale yellow, hygroscopic powder or friable mass;
Solvent: clear, colorless liquid, free from particles.
Pharmacotherapeutic group. Antihemorrhagics. Human fibrinogen.
ATC code B02B B01.
Pharmacological properties.
Pharmacodynamics.
Human fibrinogen (coagulation factor I) is converted into a stable and elastic three-dimensional (volume) fibrin hemostatic clot in the presence of thrombin, activated coagulation factor XIII (FXIIIa), and calcium ions.
Administration of human fibrinogen provides an increase in plasma fibrinogen levels and may temporarily correct blood coagulation disorders in patients with fibrinogen deficiency.
An open-label, prospective, randomized, controlled, cross-over Phase II pharmacokinetic study with two parallel groups involving 22 patients with congenital fibrinogen deficiency (afibrinogenemia) also evaluated maximum clot firmness (MCF) as a surrogate marker of hemostatic efficacy (FORMA-01). MCF was determined by thromboelastometry (ROTEM). For each patient, MCF was measured before (baseline value) and one hour after a single administration of the drug. MCF values were significantly higher after administration of FIBRIGA than at baseline (see Table 1).
Table 1
Maximum clot firmness MCF [mm] (ITT population), n = 22
| Time point |
Mean ± SD |
Median (range) |
| Before infusion |
0 ± 0 |
0 (0–0) |
| 1 hour after infusion |
9.7 ± 3.0 |
10.0 (4.0–16.0) |
| Change from baseline (primary analysis)* |
9.7 ± 3.0 |
10.0 (4.0–16.0) |
FCS – maximum clot stability; ITT – all patients who were enrolled in the study and initiated treatment; SD – standard deviation.
*p ˂ 0.0001 (95 % confidence interval 8.37; 10.99).
An interim analysis was conducted of an ongoing prospective, open-label, uncontrolled, multicenter phase III study (FORMA-02) involving 13 patients with congenital fibrinogen deficiency (afibrinogenemia and hypofibrinogenemia), aged 13 to 53 years (2 adolescents, 11 adults). The study included treatment of 23 bleeding episodes and 4 surgical procedures. Significant changes from baseline were observed in FCS, as determined by ROTEM, and in plasma fibrinogen levels. All treated bleeding episodes and surgical procedures were rated as successful (rate of good or excellent efficacy) by both the investigator and an independent expert committee using an objective assessment scale.
In the prospective, randomized, controlled FORMA-05 study, the hemostatic efficacy and safety of FIBRIGA were evaluated in comparison with cryoprecipitate as a supplemental source of fibrinogen in patients who developed acquired fibrinogen deficiency during cytoreductive surgery for extensive malignant pseudomyxoma peritonei. The study included 43 adult patients in the per-protocol analysis population (PP): 21 patients received FIBRIGA treatment and 22 patients received cryoprecipitate therapy. Intraoperative fibrinogen supplementation was administered prophylactically (i.e., 60–90 minutes after the start of surgery, when significant blood loss was observed but not exceeding 2 liters) at a dose of 4 g of FIBRIGA or 2 pools of 5 units of cryoprecipitate, repeated as necessary. During 7.8 + 1.7 hours of surgery, 6.5 + 3 g of FIBRIGA (89 + 39 mg/kg body weight) and 4.1 + 2.2 pools of 5 units of cryoprecipitate were used. On average, 1 unit and 0.5 units of red blood cells (RBC) were administered intraoperatively to patients receiving FIBRIGA and cryoprecipitate, respectively, and an average of 0 units of RBCs were used during the first 24 hours post-surgery in both groups (see Table 2). Transfusions of fresh frozen plasma or platelet concentrates were not performed during the study. Hemostatic therapy based on fibrinogen supplementation was rated as successful in 100% of surgical procedures in both groups by an independent expert committee using an objective assessment scale.
Table 2
RBC* transfusion [units] intraoperatively and during the first 24 hours post-surgery (PP population)
| Time period |
FIBRIGA-treated group (n = 21) Median value (range) |
Cryoprecipitate-treated group (n = 22) Median value (range) |
| Intraoperatively |
1 (0–4) |
0.5 (0–5) |
| During the first 24 hours after surgery |
0 (0–2) |
0 (0–2) |
RBC – red blood cell concentrates; PP – per-protocol patient population.
*No transfusion of other allogeneic blood products, such as fresh frozen plasma or platelet concentrates, was performed.
Pediatric patients
FIBRIGA was administered to 8 patients aged 12 to 18 years in two clinical trials. The European Medicines Agency has deferred the obligation to submit the results of studies on the use of FIBRIGA for the treatment of congenital fibrinogen deficiency in patients under 12 years of age (information on use in pediatric patients can be found in section "Posology and method of administration").
Pharmacokinetics
Human fibrinogen is a normal component of human plasma and acts as endogenous fibrinogen. In plasma, the biological half-life of fibrinogen is 3–4 days. FIBRIGA is administered intravenously and immediately appears in plasma at a concentration corresponding to the administered dose. In an open-label, prospective, randomized, controlled, two-period crossover Phase II study with two parallel groups involving 22 patients with congenital fibrinogen deficiency (afibrinogenemia), aged 12 to 53 years (6 adolescents, 16 adults), the pharmacokinetic properties of FIBRIGA were compared with those of another commercially available fibrinogen concentrate in the same patients (FORMA-01). Each patient received a single intravenous dose of 70 mg/kg of FIBRIGA and the comparator product. Blood samples were collected to determine fibrinogen activity at baseline and over 14 days following infusion. Pharmacokinetic parameters of FIBRIGA in the per-protocol analysis population (PP*) (n = 21) are presented in Table 3.
Table 3
Pharmacokinetic parameters (n = 21) of fibrinogen activity (PP population*)
| Parameter |
Mean value ± SD |
Range |
| Half-life [hours] |
75.9 ± 23.8 |
40.0–157.0 |
| Cmax [mg/dL] |
139.0 ± 36.9 |
83.0–216.0 |
| AUCnorm for dose 70 mg/kg [mg·hour/mL] |
113.7 ± 31.5 |
59.7–175.5 |
| Clearance [mL/hour/kg] |
0.67 ± 0.2 |
0.4–1.2 |
| Mean residence time [hours] |
106.3 ± 30.9 |
58.7–205.5 |
| Volume of distribution at steady state [mL/kg] |
70.2 ± 29.9 |
36.9–149.1 |
*One patient was excluded from the per-protocol (PP) patient population due to receiving < 90% of the planned dose of FIBRIGA and the comparator drug.
Cmax – maximum plasma concentration; AUCnorm – area under the curve normalized to the administered dose; SD – standard deviation.
Gradual recovery of parameters (in vivo) was determined from levels measured during the 4 hours following infusion. Median in vivo recovery was
1.8 mg/dl (range 1.08–2.62 mg/dl) per mg/kg. Median in vivo recovery indicates that a dose of 70 mg/kg will increase plasma fibrinogen concentration by approximately 125 mg/dl.
Pharmacokinetics in special patient populations
No statistically significant difference in fibrinogen activity was observed between male and female study participants. According to the per-protocol analysis, a minor difference in elimination half-life was observed in patients under 18 years of age (n = 5), which was 72.8 + 16.5 hours, compared to 76.9 + 26.1 hours in the adult group (n = 16). Clearance was nearly the same in both age groups, at 0.68 + 0.18 ml/hour/kg and 0.66 + 0.21 ml/hour/kg, respectively.
Pediatric patients
Pharmacokinetic data in pediatric patients under 12 years of age are not available.
Clinical characteristics.
Indications.
Treatment of bleeding and perioperative prophylaxis in patients with congenital hypofibrinogenemia or afibrinogenemia who have a tendency to bleed.
As an adjunctive therapy in the treatment of uncontrolled severe bleeding in patients with acquired hypofibrinogenemia during surgical procedures.
Contraindications.
Hypersensitivity to the active substance or to any of the excipients.
Interaction with other medicinal products and other forms of interaction.
There is no information available on interactions between human fibrinogen and other medicinal products.
Special precautions for use.
One vial of FIBRIGA contains 1 g of human fibrinogen. After reconstitution with the solvent, 50 mL of water for injections, FIBRIGA contains approximately 20 mg/mL of human fibrinogen.
The content of protein capable of forming clots is determined according to the European Pharmacopoeia using human fibrinogen. The product is manufactured from plasma obtained from human donors.
Thromboembolism
There is a risk of thrombosis when treating patients with congenital or acquired deficiency of human fibrinogen, especially when high or repeated doses are administered. Patients receiving human fibrinogen should be carefully monitored for signs or symptoms of thrombosis.
In patients with ischemic heart disease or history of myocardial infarction, in patients with liver disease, peri- and postoperative patients, neonates, and patients at risk of thromboembolic complications or disseminated intravascular coagulation (DIC), the anticipated benefit of treatment with human fibrinogen should be weighed against the risk of thromboembolic complications. Caution should be exercised and careful monitoring of such patients is required.
Acquired hypofibrinogenemia is associated with low plasma concentrations of all coagulation factors (not only fibrinogen) and the presence of inhibitors; therefore, treatment with blood products containing coagulation factors should be considered. Careful monitoring of the coagulation system is necessary.
Allergic or anaphylactic reactions
If allergic or anaphylactic reactions occur, the injection/infusion should be stopped immediately. In case of anaphylactic shock, standard medical treatment for shock should be initiated.
Sodium content
FIBRIGA contains up to 132 mg (5.8 mmol) of sodium per vial, equivalent to 6.6% of the WHO recommended maximum daily intake of 2 g sodium for adults. This should be taken into account for patients on a controlled low-sodium diet.
Viral safety
Standard precautions to prevent infections from medicinal products manufactured from human blood or plasma include donor selection, screening of blood from individual donors and plasma pools for specific markers of infection, and inclusion of effective steps in the manufacturing process to inactivate/remove viruses. Nevertheless, when medicinal products made from human blood or plasma are administered, the possibility of transmitting infectious agents cannot be completely excluded. This also applies to unknown or new viruses and other pathogenic microorganisms.
The measures taken are considered effective against enveloped viruses such as human immunodeficiency virus (HIV), hepatitis B virus (HBV), and hepatitis C virus (HCV), as well as against non-enveloped hepatitis A virus (HAV). The measures taken may have limited effectiveness against non-enveloped viruses such as parvovirus B19. Parvovirus B19 infection may be dangerous for pregnant women (fetal infection) and for individuals with immunodeficiency or increased erythropoiesis (e.g., hemolytic anemia).
Appropriate vaccination (hepatitis A and B) should be considered for patients who receive repeated or long-term treatment with products manufactured from human plasma.
It is strongly recommended that each time FIBRIGA is administered, the name and batch number of the product be recorded to allow traceability between the patient's condition and the administration of a specific batch.
Immunogenicity
Antibody responses have been observed during replacement therapy with coagulation factors in other congenital deficiencies; however, there are currently no data available regarding fibrinogen concentrate.
Special precautions for disposal and further processing
General instructions
The reconstituted solution should be almost colorless and slightly opalescent.
Solutions that are cloudy or contain a precipitate must not be used.
Reconstitution
- Warm the vials containing the powder (FIBRIGA) and the solvent (water for injections) to room temperature while keeping them closed. Room temperature should also be maintained during solution preparation. If a water bath is used for warming, care must be taken to prevent water from contacting the rubber stoppers or caps of the vials. The water bath temperature must not exceed +37°C.
- Remove the caps from the vials containing the powder (FIBRIGA) and the solvent to expose the central portion of the stoppers for injection. Wipe the rubber stoppers with an alcohol-impregnated swab and allow them to dry.
- Remove the cover from the outer packaging of the transfer device Octajet. To maintain sterility, keep/place the Octajet device in the clean outer packaging.
|
Figure 1 |
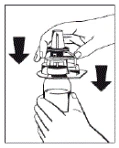
|
Figure 2 |
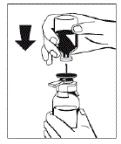
|
Figure 3 Figure 4 |
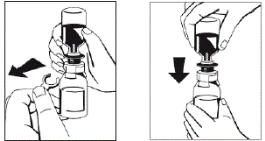
- Once the solvent has completely entered the vial, gently rotate the vial containing the medicinal product until the powder is completely dissolved. Do not shake the vial to avoid foaming. The powder should be completely dissolved within approximately 5 minutes. Usually, dissolution of the powder takes no longer than 20 minutes. If the powder has not dissolved within 20 minutes, the preparation should be discarded.
In rare cases, when undissolved powder in the form of a friable mass floats during transfer of the solvent or the dissolution time unexpectedly prolongs, avoid vertical shaking to prevent leakage of liquid through the vial connector. The dissolution process may be facilitated by more vigorous horizontal mixing of the vial contents. Once the friable mass of powder is completely dissolved, reconstitution will proceed within the expected timeframe.
|
Figure 5 |
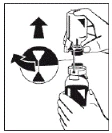
Draw the solution into the syringe through the filter (Figure 8). |
|
Figure 6 Figure 7 Figure 8 |
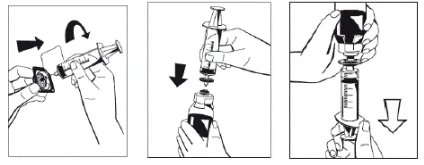
- Disconnect the filled syringe from the filter and discard the empty vial.
A standard intravenous infusion set is recommended for administration of the diluted solution at room temperature.
Any unused medicinal product or waste material should be disposed of in accordance with local regulatory requirements.
Use during pregnancy or breastfeeding.
Pregnancy
Reproductive studies in animals have not been conducted with FIBRIGA. Since the active substance is of human origin, it is catabolized similarly to the patient's own proteins. It is not expected that these physiological components of human blood may exert adverse effects on reproductive function or on the fetus.
The safety of FIBRIGA for use during pregnancy has not been established in controlled clinical trials.
Clinical experience with fibrinogen preparations in the treatment of obstetric complications suggests that harmful effects on the course of pregnancy or on the health of the fetus or newborn are not to be expected.
Lactation
It is unknown whether FIBRIGA is excreted in human breast milk. The use of FIBRIGA in women who are breastfeeding has not been studied in clinical trials.
Fertility
There are no data on the effect on fertility.
Ability to affect reaction speed when driving vehicles or operating other machinery.
FIBRIGA does not affect the ability to drive vehicles or operate machinery.
Method of Administration and Dosage
Treatment should be carried out under the supervision of a physician experienced in the management of bleeding disorders.
Doses
The dose and duration of replacement therapy depend on the severity of the deficiency, the location and extent of bleeding, and the patient's clinical condition.
The (functional) fibrinogen level should be determined to calculate the individual dose. The amount and frequency of administration must be individually adjusted for each patient based on continuous monitoring of plasma fibrinogen levels, the patient’s clinical status, and any concomitant replacement therapies used.
In the case of major surgical procedures, careful monitoring of replacement therapy using hemostasis blood tests (coagulation activity assays) is essential.
Prophylaxis in patients with congenital hypofibrinogenemia or afibrinogenemia and known bleeding tendency
To prevent excessive bleeding during surgical procedures, prophylactic treatment is recommended to increase fibrinogen levels to 1 g/L and maintain levels at this concentration until hemostasis is achieved, and above 0.5 g/L until complete wound healing.
For surgical procedures or treatment of bleeding, the dose should be calculated as follows:
Dose (mg/kg body weight) = [target level (g/L) – measured level (g/L)]
0.018 (g/L per mg/kg body weight)
Subsequent dosing (dose amounts and frequency of injections) should be adjusted according to the patient’s clinical condition and laboratory test results.
The biological half-life of fibrinogen is 3–4 days. Therefore, in the absence of fibrinogen consumption during surgery, repeat treatment with human fibrinogen is usually not required. In cases of accumulation due to repeated administration for prophylactic purposes, the dose and frequency should be determined according to the physician’s therapeutic goals for the individual patient.
Dosing in Specific Patient Populations
Pediatric Patients
Current data are described in the sections “Adverse Reactions” and “Pharmacodynamics,” but dosage recommendations for children are not currently available.
Elderly Patients
Clinical studies with FIBRIGA have not included patients aged 65 years and older to provide sufficient evidence regarding their response to treatment compared to younger patients.
Treatment of Bleeding
Bleeding in patients with congenital hypofibrinogenemia or afibrinogenemia
Bleeding should be treated to achieve the recommended target plasma fibrinogen level of 1 g/L. This level should be maintained until hemostasis is achieved.
Bleeding in patients with acquired fibrinogen deficiency
Adults
Typically, an initial dose of 1–2 g of the drug is administered, followed by additional infusions as needed.
In cases of severe bleeding, such as during major surgical procedures, higher doses of fibrinogen (4–8 g) may be required.
Method of Administration
Intravenous infusion or injection.
FIBRIGA should be administered slowly by intravenous infusion at a recommended maximum rate of 5 mL per minute in patients with congenital hypofibrinogenemia or afibrinogenemia, and at a recommended maximum rate of 10 mL per minute in patients with acquired fibrinogen deficiency.
For information on dilution of the medicinal product prior to administration, refer to the section “Special Instructions.”
Children
The dose should be determined according to body weight and clinical need. It is typically 20–30 mg/kg.
Overdose
To prevent overdose, continuous monitoring of plasma fibrinogen levels during treatment is recommended.
In the event of overdose, the risk of thromboembolic complications increases.
Adverse reactions.
Summary of safety profile
There are no reliable data on the frequency of adverse reactions obtained from clinical trials of this medicinal product.
The following adverse reactions have been reported in clinical trials: moderate fever observed in one patient, and drug-induced dermatitis manifested as mild skin reaction – itching and redness – after administration of the product, observed also in one patient.
Adverse reactions reported with the use of FIBRIGA and other fibrinogen concentrates are listed in Table 4.
Table 4
| Organ system classes |
Adverse reaction |
Frequency |
| Immune system disorders: |
Allergic or anaphylactic reactions Skin reactions |
Unknown |
| Vascular disorders |
Thromboembolic complications (including myocardial infarction and pulmonary embolism) Thrombophlebitis |
Unknown |
| General disorders and administration site reactions |
Increased body temperature (pyrexia) |
Unknown |
Information on the risk of transmission of infectious agents can be found in section "Special precautions for use".
Paediatric population
Eight patients aged 12 to 18 years with congenital fibrinogen deficiency were included in the safety study.
The overall safety profile does not differ between adults and adolescents.
There are no data on the use of FIBRIGA in paediatric patients with acquired fibrinogen deficiency.
Suspected adverse reactions
Reporting suspected adverse reactions after authorization of the medicinal product is important. It allows continuous monitoring of the benefit-risk balance of the medicinal product. Healthcare professionals are asked to report any suspected adverse reactions.
Shelf life.
2 years.
Chemical and physical in-use stability has been demonstrated for 24 hours at room temperature (not above 25 °C). From a microbiological point of view, the product should be used immediately after reconstitution. If not used immediately, the storage time and conditions after opening are the responsibility of the user. The reconstituted solution must not be frozen or stored in a refrigerator. Partially used vials must be discarded.
Storage conditions.
Store below 25 °C. Do not freeze.
Keep the vial in the outer carton to protect from light.
Keep out of the reach of children.
For storage conditions after reconstitution of the medicinal product, see section “Shelf life”.
Incompatibilities.
This medicinal product must not be mixed with other medicinal products.
Packaging.
1 g of powder for solution for injection/infusion in a glass vial stoppered with a rubber stopper and sealed with an aluminium crimp cap; 50 mL of solvent (water for injections) in a glass vial stoppered with a rubber stopper and sealed with an aluminium crimp cap. One vial of powder, one vial of solvent, one Octajet transfer device, and one filter in a cardboard box.
Prescription status.
Prescription only.
Manufacturer.
- Octapharma AB.
- Octapharma Pharmazeutika Produktionsges.m.b.H.
Manufacturer's address and place of business.
- Lars Forssells gata 23, Stockholm, 11275, Sweden.
- Oberlaaer Strasse 235, 1100 Vienna, Austria.