Enzertu
UkrainaSpis treści
INSTRUKCJA DOTYCZĄCA STOSOWANIA LEKU ENZERTU (ENHERTU®)
Skład:
substancja czynna: trastuzumab deruxtecan (trastuzumab deruxtecan);
1 fiolka zawiera 107 mg liofilizowanego, sterylnego proszku do sporządzenia stężonego roztworu do wlewania dożylnego, co odpowiada nominalnej ilości 100 mg w 5 ml roztworu po rekonstytucji; po rekonstytucji 1 fiolka o pojemności 5 ml zawiera 20 mg/ml trastuzumabu deruxtecanu (patrz sekcja „Sposób stosowania i dawki”);
substancje pomocnicze: sacharoza, L-histydyna, L-histydyny chlorowodorek monohydrat, polisorbat 80.
Postać leku
Proszek do sporządzenia stężonego roztworu do wlewania dożylnego.
Główne właściwości fizykochemiczne: liofilizat od białego do żółtobiałego koloru.
Grupa farmakoterapeutyczna
Leki przeciwnowotworowe. Przeciwciała monoklonalne i koniugaty przeciwciał z lekiem. Inhibitory HER2 (człowieka receptor 2 czynnika wzrostu nabłonka). Trastuzumab deruxtecan. Kod ATC L01F D04.
Właściwości farmakodynamiczne
Mechanizm działania
Lek Enzertu jest koniugatem przeciwciała przeciwko receptorowi HER2 z lekiem. Przeciwciało stanowi ludzką przeciwciało IgG1 przeciwko receptorowi HER2, połączone z deruksytekanem, inhibitorem topoizomerazy I (DXd), za pomocą rozkładalnego łącznika opartego na tetrapeptydzie. W osoczu krwi koniugat pozostaje stabilny. Część przeciwciała ma za zadanie wiązanie się z HER2, który jest ekspresowany na powierzchni niektórych komórek nowotworowych. Po połączeniu się z receptorami na komórkach nowotworowych trastuzumab deruksytekan ulega internaryzacji oraz wewnątrzkomórkowemu rozszczepieniu łącznika przez enzymy lizosomalne, które są aktywowane w komórkach nowotworowych. Po uwolnieniu przenikający przez błonę DXd powoduje uszkodzenie kwasu deoksyrybonukleinowego (DNA) oraz apoptozę komórek nowotworowych. DXd, pochodna eksatekanu, jest około 10 razy skuteczniejszy niż SN-38, aktywny metabolit irynotekanu.
Badania in vitro wykazują, że część przeciwciała trastuzumabu deruksytekanu, która ma tę samą sekwencję aminokwasową co trastuzumab, wiąże się również z FcγRIIIa oraz dopełniaczem C1q. Przeciwciało pośredniczy cytotoksyczność komórkową zależną od przeciwciał (ADCC) w komórkach raka piersi u ludzi, które nadmiernie ekspresują HER2. Ponadto przeciwciało hamuje przekazywanie sygnałów przez szlak fosfatydyloinozytol-3-kinazy (PI3-K) w komórkach raka piersi u ludzi, które nadmiernie ekspresują HER2.
Skuteczność kliniczna
Pozatywny HER2 rak piersi
Badanie DESTINY-Breast03 (NCT03529110)
Skuteczność i bezpieczeństwo stosowania leku Enzertu oceniano w badaniu DESTINY-Breast03, wieloośrodkowym, randomizowanym, otwartym badaniu fazy III z aktywnym kontrolowaniem, prowadzonym w dwóch grupach. W badaniu uczestniczyli pacjenci z pozatywnym HER2 nieroztężalnym lub przerzutowym rakiem piersi, którzy wcześniej otrzymywali terapię trastuzumabem i taksanami w leczeniu choroby przerzutowej lub u których doszło do nawrotu choroby podczas terapii adiuwantnej lub w ciągu 6 miesięcy po jej zakończeniu.
Kryterium włączenia była obecność archiwalnych próbek tkanki nowotworowej piersi w celu potwierdzenia statusu HER2-pozatywnego, określonego na podstawie wyników immunohistochemicznego badania jako 3+ (HER2 immunohistochemical (IHC) 3+), lub pozytywny status na podstawie badania hybrydyzacji in situ (in situ hybridization (ISH)-positive). Do badania nie włączano pacjentów z wywiadem leczenia glikokortykosteroidami z powodu choroby interpłucnej/ zapalenia płuc (ILD/pneumonia) lub z obecnością ILD/pneumonia w trakcie skriningu, pacjentów z nieleczonymi przerzutami do mózgu lub z przerzutami do mózgu z objawami klinicznymi oraz pacjentów z wywiadem klinicznie istotnej choroby serca, którzy wcześniej otrzymali leczenie koniugatem przeciwciało-lek przeciwko HER2 z powodu choroby przerzutowej. Pacjenci byli randomizowani w stosunku 1:1 do otrzymywania leku Enzertu w dawce 5,4 mg/kg (N = 261) lub emtanzynu trastuzumabu w dawce 3,6 mg/kg (N = 263), podawanego dożylnie w formie infuzji raz na trzy tygodnie. Randomizacja była prowadzona z uwzględnieniem stratyfikacji według statusu receptorów hormonalnych, wcześniejszego leczenia pertuzumabem oraz obecności przerzutów do narządów wewnętrznych. Leczenie kontynuowano do postępu choroby, śmierci, odwołania zgody lub rozwoju nieakceptowalnej toksyczności.
Pierwotnym kryterium skuteczności była przeżycie wolne od postępu (PFS), oceniane przez niezależną centralną ocenę w sposób ślepy (Blinded Independent Central Review (BICR)) zgodnie z kryteriami oceny odpowiedzi w nowotworach stałych (Response Evaluation Criteria In Solid Tumors (RECIST), wersja 1.1). Kluczowym wtórnym punktem końcowym skuteczności było przeżycie ogólne (OS). Punkty końcowe wtórne obejmowały PFS według oceny badacza, potwierdzoną częstotliwość odpowiedzi obiektywnej (ORR) oraz czas trwania odpowiedzi (DoR).
Dane demograficzne i wyjściowe cechy pacjentów były dobrze zrównoważone między grupami leczenia. Wyjściowe dane demograficzne i cechy choroby u 524 randomizowanych pacjentów były następujące: mediana wieku – 54 lata (zakres od 20 do 83 lat); wiek 65 lat i więcej (20,2%); płeć żeńska (99,6%); rasa mongoloidalna (59,9%); rasa europejska (27,3%); rasa negroidalna lub afroamerykańska (3,6%); status funkcjonalny 0 punktów (62,8%) lub 1 punkt (36,8%) według klasyfikacji Wschodniej Grupy Onkologicznej Kooperacyjnej (Eastern Cooperative Oncology Group (ECOG)); status receptorów hormonalnych (pozytywny – 51,9%); obecność przerzutów do narządów wewnętrznych (73,3%); obecność przerzutów do mózgu na poziomie wyjściowym (15,6%); 48,3% pacjentów otrzymało jedną linię wcześniejszej terapii systemowej z powodu choroby przerzutowej. Odsetek pacjentów, którzy wcześniej nie otrzymywali terapii z powodu choroby przerzutowej, wynosił 9,5%. Odsetek pacjentów, którzy wcześniej otrzymali terapię pertuzumabem, wynosił 61,1%.
W momencie przeprowadzenia wcześniej zaplanowanego analizy pośredniej PFS, obejmującej 245 zdarzeń (73% od całej zaplanowanej liczby zdarzeń do analizy ostatecznej), badanie wykazało statystycznie istotne poprawienie PFS według oceny BICR u pacjentów randomizowanych do grupy leku Enzertu w porównaniu z pacjentami z grupy otrzymującej emtanzyn trastuzumabu. Wyniki PFS według oceny BICR z analizy pierwotnej (data zakończenia zbierania danych – 21 maja 2021 r.) oraz zaktualizowane wyniki OS, ORR i DoR po dacie zakończenia zbierania danych 25 lipca 2022 r. przedstawiono w tabeli 1.
Tabela 1: Wyniki oceny skuteczności w badaniu DESTINY-Breast03
| Wskaźnik skuteczności |
Enzertu N = 261 |
trastuzumab emtansyne N = 263 |
| Przeżycie wolne od progresji (PFS) według oceny BICRa |
||
| Liczba zdarzeń (%) |
87 (33,3) |
158 (60,1) |
| Mediana, miesiące (95 % CI) |
ND (18,5; NPO) |
6,8 (5,6; 8,2) |
| Stosunek ryzyka (95 % CI) |
0,28 (0,22; 0,37) |
|
| Wartość p |
p < 0,000001† |
|
| Całkowite przeżycie (OS)b |
||
| Liczba zdarzeń (%) |
72 (27,6) |
97 (36,9) |
| Mediana, miesiące (95 % CI) |
ND (40,5; NPO) |
ND (34,0; NPO) |
| Stosunek ryzyka (95 % CI) |
0,64 (0,47; 0,87) |
|
| Wartość pc |
p = 0,0037 |
|
| PFS według oceny BICR (zaktualizowane)b |
||
| Liczba zdarzeń (%) |
117 (44,8) |
171 (65,0) |
| Mediana, miesiące (95 % CI) |
28,8 (22,4; 37,9) |
6,8 (5,6; 8,2) |
| Stosunek ryzyka (95 % CI) |
0,33 (0,26; 0,43) |
|
| Weryfikowana częstotliwość odpowiedzi obiektywnej (ORR) według oceny BICRb |
||
| n (%) |
205 (78,5) |
92 (35,0) |
| 95 % CI |
(73,1; 83,4) |
(29,2; 41,1) |
| Pełna odpowiedź, n (%) |
55 (21,1) |
25 (9,5) |
| Częściowa odpowiedź, n (%) |
150 (57,5) |
67 (25,5) |
| Czas trwania odpowiedzi według oceny BICRb |
||
| Mediana, miesiące (95 % CI) |
36,6 (22,4; NPO) |
23,8 (12,6; 34,7) |
CI — przedział ufności; NPO — nie podlega ocenie; ND — nie osiągnięto.
† Przedstawione z dokładnością do 6 miejsc po przecinku.
a Data zakończenia zbierania danych 21 maja 2021 r.
b Data zakończenia zbierania danych 25 lipca 2022 r. dla wcześniej zaplanowanej pośredniej analizy ZW.
c wartość p oparta na uwarstwionym teście log-rank; wyniki były wyższe niż granica skuteczności 0,013.
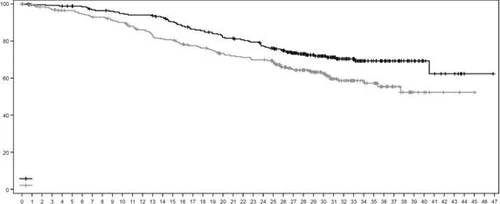
Rys. 1. Krzywa przeżycia całkowitego według Kaplana-Meiera (data zakończenia zbierania danych 25 lipca 2022 r.)
| Całkowite przeżycie, % |
|
||||||||||||||||||||||||||||||||||||||||||||||||||
| Czas, miesiące |
|||||||||||||||||||||||||||||||||||||||||||||||||||
| Liczba pacjentów w grupie ryzyka: |
|||||||||||||||||||||||||||||||||||||||||||||||||||
| Enzertu |
261 |
256 |
256 |
255 |
254 |
251 |
249 |
244 |
243 |
241 |
238 |
236 |
236 |
236 |
231 |
224 |
218 |
213 |
211 |
206 |
201 |
200 |
196 |
193 |
187 |
182 |
173 |
156 |
142 |
124 |
109 |
91 |
73 |
64 |
51 |
44 |
38 |
30 |
22 |
18 |
11 |
9 |
7 |
6 |
1 |
1 |
0 |
||||
| Trastruzumab emtanzin (263) |
263 |
257 |
252 |
248 |
243 |
242 |
237 |
233 |
232 |
227 |
224 |
217 |
211 |
203 |
199 |
197 |
191 |
186 |
183 |
179 |
172 |
169 |
167 |
164 |
164 |
158 |
140 |
129 |
117 |
106 |
90 |
70 |
59 |
45 |
41 |
38 |
27 |
20 |
15 |
8 |
7 |
4 |
3 |
3 |
1 |
0 |
|||||
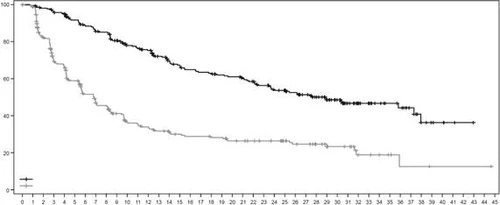
Rys. 2. Krzywa przeżycia bez progresji według Kaplana-Meiera według oceny BICR (data zakończenia zbierania danych: 25 lipca 2022 r.)
| Przeżycie bez postępu, % |
|
||||||||||||||||||||||||||||||||||||||||||||||||
| Czas, miesięcy |
|||||||||||||||||||||||||||||||||||||||||||||||||
| Liczba pacjentów w grupie ryzyka: |
|||||||||||||||||||||||||||||||||||||||||||||||||
| Enzertu |
261 |
256 |
250 |
244 |
240 |
225 |
216 |
207 |
205 |
191 |
176 |
173 |
167 |
154 |
146 |
140 |
134 |
131 |
130 |
125 |
123 |
117 |
113 |
107 |
99 |
96 |
90 |
82 |
73 |
64 |
55 |
41 |
32 |
28 |
23 |
20 |
18 |
13 |
7 |
5 |
4 |
2 |
1 |
0 |
|||||
| Trazystuzumab emtanzyń (263) |
263 |
253 |
201 |
164 |
156 |
134 |
111 |
99 |
96 |
81 |
69 |
67 |
63 |
58 |
54 |
51 |
49 |
49 |
47 |
47 |
42 |
41 |
39 |
37 |
36 |
32 |
28 |
27 |
15 |
14 |
8 |
7 |
6 |
4 |
2 |
2 |
2 |
1 |
1 |
1 |
1 |
1 |
1 |
0 |
|||||
Podobne wyniki PFS obserwowano w wcześniej określonych podgrupach, w tym podgrupach według wcześniejszej terapii pertuzumabem, statusu receptorów hormonalnych oraz obecności przerzutów do narządów wewnętrznych.
Badanie DESTINY-Breast02 (NCT03523585)
Skuteczność i bezpieczeństwo leku Enzertu oceniano w badaniu DESTINY-Breast02, wieloośrodkowym, randomizowanym, otwartym badaniu fazy III z aktywnym kontrolowaniem, w którym wzięto pod uwagę pacjentów z nierezekcyjnym lub przerzutowym rakiem piersi HER2-dodatnim, u których stwierdzono oporność lub refrakcyjność na wcześniejszą terapię T-DM1. Kryterium włączenia stanowiła dostępność archiwalnych próbek tkanki nowotworowej piersi w celu potwierdzenia statusu HER2-dodatniego, określonego metodą immunohistochemiczną jako 3+ (HER2 IHC 3+) lub pozytywny wynik testu metodą hybrydyzacji in situ (ISH-dodatni). Do badania nie włączano pacjentów z wywiadem leczenia glikokortykosteroidami z powodu ILD/ zapalenia płuc lub z ILD/ zapaleniem płuc w trakcie skriningu, pacjentów z nieleczonymi przerzutami do mózgu lub przerzutami do mózgu z objawami klinicznymi oraz pacjentów z wywiadem istotnej klinicznie choroby serca. Pacjentów randomizowano w stosunku 2:1 w celu otrzymania leku Enzertu w dawce 5,4 mg/kg (n = 406) w formie wlewu dożylnego raz na trzy tygodnie lub terapii wybranej przez lekarza (n = 202, trastuzumab plus kapacytabina lub lapatyneb plus kapacytabina). Randomizację przeprowadzono z uwzględnieniem warstw statusu receptorów hormonalnych, wcześniejszego leczenia pertuzumabem oraz obecności przerzutów do narządów wewnętrznych w wywiadzie. Leczenie kontynuowano do postępu choroby, wyniku śmiertelnego, odwołania zgody lub rozwoju nieakceptowalnej toksyczności.
Pierwotnym kryterium skuteczności była przeżycie wolne od progresji (PFS), oceniane przez niezależną centralną ocenę w sposób ślepy (BICR) zgodnie z kryteriami RECIST w wersji 1.1. Kluczowym wtórnym punktem końcowym skuteczności było przeżycie ogólne (OS). Punkty końcowe wtórne obejmowały PFS według oceny badacza, potwierdzona częstotliwość odpowiedzi obiektywnej (ORR) oraz czas trwania odpowiedzi (DoR).
Dane demograficzne pacjentów oraz wyjściowe cechy choroby były podobne we wszystkich grupach leczenia. Mediana wieku 608 pacjentów zakwalifikowanych do randomizacji wynosiła 54 lata (zakres od 22 do 88 lat); 99,2% uczestników badania stanowiły kobiety; rasa europejska – 63,2%, rasa mongolska – 29,3%, rasa negroidalna lub afroamerykańska – 2,8%; status funkcjonalny 0 punktów według skali ECOG – 57,4% pacjentów lub 1 punkt – 42,4% pacjentów; pozytytywny status receptorów hormonalnych – 58,6%; obecność przerzutów do narządów wewnętrznych – 78,3%; obecność przerzutów do mózgu na poziomie wyjściowym – 18,1%; jedna linia wcześniejszej terapii systemowej z powodu choroby przerzutowej – 4,9% pacjentów.
Wyniki oceny skuteczności podsumowano w tabeli 2 oraz na rysunkach 3 i 4.
Tabela 2: Wyniki oceny skuteczności w badaniu DESTINY-Breast02
| Wskaźnik skuteczności |
Enzertu N = 406 |
Terapia według wyboru lekarza N = 202 |
| WBP według oceny BICR |
||
| Liczba zdarzeń (%) |
200 (49,3) |
125 (61,9) |
| Mediana, miesiące (95 % CI) |
17,8 (14,3; 20,8) |
6,9 (5,5; 8,4) |
| Stosunek ryzyka (95 % CI) |
0,36 (0,28; 0,45) |
|
| Wartość p |
p < 0,000001† |
|
| Ogólna przeżycie (OP) |
||
| Liczba zdarzeń (%) |
143 (35,2) |
86 (42,6) |
| Mediana, miesiące (95 % CI) |
39,2 (32,7; NPD) |
26,5 (21,0; NPD) |
| Stosunek ryzyka (95 % CI) |
0,66 (0,50; 0,86) |
|
| Wartość pa |
p = 0,0021 |
|
| WBP według oceny badacza |
||
| Liczba zdarzeń (%) |
206 (50,7) |
152 (75,2) |
| Mediana, miesiące (95 % CI) |
16,7 (14,3; 19,6) |
5,5 (4,4; 7,0) |
| Stosunek ryzyka (95 % CI) |
0,28 (0,23; 0,35) |
|
| Potwierdzona częstotliwość odpowiedzi obiektywnej (CZO) według oceny BICR |
||
| n (%) |
283 (69,7) |
59 (29,2) |
| 95 % CI |
(65,0; 74,1) |
(23,0; 36,0) |
| Pełna odpowiedź, n (%) |
57 (14,0) |
10 (5,0) |
| Cząstkowa odpowiedź, n (%) |
226 (55,7) |
49 (24,3) |
| Trwałość odpowiedzi według oceny BICR |
||
| Mediana, miesiące (95 % CI) |
19,6 (15,9; NPD) |
8,3 (5,8; 9,5) |
CI — przedział ufności; NE — nie oceniane.
† Podano z dokładnością do 6 miejsc po przecinku.
a wartość p oparta na warstwowym teście log-rank; wyniki były wyższe niż granica skuteczności 0,004.
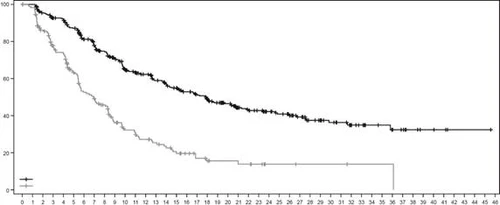
Rys. 3. Krzywa przeżycia bez postępu choroby według oceny BICR wg Kaplana-Meiera
| Przeżycie bez postępu, % |
|
||||||||||||||||||||||||||||||||||||||||||||||||
| Czas, miesięcy |
|||||||||||||||||||||||||||||||||||||||||||||||||
| Liczba pacjentów w grupie ryzyka: |
|||||||||||||||||||||||||||||||||||||||||||||||||
| Enzertu |
406 |
400 |
374 |
359 |
355 |
330 |
296 |
278 |
260 |
239 |
213 |
203 |
194 |
179 |
170 |
161 |
149 |
141 |
132 |
119 |
109 |
88 |
83 |
76 |
65 |
60 |
55 |
47 |
38 |
35 |
31 |
27 |
23 |
19 |
15 |
14 |
12 |
10 |
6 |
4 |
4 |
3 |
1 |
1 |
1 |
1 |
0 |
||
| Terapia według uznania lekarza (202) |
202 |
180 |
148 |
126 |
118 |
95 |
78 |
72 |
64 |
48 |
39 |
37 |
32 |
28 |
20 |
17 |
13 |
11 |
9 |
9 |
8 |
8 |
6 |
3 |
3 |
3 |
2 |
2 |
2 |
2 |
2 |
1 |
1 |
1 |
1 |
1 |
0 |
||||||||||||
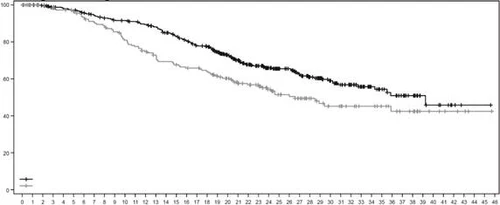
Rys. 4. Krzywa przeżycia całkowitego według Kaplana-Meiera
| Ogólna przeżywalność, % |
|
||||||||||||||||||||||||||||||||||||||||||||||||
| Czas, miesięcy |
|||||||||||||||||||||||||||||||||||||||||||||||||
| Liczba pacjentów w grupie ryzyka: |
|||||||||||||||||||||||||||||||||||||||||||||||||
| Enzertu |
406 |
404 |
400 |
390 |
385 |
382 |
374 |
366 |
357 |
352 |
350 |
346 |
339 |
331 |
317 |
306 |
295 |
282 |
277 |
257 |
234 |
215 |
196 |
83 |
160 |
144 |
139 |
122 |
104 |
93 |
82 |
72 |
63 |
51 |
40 |
34 |
29 |
25 |
19 |
10 |
8 |
6 |
3 |
1 |
1 |
1 |
0 |
||
| Terapia według wyboru lekarza (202) |
202 |
192 |
187 |
182 |
178 |
173 |
167 |
161 |
157 |
151 |
142 |
136 |
130 |
124 |
118 |
114 |
111 |
110 |
106 |
95 |
89 |
79 |
76 |
72 |
61 |
53 |
50 |
46 |
38 |
33 |
29 |
28 |
25 |
22 |
22 |
18 |
15 |
13 |
12 |
7 |
6 |
5 |
4 |
3 |
1 |
1 |
0 |
||
Badanie DESTINY-Breast01 (NCT03248492)
Skuteczność i bezpieczeństwo leku Enzertu oceniano w badaniu DESTINY-Breast01, wieloośrodkowym, otwartym badaniu fazy 2 z jedną grupą, w którym uczestniczyli pacjenci z HER2-dodatnim nieroztężalnym i/lub przerzutowym rakiem piersi, którzy otrzymali dwie lub więcej poprzednich linii terapii opartej na anty-HER2, w tym trastuzumabem emtanzinem (100 %), trastuzumabem (100 %) oraz pertuzumabem (65,8 %). Kryterium włączenia stanowiło posiadanie archiwalnych próbek tkanki nowotworowej piersi w celu potwierdzenia statusu HER2-dodatniego, określonego metodą immunohistochemiczną jako 3+ punkty (HER2 IHC 3+) lub pozytywny wynik badania metodą hybrydyzacji in situ (ISH-positive). Do badania nie włączano pacjentów z wywiadem choroby płuc typu interstycjalnego (ILD) lub z obecnością ILD w czasie skriningu, pacjentów z nieleczonymi przerzutami do mózgu lub przerzutami do mózgu z objawami klinicznymi oraz pacjentów z wywiadem istotnej klinicznie choroby serca. Pacjenci włączeni do badania mieli co najmniej jeden pomierzalny ognisko zgodnie z kryteriami RECIST, wersja 1.1. Lek Enzertu podawano dożylnie w dawce 5,4 mg/kg co trzy tygodnie aż do progresji choroby, zgonu, wycofania zgody lub wystąpienia nieakceptowalnej toksyczności. Pierwotnym kryterium skuteczności była potwierdzona częstość odpowiedzi obiektywnej (ORR) zgodnie z kryteriami RECIST, wersja 1.1, w populacji wszystkich pacjentów zrandomizowanych zgodnie z zamiarem leczenia (intent-to-treat, ITT), oceniana przez niezależne centrum oceny (independent central review, ICR). Wtórny punkt końcowy skuteczności stanowiła trwałość odpowiedzi (DoR).
Wyjściowe dane demograficzne i charakterystyka choroby 184 pacjentów włączonych do badania DESTINY-Breast01 były następujące: mediana wieku – 55 lat (zakres od 28 do 96 lat); wiek powyżej 65 lat (23,9 %); płeć żeńska (100 %); rasa kaukaska (54,9 %); rasa mongoloidalna (38,0 %); rasa czarna lub afroamerykańska (2,2 %); status funkcjonalny 0 punktów (55,4 %) lub 1 punkt (44,0 %) według klasyfikacji ECOG; pozytywny status receptorów hormonalnych (52,7 %); obecność przerzutów do narządów wewnętrznych (91,8 %); wcześniej leczone i stabilne przerzuty do mózgu (13,0 %); mediana liczby poprzednich linii terapii w przypadku przerzutów wynosiła 5 (zakres od 2 do 17); suma średnic wszystkich ognisk docelowych (< 5 cm – 42,4 %, ≥ 5 cm – 50,0 %).
Wcześniejsza analiza (mediana czasu obserwacji wyniosła 11,1 miesiąca [zakres od 0,7 do 19,9 miesiąca]) wykazała potwierdzoną częstość odpowiedzi obiektywnej na poziomie 60,9 % (95 % CI 53,4; 68,0), w tym 6,0 % pacjentów z pełną odpowiedzią i 54,9 % pacjentów z częściową odpowiedzią; 36,4 % miało chorobę stabilną, u 1,6 % doszło do progresji choroby, a dane 1,1 % nie poddano ocenie. Mediana trwałości odpowiedzi w momencie analizy wyniosła 14,8 miesiąca (95 % CI 13,8; 16,9), przy czym u 81,3 % pacjentów trwałość odpowiedzi wyniosła ≥ 6 miesięcy (95 % CI 71,9; 87,8). Wyniki oceny skuteczności w momencie aktualizacji daty zakończenia zbierania danych, przy medianie czasu obserwacji wynoszącej 20,5 miesiąca (zakres od 0,7 do 31,4 miesiąca), przedstawiono w tabeli 3.
Tabela 3: Wyniki oceny skuteczności w badaniu DESTINY-Breast01 (populacja do pełnej analizy)
| DESTINY-Breast01 N = 184 |
|
| Potwierdzona częstość obiektywnej odpowiedzi (95 % przedział ufności)*† |
61,4 % (54,0; 68,5) |
| Pełna odpowiedź (PR) |
6,5 % |
| Częściowa odpowiedź (CR) |
54,9 % |
| Trwanie odpowiedzi ‡ |
|
| Mediana, miesiące (95 % przedział ufności) |
20,8 (15,0; ND) |
| % pacjentów z trwaniem odpowiedzi ≥ 6 miesięcy (95 % przedział ufności)§ |
81,5 % (72,2; 88,0) |
95 % CI obliczone metodą Cloppera-Pearsona.
CI – przedział ufności.
95 % CI obliczone metodą Brookmeyer-Crowley.
* Potwierdzone odpowiedzi (na podstawie niezależnej oceny centralnej w sposób ślepy) definiowano jako ustaloną CR/PR potwierdzoną metodą wizualizacyjną nie wcześniej niż po 4 tygodniach od wizyty, podczas której zaobserwowano odpowiedź po raz pierwszy.
† Spośród 184 pacjentów 35,9 % miało stabilną chorobę, u 1,6 % zaobserwowano postęp choroby, a dane 1,1 % nie były ocenialne.
‡ Obejmuje 73 pacjentów z danymi cenzurowanymi.
§ Na podstawie oszacowania metodą Kaplana-Meiera.
ND – nie osiągnięto.
Stabilna aktywność przeciwnowotworowa obserwowana była w wcześniej zdefiniowanych podgrupach według wcześniejszej terapii pertuzumabem oraz statusu receptorów hormonalnych.
Rak piersi z niskim poziomem ekspresji HER2
Badanie DESTINY-Breast04 (NCT03734029)
Skuteczność i bezpieczeństwo leku Enzertu oceniano w badaniu DESTINY-Breast04, wieloośrodkowym randomizowanym otwartym badaniu fazy 3, w którym wzięło udział 557 dorosłych pacjentów z nieoperacyjnym lub przerzutowym rakiem piersi z niskim poziomem ekspresji HER2. Badanie obejmuje dwie kohorty: 494 pacjentów z pozytywnym statusem receptorów hormonalnych (HR+) oraz 63 pacjentów z negatywnym statusem receptorów hormonalnych (HR-). Niski poziom ekspresji HER2 oceniano w laboratorium centralnym jako IHC 1+ (określany jako słabe częściowe barwienie błony w ponad 10 % komórek nowotworowych) lub IHC 2+/ISH- na urządzeniu PATHWAY/VENTANA przy użyciu klonu 4B5 do określenia statusu HER2 (anti-HER2/neu (4B5)). Pacjenci mieli otrzymać chemioterapię z powodu choroby przerzutowej lub doznać nawrotu choroby podczas adiuwantowej chemioterapii lub w ciągu 6 miesięcy po jej zakończeniu. Zgodnie z kryteriami włączenia w chwili randomizacji pacjenci z HR+ musieli otrzymać co najmniej jeden schemat terapii endokrynnej lub mieć przeciwwskazania do dalszej terapii endokrynnej. Pacjentów randomizowano w stosunku 2:1 do grupy leczenia lekiem Enzertu w dawce 5,4 mg/kg (N = 373) drogą dożylną co trzy tygodnie lub chemioterapii wybranej przez lekarza (N = 184, eribulin – 51,1 %, kapacytabina – 20,1 %, gemcytabina – 10,3 %, nab-paklitaksel – 10,3 % lub paklitaksel – 8,2 %). Randomizację stratyfikowano według statusu HER2 IHC ustalonego na podstawie immunohistochemicznego badania próbek tkanki nowotworowej piersi (IHC 1+ lub IHC 2+/ISH-), liczby poprzednich linii chemioterapii w stadium przerzutowym (1 lub 2) oraz statusu HR/CDK4/6i (HR+ z wcześniejszą terapią inhibitorem CDK4/6, HR+ bez wcześniejszej terapii inhibitorem CDK4/6 lub HR-). Leczenie prowadzono do postępu choroby, zgonu, odwołania zgody lub rozwoju nieakceptowalnej toksyczności. Do badania nie włączano pacjentów z wywiadem leczenia glikokortykosteroidami z powodu ILD/ zapalenia płuc lub z obecnością ILD/ zapalenia płuc w trakcie skriningu, a także pacjentów z wywiadem istotnej klinicznie choroby serca. Do badania nie włączono również pacjentów z nieleczonymi przerzutami do mózgu lub przerzutami do mózgu z obecnością objawów klinicznych lub statusu funkcjonalnego wg skali ECOG >1 punktu.
Pierwotnym punktem końcowym skuteczności była przeżycie wolne od progresji (PFS) u pacjentów z HR+ rakiem piersi, oceniane przez BICR zgodnie z kryteriami RECIST wersja 1.1. Kluczowymi wtórnymi punktami końcowymi skuteczności były PFS oceniane przez BICR zgodnie z kryteriami RECIST wersja 1.1 w populacji ogólnej (wszyscy randomizowani pacjenci z HR+ i HR-), przeżycie całkowite (OS) u pacjentów z HR+ oraz OS w populacji ogólnej. Wtórnymi punktami końcowymi były ORR, DoR oraz wyniki zgłaszane przez pacjentów (patient-reported outcomes (PRO)).
Dane demograficzne i cechy guza na poziomie wyjściowym były porównywalne w grupach leczenia. Średni wiek 557 randomizowanych pacjentów wynosił 57 lat (zakres od 28 do 81 lat); 23,5 % miało co najmniej 65 lat; 99,6 % stanowiły kobiety, a 0,4 % mężczyźni; 47,9 % to przedstawiciele rasy europejskiej, 40,0 % – azjatyckiej oraz 1,8 % – rasy czarnej lub Afroamerykanie. Na poziomie wyjściowym pacjenci mieli status funkcjonalny wg skali ECOG 0 (54,8 %) lub 1 (45,2 %); 57,6 % miało IHC 1+; 42,4 % – IHC 2+/ISH-; 88,7 % – HR+ oraz 11,3 % – HR-; 69,8 % miało przerzuty do wątroby, 32,9 % – do płuc oraz 5,7 % – do mózgu. Odsetek pacjentów, którzy wcześniej otrzymywali antybiotyki w ramach leczenia (neo)adijuwantnego, wynosił 46,3 % oraz 19,4 % w przypadku choroby lokalnie zaawansowanej i/lub przerzutowej. W przypadku choroby przerzutowej pacjenci otrzymali średnio 3 poprzednie linie terapii systemowej (zakres od 1 do 9), przy czym 57,6 % otrzymało 1, a 40,9 % – 2 poprzednie schematy chemioterapii; u 3,9 % wystąpił wczesny postęp choroby (postęp podczas terapii neoadiuwantowej). U pacjentów z HR+ średnia liczba poprzednich linii terapii endokrynnej wynosiła 2 (zakres od 0 do 9) i 70 % wcześniej otrzymało leczenie inhibitorami CDK4/6.
Wyniki oceny skuteczności podsumowano w tabeli 4 oraz na rysunkach 5 i 6.
Tabela 4: Wyniki oceny skuteczności w badaniu DESTINY-Breast04
| Wskaźnik skuteczności |
Kohorta HR+ |
Populacja ogólna (kohorta HR+ i HR-) |
||
| Enzertu (N = 331) |
Chemioterapia (N = 163) |
Enzertu (N = 373) |
Chemioterapia (N = 184) |
|
| Przeżycie ogólne |
||||
| Liczba zdarzeń (%) |
126 (38,1) |
73 (44,8) |
149 (39,9) |
90 (48,9) |
| Mediana, miesiące (95 % CI) |
23,9 (20,8; 24,8) |
17,5 (15,2; 22,4) |
23,4 (20,0; 24,8) |
16,8 (14,5; 20,0) |
| Stosunek ryzyka (95 % CI) |
0,64 (0,48; 0,86) |
0,64 (0,49; 0,84) |
||
| Wartość p |
0,0028 |
0,001 |
||
| Przeżycie bez postępu choroby według oceny BICR |
||||
| Liczba zdarzeń (%) |
211 (63,7) |
110 (67,5) |
243 (65,1) |
127 (69,0) |
| Mediana, miesiące (95 % CI) |
10,1 (9,5; 11,5) |
5,4 (4,4; 7,1) |
9,9 (9,0; 11,3) |
5,1 (4,2; 6,8) |
| Stosunek ryzyka (95 % CI) |
0,51 (0,40; 0,64) |
0,50 (0,40; 0,63) |
||
| Wartość p |
< 0,0001 |
< 0,0001 |
||
| Potwierdzona częstość obiektywnej odpowiedzi według oceny BICR* |
||||
| n (%) |
175 (52,6) |
27 (16,3) |
195 (52,3) |
30 (16,3) |
| 95 % CI |
47,0; 58,0 |
11,0; 22,8 |
47,1; 57,4 |
11,3; 22,5 |
| Pełna odpowiedź, n (%) |
12 (3,6) |
1 (0,6) |
13 (3,5) |
2 (1,1) |
| Częściowa odpowiedź, n (%) |
164 (49,2) |
26 (15,7) |
183 (49,1) |
28 (15,2) |
| Czas trwania odpowiedzi według oceny BICR* |
||||
| Mediana, miesiące (95 % CI) |
10,7 (8,5; 13,7) |
6,8 (6,5; 9,9) |
10,7 (8,5; 13,2) |
6,8 (6,0; 9,9) |
CI — przedział ufności.
* Na podstawie danych z elektronicznych formularzy rejestracyjnych dla kohorty HR+: N = 333 w grupie leczenia lekiem Enzertu oraz N = 166 w grupie chemioterapii.
W podgrupach pacjentów wstępnie określonych na podstawie statusu HR, wcześniejszej terapii inhibitorami CDK4/6i, liczby wcześniejszych linii chemioterapii, statusu IHC1+ oraz IHC2+/ISH- zaobserwowano poprawę OS i PFS. W podgrupie pacjentów z HR- mediana OS wyniosła 18,2 miesiąca (95 % CI 13,6, nie można oszacować) w grupie leczenia lekiem Enzertu w porównaniu z 8,3 miesiąca (95 % CI 5,6; 20,6) w grupie chemioterapii, przy współczynniku ryzyka 0,48 (95 % CI 0,24; 0,95). Mediana PFS wyniosła 8,5 miesiąca (95 % CI 4,3; 11,7) u pacjentów randomizowanych do grupy leczenia lekiem Enzertu oraz 2,9 (95 % CI 1,4; 5,1) u pacjentów randomizowanych do grupy chemioterapii, przy współczynniku ryzyka 0,46 (95 % CI 0,24; 0,89).
W trakcie zaktualizowanego analizy opisowej z medianą czasu obserwacji 32 miesiące, poprawa wyników OS odpowiadała wynikom analizy pierwotnej. HR w populacji ogólnej wyniósł 0,69 (95 % CI 0,55; 0,86), mediana OS wyniosła 22,9 miesiąca (95 % CI 21,2; 24,5) w grupie leczenia lekiem Enzertu oraz 16,8 miesiąca (95 % CI 14,1; 19,5) w grupie chemioterapii. Krzywe Kaplana-Meiera dla OS na podstawie zaktualizowanej analizy przedstawiono na rysunku 5.
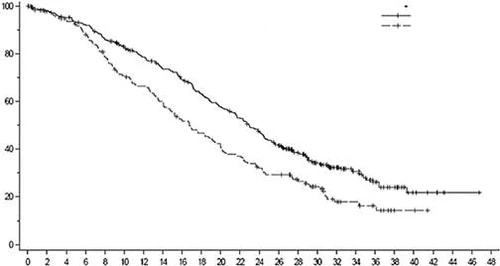
Rys. 5. Krzywa przeżycia całkowitego według Kaplana-Meiera (populacja ogólna) (zaktualizowana analiza)
| Ogólna przeżywalność, % |
|
|||||||||||||||||||||||||||||||||||||||||||||||||||
| Czas (miesięcy) |
||||||||||||||||||||||||||||||||||||||||||||||||||||
| Liczba pacjentów w grupie ryzyka |
||||||||||||||||||||||||||||||||||||||||||||||||||||
| Enzertu (373) |
373 |
366 |
363 |
366 |
350 |
342 |
337 |
325 |
314 |
306 |
256 |
235 |
276 |
269 |
257 |
254 |
240 |
231 |
217 |
206 |
199 |
191 |
182 |
168 |
160 |
148 |
137 |
122 |
107 |
94 |
81 |
75 |
62 |
52 |
48 |
39 |
26 |
21 |
18 |
11 |
7 |
6 |
5 |
3 |
1 |
1 |
1 |
0 |
||||
| Chemioterapia (184) |
184 |
170 |
164 |
160 |
156 |
152 |
145 |
137 |
127 |
119 |
113 |
107 |
106 |
100 |
96 |
88 |
81 |
76 |
73 |
69 |
64 |
59 |
58 |
53 |
49 |
45 |
45 |
44 |
37 |
33 |
27 |
18 |
15 |
12 |
12 |
10 |
8 |
5 |
2 |
2 |
2 |
1 |
0 |
|||||||||
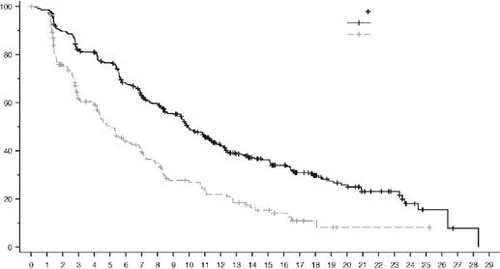
Rys. 6. Krzywa przeżycia bez postępu choroby według oceny BICR wg Kaplana-Meiera (populacja ogólna)
| Przeżycie bez progresji, % |
|
|||||||||||||||||||||||||||||||||
| Czas (miesięcy) |
||||||||||||||||||||||||||||||||||
| Liczba pacjentów w grupie ryzyka |
||||||||||||||||||||||||||||||||||
| Enzertu (373) |
373 |
365 |
325 |
295 |
290 |
272 |
238 |
217 |
201 |
183 |
156 |
142 |
118 |
100 |
88 |
81 |
71 |
53 |
42 |
35 |
32 |
21 |
18 |
15 |
8 |
4 |
4 |
1 |
1 |
0 |
||||
| Chemioterapia (184) |
184 |
166 |
119 |
93 |
90 |
73 |
60 |
51 |
45 |
34 |
32 |
29 |
26 |
22 |
15 |
13 |
9 |
5 |
4 |
3 |
1 |
1 |
1 |
1 |
1 |
1 |
0 |
|||||||
Choroba nowotworowa nieoskrzelowa raka płuc (NSCLC)
Badaanie DESTINY-Lung02 (NCT04644237)
Skuteczność i bezpieczeństwo stosowania leku Enzertu oceniano w badaniu DESTINY-Lung02 – randomizowanym badaniu fazy 2, w którym oceniano dwa poziomy dawek. Przydział dawki leczenia był prowadzony w sposób ślepy dla pacjentów i badaczy. Do badania zakwalifikowano dorosłych pacjentów z przerzutową chorobą NSCLC z mutacjami genu HER2, którzy otrzymali co najmniej jedną linię chemioterapii opartej na związkach platyny. Aktywującą mutację HER2 (ERBB2) wykrywano prospektywnie w tkance nowotworowej przez lokalne laboratoria za pomocą zwalidowanego testu, takiego jak sekwencjonowanie następnej generacji, reakcja łańcuchowa polimerazy lub spektrometria mas. Pacjenci zostali zrandomizowani w stosunku 2:1 do grupy otrzymującej lek Enzertu w dawce 5,4 mg/kg lub 6,4 mg/kg co 3 tygodnie. Randomizację przeprowadzono z uwzględnieniem warstwowania według wcześniejszej terapii przeciwciałem przeciwko białku 1 zaprogramowanej śmierci komórki (PD-1) i/lub przeciwciałem przeciwko ligandowi 1 zaprogramowanej śmierci komórki (PD-L1) (tak/nie). Leczenie kontynuowano do postępu choroby, zgonu, odwołania zgody lub rozwoju nieakceptowalnej toksyczności. Do badania nie włączano pacjentów z wywiadem leczenia glikokortykosteroidami z powodu ILD/ zapalenia płuc lub z obecnością ILD/ zapalenia płuc w trakcie skriningu, a także pacjentów z wywiadem istotnej klinicznie choroby serca. Do badania nie włączono również pacjentów z nieleczonymi przerzutami do mózgu oraz z przerzutami do mózgu z objawami klinicznymi lub ze stanem funkcjonalnym wg skali ECOG >1 punktu.
Pierwotnym kryterium skuteczności była potwierdzona ORR oceniona przez BICR zgodnie z kryteriami RECIST w wersji 1.1. Wtórny punkt końcowy oceny skuteczności to DOR.
Dane demograficzne pacjentów i wyjściowe cechy choroby u 102 uczestników badania, którzy otrzymywali dawkę 5,4 mg/kg, były następujące: średni wiek 59,4 roku (zakres od 31 do 84 lat); płeć żeńska (63,7%); rasa mongoloidalna (63,7%), rasa kaukaska (22,5%), inne (13,7%); status funkcjonalny wg skali ECOG 0 (28,4%) lub 1 (71,6%) punktu; 97,1% miało mutację w domenie kinazowej ERBB2, 2,9% – w domenie pozakomórkowej; 96,1% miało mutację HER2 w egzonie 19 lub egzonie 20; 34,3% miało stabilne przerzuty do mózgu; 46,1% wcześniej paliło, żaden z nich nie był aktualnie palaczem w trakcie badania; 21,6% wcześniej przeszło resekcję płuc. W przypadku choroby przerzutowej 32,4% pacjentów otrzymało ponad 2 poprzednie linie terapii systemowej, 100% otrzymało chemioterapię opartą na związkach platyny, 73,5% otrzymało terapię anty-PD-1/PD-L1 oraz 50,0% otrzymało chemioterapię opartą na związkach platyny i terapię anty-PD-1/PD-L1 w kombinacji.
Wyniki oceny skuteczności podsumowano w tabeli 5. Mediana czasu dalszej obserwacji wyniosła 11,5 miesiąca (data zakończenia zbierania danych – 23 grudnia 2022 r.).
Tabela 5: Wyniki oceny skuteczności w badaniu DESTINY-Lung02
| Wskaźnik skuteczności |
DESTINY-Lung02 5,4 mg/kg N = 102 |
| Udokumentowana częstość odpowiedzi obiektywnej (ORR) według oceny BICR |
|
| n (%) |
50 (49,0) |
| (95 % CI)* |
(39,0; 59,1) |
| Pełna odpowiedź (CR), n (%) |
1 (1,0) |
| Częściowa odpowiedź (PR), n (%) |
49 (48,0) |
| Trwałość odpowiedzi |
|
| Mediana, miesiące (95 % CI) † |
16,8 (6,4; NPD) |
* 95 % CI, obliczone metodą Cloppera – Pearsoна.
CI – przedział ufności, NPO – nie poddaje się ocenie.
† 95 % CI, obliczone metodą Brookmiera – Crowleya.
Rak żołądka
Badanie DESTINY-Gastric02 (NCT04014075)
Skuteczność i bezpieczeństwo leku Enzertu badano w badaniu DESTINY-Gastric02 – wieloośrodkowym, randomizowanym, otwartym, nieporównawczym badaniu fazy 2 prowadzonym w ośrodkach badawczych w Europie i Stanach Zjednoczonych. Do badania zakwalifikowano pacjentów z lokalnie zaawansowanym lub przerzutującym HER2-dodatnim rakiem gruczołowym żołądka lub przejścia żołądkowo-jelitowego (PŻJ), u których doszło do progresji choroby mimo wcześniejszej terapii opartej na trastuzumabie. Pacjenci musieli posiadać potwierdzony w centralnym laboratorium status HER2-dodatni, określony jako IHC 3+ lub IHC 2+/ISH-poz. Nie włączano do badania pacjentów z wywiadem leczenia glikokortykosteroidami z powodu ILD/ zapalenia płuc, pacjentów z obecnością ILD/ zapalenia płuc w trakcie skriningu, pacjentów z wywiadem klinicznie istotnej choroby serca oraz pacjentów z aktywnymi przerzutami do mózgu. Lek Enzertu podawano w formie dożylnej infuzji w dawce 6,4 mg/kg co trzy tygodnie aż do progresji choroby, śmiertelnego wyniku, odwołania zgody lub wystąpienia nieakceptowalnej toksyczności. Głównym kryterium skuteczności była potwierdzona ORR oceniona przez ICR zgodnie z kryteriami RECIST w wersji 1.1. Punkty końcowe wtórne obejmowały CDR i OS.
Dane demograficzne pacjentów oraz wyjściowe cechy choroby 79 uczestników badania DESTINY-Gastric02 były następujące: średni wiek – 61 lat (zakres od 20 do 78 lat); 72 % stanowili mężczyźni; 87 % należało do rasy europejskiej, 5,0 % do rasy mongolskiej oraz 1,0 % do rasy negroidalnej lub Afroamerykanów. Pacjenci mieli status ECOG wynoszący 0 (37 %) lub 1 (63 %); u 34 % stwierdzono rak gruczołowy żołądka, a u 66 % – rak gruczołowy PŻJ; 86 % miało IHC 3+, 13 % miało IHC 2+/ISH-poz., a u 63 % stwierdzono przerzuty do wątroby.
Wyniki oceny skuteczności pod względem ORR i CDR podsumowano w tabeli 6.
Tabela 6: Wyniki oceny skuteczności w badaniu DESTINY-Gastric02 (populacja do pełnej analizy*)
| Wskaźnik skuteczności |
DESTINY-Gastric02 N = 79 |
| Data zakończenia zbierania danych: 8 listopada 2021 r. |
|
| Potwierdzona częstość obiektywnej odpowiedzi† |
|
| % (95 % CI)‡ |
41,8 (30,8; 53,4) |
| Pełna odpowiedź, n (%) |
4 (5,1) |
| Cząstkowa odpowiedź, n (%) |
29 (36,7) |
| Trwanie odpowiedzi |
|
| Mediana§, miesiące (95 % CI)¶ |
8,1 (5,9; NPO) |
NPO — nie poddaje się ocenie.
* Obejmuje wszystkich pacjentów, którzy otrzymali co najmniej jedną dawkę leku Enzertu.
† Ocena niezależna, centralna, w ślepej próbie.
‡ Obliczone metodą Cloppera-Pearsona.
§ Na podstawie oszacowania metodą Kaplana-Meiera.
¶ Obliczone metodą Brookmeyer-Crowley.
Badanie DESTINY-Gastric01 (NCT03329690)
Skuteczność i bezpieczeństwo leku Enzertu oceniano w badaniu DESTINY-Gastric01 – wieloośrodkowym, randomizowanym, otwartym badaniu fazy 2 przeprowadzonym w centrach badawczych w Japonii i Korei Południowej. Do tego badania potwierdzającego zakwalifikowano dorosłych pacjentów z lokalnie zaawansowanym lub przerzutowym HER2-dodatnim rakiem żołądka lub rakiem gruczołowym złącza żołądkowo-jelitowego (GEJ), u których doszło do progresji choroby po co najmniej dwóch poprzednich schematach leczenia, w tym z zastosowaniem trastuzumabu, fluoropirydyny i leku platynowego. Pacjenci zostali losowo przydzieleni w stosunku 2:1 do grupy leczenia lekiem Enzertu (N = 126) lub chemioterapii wybranej przez lekarza: irynotekan (N = 55) lub paklitaksel (N = 7). Do badania wymagane było potwierdzone w laboratorium centralnym stanowisko HER2-dodatni, określone jako IHC 3+ lub IHC 2+/ISH-dodatni. Do badania nie zakwalifikowano pacjentów z wywiadem leczenia glikokortykosteroidami z powodu chorób autoimmunologicznych/ zapalenia płuc lub z chorobą autoimmunologiczną/ zapaleniem płuc w trakcie skriningu, pacjentów z wywiadem klinicznie istotnej choroby serca oraz pacjentów z aktywnymi przerzutami do mózgu. Leczenie prowadzono do progresji choroby, śmierci, odwołania zgody lub rozwoju nieakceptowalnej toksyczności. Głównym kryterium skuteczności była niepotwierdzona ORR oceniona przez ICR zgodnie z kryteriami RECIST w wersji 1.1. Wtórne kryteria skuteczności obejmowały całkowitą przeżycie (OS), przeżycie bez progresji (PFS), czas trwania odpowiedzi (DoR) oraz potwierdzoną ORR.
Dane demograficzne pacjentów i pierwotne charakterystyki choroby były podobne we wszystkich grupach leczenia. Mediana wieku 188 pacjentów wynosiła 66 lat (zakres od 28 do 82 lat); 76 % uczestników stanowili mężczyźni; 100 % – rasa mongoloidzka. Pacjenci mieli status wydolności według skali ECOG 0 (49 %) lub 1 (51 %); u 87 % stwierdzono adenokarcynomę żołądka, u 13 % – adenokarcynomę GEJ; 76 % miało IHC 3+, a 23 % – IHC 2+/ISH-dodatni status; u 54 % stwierdzono przerzuty do wątroby; u 29 % – przerzuty do płuc; suma średnic wszystkich ognisk docelowych była mniejsza niż 5 cm u 47 %, od ≥ 5 do < 10 cm u 30 % i ≥ 10 cm u 17 %; 55 % otrzymało dwie, a 45 % – trzy lub więcej poprzednich schematów leczenia w przypadku choroby lokalnie zaawansowanej lub przerzutowej.
Wyniki skuteczności (data zakończenia zbierania danych – 3 czerwca 2020 r.) leku Enzertu (n = 126) w porównaniu do chemioterapii wybranej przez lekarza (n = 62) wykazały potwierdzoną ORR na poziomie 39,7 % (95 % CI 31,1; 48,8) i 11,3 % (95 % CI 4,7; 21,9). Liczba pacjentów z pełną odpowiedzią wyniosła odpowiednio 7,9 % i 0 %, a z częściową odpowiedzią – 31,7 % i 11,3 %. Dodatkowe wyniki skuteczności leku Enzertu w porównaniu do chemioterapii wybranej przez lekarza obejmowały medianę DoR wynoszącą 12,5 miesiąca (95 % CI 5,6; NPO) w porównaniu do 3,9 miesiąca (95 % CI 3,0; 4,9). Mediana PFS wyniosła 5,6 miesiąca (95 % CI 4,3; 6,9) w porównaniu do 3,5 miesiąca (95 % CI 2,0; 4,3; HR = 0,47 [95 % CI 0,31; 0,71]). Analiza OS, pierwotnie zaplanowana przy 133 przypadkach śmierci, wykazała poprawę przeżycia w grupie leku Enzertu w porównaniu do grupy leczenia wybranego przez lekarza (HR = 0,60). Mediana OS wyniosła 12,5 miesiąca (95 % CI 10,3; 15,2) w grupie leku Enzertu i 8,9 miesiąca (95 % CI 6,4; 10,4) w grupie leczenia wybranego przez lekarza.
Populacja pediatryczna
Europejska Agencja Leków zgodziła się na nie przedstawienie wyników badań we wszystkich podgrupach dziecięcych z rakiem piersi, NSCLC i rakiem żołądka (informacje dotyczące stosowania leku Enzertu w populacji pediatrycznej zawarte są w sekcji „Dzieci”).
Ten lek został zarejestrowany w ramach tzw. schematu „zatwierdzenia warunkowego”. Oznacza to, że oczekuje się uzyskania dodatkowych dowodów dotyczących tego leku.
Europejska Agencja Leków będzie corocznie przeglądać wszelkie nowe informacje, które mogą się pojawić, a producent w razie potrzeby będzie aktualizował niniejszą instrukcję.
Farmakokinetyka
Wchłanianie
Trastuzumab deruxtecan podaje się dożylnie. Inne drogi podania leku nie były badane w badaniach klinicznych.
Rozkład
Na podstawie analizy populacyjnej farmakokinetyki objętość rozkładu w kompartmencie centralnym (Vc) trastuzumabu deruxtecanu i inhibitora topoisomerazy I (DXd) wyniosła odpowiednio 2,68 l i 28,0 l.
Średnie wiązanie DXd z białkami osocza krwi człowieka in vitro wyniosło około 97 %.
Stosunek stężeń DXd w krwi i osoczu in vitro wyniósł około 0,6.
Biopreparaty
Trastuzumab deruxtecan ulega wewnątrzkomórkowemu rozszczepieniu przez enzymy lizosomalne z uwolnieniem DXd.
Oczekuje się, że humanizowane monoklonalne przeciwciało IgG1 przeciwko receptorowi HER2 będzie rozszczepiane na małe peptydy i aminokwasy drogą kataboliczną, podobnie jak endogenne IgG.
Wyniki badań metabolizmu in vitro w mikrosomach wątroby człowieka wskazują, że DXd metabolizuje się głównie drogą utleniającą z udziałem CYP3A4.
Eliminacja
Na podstawie analizy populacyjnej farmakokinetyki po wstrzyknięciu dożylnym trastuzumabu deruxtecanu pacjentom z przerzutowym rakiem piersi HER2-dodatnim, rakiem piersi z niskim poziomem ekspresji HER2 lub NSCLC z mutacjami genu HER2, klirens trastuzumabu deruxtecanu wyniósł 0,4 l/dobę, a klirens DXd wyniósł 18,4 l/godz. U pacjentów z lokalnie zaawansowanym lub przerzutowym adenokarcynomą żołądka lub GEJ klirens trastuzumabu deruxtecanu był o 20 % wyższy niż u pacjentów z przerzutowym rakiem piersi HER2-dodatnim. W cyklu 3 okres półtrwania (t1/2) trastuzumabu deruxtecanu i uwolnionego DXd wyniósł około 7 dni. Obserwowano umiarkowane nagromadzenie trastuzumabu deruxtecanu (około 35 % w cyklu 3 w porównaniu do cyklu 1).
Po dożylnej podaniu DXd szczurom główną drogą eliminacji było wydzielanie z żółcią i wydalanie przez jelita. DXd był najbardziej rozpowszechnionym składnikiem wykrytym w moczu, kale i żółci. Po dożylnej podaniu małpom pojedynczej dawki trastuzumabu deruxtecanu (6,4 mg/kg) uwolniony niezmieniony DXd był najbardziej rozpowszechnionym składnikiem wykrytym w moczu i kale. Wydalanie DXd u człowieka nie było badane.
Interakcje in vitro
Wpływ leku Enzertu na farmakokinetykę innych leków
Badania in vitro wskazują, że DXd nie hamuje głównych enzymów CYP450, w tym CYP1A2, 2B6, 2C8, 2C9, 2C19, 2D6 i 3A. Badania in vitro wskazują, że DXd nie hamuje transporterów OAT1, OAT3, OCT1, OCT2, OATP1B1, OATP1B3, MATE1, MATE2-K, P-gp, BCRP i BSEP.
Wpływ innych leków na farmakokinetykę Enzertu
W warunkach in vitro DXd był substratem P-gp, OATP1B1, OATP1B3, MATE2-K, MRP1 i BCRP.
Nie oczekuje się klinicznie istotnej interakcji z lekami, które są inhibitorami transporterów MATE2-K, MRP1, P-gp, OATP1B lub BCRP (patrz sekcja „Interakcje z innymi lekami i inne rodzaje interakcji”).
Liniowość/nieliniowość
Ekspozycja na trastuzumab deruxtecan i uwolniony DXd po dożylnej podawaniu wzrastała proporcjonalnie do dawki w zakresie dawek od 3,2 mg/kg do 8,0 mg/kg (około 0,6–1,5 razy wyższa niż dawka zalecana) przy niskiej i średniej międzyosobniczej zmienności. Na podstawie analizy populacyjnej farmakokinetyki międzyosobnicza zmienność wartości klirensu trastuzumabu deruxtecanu i DXd wyniosła odpowiednio 24 % i 28 %, a objętości rozkładu centralnego – odpowiednio 16 % i 55 %. Międzyosobnicza zmienność wartości AUC (pole pod krzywą stężenie-czas) trastuzumabu deruxtecanu i DXd wyniosła odpowiednio około 8 % i 14 %.
Osobliwe grupy pacjentów
Na podstawie wyników analizy populacyjnej farmakokinetyki wiek (20–96 lat), przynależność rasowa i etniczna, płeć oraz masa ciała pacjenta nie miały klinicznie istotnego wpływu na ekspozycję na trastuzumab deruxtecan lub uwolniony DXd.
Pacjenci starsi
Analiza populacyjna PK wykazała, że wiek pacjenta (zakres 20–96 lat) nie wpływa na PK trastuzumabu deruxtecanu.
Naruszenie funkcji nerek
Specjalnych badań z udziałem pacjentów z zaburzeniami funkcji nerek nie przeprowadzono. Wyniki analizy populacyjnej farmakokinetyki z udziałem pacjentów z łagodnym (klirens kreatyniny [eGFR] ≥ 60 i < 90 ml/min) lub umiarkowanym (eGFR ≥ 30 i < 60 ml/min; wartość eGFR obliczono wg wzoru Cockcrofta-Gaulta) zaburzeniem funkcji nerek wykazały, że farmakokinetyka uwolnionego DXd nie zmienia się u pacjentów z zaburzeniem funkcji nerek od łagodnego do umiarkowanego w porównaniu z farmakokinetyką u osób z normalną funkcją nerek (eGFR ≥ 90 ml/min).
Naruszenie funkcji wątroby
Specjalnych badań z udziałem pacjentów z zaburzeniami funkcji wątroby nie przeprowadzono. Na podstawie wyników analizy populacyjnej farmakokinetyki ustalono, że wpływ na zmiany farmakokinetyki trastuzumabu deruxtecanu u pacjentów z całkowitym bilirubiną ≤ 1,5 razy WSN, niezależnie od poziomu AST, nie jest klinicznie istotny. Dane dotyczące pacjentów z całkowitym bilirubiną > 1,5–3 razy WSN, niezależnie od poziomu AST, są niewystarczające, aby wyciągnąć wnioski, a dane dotyczące pacjentów z całkowitym bilirubiną > 3 razy WSN, niezależnie od poziomu AST, są niedostępne (patrz sekcje „Sposób stosowania i dawki” oraz „Szczególne ostrzeżenia i środki ostrożności”).
Dzieci
Badania farmakokinetyki trastuzumabu deruxtecanu u dzieci nie były prowadzone.
Dane kliniczne
Wskazania
Rak piersi
Rak piersi HER2- pozytywny
Monoterapia lekiem Enzertu jest wskazana w leczeniu dorosłych pacjentów z nieoperacyjnym lub przerzutowym rakiem piersi HER2-pozatywnym, którzy wcześniej otrzymali jedną lub więcej terapii opartych na lekach przeciwbiałkowych HER2.
Rak piersi z niskim poziomem ekspresji HER2
Monoterapia lekiem Enzertu jest wskazana w leczeniu dorosłych pacjentów z nieoperacyjnym lub przerzutowym rakiem piersi z niskim poziomem ekspresji HER2, którzy wcześniej otrzymali chemioterapię w przypadku przerzutów lub u których doszło do nawrotu w trakcie lub w ciągu 6 miesięcy po zakończeniu chemioterapii adiuwantowej (patrz sekcja „Sposób stosowania i dawki”).
Gruczolakorak niedrobnokomórkowy płuca (NSCLC)
Monoterapia lekiem Enzertu jest wskazana w leczeniu dorosłych pacjentów z zaawansowanym NSCLC, u których w guzach występuje aktywująca mutacja HER2 (ERBB2), którzy wymagają terapii systemowej po chemioterapii opartej na lekach platynowych z immunoterapią lub bez niej.
Rak żołądka
Monoterapia lekiem Enzertu jest wskazana w leczeniu dorosłych pacjentów z zaawansowanym HER2-pozatywnym gruczolakorakiem żołądka lub przejścia żołądkowo-jelitowego (GEJ), którzy wcześniej otrzymali leczenie oparte na trastuzumabie.
Przeciwwskazania
Podwyższona wrażliwość na substancję czynną lub na którąkolwiek z substancji pomocniczych wymienionych w sekcji „Skład”.
Interakcje z innymi lekami i inne rodzaje interakcji
Jednoczesne stosowanie z rytonawirem, inhibitorem OATP1B, CYP3A i P-gp, lub z itrakonazolem, silnym inhibitorem CYP3A i P-gp, nie prowadziło do klinicznie istotnego (około 10–20 %) wzrostu wpływu trastuzumabu deruxtecanu lub uwalnianego inhibitora topoizomerazy I DXd. Nie jest wymagana korekta dawki przy jednoczesnym stosowaniu trastuzumabu deruxtecanu z lekami, które nie są inhibitorami CYP3A, OATP1B ani transporterów P-gp (patrz sekcja „Właściwości farmakologiczne”).
Szczególne środki ostrożności
W celu wykluczenia błędnego stosowania należy sprawdzić etykiety na fiolkach i upewnić się, że przygotowano lek Enzertu (trastuzumab deruxtecan), a nie trastuzumab ani trastuzumab emtansyn.
Śledzenie
W celu ułatwienia śledzenia leków biologicznych, należy dokładnie odnotować nazwę i numer serii zastosowanego leku w dokumentacji medycznej pacjenta.
Choroba płucna międzywistowata/zapalenie płuc
Podczas stosowania leku Enzertu odnotowano przypadki choroby płucnej międzywistowatej (IPF) i/lub zapalenia płuc (patrz sekcja „Działania niepożądane”). Zanotowano przypadki zakończone śmiercią. Pacjentom należy zalecić natychmiastowe zgłaszanie kaszlu, duszności, gorączki i/lub jakichkolwiek nowych lub nasilających się objawów ze strony układu oddechowego. Pacjentów należy obserwować pod kątem wystąpienia objawów i oznak IPF/zapalenia płuc. W przypadku wystąpienia objawów IPF/zapalenia płuc należy natychmiast przeprowadzić badanie pacjenta. Pacjentom z podejrzeniem IPF/zapalenia płuc należy wykonać rentgen klatki piersiowej, w miarę możliwości tomografię komputerową (CT). Należy rozważyć możliwość skonsultowania się z pulmonologiem. W przypadku bezobjawowego przebiegu IPF/zapalenia płuc (stopień 1 ciężkości) należy rozważyć zastosowanie kortykosteroidów (np. prednizolon w dawce ≥ 0,5 mg/kg/dobę lub inny lek w dawce równoważnej). Podawanie leku Enzertu należy wstrzymać, aż do spadku nasilenia działania niepożądanego do stopnia 0, po czym leczenie można wznowić zgodnie z zaleceniami podanymi w tabeli 8 (patrz sekcja „Sposób stosowania i dawki”). W przypadku objawowego IPF/zapalenia płuc (stopień 2 ciężkości lub wyższy) należy natychmiast rozpocząć leczenie systemowe kortykosteroidami (np. prednizolon w dawce ≥ 1 mg/kg/dobę lub inny lek w dawce równoważnej) i kontynuować je przez co najmniej 14 dni, po czym stopniowo zmniejszać dawkę przez co najmniej 4 tygodnie. W przypadku potwierdzenia diagnozy objawowego IPF/zapalenia płuc (stopień 2 ciężkości lub wyższy) należy przerwać stosowanie leku Enzertu (patrz sekcja „Sposób stosowania i dawki”). Pacjenci z wywiadem IPF/zapalenia płuc lub pacjenci z umiarkowanym lub ciężkim zaburzeniem funkcji nerek mogą mieć zwiększony ryzyko rozwoju IPF/zapalenia płuc i powinni być poddani dokładnej opiece medycznej (patrz sekcja „Sposób stosowania i dawki”).
Neutropenia
W badaniach klinicznych stosowanie leku Enzertu wiązało się z wystąpieniem neutropenii, w tym neutropenii febrilnej z letalnym skutkiem. Przed rozpoczęciem stosowania leku Enzertu oraz przed podaniem każdej kolejnej dawki, a także w razie potrzeby klinicznej, należy kontrolować wyniki ogólnego badania krwi. W zależności od stopnia ciężkości neutropenii może być konieczne przerwanie terapii lub zmniejszenie dawki leku Enzertu (patrz sekcja „Sposób stosowania i dawki”).
Obniżenie frakcji wyrzutowej lewej komory
Podczas stosowania leków przeciwno-HER2 obserwowano obniżenie frakcji wyrzutowej lewej komory (LVEF). Standardowe badanie funkcji serca (echokardiografia lub badanie MUGA [radioizotopowa wентrykulografia]) należy przeprowadzić w celu oceny LVEF przed rozpoczęciem stosowania leku Enzertu oraz w regularnych odstępach czasu podczas leczenia, zgodnie z wskazaniami klinicznymi. Obniżenie LVEF należy korygować poprzez wstrzymanie leczenia. Stosowanie leku Enzertu należy całkowicie przerwać w przypadku potwierdzenia LVEF poniżej 40% lub bezwzględnego spadku wartości o ponad 20% w porównaniu z wartością wyjściową. Stosowanie leku Enzertu należy całkowicie przerwać pacjentom z przewlekłą niewydolnością serca (CHF) z objawami (patrz tabela 8 w sekcji „Sposób stosowania i dawki”).
Toxyczność dla zarodka i płodu
Stosowanie leku Enzertu w czasie ciąży może szkodzić płodowi. Zgodnie z doniesieniami z okresu pogwarancyjnego, stosowanie trastuzumabu, blokera receptora HER2, w czasie ciąży powodowało oligohydramnios, który prowadził do śmiertelnej hipoplazji płuc, wad szkieletu i śmierci noworodków. Na podstawie danych z badań na zwierzętach oraz istniejącej wiedzy na temat mechanizmu działania leku Enzertu przewiduje się, że inhibitor topoizomerazy I DXd, będący jego składnikiem, może również szkodzić zarodkowi i płodowi po podaniu leku kobiecie w ciąży (patrz sekcja „Stosowanie w czasie ciąży lub karmienia piersią”).
Przed rozpoczęciem stosowania leku Enzertu u kobiet w wieku rozrodczym należy wykluczyć ciążę. Pacjentki powinny zostać poinformowane o potencjalnych ryzykach dla płodu. Kobietom w wieku rozrodczym należy zalecić stosowanie skutecznej antykoncepcji podczas leczenia oraz przez co najmniej 7 miesięcy po podaniu ostatniej dawki leku Enzertu. Mężczyznom prowadzącym życie seksualne z kobietami w wieku rozrodczym należy zalecić stosowanie skutecznej antykoncepcji podczas leczenia oraz przez co najmniej 4 miesiące po podaniu ostatniej dawki leku Enzertu (patrz sekcja „Stosowanie w czasie ciąży lub karmienia piersią”).
Pacjenci z umiarkowanym lub ciężkim zaburzeniem funkcji wątroby
Dane dotyczące pacjentów z umiarkowanym zaburzeniem funkcji wątroby są ograniczone, a dane dotyczące pacjentów z ciężkim zaburzeniem funkcji wątroby są nieistniejące. Ponieważ metabolizm i wydalanie z żółcią są głównymi drogami eliminacji inhibitora topoizomerazy I (DXd), lek Enzertu należy stosować z ostrożnością u pacjentów z umiarkowanym lub ciężkim zaburzeniem funkcji wątroby (patrz sekcje „Sposób stosowania i dawki” oraz „Właściwości farmakologiczne”).
Stosowanie w czasie ciąży lub karmienia piersią
Kobiety w wieku rozrodczym/antykoncepcja u mężczyzn i kobiet
Przed rozpoczęciem stosowania leku Enzertu u kobiet w wieku rozrodczym należy wykluczyć ciążę.
Kobiety w wieku rozrodczym powinny stosować skuteczną antykoncepcję podczas terapii lekiem Enzertu oraz przez co najmniej 7 miesięcy po jej zakończeniu.
Mężczyznom prowadzącym życie seksualne z kobietami w wieku rozrodczym należy zalecić stosowanie skutecznej antykoncepcji podczas terapii lekiem Enzertu oraz przez co najmniej 4 miesiące po jej zakończeniu.
Ciąża
Brak danych dotyczących stosowania leku Enzertu u kobiet w ciąży. Jednak stosowanie trastuzumabu, blokera receptora HER2, w czasie ciąży może szkodzić płodowi. Zgodnie z doniesieniami z okresu pogwarancyjnego, stosowanie trastuzumabu w czasie ciąży powodowało oligohydramnios, który w niektórych przypadkach prowadził do śmiertelnej hipoplazji płuc, wad szkieletu i śmierci noworodków. Na podstawie danych z badań na zwierzętach oraz istniejącej wiedzy na temat mechanizmu działania leku Enzertu przewiduje się, że inhibitor topoizomerazy I DXd, będący jego składnikiem, może również szkodzić zarodkowi i płodowi po podaniu leku kobiecie w ciąży.
Leku Enzertu nie zaleca się stosować kobietom w ciąży. Pacjentki powinny zostać poinformowane o potencjalnych ryzykach dla płodu przed planowaniem ciąży. W przypadku zajścia w ciążę kobieta powinna natychmiast skontaktować się z lekarzem. Jeśli pacjentka zajdzie w ciążę podczas leczenia lub w ciągu 7 miesięcy po podaniu ostatniej dawki leku Enzertu, zaleca się dokładne obserwowanie.
Karmienie piersią
Nie wiadomo, czy trastuzumab deruxtecan przechodzi do mleka matki. Ludzki IgG wydzielany jest do mleka matki, a prawdopodobieństwo jego wchłonięcia i wystąpienia poważnych działań niepożądanych u noworodka jest nieznane. Dlatego kobiety nie powinny karmić piersią podczas leczenia lekiem Enzertu oraz przez 7 miesięcy po podaniu ostatniej dawki. Decyzję o przerwaniu karmienia piersią lub przerwaniu terapii lekiem Enzertu należy podjąć, biorąc pod uwagę korzyści z karmienia piersią dla dziecka i/lub korzyści z terapii lekiem Enzertu dla kobiety.
Plodność
Nie przeprowadzono specjalnych badań dotyczących wpływu trastuzumabu deruxtecanu na płodność. Ze względu na wyniki badań toksyczności na zwierzętach lek Enzertu może powodować zaburzenia funkcji rozrodczych i płodności. Nie wiadomo, czy trastuzumab deruxtecan lub jego metabolity wydzielają się w nasieniu. Przed rozpoczęciem leczenia mężczyźni powinni otrzymać zalecenie skonsultowania się w sprawie kriokonserwacji nasienia. Mężczyźni nie powinni oddawać nasienia na kriokonserwację ani być jego dawcami przez cały okres leczenia oraz przez co najmniej 4 miesiące po podaniu ostatniej dawki leku Enzertu.
Wpływ na zdolność prowadzenia pojazdów i obsługiwanie maszyn
Lek Enzertu może mieć nieznaczny wpływ na szybkość reakcji podczas prowadzenia pojazdów lub obsługiwanie innych maszyn. Pacjentom należy zalecić zachowanie ostrożności podczas prowadzenia pojazdów i pracy z innymi maszynami, jeśli odczuwają zwiększone zmęczenie, ból głowy lub zawroty głowy podczas terapii lekiem Enzertu (patrz sekcja „Działania niepożądane”).
Sposób stosowania i dawki
Lek Enzertu powinien być przepisywany przez lekarza i stosowany pod nadzorem personelu medycznego mającego doświadczenie w stosowaniu leków przeciwnowotworowych. Aby wykluczyć błąd w stosowaniu, należy dokładnie sprawdzić etykiety na fiolkach i upewnić się, że przygotowano lek Enzertu (trastuzumab deruxtecan), a nie trastuzumab ani trastuzumab emtansyn.
Nie należy zastępować leku Enzertu trastuzumabem ani trastuzumabem emtansynem.
Selekcja pacjentów
Rak piersi HER2-pozatywny
U pacjentów otrzymujących trastuzumab deruxtecan w leczeniu raka piersi, potwierdzony status HER2-pozatywny guza powinien być potwierdzony dokumentacyjnie za pomocą immunohistochemicznego (IHC) badania z wynikiem 3+ lub badania in situ hybrydyzacji (ISH) z współczynnikiem ≥ 2,0 lub fluorescencyjnego badania hybrydyzacji in situ (FISH), ocenionego przy użyciu środka diagnostycznego in vitro (IVD) oznaczonego znakiem CE. Jeżeli środek IVD oznaczony znakiem CE nie jest dostępny, status HER2 należy ocenić za pomocą alternatywnego, zwalidowanego testu.
Rak piersi z niskim poziomem ekspresji HER2
U pacjentów otrzymujących trastuzumab deruxtecan, potwierdzony status guza z niskim poziomem ekspresji HER2 powinien być potwierdzony dokumentacyjnie za pomocą wyników IHC jako 1+ lub IHC 2+/ISH-, ocenionego przy użyciu środka diagnostycznego in vitro (IVD). Jeżeli środek IVD oznaczony znakiem CE nie jest dostępny, status HER2 należy ocenić za pomocą alternatywnego, zwalidowanego testu (patrz sekcja „Farmakodynamika”).
NSCLC
U pacjentów otrzymujących trastuzumab deruxtecan w leczeniu zaawansowanego NSCLC, w guzach powinna występować aktywująca mutacja HER2 (ERBB2), wykryta przy użyciu środka diagnostycznego in vitro (IVD) oznaczonego znakiem CE. Jeżeli środek IVD oznaczony znakiem CE nie jest dostępny, status mutacji HER2 należy ocenić za pomocą alternatywnego, zwalidowanego testu.
Rak żołądka
U pacjentów otrzymujących trastuzumab deruxtecan w leczeniu raka żołądka lub przejścia żołądkowo-jelitowego, potwierdzony status HER2-pozatywny guza powinien być potwierdzony dokumentacyjnie za pomocą immunohistochemicznego (IHC) badania z wynikiem 3+ lub badania in situ hybrydyzacji (ISH) z współczynnikiem ≥ 2 lub fluorescencyjnego badania hybrydyzacji in situ (FISH), ocenionego przy użyciu środka diagnostycznego in vitro (IVD) oznaczonego znakiem CE. Jeżeli środek IVD oznaczony znakiem CE nie jest dostępny, status HER2 należy ocenić za pomocą alternatywnego, zwalidowanego testu.
Dozowanie
Rak piersi
Zalecana dawka leku Enzertu wynosi 5,4 mg/kg, podawana jako wlewy dożylny co 3 tygodnie (cykle leczenia trwające 21 dni) aż do postępu choroby lub wystąpienia nietolerowanej toksyczności.
NSCLC
Zalecana dawka leku Enzertu wynosi 5,4 mg/kg, podawana jako wlewy dożylny co 3 tygodnie (cykle leczenia trwające 21 dni) aż do postępu choroby lub wystąpienia nietolerowanej toksyczności.
Rak żołądka
Zalecana dawka leku Enzertu wynosi 6,4 mg/kg, podawana jako wlewy dożylny co 3 tygodnie (cykle leczenia trwające 21 dni) aż do postępu choroby lub wystąpienia nietolerowanej toksyczności.
Początkową dawkę należy podawać jako wlew dożylny trwający 90 minut. Jeżeli poprzedni wlew został dobrze tolerowany, czas trwania kolejnych wlewu leku Enzertu może wynosić 30 minut.
W przypadku wystąpienia objawów związanych z wlewem należy zmniejszyć szybkość wlewu lub przerwać podawanie leku Enzertu (patrz sekcja „Efekty uboczne”). W przypadku wystąpienia ciężkich reakcji związanych z wlewem należy całkowicie przerwać podawanie leku Enzertu.
Premedykacja
Lek Enzertu jest emetogenny (patrz sekcja „Efekty uboczne”), co może prowadzić do wystąpienia późnego mdłości i/lub wymiotów. Przed każdym podaniem leku Enzertu pacjentom należy przeprowadzić premedykację kombinacją dwóch lub trzech leków (np. dexametazonem w połączeniu z antagonistą receptora 5-HT3 i/lub antagonistą receptora NK1, oraz innymi niezbędnymi lekami) w celu zapobiegania wymiotom i mdłościom spowodowanym chemioterapią.
Modyfikacje dawki
Leczenie efektów ubocznych może wymagać tymczasowego przerwania terapii, zmniejszenia dawki lub całkowitego zakończenia terapii lekiem Enzertu zgodnie z zaleceniami przedstawionymi w tabelach 7 i 8.
W przypadku zmniejszenia dawki leku Enzertu nie należy ponownie zwiększać dawki.
Tabela 7: Schemat zmniejszania dawki
| Schemat zmniejszenia dawki |
Rak piersi i NSCLC |
Rak żołądka |
| Zalecana dawka początkowa |
5,4 mg/kg |
6,4 mg/kg |
| Pierwsze zmniejszenie dawki |
4,4 mg/kg |
5,4 mg/kg |
| Drugie zmniejszenie dawki |
3,2 mg/kg |
4,4 mg/kg |
| Potrzeba dalszego zmniejszenia dawki |
Przerwanie leczenia |
Przerwanie leczenia |
Tabela 8: Dostosowanie dawki w przypadku działań niepożądanych
| Reakcja niepożądana |
Stopień ciężkości |
Korekta schematu leczenia |
|
| Choroba płucna międzybłoniowa (ILD)/zapalenie płuc |
Bezobjawowa ILD/zapalenie płuc (stopień 1) |
Wstrzymać leczenie lekiem Enzertu, aż ciężkość reakcji niepożądanej zmniejszy się do stopnia 0, następnie:
|
|
| ILD/zapalenie płuc z objawami (stopień 2 lub wyższy) |
|
||
| Neutropenia |
Stopień 3 ciężkości (mniej niż 1,0–0,5 × 10⁹/l) |
|
|
| Stopień 4 ciężkości (mniej niż 0,5 × 10⁹/l) |
|
||
| Neutropenia febrilna |
Obniżenie bezwzględnej liczby neutrofilów poniżej 1,0 × 10⁹/l oraz podwyższenie temperatury ciała powyżej 38,3 °C lub trwająca temperatura 38 °C lub wyższa przez ponad jedną godzinę. |
|
|
| Obniżenie frakcji wyrzutowej lewej komory (LVEF) |
LVEF powyżej 45 % oraz bezwzględne zmniejszenie od wartości wyjściowej o 10–20 % |
|
|
| LVEF od 40 % do 45 % |
oraz bezwzględne zmniejszenie wskaźnika o mniej niż 10 % od wartości wyjściowej |
|
|
| oraz bezwzględne zmniejszenie wskaźnika o 10–20 % od wartości wyjściowej |
|
||
| LVEF poniżej 40 % lub bezwzględne zmniejszenie od wartości wyjściowej o więcej niż 20 % |
|
||
| Chroniczne niewydolność serca (CHF) z objawami |
|
||
Stopień ciężkości toksyczności wskazano zgodnie z Ogólnymi Kryteriami Terminologii dla Niepożądanych Zdarzeń, wersja 5.0 (NCI-CTCAE, wersja 5.0).
Opóźnienie lub pominięcie dawki leku
Jeśli podanie zaplanowanej dawki zostanie opóźnione lub pominięte, należy podać ją tak szybko, jak to możliwe, bez oczekiwania na następny zaplanowany cykl. Harmonogram kolejnych podań należy dostosować, aby zachować 3-tygodniowy odstęp między dawkami. Lek należy podawać w tej samej dawce i z taką samą prędkością, z jaką pacjent dobrze tolerował ostatnie wlewanie.
Grupy specjalne pacjentów
Pacjenci w podeszłym wieku
Pacjentom w wieku 65 lat i starszym nie wymaga się dostosowania dawki leku Enzertu. Dane dotyczące stosowania leku Enzertu u pacjentów w wieku ≥ 75 lat są ograniczone.
Zaburzenia funkcji nerek
U pacjentów z zaburzeniami funkcji nerek o lekkim (klirens kreatyniny [KK] ≥ 60 i < 90 ml/min) lub średnim (KK ≥ 30 i < 60 ml/min) stopniu ciężkości nie wymaga się dostosowania dawki (patrz sekcja „Właściwości farmakologiczne”). Nie można określić potencjalnej potrzeby dostosowania dawki u pacjentów z ciężkimi zaburzeniami funkcji nerek lub w terminalnym stadium choroby nerek, ponieważ ciężkie zaburzenia funkcji nerek były kryterium wykluczającym w badaniach klinicznych. U pacjentów z zaburzeniami funkcji nerek o średnim stopniu ciężkości częściej obserwowano wystąpienie ILD/ zapalenia płuc o stopniu ciężkości 1 lub 2. U pacjentów z zaburzeniami funkcji nerek o średnim stopniu ciężkości na poziomie wyjściowym, którzy otrzymywali lek Enzertu w dawce 6,4 mg/kg, częstość poważnych reakcji niepożądanych była wyższa niż u pacjentów z prawidłową funkcją nerek. Pacjenci z zaburzeniami funkcji nerek o średnim lub ciężkim stopniu ciężkości powinni być poddawani dokładnej obserwacji pod kątem rozwoju reakcji niepożądanych, w szczególności ILD/ zapalenia płuc (patrz sekcja „Szczególne ostrzeżenia i środki ostrożności postępowania”).
Zaburzenia funkcji wątroby
U pacjentów z całkowitym stężeniem bilirubiny ≤ 1,5 razy wyższym od górnej granicy normy (GGN), niezależnie od poziomu aminotransferazy asparaginianowej (AST), nie wymaga się dostosowania dawki. Potencjalna potrzeba dostosowania dawki u pacjentów z całkowitym stężeniem bilirubiny > 1,5 razy wyższym od GGN, niezależnie od poziomu AST, nie może być określona ze względu na ograniczone dane; dlatego pacjenci ci powinni być poddawani dokładnej obserwacji (patrz sekcje „Szczególne ostrzeżenia i środki ostrożności postępowania” oraz „Właściwości farmakologiczne”).
Sposób podania
Lek Enzertu przeznaczony jest do wstrzykiwania dożylnego. Przygotowanie koncentratu, roztworu do wlewania oraz podanie tego leku w formie wlewu dożylnego powinno być wykonywane przez personel medyczny. Leku Enzertu nie wolno podawać dożylnie w formie strumienia lub bolusowo.
Instrukcje dotyczące przygotowania koncentratu roztworu do wlewania oraz rozcieńczenia leku przed podaniem
Aby wykluczyć błąd w stosowaniu, ważne jest sprawdzenie etykiet na fiolkach i upewnienie się, że przygotowano lek Enzertu (trastuzumab deruxtecan), a nie trastuzumab ani trastuzumab emtansyn.
Należy przestrzegać odpowiednich procedur przygotowywania leków chemioterapeutycznych. Należy również przestrzegać niezbędnych warunków aseptycznych podczas wykonywania poniższych procedur rekonstytucji i rozcieńczania roztworu.
Przygotowanie koncentratu roztworu do wlewania
- Przygotowanie koncentratu należy wykonywać bezpośrednio przed rozcieńczeniem.
- Aby uzyskać pełną dawkę, może być konieczne użycie więcej niż jednej fiolki ze sterylnym proszkiem liofilizowanym. Należy obliczyć dawkę (w miligramach), całkowitą objętość wymaganego koncentratu leku Enzertu oraz niezbędną liczbę fiolki leku Enzertu.
- Aby odtworzyć zawartość fiolki w celu uzyskania dawki 100 mg trastuzumabu deruxtecanu, do fiolki za pomocą sterylnego strzykawki powoli wprowadza się 5 ml wody do wstrzykiwań, aby uzyskać końcową stężenie 20 mg/ml.
- Ostrożnie wymieszać zawartość fiolki aż do całkowitego odtworzenia. Nie wstrząsać.
- Jeśli przygotowany koncentrat leku Enzertu nie zostanie natychmiast użyty, należy go przechowywać w lodówce w temperaturze od 2 do 8 °C przez maksymalnie 24 godziny od momentu przygotowania koncentratu, w miejscu chronionym przed działaniem światła. Nie zamrażać.
- Odtworzony lek nie zawiera środków konserwujących i przeznaczony jest wyłącznie do jednorazowego użycia.
Rozcieńczenie
- Obliczoną objętość koncentratu pobiera się z fiolki(i) za pomocą sterylnego strzykawki. Sprawdza się koncentrat pod kątem obecności cząstek stałych i zmiany koloru. Roztwór powinien być przezroczysty i bezbarwny lub jasnożółty. Nie należy używać roztworu, jeśli występują w nim obce cząstki lub jeśli roztwór jest mętny lub zmienił kolor.
- Obliczoną objętość koncentratu leku Enzertu rozpuszcza się w worku do wlewania zawierającym 100 ml 5% roztworu glukozy. Nie należy używać roztworu chlorku sodu (patrz sekcja „Niezgodność”). Zaleca się stosowanie worka do wlewania z poli(chlorku winylu) lub poliolefiny (kopolimer etylenu i polipropylenu).
- Ostrożnie odwraca się worek do wlewania, aby dokładnie wymieszać roztwór. Nie wstrząsać.
- Należy zakryć worek do wlewania w celu ochrony przed działaniem światła.
- Jeśli przygotowany roztwór nie zostanie natychmiast użyty, należy go przechowywać w temperaturze pokojowej przez maksymalnie 4 godziny, włącznie z czasem przygotowania koncentratu i czasem wlewania, lub w lodówce w temperaturze od 2 do 8 °C przez maksymalnie 24 godziny w miejscu chronionym przed działaniem światła. Nie zamrażać.
- Nieużywaną część pozostałą w fiolce należy zutylizować.
Podanie
- Jeśli przygotowany roztwór do wlewania był przechowywany w lodówce (w temperaturze od 2 do 8 °C), przed podaniem zaleca się doprowadzenie roztworu do temperatury pokojowej w miejscu chronionym przed działaniem światła.
- Lek Enzertu należy podawać w formie wlewu dożylnego wyłącznie za pomocą wbudowanego filtra (0,20 lub 0,22 μm) z polietersulfonu (PES) lub polisulfonu (PS).
- Pierwszą dawkę należy podawać w formie wlewu dożylnego trwającego 90 minut. Jeśli poprzednie wlewanie zostało dobrze tolerowane, czas kolejnych wlewów leku Enzertu może wynosić 30 minut. Leku nie można podawać dożylnie w formie strumienia lub bolusowo.
- Należy zakryć worek do wlewania w celu ochrony przed działaniem światła.
- Nie wolno mieszać Enzertu z innymi lekami, a także nie należy podawać innych leków za pomocą tego samego systemu do wlewania dożylnego.
Utylizacja
Każdy nieużywany lek i odpady należy utylizować zgodnie z lokalnymi wymaganiami.
Dzieci
Bezpieczeństwo i skuteczność stosowania leku Enzertu u dzieci (do 18 roku życia) nie zostały ustalone. Brak danych.
Przedawkowanie
Maksymalna dawka tolerowana trastuzumabu deruxtecanu nie została określona. Dawek pojedynczych przekraczających 8,0 mg/kg nie badano w badaniach klinicznych. W przypadku przedawkowania pacjenci powinni być poddawani ścisłej obserwacji medycznej w celu wykrycia możliwych objawów lub symptomów reakcji niepożądanych i, w razie potrzeby, otrzymywać leczenie objawowe.
Niepożądane reakcje
▼ Ten lek jest objęty dodatkowym nadzorem. Umożliwia to szybkie gromadzenie nowych informacji dotyczących bezpieczeństwa. Osoby pracujące w ochronie zdrowia proszone są o zgłaszanie wszelkich podejrzewanych niepożądanych reakcji.
Podsumowanie profilu bezpieczeństwa
Enzertu 5,4 mg/kg
Bezpieczeństwo leku oceniano w połączonej grupie pacjentów, którzy otrzymali co najmniej jedną dawkę leku Enzertu w dawce 5,4 mg/kg (n = 1449) w badaniach klinicznych różnych typów nowotworów. Mediana czasu trwania leczenia w tej połączonej grupie wyniosła 9,8 miesiąca (zakres: 0,7–45,1 miesiąca).
Najczęstsze niepożądane reakcje to nudności (75,0%), zmęczenie (57,3%), wymioty (42,1%), wypadanie włosów (37,6%), neutropenia (35,2%), zaparcia (35,0%), anemia (34,4%), osłabienie apetytu (33,1%), biegunka (28,8%), podwyższenie poziomu transaminaz (26,5%), ból mięśniowo-szkieletny (26,2%), trombocytopenia (24,5%) oraz leukopenia (23,7%).
Najczęstsze niepożądane reakcje o nasileniu 3 lub 4 stopnia według Ogólnych Kryteriów Terminologii Oceny Niepożądanych Zdarzeń Klinicznych Narodowego Instytutu Onkologii USA (NCI-CTCAE wersja 5.0) to: neutropenia (17,0%), anemia (9,5%), zmęczenie (8,4%), leukopenia (6,4%), nudności (5,9%), trombocytopenia (5,0%), limfopenia (4,8%), hipokaliemia (3,8%), podwyższenie poziomu transaminaz (3,6%), wymioty (2,7%), biegunka (2,0%), osłabienie apetytu (1,7%), zapalenie płuc (1,4%) oraz obniżenie frakcji wyrzutowej (1,1%). Niepożądane reakcje 5 stopnia wystąpiły u 1,4% pacjentów, w tym choroba płucna pośredniczona przez lek (ang. interstitial lung disease – ILD) (1,0%).
Tymczasowe wstrzymanie leczenia z powodu niepożądanych reakcji wystąpiło u 34,3% pacjentów otrzymujących lek Enzertu. Najczęstsze niepożądane reakcje związane z tymczasowym wstrzymaniem leczenia to: neutropenia (13,3%), zmęczenie (5,0%), anemia (4,7%), leukopenia (3,7%), trombocytopenia (3,0%), infekcja dróg oddechowych górnych (2,7%) oraz ILD (2,6%). Dawkę obniżono u 20,6% pacjentów otrzymujących lek Enzertu. Najczęstsze niepożądane reakcje związane z obniżeniem dawki to: zmęczenie (5,0%), nudności (4,9%), neutropenia (3,5%) oraz trombocytopenia (2,1%). Stałe zakończenie leczenia z powodu niepożądanych reakcji wystąpiło u 13,0% pacjentów otrzymujących lek Enzertu. Najczęstszą niepożądaną reakcją związaną ze stałym zakończeniem leczenia była ILD (9,2%).
Enzertu 6,4 mg/kg
W połączonej grupie do oceny bezpieczeństwa oceniano pacjentów, którzy otrzymali co najmniej jedną dawkę leku Enzertu w dawce 6,4 mg/kg (n = 669) w badaniach klinicznych różnych typów nowotworów. Mediana czasu trwania leczenia w tej połączonej grupie wyniosła 5,7 miesiąca (zakres: 0,7–41,0 miesiąca).
Najczęstsze niepożądane reakcje to: nudności (72,2%), zmęczenie (58,4%), osłabienie apetytu (53,5%), anemia (44,7%), neutropenia (43,5%), wymioty (40,1%), biegunka (35,9%), wypadanie włosów (35,4%), zaparcia (32,3%), trombocytopenia (30,8%), leukopenia (29,3%) oraz podwyższenie poziomu transaminaz (24,2%).
Najczęstsze niepożądane reakcje o nasileniu 3 lub 4 stopnia według Ogólnych Kryteriów Terminologii Oceny Niepożądanych Zdarzeń Klinicznych Narodowego Instytutu Onkologii USA (NCI-CTCAE, wersja 5.0) to: neutropenia (28,7%), anemia (22,6%), leukopenia (13,3%), trombocytopenia (9,1%), zmęczenie (8,4%), osłabienie apetytu (7,8%), limfopenia (6,9%), nudności (5,8%), podwyższenie poziomu transaminaz (4,3%), hipokaliemia (4,3%), zapalenie płuc (3,1%), neutropenia gorączkowa (2,8%), wymioty (2,4%), biegunka (2,2%), utrata masy ciała (1,9%), podwyższenie poziomu fosfatazy alkalicznej we krwi (1,6%), choroba płuc pośredniczona przez lek (ILD, 1,5%), duszność (1,2%), obniżenie frakcji wyrzutowej (1,2%) oraz podwyższenie poziomu bilirubiny we krwi (1,2%). Niepożądane reakcje 5 stopnia, w tym ILD (2,1%), wystąpiły u 2,7% pacjentów.
Tymczasowe wstrzymanie leczenia z powodu niepożądanych reakcji wystąpiło u 40,7% pacjentów otrzymujących lek Enzertu. Najczęstsze niepożądane reakcje związane z tymczasowym wstrzymaniem leczenia to: neutropenia (16,6%), anemia (7,8%), zmęczenie (5,7%), ILD (4,8%), leukopenia (4,2%), osłabienie apetytu (3,7%), zapalenie płuc (3,6%), infekcja dróg oddechowych górnych (3,4%) oraz trombocytopenia (3,1%). Dawkę obniżono u 31,1% pacjentów otrzymujących lek Enzertu. Najczęstsze niepożądane reakcje związane z obniżeniem dawki to: zmęczenie (10,6%), neutropenia (6,6%), nudności (6,4%), osłabienie apetytu (5,4%) oraz trombocytopenia (3,0%). Stałe zakończenie leczenia z powodu niepożądanych reakcji wystąpiło u 17,6% pacjentów otrzymujących lek Enzertu. Najczęstszą niepożądaną reakcją związaną ze stałym zakończeniem leczenia była ILD (12,9%).
25,3% pacjentów z rakiem żołądka, którzy otrzymywali lek Enzertu w dawce 6,4 mg/kg (n = 229), otrzymało przetoczenie krwi w ciągu 28 dni od momentu rozpoznania anemii lub trombocytopenii. Przetoczenia przeprowadzano głównie z powodu anemii.
Lista niepożądanych reakcji przedstawiona w formie tabeli
Niepożądane reakcje u pacjentów, którzy otrzymali co najmniej jedną dawkę leku Enzertu w badaniach klinicznych, przedstawiono w tabeli 9. Niepożądane reakcje pogrupowano według klas układów narządów (SOC) zgodnie z Medycznym Słownikiem Terminologii Działalności Regulacyjnej (MedDRA) oraz częstości występowania. Częstość występowania sklasyfikowano następująco: bardzo często (≥ 1/10), często (≥ 1/100 do < 1/10), rzadko (≥ 1/1000 do < 1/100), niezwykle rzadko (≥ 1/10 000 do < 1/1000), bardzo rzadko (< 1/10 000) oraz nieznana (nie można określić na podstawie dostępnych danych). W ramach każdej grupy niepożądanych reakcji uporządkowano je według malejącego nasilenia.
Tabela 9: Niepożądane reakcje u pacjentów, którzy otrzymywali trastuzumab deruxtecan w dawce 5,4 mg/kg oraz 6,4 mg/kg w różnych typach nowotworów
| Klasa układów narządów |
5,4 mg/kg |
6,4 mg/kg |
| Kategoria częstości |
Reakcja niepożądana |
Reakcja niepożądana |
| Infekcje i choroby pasożytnicze |
||
| Bardzo często |
infekcja dróg oddechowych górnycha |
zapalenie płuc, infekcja dróg oddechowych górnycha |
| Często |
zapalenie płuc |
|
| Zaburzenia układu krwiotwórczego i chłonnego |
||
| Bardzo często |
anemiab, neutropeniac, trombocytopeniaf, leukopeniag, limfopeniad |
anemiab, neutropeniac, trombocytopeniaf, leukopeniag, limfopeniad |
| Często |
gorączkowa neutropenia |
|
| Nieczęsto |
gorączkowa neutropenia |
|
| Zaburzenia metaboliczne i odżywienia |
||
| Bardzo często |
hipokaliemiad, spadek apetytu |
hipokaliemiad, spadek apetytu |
| Często |
odwodnienie |
odwodnienie |
| Zaburzenia układu nerwowego |
||
| Bardzo często |
ból głowyę, zawroty głowy |
ból głowyę, dysgezja |
| Często |
dysgezja |
zawroty głowy |
| Zaburzenia narządu wzroku |
||
| Często |
susza oczu, nieostre widzenież |
susza oczu, nieostre widzenież |
| Zaburzenia układu oddechowego, organów klatki piersiowej i śródpiersia |
||
| Bardzo często |
choroba płuc śródmiąższową, duszność, kaszel, krwawienie z nosa |
choroba płuc śródmiąższową, duszność, kaszel |
| Często |
krwawienie z nosa |
|
| Zaburzenia układu pokarmowego |
||
| Bardzo często |
nudności, wymioty, zaparcia, biegunka, ból brzucha i, zapalenie błony śluzowej jamy ustnej l, dyspepsja |
nudności, wymioty, biegunka, zaparcia, ból brzucha i, zapalenie błony śluzowej jamy ustnej l |
| Często |
wzdęcia, zapalenie błony śluzowej żołądka, meteorizm |
dyspepsja, wzdęcia, zapalenie błony śluzowej żołądka, meteorizm |
| Zaburzenia wątroby i dróg żółciowych |
||
| Bardzo często |
wzrost poziomu transaminazę |
wzrost poziomu transaminazę |
| Zaburzenia skóry i tkanek podskórnych |
||
| Bardzo często |
alopecia |
alopecia |
| Często |
wysypka j, świąd, hiperpigmentacja skóry k |
wysypka j, świąd, hiperpigmentacja skóry k |
| Zaburzenia układu mięśniowo-szkieletowego i tkanki łącznej |
||
| Bardzo często |
ból mięśniowo-szkieletowy l |
ból mięśniowo-szkieletowy l |
| Zaburzenia ogólne i reakcje w miejscu podania |
||
| Bardzo często |
zmęczenie m, gorączka |
zmęczenie m, gorączka, obrzęk obwodowy |
| Często |
obrzęk obwodowy |
|
| Wyniki badań laboratoryjnych i instrumentalnych |
||
| Bardzo często |
spadek frakcji wyrzutowej n, spadek masy ciała |
spadek frakcji wyrzutowej n, spadek masy ciała |
| Często |
wzrost poziomu fosfatazy alkalicznej we krwi, wzrost poziomu bilirubiny we krwi o, wzrost poziomu kreatyniny we krwi |
wzrost poziomu fosfatazy alkalicznej we krwi, wzrost poziomu bilirubiny we krwi o, wzrost poziomu kreatyniny we krwi |
| Urazy, zatrucia i komplikacje procedur |
||
| Często |
reakcje infuzyjne p |
reakcje infuzyjne p |
а W tym objawy grypy, stany grypopodobne, zapalenie gardła i nosa, zapalenie gardła, zapalenie zatok, zapalenie nosa, zapalenie krtani oraz infekcje górnych dróg oddechowych.
b U wszystkich typów nowotworów przy dawce 5,4 mg/kg występuje anemia, obniżenie stężenia hemoglobiny, zmniejszenie liczby erytrocytów oraz obniżenie hematokrytu. U wszystkich typów nowotworów przy dawce 6,4 mg/kg występuje anemia, obniżenie stężenia hemoglobiny oraz zmniejszenie liczby erytrocytów.
c W tym obejmuje się neutropenię oraz zmniejszenie liczby neutrofili.
d W tym obejmuje się trombocytopenię oraz zmniejszenie liczby płytek krwi.
e W tym obejmuje się leukopenię oraz zmniejszenie liczby białych krwinek.
f W tym obejmuje się limfopenię oraz zmniejszenie liczby limfocytów.
g W tym obejmuje się hipokaliemię oraz obniżenie stężenia potasu we krwi.
h U wszystkich typów nowotworów przy dawce 5,4 mg/kg obejmuje bóle głowy, ból zatokowy oraz migrenę. U wszystkich typów nowotworów przy dawce 6,4 mg/kg obejmuje bóle głowy oraz migrenę.
i W tym obejmuje się nieostrość widzenia oraz zaburzenia wzroku.
j U wszystkich typów nowotworów przy dawce 5,4 mg/kg choroba śródmiąższowa płuc obejmuje zjawiska uznane za CHPL: zapalenie płuc (n = 88), choroba śródmiąższowa płuc (n = 72), zapalenie płuc organizujące (n = 6), zapalenie płuc (n = 4), niewydolność oddechowa (n = 5), zapalenie promieniowe płuc (n = 2), alwegolit (n = 2), toksyczność płuc (n = 2), grzybicze zapalenie płuc (n = 1), guz płuc (n = 1), ostra niewydolność oddechowa (n = 1), infiltracja płuc (n = 1), limfangiit (n = 1), włóknienie płuc (n = 1), idiopatyczne zapalenie śródmiąższowe płuc (n = 1), choroba płuc (n = 1), nadwrażliwość płuc (n = 1) oraz zacienienie w płucach (n = 1). U wszystkich typów nowotworów przy dawce 6,4 mg/kg choroba śródmiąższowa płuc obejmuje zjawiska uznane za CHPL: zapalenie płuc (n = 75), choroba śródmiąższowa płuc (n = 39), zapalenie płuc organizujące (n = 4), niewydolność oddechowa (n = 4), zacienienie w płucach (n = 2), zapalenie płuc (n = 1) oraz zapalenie promieniowe płuc (n = 1).
k W tym obejmuje się dyskomfort brzucha, ból w przewodzie pokarmowym, ból brzucha, ból w dolnej części brzucha oraz ból w górnej części brzucha.
l U wszystkich typów nowotworów przy dawce 5,4 mg/kg obejmuje się stomatytę, owrzodzenie aftowe, owrzodzenie błony śluzowej jamy ustnej, erozję błony śluzowej jamy ustnej oraz wysypkę na błonie śluzowej jamy ustnej. U wszystkich typów nowotworów przy dawce 6,4 mg/kg obejmuje się wyłącznie stomatytę.
m W tym obejmuje się podwyższenie stężenia transaminaz, podwyższenie stężenia alaninotransaminazy, podwyższenie stężenia aspартaminotransferazy, podwyższenie stężenia gamma-glutamylotransferazy, zaburzenia funkcji wątroby, patologiczne wyniki testów czynności wątroby, podwyższone wyniki testów czynności wątroby oraz hipertransaminazemię.
n U wszystkich typów nowotworów przy dawce 5,4 mg/kg obejmuje się wysypkę, wysypkę pustulopną, wysypkę makulopapularną, wysypkę papularną, wysypkę makularną oraz wysypkę swędzącą. U wszystkich typów nowotworów przy dawce 6,4 mg/kg obejmuje się wysypkę, wysypkę pustulopną, wysypkę makulopapularną oraz wysypkę swędzącą.
o U wszystkich typów nowotworów przy dawce 5,4 mg/kg obejmuje się hiperpigmentację skóry, zmianę koloru skóry oraz zaburzenia pigmentacji. U wszystkich typów nowotworów przy dawce 6,4 mg/kg obejmuje się hiperpigmentację skóry oraz zaburzenia pigmentacji.
p W tym obejmuje się ból pleców, mialgię, ból kończyn, ból mięśniowo-szkieletowy, skurcze mięśni, ból kości, ból szyi, ból mięśniowo-szkieletowy w klatce piersiowej oraz dyskomfort kończyn.
q W tym obejmuje się osłabienie, zmęczenie, niedobór samopoczucia oraz senność.
r U wszystkich typów nowotworów przy dawce 5,4 mg/kg termin „obniżenie frakcji wyrzutowej” obejmuje laboratoryjne oznaczenia obniżenia FVLP (n = 214) i/lub głównie następujące terminy: obniżenie frakcji wyrzutowej (n = 52), niewydolność serca (n = 3), niewydolność serca zastoinowego (n = 1) oraz dysfunkcja lewej komory (n = 2). U wszystkich typów nowotworów przy dawce 6,4 mg/kg termin „obniżenie frakcji wyrzutowej” obejmuje laboratoryjne oznaczenia obniżenia FVLP (n = 97) i/lub głównie następujące terminy: obniżenie frakcji wyrzutowej (n = 11) oraz dysfunkcja lewej komory (n = 1).
s U wszystkich typów nowotworów przy dawce 5,4 mg/kg obejmuje się podwyższenie stężenia bilirubiny we krwi, hiperbilirubinemię, podwyższenie stężenia bilirubiny sprzężonej oraz podwyższenie stężenia bilirubiny niesprzężonej we krwi. U wszystkich typów nowotworów przy dawce 6,4 mg/kg obejmuje się podwyższenie stężenia bilirubiny we krwi, hiperbilirubinemię oraz podwyższenie stężenia bilirubiny sprzężonej.
t U wszystkich typów nowotworów przy dawce 5,4 mg/kg przypadki reakcji infuzyjnych obejmują reakcję infuzyjną (n = 16) oraz nadwrażliwość (n = 2). U wszystkich typów nowotworów przy dawce 6,4 mg/kg przypadki reakcji infuzyjnych obejmują reakcję infuzyjną (n = 6) oraz nadwrażliwość (n = 1). Wszystkie przypadki reakcji infuzyjnych miały stopień ciężkości 1 lub 2.
Opis wybranych działań niepożądanych
Choroba śródmiąższowa płuc/zapalenie płuc
W badaniach klinicznych z udziałem kilku typów nowotworów (n = 1449) po zastosowaniu leku Enzertu w dawce 5,4 mg/kg CHPL wystąpiła u 12,5 % pacjentów. Większość przypadków CHPL miała stopień ciężkości 1 (3,2 %) i 2 (7,4 %). Przypadki o stopniu ciężkości 3 zaobserwowano u 0,8 % pacjentów, a przypadków o stopniu 4 nie odnotowano. Zjawiska o stopniu 5 (śmiertelne) wystąpiły u 1,0 % pacjentów. Mediana czasu do wystąpienia pierwszego zjawiska wynosiła 5,5 miesiąca (zakres od 26 dni do 31,5 miesiąca) (patrz rozdziały „Sposób stosowania i dawki” oraz „Szczególne wskazówki dotyczące stosowania”).
W badaniach klinicznych z udziałem kilku typów nowotworów (n = 669) po zastosowaniu leku Enzertu w dawce 6,4 mg/kg CHPL wystąpiła u 17,9 % pacjentów. Większość przypadków CHPL miała stopień ciężkości 1 (4,9 %) i 2 (9,4 %). Przypadki o stopniu ciężkości 3 zaobserwowano u 1,3 % pacjentów, a przypadki o stopniu 4 – u 0,1 % pacjentów. Zjawiska o stopniu 5 (śmiertelne) wystąpiły u 2,1 % pacjentów. U jednego pacjenta wystąpiła CHPL, która nasiliła się po leczeniu i doprowadziła do CHPL o stopniu 5 (śmiertelna). Mediana czasu do wystąpienia pierwszego zjawiska wynosiła 4,2 miesiąca (zakres od -0,5 do 21,0) (patrz rozdziały „Sposób stosowania i dawki” oraz „Szczególne wskazówki dotyczące stosowania”).
Neutropenia
W badaniach klinicznych (n = 1449) z udziałem kilku typów nowotworów po zastosowaniu leku Enzertu w dawce 5,4 mg/kg neutropenia występowała u 35,2 % pacjentów, a u 17,0 % – zjawiska miały stopień ciężkości 3 lub 4. Mediana czasu do wystąpienia neutropenii wynosiła 43 dni (zakres od 1 dnia do 31,9 miesiąca), a mediana trwania pierwszego zjawiska wynosiła 22 dni (zakres od 1 dnia do 17,1 miesiąca). Gorączkową neutropenię odnotowano u 0,9 % pacjentów, a 0,1 % miało stopień 5 (patrz rozdział „Sposób stosowania i dawki”).
W badaniach klinicznych (n = 669) z udziałem kilku typów nowotworów po zastosowaniu leku Enzertu w dawce 6,4 mg/kg neutropenia występowała u 43,5 % pacjentów, a u 28,7 % – zjawiska miały stopień ciężkości 3 lub 4. Mediana czasu do wystąpienia zjawiska wynosiła 16 dni (zakres od 1 dnia do 24,8 miesiąca), a mediana trwania pierwszego zjawiska wynosiła 9 dni (zakres od 2 dni do 17,2 miesiąca). Gorączkową neutropenię odnotowano u 3,0 % pacjentów, a 0,1 % miało zjawiska o stopniu 5 (patrz rozdział „Sposób stosowania i dawki”).
Obniżenie frakcji wyrzutowej lewej komory
W badaniach klinicznych z udziałem kilku typów nowotworów (n = 1449) po zastosowaniu leku Enzertu w dawce 5,4 mg/kg obniżenie FVLP odnotowano u 57 pacjentów (3,9 %), z których u 10 (0,7 %) zjawisko to miało stopień ciężkości 1, u 40 (2,8 %) – stopień 2, a u 7 (0,5 %) – stopień 3. Według danych laboratoryjnych (echokardiografia lub badanie MUGA) częstość przypadków obniżenia FVLP o stopniu 2 wynosiła 202/1341 (15,1 %), a o stopniu 3 – 12/1341 (0,9 %). Stosowanie leku Enzertu pacjentom, u których przed rozpoczęciem leczenia FVLP była niższa niż 50 %, nie było badane (patrz rozdział „Sposób stosowania i dawki”).
W badaniach klinicznych z udziałem kilku typów nowotworów (n = 669) po zastosowaniu leku Enzertu w dawce 6,4 mg/kg obniżenie FVLP odnotowano u 12 pacjentów (1,8 %), z których u 1 (0,1 %) zjawisko to miało stopień ciężkości 1, u 8 (1,2 %) – stopień 2, a u 3 (0,4 %) – stopień 3. Według danych laboratoryjnych (echokardiografia lub badanie MUGA) częstość przypadków obniżenia FVLP o stopniu 2 wynosiła 89/597 (14,9 %), a o stopniu 3 – 8/597 (1,3 %).
Reakcje infuzyjne
W badaniach klinicznych (n = 1449) z udziałem kilku typów nowotworów po zastosowaniu leku Enzertu w dawce 5,4 mg/kg reakcje infuzyjne występowały u 18 pacjentów (1,2 %), wszystkie zjawiska miały stopień ciężkości 1 lub 2. Nie odnotowano zjawisk o stopniu ciężkości 3. W trzech przypadkach (0,2 %) reakcje infuzyjne doprowadziły do tymczasowego przerwania leczenia, a zjawisk prowadzących do całkowitego przerwania leczenia nie odnotowano.
W badaniach klinicznych (n = 669) z udziałem kilku typów nowotworów po zastosowaniu leku Enzertu w dawce 6,4 mg/kg reakcje infuzyjne występowały u 7 pacjentów (1,0 %), wszystkie zjawiska miały stopień ciężkości 1 lub 2. Nie odnotowano zjawisk o stopniu ciężkości 3. W jednym przypadku (0,1 %) reakcja infuzyjna doprowadziła do tymczasowego przerwania leczenia, a zjawisk prowadzących do całkowitego przerwania leczenia nie odnotowano.
Immunogenność
Podobnie jak wszystkie leki białkowe, Enzertu może wywoływać odpowiedź immunologiczną. W całym zakresie dawek ocenianych w badaniach klinicznych, po zastosowaniu leku Enzertu u 2,1 % (47/2213) pacjentów wytworzyły się przeciwciała przeciwko trastuzumabu deruxtecanowi. Częstość występowania przeciwciał neutralizujących przeciwko trastuzumabu deruxtecanowi podczas leczenia wynosiła 0,1 % (2/2213). Nie stwierdzono związku między powstawaniem przeciwciał a reakcjami alergicznymi.
Populacja pediatryczna
Bezpieczeństwo stosowania leku Enzertu w tej populacji nie zostało ustalone.
Pacjenci starsi
W badaniach klinicznych (n = 1449) z udziałem kilku typów nowotworów, w których lek Enzertu stosowano w dawce 5,4 mg/kg, 24,2 % pacjentów miało 65 lat i więcej, a 4,3 % – 75 lat i więcej. U pacjentów w wieku 65 lat i więcej (50,0 %) obserwowano wyższą częstość działań niepożądanych o stopniu ciężkości 3–4 w porównaniu z pacjentami poniżej 65 roku życia (42,7 %), co prowadziło do większej liczby przypadków przerwania leczenia z powodu działań niepożądanych.
Spośród 669 pacjentów z kilkoma typami nowotworów, którzy brali udział w badaniach klinicznych stosowania leku Enzertu w dawce 6,4 mg/kg, 39,2 % miało 65 lat i więcej, a 7,6 % – 75 lat i więcej. Częstość działań niepożądanych o stopniu ciężkości 3–4 obserwowanych u pacjentów w wieku 65 lat i więcej wynosiła 59,9 %, a u młodszych pacjentów – 62,9 %. Częstość działań niepożądanych o stopniu ciężkości 3–4 obserwowanych u pacjentów w wieku 75 lat wynosiła 64,7 %, a u pacjentów poniżej 75 roku życia – 61,5 %. U pacjentów w wieku 75 lat i więcej obserwowano wyższą częstość poważnych działań niepożądanych (37,3 %) i zgonów (7,8 %) w porównaniu z pacjentami poniżej 75 roku życia (20,7 % i 2,3 %). Dane dotyczące bezpieczeństwa stosowania u pacjentów w wieku 75 lat i więcej są ograniczone.
Różnice etniczne
W badaniach klinicznych nie zaobserwowano istotnych różnic w działaniu lub skuteczności tego leku między pacjentami z różnych grup etnicznych. U pacjentów pochodzenia azjatyckiego, którzy otrzymywali lek Enzertu w dawce 6,4 mg/kg, obserwowano wyższą częstość (różnica ≥ 10 %) neutropenii (58,1 % w porównaniu z 18,6 %), anemii (51,1 % w porównaniu z 32,4 %), leukopenii (42,7 % w porównaniu z 6,9 %), trombocytopenii (40,5 % w porównaniu z 15,4 %) oraz limfopenii (17,6 % w porównaniu z 7,3 %) niż u pacjentów innych grup etnicznych. U 4,3 % pacjentów pochodzenia azjatyckiego obserwowano krwawienia w ciągu 14 dni po wystąpieniu trombocytopenii w porównaniu z 1,6 % pacjentów innych grup etnicznych.
Zgłaszanie działań niepożądanych
Zgłaszanie działań niepożądanych po rejestracji leku ma istotne znaczenie. Umożliwia to monitorowanie stosunku korzyści do ryzyka stosowania tego leku. Personel medyczny i farmaceutyczny, a także pacjenci lub ich prawni przedstawiciele powinni zgłaszać wszystkie przypadki podejrzewanych działań niepożądanych oraz braku skuteczności leku poprzez Automatyczny System Informacyjny do Nadzoru Farmakologicznego pod adresem: https://aisf.dec.gov.ua.
Okres ważności
48 miesięcy.
Proponowany okres ważności (po odtworzeniu i rozcieńczeniu)
Odtworzony roztwór
Stabilność chemiczna i fizyczna w warunkach użytkowania została wykazana przez 24 godziny w temperaturze od 2 do 8 °C.
Z mikrobiologicznego punktu widzenia lek należy użyć natychmiast po odtworzeniu zawartości fiolki. Jeśli nie został użyty natychmiast, odpowiedzialność za czas i warunki przechowywania przed rozpoczęciem stosowania oraz podczas stosowania ponosi użytkownik. Jeśli odtworzenie roztworu nie odbywało się w miejscu z kontrolowanymi i walidowanymi warunkami aseptycznymi, czas przechowywania nie powinien przekraczać 24 godzin w temperaturze od 2 do 8 °C.
Rozcieńczony roztwór
Rozcieńczony roztwór zaleca się użyć natychmiast. Jeśli nie został użyty natychmiast, odtworzony roztwór rozcieńczony w workach do infuzji z 5 % roztworem glukozy można przechowywać w temperaturze pokojowej (≤ 30 °C) przez 4 godziny lub w lodówce w temperaturze od 2 do 8 °C przez 24 godziny, w miejscu chronionym przed światłem. Wskazane okresy przechowywania zaczynają się od momentu odtworzenia roztworu.
Warunki przechowywania
Przechowywać w lodówce w temperaturze od 2 do 8 °C.
Nie zamrażać.
Przechowywać w miejscu niedostępnym dla dzieci.
Niezgodność
Brak danych dotyczących zgodności Enzertu z innymi lekami, dlatego nie należy mieszać tego leku z innymi lekami, z wyjątkiem tych, które są wymienione w rozdziale „Sposób stosowania i dawki”.
Nie można stosować roztworu chlorku sodu do wlewów do przygotowania roztworu lub rozcieńczania, ponieważ może to prowadzić do powstawania cząstek stałych.
Opakowanie
Sterylny proszek liofilizowany do koncentratu do roztworu do wlewów do jednorazowego użytku w szklanej fiolce, zamkniętej butelkowym korkiem gumowym z metalową pokrywką typu „flip-off crimp cap”; po 1 fiolce w pudełku kartonowym z oznaczeniem w języku ukraińskim.
Kategoria wydawania
Na receptę.
Producent
Producent: Daichi Sankyo Europe GmbH / Daiichi Sankyo Europe GmbH.
Siedziba producenta i adres miejsca prowadzenia działalności
Producent: Luitpoldstrasse 1, 85276 Pfaffenhofen, Niemcy / Luitpoldstrasse 1, 85276 Pfaffenhofen, Germany.
Wnioskodawca
AstraZeneca UK Limited / AstraZeneca UK LIMITED
Siedziba wnioskodawcy
1 Francis Crick Avenue, Cambridge Biomedical Campus, Cambridge, CB2 0AA, Wielka Brytania / 1 Francis Crick Avenue, Cambridge Biomedical Campus, Cambridge, CB2 0AA, United Kingdom.