Enhertu
UkraineTable of Contents
INSTRUCTION FOR MEDICAL USE OF THE MEDICINAL PRODUCT ENHERTU (ENHERTU®)
Composition:
Active substance: trastuzumab deruxtecan;
One vial contains 107 mg of trastuzumab deruxtecan as a sterile lyophilized powder for concentrate for solution for infusion, corresponding to a nominal amount of 100 mg in 5 mL of reconstituted solution; after reconstitution, one 5 mL vial contains 20 mg/mL of trastuzumab deruxtecan (see section "Dosage and method of administration");
Excipients: sucrose, L-histidine, L-histidine hydrochloride monohydrate, polysorbate 80.
Pharmaceutical form
Powder for concentrate for solution for infusion.
Main physicochemical characteristics: lyophilized mass of white to yellowish-white color.
Pharmacotherapeutic group
Antineoplastic agents. Monoclonal antibodies and antibody-drug conjugates. HER2 (human epidermal growth factor receptor type 2) inhibitors. Trastuzumab deruxtecan. ATC code L01F D04.
Pharmacological Properties
Pharmacodynamics
Mechanism of Action
The medicinal product Enhertu is an antibody-drug conjugate targeting the HER2 receptor. The antibody component is a humanized IgG1 monoclonal antibody directed against the HER2 receptor, linked to deruxtecan, a topoisomerase I inhibitor (DXd), via a cleavable tetrapeptide-based linker. The conjugate remains stable in blood plasma. The function of the antibody portion is to bind to HER2 expressed on the surface of certain tumor cells. After binding to HER2 receptors on tumor cells, trastuzumab deruxtecan undergoes internalization and intracellular cleavage of the linker by lysosomal enzymes active within tumor cells. Upon release, the membrane-permeable DXd causes deoxyribonucleic acid (DNA) damage and tumor cell apoptosis. DXd, a derivative of exatecan, is approximately 10-fold more potent than SN-38, the active metabolite of irinotecan.
In vitro studies show that the antibody portion of trastuzumab deruxtecan, which has the same amino acid sequence as trastuzumab, also binds to FcγRIIIa and complement C1q. The antibody mediates antibody-dependent cellular cytotoxicity (ADCC) in human breast cancer cells that overexpress HER2. Additionally, the antibody inhibits signaling through the phosphatidylinositol-3-kinase (PI3-K) pathway in human breast cancer cells that overexpress HER2.
Clinical Efficacy
HER2-positive Breast Cancer
DESTINY-Breast03 Study (NCT03529110)
The efficacy and safety of the medicinal product Enhertu were evaluated in the DESTINY-Breast03 trial, a multicenter, randomized, open-label, phase 3 study with active control conducted in two groups. The study enrolled patients with HER2-positive unresectable or metastatic breast cancer who had previously received trastuzumab- and taxane-based therapy for metastatic disease, or who experienced disease recurrence during or within 6 months after completing adjuvant therapy.
Eligibility required archived tumor tissue samples from breast cancer for confirmation of HER2-positive status, defined as immunohistochemical (IHC) score of 3+ or positive in situ hybridization (ISH) result. Patients with a history of glucocorticoid-treated interstitial lung disease (ILD)/pneumonitis, or with ILD/pneumonitis at screening, were excluded. Also excluded were patients with untreated brain metastases or brain metastases with clinical symptoms, and those with a history of clinically significant cardiac disease. Patients previously treated with an anti-HER2 antibody-drug conjugate for metastatic disease were also excluded. Patients were randomized in a 1:1 ratio to receive either Enhertu at a dose of 5.4 mg/kg (N = 261) or trastuzumab emtansine at 3.6 mg/kg (N = 263), administered as intravenous infusions once every three weeks. Randomization was stratified by hormone receptor status, prior pertuzumab treatment, and history of visceral metastases. Treatment continued until disease progression, death, withdrawal of consent, or development of unacceptable toxicity.
The primary efficacy endpoint was progression-free survival (PFS), assessed by blinded independent central review (BICR) according to Response Evaluation Criteria in Solid Tumors (RECIST) version 1.1. A key secondary efficacy endpoint was overall survival (OS). Secondary endpoints included investigator-assessed PFS, confirmed objective response rate (ORR), and duration of response (DoR).
Demographic data and baseline characteristics were well balanced between treatment groups. Baseline demographic and disease characteristics of the 524 randomized patients were as follows: median age 54 years (range 20–83 years); 20.2% were aged 65 years or older; 99.6% were female; 59.9% were of Mongoloid race; 27.3% were of Caucasian race; 3.6% were of Black or African American race; ECOG performance status 0 (62.8%) or 1 (36.8%); hormone receptor status positive (51.9%); presence of visceral metastases (73.3%); presence of brain metastases at baseline (15.6%); 48.3% of patients had received one prior line of systemic therapy for metastatic disease. The proportion of patients who had not received prior therapy for metastatic disease was 9.5%. The proportion of patients previously treated with pertuzumab was 61.1%.
At the time of the pre-specified interim analysis for PFS, which included 245 events (73% of the total number of events planned for the final analysis), the study demonstrated a statistically significant improvement in PFS per BICR in patients randomized to Enhertu compared to those in the trastuzumab emtansine group. PFS per BICR from the primary analysis (data cutoff date: May 21, 2021) and updated OS, ORR, and DoR results after the data cutoff date of July 25, 2022, are presented in Table 1.
Table 1: Efficacy Results from the DESTINY-Breast03 Study
| Measure of efficacy |
Enhertu N = 261 |
trastuzumab emtansine N = 263 |
| Progression-free survival (PFS) by BICR assessment |
||
| Number of events (%) |
87 (33.3) |
158 (60.1) |
| Median, months (95% CI) |
NR (18.5; NE) |
6.8 (5.6; 8.2) |
| Hazard ratio (95% CI) |
0.28 (0.22; 0.37) |
|
| p-value |
p < 0.000001† |
|
| Overall survival (OS)b |
||
| Number of events (%) |
72 (27.6) |
97 (36.9) |
| Median, months (95% CI) |
NR (40.5; NE) |
NR (34.0; NE) |
| Hazard ratio (95% CI) |
0.64 (0.47; 0.87) |
|
| p-valuec |
p = 0.0037 |
|
| Updated PFS by BICR assessmentb |
||
| Number of events (%) |
117 (44.8) |
171 (65.0) |
| Median, months (95% CI) |
28.8 (22.4; 37.9) |
6.8 (5.6; 8.2) |
| Hazard ratio (95% CI) |
0.33 (0.26; 0.43) |
|
| Confirmed objective response rate (ORR) by BICRb |
||
| n (%) |
205 (78.5) |
92 (35.0) |
| 95% CI |
(73.1; 83.4) |
(29.2; 41.1) |
| Complete response, n (%) |
55 (21.1) |
25 (9.5) |
| Partial response, n (%) |
150 (57.5) |
67 (25.5) |
| Duration of response by BICR assessmentb |
||
| Median, months (95% CI) |
36.6 (22.4; NE) |
23.8 (12.6; 34.7) |
CI — confidence interval; NE — not estimable; NR — not reached.
† Presented with 6 decimal places.
a Data cutoff date May 21, 2021.
b Data cutoff date July 25, 2022, for the pre-planned interim OS analysis.
c p-value based on stratified log-rank test; results exceeded the efficacy boundary of 0.013.
Fig. 1. Kaplan–Meier curve of overall survival (data cutoff date July 25, 2022)
| Overall survival, % |
|
||||||||||||||||||||||||||||||||||||||||||||||||||
| Time, months |
|||||||||||||||||||||||||||||||||||||||||||||||||||
| Number of patients at risk: |
|||||||||||||||||||||||||||||||||||||||||||||||||||
| EnherTu |
261 |
256 |
256 |
255 |
254 |
251 |
249 |
244 |
243 |
241 |
238 |
236 |
236 |
236 |
231 |
224 |
218 |
213 |
211 |
206 |
201 |
200 |
196 |
193 |
187 |
182 |
173 |
156 |
142 |
124 |
109 |
91 |
73 |
64 |
51 |
44 |
38 |
30 |
22 |
18 |
11 |
9 |
7 |
6 |
1 |
1 |
0 |
||||
| Trastuzumab emtansine (263) |
263 |
257 |
252 |
248 |
243 |
242 |
237 |
233 |
232 |
227 |
224 |
217 |
211 |
203 |
199 |
197 |
191 |
186 |
183 |
179 |
172 |
169 |
164 |
164 |
158 |
140 |
129 |
117 |
106 |
90 |
70 |
59 |
45 |
41 |
38 |
27 |
20 |
15 |
8 |
7 |
4 |
3 |
3 |
1 |
0 |
||||||
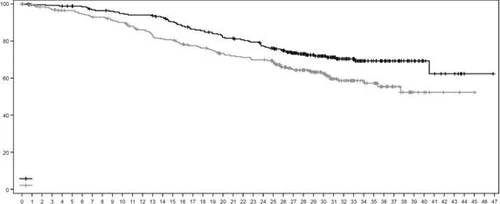
Fig. 2. Kaplan-Meier progression-free survival curve according to BICR assessment (data cutoff date July 25, 2022)
| Progression-free survival, % |
|
|||||||||||||||||||||||||||||||||||||||||||||||
| Time, months |
||||||||||||||||||||||||||||||||||||||||||||||||
| Number of patients at risk: |
||||||||||||||||||||||||||||||||||||||||||||||||
| Enhortu |
261 |
256 |
250 |
244 |
240 |
225 |
216 |
207 |
205 |
191 |
176 |
173 |
167 |
154 |
146 |
140 |
134 |
131 |
130 |
125 |
123 |
117 |
113 |
107 |
99 |
96 |
90 |
82 |
73 |
64 |
55 |
41 |
32 |
28 |
23 |
20 |
18 |
13 |
7 |
5 |
4 |
2 |
1 |
0 |
||||
| Trastuzumab emtansine (263) |
263 |
253 |
201 |
164 |
156 |
134 |
111 |
99 |
96 |
81 |
69 |
67 |
63 |
58 |
54 |
51 |
49 |
49 |
47 |
47 |
42 |
41 |
39 |
37 |
36 |
32 |
28 |
27 |
22 |
19 |
15 |
14 |
8 |
7 |
6 |
4 |
2 |
2 |
2 |
1 |
1 |
1 |
1 |
1 |
1 |
0 |
||
Similar PFS results were observed in predefined subgroups, including subgroups by prior pertuzumab therapy, hormone receptor status, and presence of visceral metastases.
Study DESTINY-Breast02 (NCT03523585)
The efficacy and safety of the medicinal product Enhertu were evaluated in the DESTINY-Breast02 study, a multicenter, randomized, open-label, phase 3 trial with active control, which included patients with unresectable or metastatic HER2-positive breast cancer who had resistance or refractoriness to prior T-DM1 therapy. Eligibility required archived tumor tissue samples from breast cancer for confirmation of HER2-positive status, defined by immunohistochemical analysis as 3+ score (HER2 IHC 3+), or positive status by in situ hybridization assay (ISH-positive). Patients with a history of glucocorticoid treatment for ILD/pneumonitis or with ILD/pneumonitis at screening, patients with untreated brain metastases or brain metastases with clinical symptoms, and patients with a history of clinically significant heart disease were excluded from the study. Patients were randomized in a 2:1 ratio to receive Enhertu at a dose of 5.4 mg/kg (n = 406) via intravenous infusion once every three weeks or physician’s choice of therapy (n = 202; trastuzumab plus capecitabine or lapatinib plus capecitabine). Randomization was stratified by hormone receptor status, prior pertuzumab treatment, and presence of visceral metastases in the medical history. Treatment continued until disease progression, death, withdrawal of consent, or occurrence of unacceptable toxicity.
The primary efficacy endpoint was progression-free survival (PFS), assessed by independent central review in a blinded manner (BICR) according to RECIST criteria, version 1.1. A key secondary efficacy endpoint was overall survival (OS). Secondary endpoints included investigator-assessed PFS, confirmed objective response rate (ORR), and duration of response (DoR).
Demographic characteristics and baseline disease characteristics were balanced across treatment groups. The median age of the 608 randomized patients was 54 years (range: 22 to 88 years); 99.2% of study participants were female; 63.2% were of Caucasian race, 29.3% were of Asian race, 2.8% were of Black or African American race; 57.4% of patients had an ECOG performance status of 0 and 42.4% had a status of 1; 58.6% had positive hormone receptor status; 78.3% had visceral metastases; 18.1% had brain metastases at baseline; 4.9% of patients had received one prior line of systemic therapy for metastatic disease.
Efficacy results are summarized in Table 2 and Figures 3 and 4.
Table 2: Efficacy results in the DESTINY-Breast02 study
| Measure of Efficacy |
Enhertu N = 406 |
Physician’s Choice Therapy N = 202 |
| PFS by BICR assessment |
||
| Number of events (%) |
200 (49.3) |
125 (61.9) |
| Median, months (95% CI) |
17.8 (14.3; 20.8) |
6.9 (5.5; 8.4) |
| Hazard ratio (95% CI) |
0.36 (0.28; 0.45) |
|
| p-value |
p < 0.000001† |
|
| Overall Survival (OS) |
||
| Number of events (%) |
143 (35.2) |
86 (42.6) |
| Median, months (95% CI) |
39.2 (32.7; NE) |
26.5 (21.0; NE) |
| Hazard ratio (95% CI) |
0.66 (0.50; 0.86) |
|
| p-valuea |
p = 0.0021 |
|
| PFS by investigator assessment |
||
| Number of events (%) |
206 (50.7) |
152 (75.2) |
| Median, months (95% CI) |
16.7 (14.3; 19.6) |
5.5 (4.4; 7.0) |
| Hazard ratio (95% CI) |
0.28 (0.23; 0.35) |
|
| Confirmed Objective Response Rate (ORR) by BICR assessment |
||
| n (%) |
283 (69.7) |
59 (29.2) |
| 95% CI |
(65.0; 74.1) |
(23.0; 36.0) |
| Complete response, n (%) |
57 (14.0) |
10 (5.0) |
| Partial response, n (%) |
226 (55.7) |
49 (24.3) |
| Duration of Response by BICR assessment |
||
| Median, months (95% CI) |
19.6 (15.9; NE) |
8.3 (5.8; 9.5) |
CI — confidence interval; NE — not evaluable.
† Presented with 6 decimal places.
a p-value based on stratified log-rank test; results were above the efficacy boundary of 0.004.
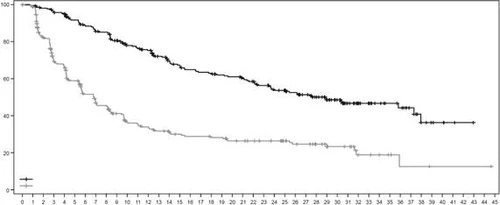
Fig. 3. Kaplan–Meier curve of progression-free survival as assessed by BICR
| Progression-free survival, % |
|
|||||||||||||||||||||||||||||||||||||||||||||||||
| Time, months |
||||||||||||||||||||||||||||||||||||||||||||||||||
| Number of patients at risk: |
||||||||||||||||||||||||||||||||||||||||||||||||||
| Enheritu |
406 |
400 |
374 |
359 |
355 |
330 |
296 |
278 |
260 |
239 |
213 |
203 |
194 |
179 |
170 |
161 |
149 |
141 |
132 |
119 |
109 |
88 |
83 |
76 |
65 |
60 |
55 |
47 |
38 |
35 |
31 |
27 |
23 |
19 |
15 |
14 |
12 |
10 |
6 |
4 |
4 |
3 |
1 |
1 |
1 |
1 |
0 |
|||
| Physician’s choice therapy (202) |
202 |
180 |
148 |
126 |
118 |
95 |
78 |
72 |
64 |
48 |
39 |
37 |
32 |
28 |
24 |
20 |
17 |
13 |
11 |
9 |
9 |
8 |
8 |
6 |
3 |
3 |
3 |
2 |
2 |
2 |
2 |
2 |
1 |
1 |
1 |
1 |
1 |
0 |
||||||||||||
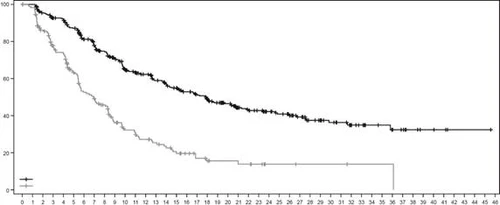
Fig. 4. Kaplan-Meier overall survival curve
| Overall survival, % |
|
||||||||||||||||||||||||||||||||||||||||||||||||
| Time, months |
|||||||||||||||||||||||||||||||||||||||||||||||||
| Number of patients at risk: |
|||||||||||||||||||||||||||||||||||||||||||||||||
| Enhertu |
406 |
404 |
400 |
390 |
385 |
382 |
374 |
366 |
357 |
352 |
350 |
346 |
339 |
331 |
317 |
306 |
295 |
282 |
277 |
257 |
234 |
215 |
196 |
83 |
160 |
144 |
139 |
122 |
104 |
93 |
82 |
72 |
63 |
51 |
40 |
34 |
29 |
25 |
19 |
10 |
8 |
6 |
3 |
1 |
1 |
1 |
0 |
||
| Physician’s choice therapy (202) |
202 |
192 |
187 |
182 |
178 |
173 |
167 |
161 |
157 |
151 |
142 |
136 |
130 |
124 |
118 |
114 |
111 |
110 |
106 |
95 |
89 |
79 |
76 |
72 |
61 |
53 |
50 |
46 |
38 |
33 |
29 |
28 |
25 |
22 |
22 |
18 |
15 |
13 |
12 |
7 |
6 |
5 |
4 |
3 |
1 |
1 |
0 |
||
DESTINY-Breast01 Study (NCT03248492)
The efficacy and safety of the medicinal product Enhertu were evaluated in the DESTINY-Breast01 study, a multicenter, open-label, single-arm phase 2 study involving patients with HER2-positive unresectable and/or metastatic breast cancer who had received two or more prior anti-HER2-based regimens, including trastuzumab emtansine (100%), trastuzumab (100%), and pertuzumab (65.8%). Inclusion criteria included availability of archived breast tumor tissue samples to confirm HER2-positive status, defined as immunohistochemical (IHC) score of 3+ (HER2 IHC 3+), or positive in situ hybridization (ISH-positive). Patients with a history of interstitial lung disease (ILD) or with ILD at screening, patients with untreated brain metastases or brain metastases with clinical symptoms, and patients with a history of clinically significant heart disease were excluded from the study. Patients enrolled in the study had at least one measurable lesion according to RECIST version 1.1. The medicinal product Enhertu was administered via intravenous infusion at a dose of 5.4 mg/kg once every three weeks until disease progression, death, withdrawal of consent, or occurrence of unacceptable toxicity. The primary efficacy endpoint was confirmed objective response rate (ORR) according to RECIST version 1.1 in the intent-to-treat (ITT) population of all randomized patients, assessed by independent central review (ICR). A secondary efficacy endpoint was duration of response (DoR).
Baseline demographic and disease characteristics of the 184 patients enrolled in the DESTINY-Breast01 study were as follows: median age 55 years (range 28 to 96 years); age ≥65 years (23.9%); female sex (100%); Caucasian (54.9%); Asian (38.0%); Black or African American (2.2%); ECOG performance status 0 (55.4%) or 1 (44.0%); hormone receptor-positive status (52.7%); presence of visceral metastases (91.8%); previously treated and stable brain metastases (13.0%); median number of prior lines of therapy for metastatic disease was 5 (range 2 to 17); sum of diameters of all target lesions (<5 cm – 42.4%, ≥5 cm – 50.0%).
A previous analysis (median follow-up duration 11.1 months [range 0.7 to 19.9 months]) showed a confirmed objective response rate of 60.9% (95% CI 53.4; 68.0), including 6.0% of patients with complete response and 54.9% with partial response; 36.4% had stable disease, 1.6% experienced disease progression, and 1.1% were non-evaluable. The median duration of response at time of analysis was 14.8 months (95% CI 13.8; 16.9), with 81.3% of patients having a response duration of ≥6 months (95% CI 71.9; 87.8). Efficacy results at the updated data cutoff date with a median follow-up duration of 20.5 months (range 0.7 to 31.4 months) are presented in Table 3.
Table 3: Efficacy results in the DESTINY-Breast01 study (intent-to-treat population)
| DESTINY-Breast01 N = 184 |
|
| Confirmed objective response rate (95% CI)*† |
61.4% (54.0; 68.5) |
| Complete response (CR) |
6.5% |
| Partial response (PR) |
54.9% |
| Duration of response ‡ |
|
| Median, months (95% CI) |
20.8 (15.0; NE) |
| % of patients with duration of response ≥ 6 months (95% CI)§ |
81.5% (72.2; 88.0) |
95 % CI, calculated using the Clopper-Pearson method.
CI – confidence interval.
95 % CI, calculated using the Brookmeyer-Crowley method.
* Confirmed responses (based on independent central review under blinded conditions) were defined as documented CR/PR, confirmed by imaging method no less than 4 weeks after the visit at which response was first observed.
† Of 184 patients, 35.9 % had stable disease, 1.6 % had disease progression, and 1.1 % were unevaluable.
‡ Includes 73 patients with censored data.
§ Based on Kaplan-Meier estimation.
NE — not reached.
Stable antitumor activity was observed in prespecified subgroups based on prior pertuzumab therapy and hormone receptor status.
Breast cancer with low HER2 expression
Study DESTINY-Breast04 (NCT03734029)
The efficacy and safety of the medicinal product Enhertu were evaluated in the DESTINY-Breast04 study, a multicenter, randomized, open-label, phase 3 trial that included 557 adult patients with unresectable or metastatic breast cancer with low HER2 expression. The study included 2 cohorts: 494 patients with positive hormone receptor status (HR+) and 63 patients with negative hormone receptor status (HR-). Low HER2 expression was assessed at a central laboratory as IHC 1+ (defined as weak partial membrane staining in more than 10 % of cancer cells) or IHC 2+/ISH- using the PATHWAY/VENTANA system with clone 4B5 for HER2 status determination (anti-HER2/neu (4B5)). Patients were required to have received chemotherapy for metastatic disease or to have experienced disease recurrence during or within 6 months after adjuvant chemotherapy. According to inclusion criteria at randomization, patients with HR+ disease were required to have received at least one endocrine therapy regimen or to have contraindications to further endocrine therapy. Patients were randomized in a 2:1 ratio to receive Enhertu at a dose of 5.4 mg/kg (N = 373) administered intravenously once every 3 weeks or physician’s choice chemotherapy (N = 184, eribulin – 51.1 %, capecitabine – 20.1 %, gemcitabine – 10.3 %, nab-paclitaxel – 10.3 %, or paclitaxel – 8.2 %). Randomization was stratified by HER2 IHC status determined by immunohistochemical analysis of breast tumor tissue samples (IHC 1+ or IHC 2+/ISH-), number of prior lines of chemotherapy for metastatic disease (1 or 2), and HR status/CDK4/6i prior (HR+ with prior CDK4/6 inhibitor treatment, HR+ without prior CDK4/6 inhibitor treatment, or HR-). Treatment continued until disease progression, death, withdrawal of consent, or development of unacceptable toxicity. Patients with a history of glucocorticoid treatment for ILD/pneumonitis or with ILD/pneumonitis at screening were excluded from the study, as were patients with a history of clinically significant heart disease. Patients with untreated brain metastases or brain metastases with clinical symptoms or ECOG performance status >1 were also excluded from the study.
The primary efficacy endpoint was progression-free survival (PFS) in patients with HR+ breast cancer, assessed by BICR according to RECIST criteria, version 1.1. Key secondary efficacy endpoints were PFS assessed by BICR according to RECIST criteria, version 1.1, in the overall population (all randomized patients with HR+ and HR-), overall survival (OS) in patients with HR+ disease, and OS in the overall population. Secondary endpoints included ORR, DOR, and patient-reported outcomes (PRO).
Demographic data and tumor characteristics at baseline were comparable between treatment groups. The median age of the 557 randomized patients was 57 years (range 28 to 81 years); 23.5 % were aged 65 years or older; 99.6 % were female and 0.4 % were male; 47.9 % were White, 40.0 % were Asian, and 1.8 % were Black or African American. At baseline, patients had an ECOG performance status of 0 (54.8 %) or 1 (45.2 %); 57.6 % had IHC 1+; 42.4 % had IHC 2+/ISH-; 88.7 % were HR+ and 11.3 % were HR-; 69.8 % had liver metastases, 32.9 % had lung metastases, and 5.7 % had brain metastases. The proportion of patients who had previously received anthracyclines in (neo)adjuvant therapy was 46.3 %, and 19.4 % in locally advanced and/or metastatic setting. For metastatic disease, patients had received a median of 3 prior lines of systemic therapy (range 1 to 9), with 57.6 % having received 1 and 40.9 % having received 2 prior chemotherapy regimens; 3.9 % had early disease progression (progression during neoadjuvant or adjuvant therapy). Among patients with HR+, the median number of prior endocrine therapy lines was 2 (range 0 to 9), and 70 % had previously received CDK4/6 inhibitors.
The efficacy assessment results are summarized in Table 4 and Figures 5 and 6.
Table 4: Efficacy assessment results from the DESTINY-Breast04 study
| Measure of efficacy |
HR+ cohort |
Overall population (HR+ and HR- cohorts) |
||
| Enhertu (N = 331) |
Chemotherapy (N = 163) |
Enhertu (N = 373) |
Chemotherapy (N = 184) |
|
| Overall survival |
||||
| Number of events (%) |
126 (38.1) |
73 (44.8) |
149 (39.9) |
90 (48.9) |
| Median, months (95% CI) |
23.9 (20.8; 24.8) |
17.5 (15.2; 22.4) |
23.4 (20.0; 24.8) |
16.8 (14.5; 20.0) |
| Hazard ratio (95% CI) |
0.64 (0.48; 0.86) |
0.64 (0.49; 0.84) |
||
| p-value |
0.0028 |
0.001 |
||
| BICR-assessed progression-free survival |
||||
| Number of events (%) |
211 (63.7) |
110 (67.5) |
243 (65.1) |
127 (69.0) |
| Median, months (95% CI) |
10.1 (9.5; 11.5) |
5.4 (4.4; 7.1) |
9.9 (9.0; 11.3) |
5.1 (4.2; 6.8) |
| Hazard ratio (95% CI) |
0.51 (0.40; 0.64) |
0.50 (0.40; 0.63) |
||
| p-value |
< 0.0001 |
< 0.0001 |
||
| Confirmed objective response rate by BICR* |
||||
| n (%) |
175 (52.6) |
27 (16.3) |
195 (52.3) |
30 (16.3) |
| 95% CI |
47.0; 58.0 |
11.0; 22.8 |
47.1; 57.4 |
11.3; 22.5 |
| Complete response, n (%) |
12 (3.6) |
1 (0.6) |
13 (3.5) |
2 (1.1) |
| Partial response, n (%) |
164 (49.2) |
26 (15.7) |
183 (49.1) |
28 (15.2) |
| Duration of response by BICR* |
||||
| Median, months (95% CI) |
10.7 (8.5; 13.7) |
6.8 (6.5; 9.9) |
10.7 (8.5; 13.2) |
6.8 (6.0; 9.9) |
CI — confidence interval.
* Based on electronic individual case forms for the HR+ cohort: N = 333 in the Enhertu treatment group and N = 166 in the chemotherapy group.
In predefined patient subgroups based on HR status, prior CDK4/6i inhibitor therapy, number of prior chemotherapy lines, and IHC1+ and IHC2+/ISH- status, improvements in OS and PFS were demonstrated. In the HR- subgroup, median OS was 18.2 months (95% CI 13.6, not estimable) in the Enhertu treatment group compared to 8.3 months (95% CI 5.6; 20.6) in the chemotherapy group, with a hazard ratio of 0.48 (95% CI 0.24; 0.95). Median PFS was 8.5 months (95% CI 4.3; 11.7) in patients randomized to the Enhertu treatment group and 2.9 months (95% CI 1.4; 5.1) in patients randomized to the chemotherapy group, with a hazard ratio of 0.46 (95% CI 0.24; 0.89).
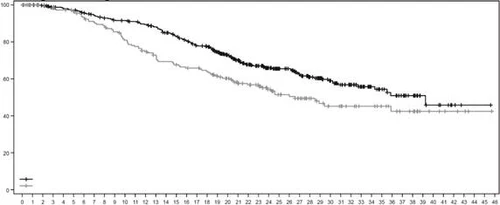
In an updated descriptive analysis with a median follow-up of 32 months, the improvement in OS outcomes was consistent with the results of the primary analysis. HR in the overall population was 0.69 (95% CI 0.55; 0.86), with median OS of 22.9 months (95% CI 21.2; 24.5) in the Enhertu treatment group versus 16.8 months (95% CI 14.1; 19.5) in the chemotherapy group. Kaplan–Meier curves for OS from the updated analysis are shown in Figure 5.
Figure 5. Kaplan–Meier overall survival curve (overall population) (updated analysis)
| Overall survival, % |
|
|||||||||||||||||||||||||||||||||||||||||||||||||||
| Time (months) |
||||||||||||||||||||||||||||||||||||||||||||||||||||
| Number of patients at risk |
||||||||||||||||||||||||||||||||||||||||||||||||||||
| Enhertu (373) |
373 |
366 |
363 |
366 |
350 |
342 |
337 |
325 |
314 |
306 |
256 |
235 |
276 |
269 |
257 |
254 |
240 |
231 |
217 |
206 |
199 |
191 |
182 |
168 |
160 |
148 |
137 |
122 |
107 |
94 |
81 |
75 |
62 |
52 |
48 |
39 |
26 |
21 |
18 |
11 |
7 |
6 |
5 |
3 |
1 |
1 |
1 |
0 |
||||
| Chemotherapy (184) |
184 |
170 |
164 |
160 |
156 |
152 |
145 |
137 |
127 |
119 |
113 |
107 |
106 |
100 |
96 |
88 |
81 |
76 |
73 |
69 |
64 |
59 |
58 |
53 |
49 |
45 |
45 |
44 |
37 |
33 |
27 |
18 |
15 |
12 |
12 |
10 |
8 |
5 |
2 |
2 |
2 |
1 |
0 |
|||||||||
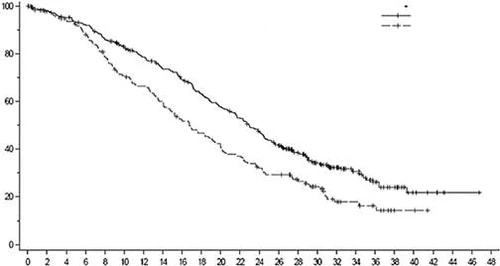
Fig. 6. Kaplan-Meier curve of progression-free survival according to BICR assessment (overall population)
| Progression-free survival, % |
|
|||||||||||||||||||||||||||||||||
| Time (months) |
||||||||||||||||||||||||||||||||||
| Number of patients at risk |
||||||||||||||||||||||||||||||||||
| Enheritu (373) |
373 |
365 |
325 |
295 |
290 |
272 |
238 |
217 |
201 |
183 |
156 |
142 |
118 |
100 |
88 |
81 |
71 |
53 |
42 |
35 |
32 |
21 |
18 |
15 |
8 |
4 |
4 |
1 |
1 |
0 |
||||
| Chemotherapy (184) |
184 |
166 |
119 |
93 |
90 |
73 |
60 |
51 |
45 |
34 |
32 |
29 |
26 |
22 |
15 |
13 |
9 |
5 |
4 |
3 |
1 |
1 |
1 |
1 |
1 |
1 |
0 |
|||||||
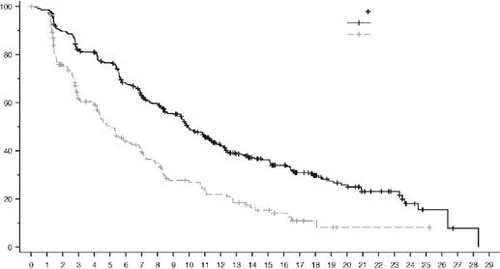
NSCLC (Non-Small Cell Lung Cancer)
DESTINY-Lung02 Study (NCT04644237)
The efficacy and safety of Enhertu were evaluated in the DESTINY-Lung02 study—a phase 2, randomized trial assessing two dose levels. Treatment assignment was blinded to both patients and investigators. The study enrolled adult patients with metastatic NSCLC harboring HER2 gene mutations who had received at least one platinum-based chemotherapy regimen. Activating HER2 (ERBB2) mutations were prospectively identified in tumor tissue by local laboratories using a validated test such as next-generation sequencing, polymerase chain reaction, or mass spectrometry. Patients were randomized in a 2:1 ratio to receive Enhertu at a dose of 5.4 mg/kg or 6.4 mg/kg every 3 weeks. Randomization was stratified by prior treatment with a programmed death protein 1 (PD-1) and/or programmed death-ligand 1 (PD-L1) antibody (yes or no). Treatment continued until disease progression, death, withdrawal of consent, or development of unacceptable toxicity. Patients with a history of glucocorticoid treatment for ILD/pneumonitis or with ILD/pneumonitis at screening, as well as those with a history of clinically significant heart disease, were excluded. Patients with untreated brain metastases or brain metastases associated with clinical symptoms or an ECOG performance status >1 were also excluded.
The primary efficacy endpoint was confirmed ORR assessed by BICR according to RECIST criteria, version 1.1. A secondary efficacy endpoint was DOR.
Demographic and baseline disease characteristics of the 102 participants receiving the 5.4 mg/kg dose were as follows: median age 59.4 years (range 31 to 84 years); female gender (63.7%); race: Mongoloid (63.7%), Caucasian (22.5%), other (13.7%); ECOG performance status of 0 (28.4%) or 1 (71.6%); 97.1% had a mutation in the ERBB2 kinase domain, 2.9% in the extracellular domain; 96.1% had a HER2 mutation in exon 19 or exon 20; 34.3% had stable brain metastases; 46.1% were former smokers, none were current smokers at study entry; 21.6% had previously undergone lung resection. Among patients with metastatic disease, 32.4% had received more than two prior lines of systemic therapy, 100% had received platinum-based chemotherapy, 73.5% had received anti-PD-1/PD-L1 therapy, and 50.0% had received platinum-based chemotherapy combined with anti-PD-1/PD-L1 therapy.
The efficacy results are summarized in Table 5. The median duration of follow-up was 11.5 months (data cutoff date: December 23, 2022).
Table 5: Efficacy Results in the DESTINY-Lung02 Study
| Measure of efficacy |
DESTINY-Lung02 5.4 mg/kg N = 102 |
| Confirmed objective response rate (ORR) by BICR assessment |
|
| n (%) |
50 (49.0) |
| (95 % CI)* |
(39.0; 59.1) |
| Complete response (CR), n (%) |
1 (1.0) |
| Partial response (PR), n (%) |
49 (48.0) |
| Duration of response |
|
| Median, months (95 % CI) † |
16.8 (6.4; NE) |
* 95% CI, calculated using the Clopper-Pearson method.
CI — confidence interval, NR — not reached.
† 95% CI, calculated using the Brookmeyer-Crowley method.
Gastric Cancer
DESTINY-Gastric02 Study (NCT04014075)
The efficacy and safety of the medicinal product Enhertu were evaluated in the DESTINY-Gastric02 study—a multicenter, randomized, open-label, non-comparative phase 2 trial conducted at investigational sites in Europe and the United States. The study enrolled patients with locally advanced or metastatic HER2-positive adenocarcinoma of the stomach or gastroesophageal junction (GEJ) who had experienced disease progression on prior trastuzumab-based therapy. Patients were required to have centrally confirmed HER2-positive status defined as IHC 3+ or IHC 2+/ISH-positive. Patients with a history of glucocorticoid treatment for ILD/pneumonitis or with ILD/pneumonitis at screening, patients with a history of clinically significant heart disease, and patients with active brain metastases were excluded from the study. Enhertu was administered as an intravenous infusion at a dose of 6.4 mg/kg once every three weeks until disease progression, death, withdrawal of consent, or development of unacceptable toxicity. The primary efficacy endpoint was confirmed ORR by ICR assessment according to RECIST criteria, version 1.1. Secondary endpoints included DOR and PFS.
Demographic characteristics and baseline disease characteristics of the 79 participants in the DESTINY-Gastric02 study were as follows: median age 61 years (range 20 to 78 years); 72% of patients were male; 87% were White, 5.0% were Asian, and 1.0% were Black or African American. Patients had an ECOG performance status of 0 (37%) or 1 (63%); 34% had gastric adenocarcinoma and 66% had GEJ adenocarcinoma; 86% had IHC 3+, 13% had IHC 2+/ISH-positive, and 63% had liver metastases.
Efficacy results based on ORR and DOR are summarized in Table 6.
Table 6: Efficacy results in the DESTINY-Gastric02 study (full analysis set*)
| Objective response rate |
DESTINY-Gastric02 N = 79 |
| Data cutoff date: November 8, 2021 |
|
| Confirmed objective response rate† |
|
| % (95 % CI)‡ |
41.8 (30.8; 53.4) |
| Complete response, n (%) |
4 (5.1) |
| Partial response, n (%) |
29 (36.7) |
| Duration of response |
|
| Median§, months (95 % CI)¶ |
8.1 (5.9; NE) |
NPO — not evaluable.
* Includes all patients who received at least one dose of Enhertu.
† By independent central review under blinded conditions.
‡ Calculated using the Clopper–Pearson method.
§ Based on Kaplan–Meier estimation.
¶ Calculated using the Breslow–Crowley method.
Study DESTINY-Gastric01 (NCT03329690)
The efficacy and safety of Enhertu were evaluated in study DESTINY-Gastric01, a multicenter, randomized, open-label, phase 2 study conducted at investigational sites in Japan and South Korea. This confirmatory study included adult patients with locally advanced or metastatic HER2-positive adenocarcinoma of the stomach or gastroesophageal junction (GEJ), whose disease had progressed on at least two prior regimens, including trastuzumab, a fluoropyrimidine, and a platinum-containing agent. Patients were randomized in a 2:1 ratio to receive Enhertu (N = 126) or chemotherapy of physician’s choice: irinotecan (N = 55) or paclitaxel (N = 7). Tumor samples were required to have confirmed HER2-positive status at a central laboratory, defined as IHC 3+ or IHC 2+/ISH-positive. Patients with a history of glucocorticoid treatment for ILD/pneumonitis or with ILD/pneumonitis at screening, patients with a history of clinically significant heart disease, and patients with active brain metastases were excluded from the study. Treatment continued until disease progression, death, withdrawal of consent, or development of unacceptable toxicity. The primary efficacy endpoint was investigator-assessed ORR according to RECIST criteria, version 1.1. Secondary efficacy endpoints included overall survival (OS), progression-free survival (PFS), duration of response (DoR), and confirmed ORR.
Demographic and baseline disease characteristics were similar across treatment groups. The median age of the 188 patients was 66 years (range 28 to 82 years); 76% of participants were male; 100% were of Mongoloid race. Patients had an ECOG performance status of 0 (49%) or 1 (51%); 87% had gastric adenocarcinoma, 13% had GEJ adenocarcinoma; 76% had IHC 3+ and 23% had IHC 2+/ISH-positive status; 54% had liver metastases; 29% had lung metastases; sum of diameters of all target lesions was < 5 cm in 47%, ≥ 5 to < 10 cm in 30%, and ≥ 10 cm in 17%; 55% had received two, and 45% had received three or more prior regimens for locally advanced or metastatic disease.
Efficacy results (data cutoff date June 3, 2020) showed confirmed ORR of 39.7% (95% CI 31.1; 48.8) for Enhertu (n = 126) compared to 11.3% (95% CI 4.7; 21.9) for chemotherapy of physician’s choice (n = 62). The number of patients with complete response was 7.9% and 0%, and with partial response was 31.7% and 11.3%, respectively. Additional efficacy results for Enhertu compared to chemotherapy of physician’s choice included median DoR of 12.5 months (95% CI 5.6; NPO) versus 3.9 months (95% CI 3.0; 4.9). Median PFS was 5.6 months (95% CI 4.3; 6.9) versus 3.5 months (95% CI 2.0; 4.3; hazard ratio = 0.47 [95% CI 0.31; 0.71]). An OS analysis, pre-specified at 133 death events, showed improved survival in the Enhertu group compared to the chemotherapy of physician’s choice group (hazard ratio = 0.60). Median OS was 12.5 months (95% CI 10.3; 15.2) in the Enhertu group and 8.9 months (95% CI 6.4; 10.4) in the chemotherapy of physician’s choice group.
Pediatric population
The European Medicines Agency has waived the obligation to submit the results of studies in all pediatric subgroups with breast cancer, NSCLC, and gastric cancer (information on the use of the medicinal product in the pediatric population is provided in the section «Paediatric population»).
This medicinal product has been authorized under a so-called conditional approval scheme. This means that additional evidence for this medicinal product is expected.
The European Medicines Agency will review any new information annually, and the manufacturer will update this information as necessary.
Pharmacokinetics
Absorption
Trastuzumab deruxtecan is administered intravenously. Other routes of administration have not been studied.
Distribution
Population pharmacokinetic analysis estimated the central compartment volume of distribution (Vc) of trastuzumab deruxtecan and the topoisomerase I inhibitor (DXd) to be 2.68 L and 28.0 L, respectively.
The mean in vitro plasma protein binding of DXd in human plasma was approximately 97%.
The in vitro blood-to-plasma concentration ratio of DXd was approximately 0.6.
Biotransformation
Trastuzumab deruxtecan undergoes intracellular cleavage by lysosomal enzymes, releasing DXd.
The humanized IgG1 monoclonal antibody to the HER2 receptor is expected to be degraded into small peptides and amino acids via catabolic pathways similar to endogenous IgG.
In vitro metabolism studies in human liver microsomes indicate that DXd is predominantly metabolized via oxidative pathways involving CYP3A4.
Elimination
Population pharmacokinetic analysis shows that after intravenous administration of trastuzumab deruxtecan to patients with metastatic HER2-positive breast cancer, breast cancer with low HER2 expression, or HER2-mutated NSCLC, the clearance of trastuzumab deruxtecan was 0.4 L/day and the clearance of DXd was 18.4 L/hour. In patients with locally advanced or metastatic gastric or GEJ adenocarcinoma, the clearance of trastuzumab deruxtecan was 20% higher than in patients with metastatic HER2-positive breast cancer. During cycle 3, the elimination half-life (t1/2) of trastuzumab deruxtecan and released DXd was approximately 7 days. Moderate accumulation of trastuzumab deruxtecan was observed (approximately 35% higher during cycle 3 compared to cycle 1).
After intravenous administration of DXd in rats, the primary route of elimination was biliary excretion with fecal elimination. DXd was the predominant component detected in urine, feces, and bile. After intravenous administration of a single dose of trastuzumab deruxtecan (6.4 mg/kg) to monkeys, unchanged DXd was the predominant component detected in urine and feces. The elimination of DXd in humans has not been studied.
Interactions in vitro
Effect of Enhertu on the pharmacokinetics of other medicinal products
In vitro studies show that DXd does not inhibit major CYP450 enzymes, including CYP1A2, 2B6, 2C8, 2C9, 2C19, 2D6, and 3A. In vitro studies show that DXd does not inhibit transporters OAT1, OAT3, OCT1, OCT2, OATP1B1, OATP1B3, MATE1, MATE2-K, P-gp, BCRP, and BSEP.
Effect of other medicinal products on the pharmacokinetics of Enhertu
In vitro, DXd was a substrate of P-gp, OATP1B1, OATP1B3, MATE2-K, MRP1, and BCRP.
Clinically significant interactions with medicinal products that are inhibitors of MATE2-K, MRP1, P-gp, OATP1B, or BCRP transporters are not expected (see section «Interaction with other medicinal products and other forms of interaction»).
Linearity/non-linearity
Exposure to trastuzumab deruxtecan and released DXd increased proportionally with dose following intravenous administration over the dose range of 3.2 mg/kg to 8.0 mg/kg (approximately 0.6 to 1.5 times the recommended dose), with low to moderate inter-individual variability. Population pharmacokinetic analysis showed inter-individual variability in clearance values of trastuzumab deruxtecan and DXd to be 24% and 28%, respectively, and in central volume of distribution to be 16% and 55%, respectively. Inter-individual variability in AUC (area under the concentration-time curve) values of trastuzumab deruxtecan and DXd was approximately 8% and 14%, respectively.
Special patient groups
Based on population pharmacokinetic analysis, age (20–96 years), race and ethnicity, sex, and patient body weight had no clinically significant effect on exposure to trastuzumab deruxtecan or released DXd.
Elderly patients
Population PK analysis showed that patient age (range 20–96 years) did not affect the PK of trastuzumab deruxtecan.
Renal impairment
No dedicated studies in patients with renal impairment were conducted. Results from population pharmacokinetic analysis including patients with mild (creatinine clearance [CrCl] ≥ 60 to < 90 mL/min) or moderate (CrCl ≥ 30 to < 60 mL/min; CrCl calculated using the Cockcroft–Gault formula) renal impairment showed that the pharmacokinetics of released DXd were not altered in patients with mild to moderate renal impairment compared to those with normal renal function (CrCl ≥ 90 mL/min).
Hepatic impairment
No dedicated studies in patients with hepatic impairment were conducted. Based on population pharmacokinetic analysis, the effect on changes in pharmacokinetics of trastuzumab deruxtecan in patients with total bilirubin ≤ 1.5 times ULN, regardless of AST level, is not clinically significant. Data in patients with total bilirubin > 1.5 to 3 times ULN, regardless of AST level, are insufficient to draw conclusions, and no data are available in patients with total bilirubin > 3 times ULN, regardless of AST level (see sections «Posology and method of administration» and «Special warnings and precautions for use»).
Children
Pharmacokinetic studies of trastuzumab deruxtecan in children have not been conducted.
Clinical characteristics
Indications
Breast cancer
HER2-positive breast cancer
Monotherapy with the medicinal product Enhertu is indicated for the treatment of adult patients with unresectable or metastatic HER2-positive breast cancer who have previously received one or more anti-HER2-based regimens.
Breast cancer with low HER2 expression
Monotherapy with the medicinal product Enhertu is indicated for the treatment of adult patients with unresectable or metastatic breast cancer with low HER2 expression who have previously received chemotherapy for metastatic disease or who have developed recurrence during or within 6 months after completing adjuvant chemotherapy (see section "Dosage and administration").
Non-small cell lung cancer (NSCLC)
Monotherapy with the medicinal product Enhertu is indicated for the treatment of adult patients with advanced NSCLC whose tumours have HER2 (ERBB2) activating mutations and who require systemic therapy following platinum-based chemotherapy with or without immunotherapy.
Gastric cancer
Monotherapy with the medicinal product Enhertu is indicated for the treatment of adult patients with advanced HER2-positive adenocarcinoma of the stomach or gastroesophageal junction (GEJ) who have previously been treated with a trastuzumab-based regimen.
Contraindications
Hypersensitivity to the active substance or to any of the excipients listed in the section "Composition".
Interaction with other medicinal products and other forms of interaction
Concomitant administration with ritonavir, an inhibitor of OATP1B, CYP3A and P-gp, or with itraconazole, a strong inhibitor of CYP3A and P-gp, did not result in a clinically relevant (approximately 10–20%) increase in exposure to trastuzumab deruxtecan or the released topoisomerase I inhibitor DXd. Dose adjustment is not required when trastuzumab deruxtecan is administered concomitantly with medicinal products that are not inhibitors of CYP3A, OATP1B, or P-gp transporters (see section "Pharmacological properties").
Special precautions for use
To avoid medication errors, it is important to check the labels on the vials and ensure that the medicinal product prepared for administration is Enhertu (trastuzumab deruxtecan) and not trastuzumab or trastuzumab emtansine.
Traceability
In order to facilitate the traceability of biological medicinal products, it is necessary to clearly record the name and batch number of the administered medicinal product in the patient's medical documentation.
Interstitial lung disease/Pneumonitis
Cases of interstitial lung disease (ILD) and/or pneumonitis have been reported with the use of Enhertu (see section "Adverse reactions"). Fatal outcomes have been reported. Patients should be advised to immediately report symptoms such as cough, dyspnea, fever, and/or any new or worsening respiratory symptoms. Patients should be monitored for signs and symptoms of ILD/pneumonitis. Prompt evaluation should be performed if signs of ILD/pneumonitis are present. Patients suspected of having ILD/pneumonitis should undergo chest X-ray and preferably computed tomography (CT). Consultation with a pulmonologist should be considered. For asymptomatic ILD/pneumonitis (Grade 1 severity), corticosteroid therapy (e.g., prednisolone at a dose of ≥ 0.5 mg/kg/day or equivalent) should be considered. Administration of Enhertu should be withheld until the adverse reaction resolves to Grade 0, after which treatment may be resumed according to the recommendations in Table 8 (see section "Dosage and administration"). For symptomatic ILD/pneumonitis (Grade 2 or higher severity), systemic corticosteroid therapy should be initiated immediately (e.g., prednisolone at a dose of ≥ 1 mg/kg/day or equivalent) and continued for at least 14 days, followed by a gradual taper over at least 4 weeks. Upon confirmed diagnosis of symptomatic ILD/pneumonitis (Grade 2 or higher severity), Enhertu should be permanently discontinued (see section "Dosage and administration"). Patients with a history of ILD/pneumonitis or those with moderate or severe renal impairment may have an increased risk of developing ILD/pneumonitis and should be under close medical supervision (see section "Dosage and administration").
Neutropenia
In clinical trials, neutropenia, including febrile neutropenia with fatal outcomes, has been reported with the use of Enhertu. Complete blood counts should be monitored before initiating Enhertu, prior to each subsequent dose, and as clinically indicated. Depending on the severity of neutropenia, treatment interruption or dose reduction of Enhertu may be required (see section "Dosage and administration").
Left ventricular ejection fraction reduction
Decreased left ventricular ejection fraction (LVEF) has been observed with anti-HER2 medicinal products. Standard cardiac function assessment (echocardiogram or MUGA scan [radionuclide ventriculography]) should be performed to evaluate LVEF before initiating Enhertu and at regular intervals during treatment, as clinically indicated. Decreased LVEF should be managed by interrupting treatment. Enhertu should be permanently discontinued if LVEF is confirmed to be less than 40% or if there is an absolute decrease in LVEF of more than 20% from baseline. Enhertu should be permanently discontinued in patients with symptomatic congestive heart failure (CHF) (see Table 8 in section "Dosage and administration").
Embryo-fetal toxicity
Administration of Enhertu during pregnancy can cause harm to the fetus. Post-marketing reports indicate that the use of trastuzumab, a HER2 receptor blocker, during pregnancy has resulted in oligohydramnios, which led to fatal pulmonary hypoplasia, skeletal abnormalities, and neonatal death. Based on animal studies and the known mechanism of action of Enhertu, the topoisomerase I inhibitor DXd component is also expected to cause harm to the embryo and fetus when administered to a pregnant woman (see section "Pregnancy and breastfeeding").
Pregnancy should be excluded before initiating Enhertu in women of reproductive potential. Patients should be informed of the potential risks to the fetus. Women of reproductive potential should be advised to use effective contraception during treatment and for at least 7 months after the last dose of Enhertu. Men receiving Enhertu who are sexually active with women of reproductive potential should be advised to use effective contraception during treatment and for at least 4 months after the last dose of Enhertu (see section "Pregnancy and breastfeeding").
Patients with moderate or severe hepatic impairment
Data in patients with moderate hepatic impairment are limited, and data in patients with severe hepatic impairment are lacking. Since metabolism and biliary excretion are the primary elimination pathways of the topoisomerase I inhibitor (DXd), Enhertu should be used with caution in patients with moderate or severe hepatic impairment (see sections "Dosage and administration" and "Pharmacological properties").
Pregnancy and breastfeeding
Women of reproductive potential/Contraception in men and women
Pregnancy should be excluded before initiating Enhertu in women of reproductive potential.
Women of reproductive potential must use effective contraception during therapy with Enhertu and for at least 7 months after completion of therapy.
Men receiving Enhertu who are sexually active with women of reproductive potential should be advised to use effective contraception during therapy and for at least 4 months after completion of therapy.
Pregnancy
There are no data on the use of Enhertu in pregnant women. However, the use of trastuzumab, a HER2 receptor blocker, during pregnancy may cause harm to the fetus. Post-marketing reports indicate that the use of trastuzumab during pregnancy has resulted in oligohydramnios, which in some cases led to fatal pulmonary hypoplasia, skeletal abnormalities, and neonatal death. Based on animal studies and the known mechanism of action of Enhertu, the topoisomerase I inhibitor DXd component is also expected to cause harm to the embryo and fetus when administered to a pregnant woman.
Enhertu is not recommended for use in pregnant women. Patients should be informed of the potential risks to the fetus before planning pregnancy. If pregnancy occurs, the woman should immediately consult her physician. If a patient becomes pregnant during treatment or within 7 months after the last dose of Enhertu, close monitoring is recommended.
Breastfeeding
It is unknown whether trastuzumab deruxtecan is excreted in human breast milk. Human IgG is excreted in human breast milk, and the likelihood of absorption and serious adverse reactions in the infant is unknown. Therefore, women should not breastfeed during treatment with Enhertu and for 7 months after the last dose. The decision on whether to discontinue breastfeeding or to discontinue therapy with Enhertu should be made, taking into account the benefits of breastfeeding for the child and/or the benefits of therapy with Enhertu for the woman.
Fertility
No specific fertility studies with trastuzumab deruxtecan have been conducted. Based on animal toxicity studies, Enhertu may impair reproductive function and fertility. It is unknown whether trastuzumab deruxtecan or its metabolites are excreted in semen. Men should be advised to seek counseling regarding sperm cryopreservation prior to starting treatment. Men should not donate sperm for cryopreservation or act as donors during the entire treatment period and for at least 4 months after the last dose of Enhertu.
Ability to drive and use machines
Enhertu may have a minor influence on the ability to drive and use machines. Patients should be advised to exercise caution when driving or operating machinery if they experience increased fatigue, headache, or dizziness during treatment with Enhertu (see section "Adverse reactions").
Method of Administration and Dosage
The medicinal product Enhertu must be prescribed by a physician and administered under the supervision of a healthcare professional experienced in the use of antineoplastic medicinal products. To prevent medication errors, it is essential to check the labels on the vials and confirm that the medicinal product prepared for administration is Enhertu (trastuzumab deruxtecan), not trastuzumab or trastuzumab emtansine.
Enhertu must not be substituted with trastuzumab or trastuzumab emtansine.
Patient Selection
HER2-positive Breast Cancer
Patients receiving trastuzumab deruxtecan for the treatment of breast cancer must have documented HER2-positive tumor status, defined by immunohistochemical (IHC) assay as 3+ or by in situ hybridization (ISH) assay with a ratio of ≥ 2.0, or by fluorescence in situ hybridization (FISH), assessed using an in vitro diagnostic (IVD) medical device with CE marking. If a CE-marked IVD is unavailable, HER2 status should be evaluated using an alternative validated test.
Breast Cancer with Low HER2 Expression
Patients receiving trastuzumab deruxtecan must have documented tumor status with low HER2 expression, defined by IHC as 1+ or IHC 2+/ISH-negative, assessed using an IVD medical device. If a CE-marked IVD is unavailable, HER2 status should be evaluated using an alternative validated test (see section "Pharmacodynamics").
NSCLC
Patients receiving trastuzumab deruxtecan for the treatment of metastatic NSCLC must have tumors harboring an activating HER2 (ERBB2) mutation, detected using a CE-marked in vitro diagnostic (IVD) medical device. If a CE-marked IVD is unavailable, HER2 mutation status should be evaluated using an alternative validated test.
Gastric Cancer
Patients receiving trastuzumab deruxtecan for the treatment of gastric or gastroesophageal junction cancer must have documented HER2-positive tumor status, defined by immunohistochemical (IHC) assay as 3+ or by in situ hybridization (ISH) assay with a ratio of ≥ 2.0, or by fluorescence in situ hybridization (FISH), assessed using an in vitro diagnostic (IVD) medical device with CE marking. If a CE-marked IVD is unavailable, HER2 status should be evaluated using an alternative validated test.
Dosage
Breast Cancer
The recommended dose of Enhertu is 5.4 mg/kg administered as an intravenous infusion every 3 weeks (21-day treatment cycles) until disease progression or occurrence of intolerable toxicity.
NSCLC
The recommended dose of Enhertu is 5.4 mg/kg administered as an intravenous infusion every 3 weeks (21-day treatment cycles) until disease progression or occurrence of intolerable toxicity.
Gastric Cancer
The recommended dose of Enhertu is 6.4 mg/kg administered as an intravenous infusion every 3 weeks (21-day treatment cycles) until disease progression or occurrence of intolerable toxicity.
The initial dose should be administered as an intravenous infusion over 90 minutes. If the previous infusion was well tolerated, subsequent infusions of Enhertu may be given over 30 minutes.
If infusion-related symptoms occur, the infusion rate should be reduced or administration of Enhertu discontinued (see section "Adverse Reactions"). In the event of severe infusion-related reactions, administration of Enhertu should be permanently discontinued.
Pre-medication
Enhertu is emetogenic (see section "Adverse Reactions"), with potential for delayed nausea and/or vomiting. Prior to each administration of Enhertu, patients should receive premedication with a combination of two or three agents (e.g., dexamethasone in combination with a 5-HT3 receptor antagonist and/or an NK1 receptor antagonist, as well as other necessary medications) to prevent chemotherapy-induced nausea and vomiting.
Dose Modifications
Management of adverse reactions may require temporary interruption of treatment, dose reduction, or discontinuation of Enhertu therapy according to the recommendations outlined in Tables 7 and 8.
Once a dose reduction of Enhertu has been implemented, the dose should not be re-escalated.
Table 7: Dose Reduction Scheme
| Dose reduction schedule |
Breast cancer and gastric cancer |
Gastric cancer |
| Recommended initial dose |
5.4 mg/kg |
6.4 mg/kg |
| First dose reduction |
4.4 mg/kg |
5.4 mg/kg |
| Second dose reduction |
3.2 mg/kg |
4.4 mg/kg |
| Need for further dose reduction |
Discontinuation of treatment |
Discontinuation of treatment |
Table 8: Dose adjustment in case of adverse reactions
| Adverse reaction |
Severity grade |
Treatment modification |
|
| Interstitial lung disease (ILD)/pneumonitis |
Asymptomatic ILD/pneumonitis (Grade 1) |
Withhold ENHERTU therapy until the severity of the adverse reaction decreases to Grade 0, then:
|
|
| Symptomatic ILD/pneumonitis (Grade 2 or higher) |
|
||
| Neutropenia |
Grade 3 (less than 1.0–0.5 × 10⁹/L) |
|
|
| Grade 4 (less than 0.5 × 10⁹/L) |
|
||
| Febrile neutropenia |
Absolute neutrophil count less than 1.0 × 10⁹/L and body temperature above 38.3°C or sustained temperature ≥38°C for more than one hour. |
|
|
| Left ventricular ejection fraction (LVEF) decrease |
LVEF >45% and an absolute decrease from baseline of 10–20% |
|
|
| LVEF 40% to 45% |
and absolute decrease from baseline of less than 10% |
|
|
| and absolute decrease from baseline of 10–20% |
|
||
| LVEF <40% or absolute decrease from baseline >20% |
|
||
| Symptomatic congestive heart failure (CHF) |
|
||
The severity of toxicity is indicated according to the Common Terminology Criteria for Adverse Events, version 5.0 (NCI-CTCAE, version 5.0).
Dose delay or missed dose
If administration of the scheduled dose is delayed or missed, it should be administered as soon as possible, without waiting for the next planned cycle. The schedule of subsequent doses should be adjusted to maintain a 3-week interval between doses. The medicinal product should be administered at the same dose and infusion rate as the patient last tolerated well.
Special patient populations
Elderly patients
No dose adjustment of Enhertu is required for patients aged 65 years and older. Limited data are available on the use of Enhertu in patients aged ≥ 75 years.
Renal impairment
No dose adjustment is required for patients with mild (creatinine clearance [CrCl] ≥ 60 and < 90 mL/min) or moderate (CrCl ≥ 30 and < 60 mL/min) renal impairment (see section “Pharmacological properties”). The potential need for dose adjustment in patients with severe renal impairment or end-stage renal disease cannot be determined, as severe renal impairment was an exclusion criterion in clinical studies. Patients with moderate renal impairment more frequently experienced ILD/pneumonitis of grade 1 or 2 severity. In patients with moderate baseline renal impairment receiving Enhertu at a dose of 6.4 mg/kg, the frequency of serious adverse reactions was higher than in patients with normal renal function. Patients with moderate or severe renal impairment should be closely monitored for the development of adverse reactions, particularly ILD/pneumonitis (see section “Special precautions for use”).
Hepatic impairment
No dose adjustment is required for patients with total bilirubin concentration ≤ 1.5 times the upper limit of normal (ULN), regardless of aspartate aminotransferase (AST) levels. The potential need for dose adjustment in patients with total bilirubin concentration > 1.5 times ULN, regardless of AST levels, cannot be determined due to limited data; therefore, these patients should be closely monitored (see sections “Special precautions for use” and “Pharmacological properties”).
Method of administration
Enhertu is intended for intravenous administration. Preparation of the concentrate, dilution, and administration of this medicinal product as an intravenous infusion must be performed by a healthcare professional. Enhertu must not be administered as an intravenous push or bolus injection.
Instructions for preparation of the concentrate and dilution of the medicinal product prior to administration
To avoid medication errors, it is essential to check the labels on the vials and confirm that Enhertu (trastuzumab deruxtecan) is being prepared, not trastuzumab or trastuzumab emtansine.
Appropriate procedures for handling cytotoxic medicinal products should be followed. Necessary aseptic conditions must also be maintained during the reconstitution and dilution procedures described below.
Preparation of the concentrate for infusion solution
- Reconstitution should be performed immediately before dilution.
- More than one vial of sterile lyophilized powder may be required to achieve the full dose. Calculate the dose (in milligrams), the total volume of Enhertu concentrate needed, and the number of Enhertu vials required.
- To reconstitute the vial to obtain a dose of 100 mg trastuzumab deruxtecan, slowly add 5 mL of water for injections to the vial using a sterile syringe to achieve a final concentration of 20 mg/mL.
- Gently swirl the vial contents until complete reconstitution is achieved. Do not shake.
- If the prepared Enhertu concentrate is not used immediately, it should be stored in a refrigerator at 2–8°C for up to 24 hours from the time of reconstitution, protected from light. Do not freeze.
- The reconstituted medicinal product contains no preservatives and is intended for single use only.
Dilution
- The calculated volume of concentrate should be withdrawn from the vial(s) using a sterile syringe. Inspect the concentrate for the presence of particulate matter and discoloration. The solution should be clear and colorless or pale yellow. Do not use the solution if particulate matter is present or if the solution appears cloudy or discolored.
- The calculated volume of Enhertu concentrate should be diluted in an infusion bag containing 100 mL of 5% glucose solution. Do not use sodium chloride solution (see section “Incompatibilities”). It is recommended to use an infusion bag made of polyvinyl chloride or polyolefin (ethylene and polypropylene copolymer).
- Gently invert the infusion bag to ensure thorough mixing. Do not shake.
- The infusion bag should be covered to protect from light.
- If the prepared solution is not used immediately, it should be stored at room temperature for no more than 4 hours, including the time for concentrate preparation and infusion, or in a refrigerator at 2–8°C for up to 24 hours, protected from light. Do not freeze.
- Any unused portion remaining in the vial should be discarded.
Administration
- If the prepared infusion solution has been stored in a refrigerator (2–8°C), it is recommended to allow the solution to reach room temperature before administration, protected from light.
- Enhertu should be administered as an intravenous infusion using only an in-line filter (0.20 or 0.22 µm) made of polyethersulfone (PES) or polysulfone (PS).
- The initial dose should be administered as a 90-minute intravenous infusion. If the previous infusion was well tolerated, subsequent infusions of Enhertu may be administered over 30 minutes. Enhertu must not be administered as an intravenous push or bolus injection.
- The infusion bag should be covered to protect from light.
- Enhertu must not be mixed with other medicinal products, and other medicinal products should not be administered through the same intravenous infusion system.
Disposal
Any unused medicinal product and waste materials should be disposed of in accordance with local requirements.
Paediatric population
The safety and efficacy of Enhertu in children (under 18 years of age) have not yet been established. No data are available.
Overdose
The maximum tolerated dose of trastuzumab deruxtecan has not been determined. Single doses exceeding 8.0 mg/kg have not been studied in clinical trials. In the event of overdose, patients should be closely monitored for possible signs or symptoms of adverse reactions and, if necessary, receive symptomatic treatment.
Adverse Reactions
▼ This medicinal product is subject to additional monitoring. This will allow for rapid identification of new safety information. Healthcare professionals are asked to report any suspected adverse reactions.
Summary of safety profile
Enhertu 5.4 mg/kg
The safety of the medicinal product was evaluated in the pooled population of patients who received at least one dose of Enhertu 5.4 mg/kg (n = 1449) in clinical trials across various tumor types. The median duration of treatment in this pooled population was 9.8 months (range: 0.7–45.1 months).
The most common adverse reactions were nausea (75.0%), fatigue (57.3%), vomiting (42.1%), alopecia (37.6%), neutropenia (35.2%), constipation (35.0%), anemia (34.4%), decreased appetite (33.1%), diarrhea (28.8%), increased transaminase levels (26.5%), musculoskeletal pain (26.2%), thrombocytopenia (24.5%), and leukopenia (23.7%).
The most common adverse reactions of Grade 3 or 4 severity according to the National Cancer Institute Common Terminology Criteria for Adverse Events (NCI-CTCAE version 5.0) were neutropenia (17.0%), anemia (9.5%), fatigue (8.4%), leukopenia (6.4%), nausea (5.9%), thrombocytopenia (5.0%), lymphopenia (4.8%), hypokalemia (3.8%), increased transaminase levels (3.6%), vomiting (2.7%), diarrhea (2.0%), decreased appetite (1.7%), pneumonia (1.4%), and decreased ejection fraction (1.1%). Grade 5 adverse reactions occurred in 1.4% of patients, including ILD (1.0%).
Temporary discontinuation of treatment due to adverse reactions occurred in 34.3% of patients receiving Enhertu. The most common adverse reactions leading to temporary discontinuation were neutropenia (13.3%), fatigue (5.0%), anemia (4.7%), leukopenia (3.7%), thrombocytopenia (3.0%), upper respiratory tract infection (2.7%), and ILD (2.6%). Dose reductions occurred in 20.6% of patients receiving Enhertu. The most common adverse reactions leading to dose reduction were fatigue (5.0%), nausea (4.9%), neutropenia (3.5%), and thrombocytopenia (2.1%). Permanent discontinuation of treatment due to adverse reactions occurred in 13.0% of patients receiving Enhertu. The most common adverse reaction leading to permanent discontinuation was ILD (9.2%).
Enhertu 6.4 mg/kg
The safety population included patients who received at least one dose of Enhertu 6.4 mg/kg (n = 669) in clinical trials across various tumor types. The median duration of treatment in this pooled population was 5.7 months (range: 0.7–41.0 months).
The most common adverse reactions were nausea (72.2%), fatigue (58.4%), decreased appetite (53.5%), anemia (44.7%), neutropenia (43.5%), vomiting (40.1%), diarrhea (35.9%), alopecia (35.4%), constipation (32.3%), thrombocytopenia (30.8%), leukopenia (29.3%), and increased transaminase levels (24.2%).
The most common adverse reactions of Grade 3 or 4 severity according to the National Cancer Institute Common Terminology Criteria for Adverse Events (NCI-CTCAE version 5.0) were neutropenia (28.7%), anemia (22.6%), leukopenia (13.3%), thrombocytopenia (9.1%), fatigue (8.4%), decreased appetite (7.8%), lymphopenia (6.9%), nausea (5.8%), increased transaminase levels (4.3%), hypokalemia (4.3%), pneumonia (3.1%), febrile neutropenia (2.8%), vomiting (2.4%), diarrhea (2.2%), weight loss (1.9%), increased blood alkaline phosphatase levels (1.6%), interstitial lung disease (ILD, 1.5%), dyspnea (1.2%), decreased ejection fraction (1.2%), and increased blood bilirubin levels (1.2%). Grade 5 adverse reactions, including ILD (2.1%), occurred in 2.7% of patients.
Temporary discontinuation of treatment due to adverse reactions occurred in 40.7% of patients receiving Enhertu. The most common adverse reactions leading to temporary discontinuation were neutropenia (16.6%), anemia (7.8%), fatigue (5.7%), ILD (4.8%), leukopenia (4.2%), decreased appetite (3.7%), pneumonia (3.6%), upper respiratory tract infection (3.4%), and thrombocytopenia (3.1%). Dose reductions occurred in 31.1% of patients receiving Enhertu. The most common adverse reactions leading to dose reduction were fatigue (10.6%), neutropenia (6.6%), nausea (6.4%), decreased appetite (5.4%), and thrombocytopenia (3.0%). Permanent discontinuation of treatment due to adverse reactions occurred in 17.6% of patients receiving Enhertu. The most common adverse reaction leading to permanent discontinuation was ILD (12.9%).
25.3% of patients with gastric cancer who received Enhertu at a dose of 6.4 mg/kg (n = 229) received blood transfusions within 28 days following the onset of anemia or thrombocytopenia. Transfusions were primarily administered for anemia.
List of adverse reactions presented in tabular form
Adverse reactions observed in patients who received at least one dose of Enhertu in clinical trials are presented in Table 9. Adverse reactions are categorized by system organ classes (SOC) according to the Medical Dictionary for Regulatory Activities (MedDRA) and by frequency. Frequency is defined as follows: very common (≥ 1/10), common (≥ 1/100 to < 1/10), uncommon (≥ 1/1,000 to < 1/100), rare (≥ 1/10,000 to < 1/1,000), very rare (< 1/10,000), and not known (cannot be estimated from available data). Within each frequency grouping, adverse reactions are presented in order of decreasing severity.
Table 9: Adverse reactions in patients receiving trastuzumab deruxtecan at doses of 5.4 mg/kg and 6.4 mg/kg in various tumor types
| System Organ Class |
5.4 mg/kg |
6.4 mg/kg |
| Frequency category |
Adverse reaction |
Adverse reaction |
| Infections and infestations |
||
| Very common |
upper respiratory tract infectiona |
pneumonia, upper respiratory tract infectiona |
| Common |
pneumonia |
|
| Blood and lymphatic system disorders |
||
| Very common |
anemiab, neutropeniac, thrombocytopeniad, leukopeniaе, lymphopeniaf |
anemiab, neutropeniac, thrombocytopeniad, leukopeniaе, lymphopeniaf |
| Common |
febrile neutropenia |
|
| Uncommon |
febrile neutropenia |
|
| Metabolism and nutrition disorders |
||
| Very common |
hypokalemiaе, decreased appetite |
hypokalemiaе, decreased appetite |
| Common |
dehydration |
dehydration |
| Nervous system disorders |
||
| Very common |
headacheg, dizziness |
headacheg, dysgeusia |
| Common |
dysgeusia |
dizziness |
| Eye disorders |
||
| Common |
dry eyes, blurred visionh |
dry eyes, blurred visionh |
| Respiratory, thoracic and mediastinal disorders |
||
| Very common |
interstitial lung diseasei, dyspnea, cough, epistaxis |
interstitial lung diseasei, dyspnea, cough |
| Common |
epistaxis |
|
| Gastrointestinal disorders |
||
| Very common |
nausea, vomiting, constipation, diarrhea, abdominal painj, stomatitisк, dyspepsia |
nausea, vomiting, diarrhea, constipation, abdominal painj, stomatitisк |
| Common |
abdominal distension, gastritis, flatulence |
dyspepsia, abdominal distension, gastritis, flatulence |
| Hepatobiliary disorders |
||
| Very common |
increased transaminase levels |
increased transaminase levels |
| Skin and subcutaneous tissue disorders |
||
| Very common |
alopecia |
alopecia |
| Common |
rashl, pruritus, skin hyperpigmentationm |
rashl, pruritus, skin hyperpigmentationm |
| Musculoskeletal and connective tissue disorders |
||
| Very common |
musculoskeletal painn |
musculoskeletal painn |
| General disorders and administration site conditions |
||
| Very common |
fatigueo, pyrexia |
fatigueo, pyrexia, peripheral edema |
| Common |
peripheral edema |
|
| Investigations |
||
| Very common |
decreased ejection fractionp, decreased body weight |
decreased ejection fractionp, decreased body weight |
| Common |
increased blood alkaline phosphatase, increased blood bilirubin, increased blood creatinine |
increased blood alkaline phosphatase, increased blood bilirubin, increased blood creatinine |
| Injury, poisoning and procedural complications |
||
| Common |
infusion reactionsq |
infusion reactionsq |
a Includes influenza, influenza-like conditions, nasopharyngitis, pharyngitis, sinusitis, rhinitis, laryngitis, and infections of the upper respiratory tract.
b For all types of tumors at a dose of 5.4 mg/kg, anemia, decreased hemoglobin levels, decreased erythrocyte count, and decreased hematocrit levels were observed. For all types of tumors at a dose of 6.4 mg/kg, anemia, decreased hemoglobin levels, and decreased erythrocyte count were observed.
c Includes neutropenia and decreased neutrophil count.
d Includes thrombocytopenia and decreased platelet count.
e Includes leukopenia and decreased leukocyte count.
f Includes lymphopenia and decreased lymphocyte count.
g Includes hypokalemia and decreased blood potassium levels.
h For all types of tumors at a dose of 5.4 mg/kg, headache, sinus headache, and migraine were observed. For all types of tumors at a dose of 6.4 mg/kg, headache and migraine were observed.
i Includes blurred vision and visual disturbances.
j For all types of tumors at a dose of 5.4 mg/kg, interstitial lung disease includes events considered as ILD: pneumonitis (n = 88), interstitial lung disease (n = 72), organizing pneumonia (n = 6), pneumonia (n = 4), respiratory failure (n = 5), radiation pneumonitis (n = 2), alveolitis (n = 2), pulmonary toxicity (n = 2), fungal pneumonia (n = 1), lung tumor (n = 1), acute respiratory failure (n = 1), lung infiltration (n = 1), lymphangitis (n = 1), pulmonary fibrosis (n = 1), idiopathic interstitial pneumonia (n = 1), lung disease (n = 1), hypersensitivity pneumonitis (n = 1), and lung opacities (n = 1). For all types of tumors at a dose of 6.4 mg/kg, interstitial lung disease includes events considered as ILD: pneumonitis (n = 75), interstitial lung disease (n = 39), organizing pneumonia (n = 4), respiratory failure (n = 4), lung opacities (n = 2), pneumonia (n = 1), and radiation pneumonitis (n = 1).
k Includes abdominal discomfort, gastrointestinal pain, abdominal pain, lower abdominal pain, and upper abdominal pain.
l For all types of tumors at a dose of 5.4 mg/kg, stomatitis, aphthous ulcer, oral mucosal ulcer, erosion of the oral mucosa, and oral mucosal rash were observed. For all types of tumors at a dose of 6.4 mg/kg, only stomatitis was observed.
m Includes increased transaminases, increased alanine aminotransferase levels, increased aspartate aminotransferase levels, increased gamma-glutamyl transferase levels, liver function disorders, abnormal liver function test results, elevated liver function test parameters, and hypertransaminasemia.
n For all types of tumors at a dose of 5.4 mg/kg, rash, pustular rash, maculopapular rash, papular rash, macular rash, and pruritic rash were observed. For all types of tumors at a dose of 6.4 mg/kg, rash, pustular rash, maculopapular rash, and pruritic rash were observed.
o For all types of tumors at a dose of 5.4 mg/kg, skin hyperpigmentation, skin color changes, and pigmentary disturbances were observed. For all types of tumors at a dose of 6.4 mg/kg, skin hyperpigmentation and pigmentary disturbances were observed.
p Includes back pain, myalgia, limb pain, musculoskeletal pain, muscle spasms, bone pain, neck pain, chest musculoskeletal pain, and limb discomfort.
q Includes asthenia, fatigue, malaise, and somnolence.
r For all types of tumors at a dose of 5.4 mg/kg, the term "decreased ejection fraction" includes laboratory findings of decreased LVEF (n = 214) and/or the following preferred terms: decreased ejection fraction (n = 52), heart failure (n = 3), congestive heart failure (n = 1), and left ventricular dysfunction (n = 2). For all types of tumors at a dose of 6.4 mg/kg, the term "decreased ejection fraction" includes laboratory findings of decreased LVEF (n = 97) and/or the following preferred terms: decreased ejection fraction (n = 11) and left ventricular dysfunction (n = 1).
s For all types of tumors at a dose of 5.4 mg/kg, increased blood bilirubin levels, hyperbilirubinemia, increased conjugated bilirubin levels, and increased unconjugated bilirubin levels were observed. For all types of tumors at a dose of 6.4 mg/kg, increased blood bilirubin levels, hyperbilirubinemia, and increased conjugated bilirubin levels were observed.
t For all types of tumors at a dose of 5.4 mg/kg, infusion reactions included infusion reaction (n = 16) and hypersensitivity (n = 2). For all types of tumors at a dose of 6.4 mg/kg, infusion reactions included infusion reaction (n = 6) and hypersensitivity (n = 1). All infusion reactions were of Grade 1 or 2 severity.
Description of selected adverse reactions
Interstitial lung disease/Pneumonitis
In clinical studies involving multiple tumor types (n = 1449) with EnherTu administered at a dose of 5.4 mg/kg, ILD occurred in 12.5% of patients. Most cases of ILD were Grade 1 (3.2%) and Grade 2 (7.4%). Grade 3 cases were observed in 0.8% of patients, and no Grade 4 cases were reported. Grade 5 (fatal) events occurred in 1.0% of patients. The median time to first event was 5.5 months (range: 26 days to 31.5 months) (see sections "Dosage and administration" and "Special instructions").
In clinical studies involving multiple tumor types (n = 669) with EnherTu administered at a dose of 6.4 mg/kg, ILD occurred in 17.9% of patients. Most cases of ILD were Grade 1 (4.9%) and Grade 2 (9.4%). Grade 3 cases were observed in 1.3% of patients, and Grade 4 cases in 0.1% of patients. Grade 5 (fatal) events occurred in 2.1% of patients. One patient had pre-existing ILD that worsened during treatment and progressed to Grade 5 ILD (fatal). The median time to first event was 4.2 months (range: -0.5 to 21.0 months) (see sections "Dosage and administration" and "Special instructions").
Neutropenia
In clinical studies (n = 1449) involving multiple tumor types with EnherTu administered at a dose of 5.4 mg/kg, neutropenia was observed in 35.2% of patients, with 17.0% experiencing Grade 3 or 4 events. The median time to onset of neutropenia was 43 days (range: 1 day to 31.9 months), and the median duration of the first event was 22 days (range: 1 day to 17.1 months). Febrile neutropenia was reported in 0.9% of patients, and 0.1% had Grade 5 events (see section "Dosage and administration").
In clinical studies (n = 669) involving multiple tumor types with EnherTu administered at a dose of 6.4 mg/kg, neutropenia was observed in 43.5% of patients, with 28.7% experiencing Grade 3 or 4 events. The median time to onset was 16 days (range: 1 day to 24.8 months), and the median duration of the first event was 9 days (range: 2 days to 17.2 months). Febrile neutropenia was reported in 3.0% of patients, and 0.1% had Grade 5 events (see section "Dosage and administration").
Decreased left ventricular ejection fraction
In clinical studies involving multiple tumor types (n = 1449) with EnherTu administered at a dose of 5.4 mg/kg, decreased LVEF was reported in 57 patients (3.9%), of whom 10 (0.7%) had Grade 1 events, 40 (2.8%) had Grade 2, and 7 (0.5%) had Grade 3. Based on laboratory findings (echocardiogram or MUGA scan), the incidence of Grade 2 LVEF decrease was 202/1341 (15.1%), and Grade 3 was 12/1341 (0.9%). The use of EnherTu in patients with LVEF < 50% prior to treatment initiation has not been studied (see section "Dosage and administration").
In clinical studies involving multiple tumor types (n = 669) with EnherTu administered at a dose of 6.4 mg/kg, decreased LVEF was reported in 12 patients (1.8%), of whom 1 (0.1%) had Grade 1, 8 (1.2%) had Grade 2, and 3 (0.4%) had Grade 3. Based on laboratory findings (echocardiogram or MUGA scan), the incidence of Grade 2 LVEF decrease was 89/597 (14.9%), and Grade 3 was 8/597 (1.3%).
Infusion reactions
In clinical studies (n = 1449) involving multiple tumor types with EnherTu administered at a dose of 5.4 mg/kg, infusion reactions occurred in 18 patients (1.2%), all of which were Grade 1 or 2. No Grade 3 events were reported. In three cases (0.2%), infusion reactions led to temporary discontinuation of treatment; no events led to permanent discontinuation.
In clinical studies (n = 669) involving multiple tumor types with EnherTu administered at a dose of 6.4 mg/kg, infusion reactions occurred in 7 patients (1.0%), all of which were Grade 1 or 2. No Grade 3 events were reported. In one case (0.1%), an infusion reaction led to temporary discontinuation of treatment; no events led to permanent discontinuation.
Immunogenicity
As with all medicinal products of protein origin, EnherTu may be immunogenic. Across the dose range evaluated in clinical studies, anti-trastuzumab deruxtecan antibodies developed in 2.1% (47/2213) of patients after administration of EnherTu. The incidence of neutralizing antibodies to trastuzumab deruxtecan during treatment was 0.1% (2/2213). No correlation between antibody formation and allergic reactions was observed.
Pediatric population
The safety of EnherTu in this population has not been established.
Elderly patients
In clinical studies (n = 1449) involving multiple tumor types with EnherTu administered at a dose of 5.4 mg/kg, 24.2% of patients were aged 65 years or older, and 4.3% were aged 75 years or older. In patients aged 65 years or older (50.0%), a higher incidence of Grade 3–4 adverse reactions was observed compared to patients under 65 years (42.7%), leading to a higher number of treatment discontinuations due to adverse reactions.
Among 669 patients with multiple tumor types enrolled in clinical studies receiving EnherTu at a dose of 6.4 mg/kg, 39.2% were aged 65 years or older, and 7.6% were aged 75 years or older. The incidence of Grade 3–4 adverse reactions in patients aged 65 years or older was 59.9%, compared to 62.9% in younger patients. The incidence of Grade 3–4 adverse reactions was 64.7% in patients aged 75 years and 61.5% in patients under 75 years. Patients aged 75 years or older experienced a higher incidence of serious adverse reactions (37.3%) and fatal outcomes (7.8%) compared to patients under 75 years (20.7% and 2.3%, respectively). Limited safety data are available for patients aged 75 years or older.
Ethnic differences
No major differences in the effect or efficacy of this medicinal product were observed between patients of different ethnic groups in clinical studies. In Asian patients receiving EnherTu at a dose of 6.4 mg/kg, a higher incidence (difference ≥ 10%) of neutropenia (58.1% vs. 18.6%), anemia (51.1% vs. 32.4%), leukopenia (42.7% vs. 6.9%), thrombocytopenia (40.5% vs. 15.4%), and lymphopenia (17.6% vs. 7.3%) was observed compared to patients of other ethnic groups. Bleeding events within 14 days after onset of thrombocytopenia occurred in 4.3% of Asian patients compared to 1.6% of patients from other ethnic groups.
Reporting of adverse reactions
Reporting of adverse reactions after marketing authorization is of great importance. It allows continuous monitoring of the benefit-risk ratio of the medicinal product. Healthcare and pharmacy professionals, as well as patients or their legal representatives, should report all suspected adverse reactions and lack of efficacy through the Automated Pharmacovigilance Information System at: https://aisf.dec.gov.ua.
Shelf life
48 months.
Proposed shelf life (after reconstitution and dilution)
Reconstituted solution
Chemical and physical in-use stability has been demonstrated for 24 hours at 2 to 8 °C.
From a microbiological standpoint, the medicinal product should be used immediately after reconstitution of the vial contents. If not used immediately, responsibility for the duration and conditions of storage prior to use lies with the user. If reconstitution was not performed under controlled and validated aseptic conditions, the storage time should generally not exceed 24 hours at 2 to 8 °C.
Diluted solution
The diluted solution should preferably be used immediately. If not used immediately, the reconstituted solution diluted in infusion bags with 5% glucose solution may be stored at room temperature (≤ 30 °C) for up to 4 hours or in a refrigerator at 2 to 8 °C for up to 24 hours, protected from light. The specified storage periods begin from the time of reconstitution.
Storage conditions
Store in a refrigerator at 2 to 8 °C.
Do not freeze.
Keep out of reach of children.
Incompatibilities
Data on compatibility of EnherTu with other medicinal products are lacking; therefore, this medicinal product should not be mixed with other medicinal products except those specified in the section "Dosage and administration."
Sodium chloride infusion solution should not be used for reconstitution or dilution, as it may cause formation of particulate matter.
Packaging
Sterile lyophilized powder for concentrate for infusion solution for single use in a glass vial stoppered with a rubber plug and sealed with a flip-off crimp cap; 1 vial per cardboard box labeled in Ukrainian.
Prescription status
Prescription only.
Manufacturer
Manufacturer: Daiichi Sankyo Europe GmbH.
Manufacturer's location and address of place of business
Manufacturer: Luitpoldstrasse 1, 85276 Pfaffenhofen, Germany.
Marketing Authorization Holder
AstraZeneca UK Limited
Address of Marketing Authorization Holder
1 Francis Crick Avenue, Cambridge Biomedical Campus, Cambridge, CB2 0AA, United Kingdom.