Dekspemedetomidyna Kalcekс
UkrainaSpis treści
INSTRUKCJA DOTYCZĄCA STOSOWANIA LEKU DEKSPMEDETOMIDYNA KALCEKS (DEXMEDETOMIDINE KALCEKS)
Skład:
substancja czynna: dexmedetomidine;
1 ml roztworu zawiera 100 µg deksmedetomidyny (w postaci chlorku deksmedetomidyny 118 µg);
substancje pomocnicze: natrium chloridum, aqua pro injectione.
Postać leku. Stężony roztwór do sporządzenia roztworu do infuzji.
Główne właściwości fizyko-chemiczne: przezroczysty bezbarwny lub żółtawy roztwór.
Grupa farmakoterapeutyczna. Leki przeciwlękowe. Inne leki nasenne i uspokajające. Dekspmedetomidyna. Kod ATC N05C M18.
Właściwości farmakologiczne.
Farmakodynamika.
Dekspemedetomidyna Kalcekс jest selektywnym agonistą receptorów α2-adrenergicznych, charakteryzującym się szerokim spektrum właściwości farmakologicznych. Wykazuje działanie sympatolityczne poprzez zmniejszenie uwalniania noradrenaliny z końcówek nerwów współczulnych. Działanie uspokajające wynika ze zmniejszenia pobudzenia w locus coeruleus w pniu mózgu (jądro z przewagą neuronów noradrenergicznych).
Dekspemedetomidyna Kalcekс wykazuje działanie przeciwbólowe oraz działanie oszczędzające środki znieczulające i przeciwbólowe. Działania na układ sercowo-naczyniowy mają charakter zależny od dawki. Przy niskiej szybkości infuzji przeważają efekty centralne, prowadzące do obniżenia częstości skurczów serca i ciśnienia tętniczego. W przypadku stosowania wyższych dawek dominuje obwodowe zwężenie naczyń, prowadzące do wzrostu ogólnego oporu naczyniowego i ciśnienia tętniczego oraz dalszego nasilenia bradykardii. Dekspemedetomidyna Kalcekс wywiera minimalny wpływ depresyjny na układ oddechowy podczas monoterapii u zdrowych ochotników.
Wskazanie 1. Uspokojenie dorosłych pacjentów przebywających w oddziale intensywnej terapii (OIT).
W badaniach z grupą placebo u pacjentów przebywających pooperacyjnie w OIT, wcześniej intubowanych i uspokajanych za pomocą midazolamu lub propofolu, dekspemedetomidyna Kalcekс istotnie zmniejszała potrzebę dodatkowego uspokojenia (midazolam lub propofol) i opioidów w ciągu 24 godzin. Większość pacjentów otrzymujących dekspemedetomidynę Kalcekс nie wymagała dodatkowego uspokojenia. Pacjenci mogli być pomyślnie ekstubowani bez przerywania infuzji dekspemedetomidyny Kalcekс. Badania przeprowadzone poza OIT potwierdziły, że dekspemedetomidynę Kalcekс można bezpiecznie podawać pacjentom nieintubowanym, pod warunkiem zapewnienia odpowiedniego monitorowania.
Dekspemedetomidyna Kalcekс była porównywalna do midazolamu (stosunek ryzyka 1,07; 95 % przedział ufności (PU) 0,971; 1,176) i propofolu (stosunek ryzyka 1,00; 95 % PU 0,922; 1,075) pod względem czasu przebywania w celowym zakresie uspokojenia głównie u pacjentów terapeutycznych w OIT wymagających długotrwałego uspokojenia o lekkim do umiarkowanym nasileniu (od 0 do -3 punktów według skali pobudzenia-uspokojenia Richmond (RASS)) przez okres do 14 dni; skracała czas sztucznej wentylacji płuc w porównaniu z midazolamem oraz zmniejszała czas do ekstubacji w porównaniu z midazolamem i propofolem. Pacjenci otrzymujący dekspemedetomidynę Kalcekс łatwiej się budzili, lepiej współpracowali z personelą medycznym i lepiej informowali o nasileniu bólu w porównaniu z pacjentami otrzymującymi midazolam lub propofol.
U pacjentów otrzymujących dekspemedetomidynę Kalcekс częściej występowały hipotensja tętnicza i bradykardia (rzadziej tachykardia) w porównaniu z pacjentami otrzymującymi midazolam, natomiast częściej występowała tachykardia, ale częstość występowania hipotensji tętniczej była podobna w porównaniu z pacjentami otrzymującymi propofol.
W porównaniu z grupą otrzymującą propofol, częstość występowania tachykardii u pacjentów otrzymujących dekspemedetomidynę Kalcekс była wyższa, natomiast częstość występowania hipotensji tętniczej była mniej więcej taka sama. Ocena według skali CAM-ICU wykazała, że częstość występowania delirium u pacjentów otrzymujących dekspemedetomidynę Kalcekс była niższa w porównaniu z midazolamem, a niepożądane zdarzenia związane z delirium występowały rzadziej w grupie dekspemedetomidyny Kalcekс w porównaniu z propofolem. Pacjenci, u których leczenie dekspemedetomidyną Kalcekс zostało przerwane z powodu niewystarczającej głębokości uspokojenia, zostali przekwalifikowani na propofol lub midazolam. Ryzyko niewystarczającego poziomu uspokojenia było wyższe u pacjentów, których trudno było uspokoić standardowymi środkami tuż przed przełączeniem na inną metodę uspokojenia.
Dane skuteczności w grupie pediatrycznej uzyskano w badaniu kontrolowanym dawkowanie w OIT na dużej populacji pooperacyjnej w wieku od 1 miesiąca do 17 lat. Około 50 % pacjentów otrzymujących dekspemedetomidynę Kalcekс nie wymagało dodatkowego uspokojenia za pomocą midazolamu w ciągu okresu leczenia trwającego średnio 20,3 godziny, ale nie dłużej niż 24 godziny. Brak danych dotyczących leczenia dzieci przez okres dłuższy niż 24 godziny. Informacje dotyczące stosowania leku u noworodków (po 28–44 tygodniach ciąży) są bardzo ograniczone i dotyczą wyłącznie niskich dawek (≤ 0,2 μg/kg/godz). Noworodki mogą być szczególnie wrażliwe na działanie bradykardyzujące dekspemedetomidyny Kalcekс w przypadku hipotermii i w stanach, w których rzut serca zależy od częstości skurczów serca.
W podwójnych ślepych badaniach kontrolowanych częstość występowania stłumienia kortyzolu u pacjentów otrzymujących dekspemedetomidynę Kalcekс (n=778) wynosiła 0,5 % w porównaniu z 0 % u pacjentów otrzymujących midazolam (n=338) lub propofol (n=275). Ten efekt występował jako łagodny w jednym przypadku i umiarkowany w trzech przypadkach.
Wskazanie 2. Uspokojenie procedur bez utraty przytomności.
Bezpieczeństwo i skuteczność stosowania dekspemedetomidyny Kalcekс w celu uspokojenia pacjentów nieintubowanych przed i/lub podczas zabiegów chirurgicznych i diagnostycznych oceniano w ramach dwóch randomizowanych, podwójnych ślepych, placebo-kontrolowanych, wieloośrodkowych badań.
Badanie 1. W badaniu 1 pacjenci, u których wykonano określone operacje/zabiegi pod kontrolowaną analgezją i/lub z zastosowaniem znieczynienia miejscowego/regionalnego, zostali zrandomizowani do otrzymywania dawki wstępnej dekspemedetomidyny Kalcekс 1 μg/kg (n=129) lub 0,5 μg/kg (n=134), lub placebo (roztwór fizjologiczny) (n=63) przez ponad 10 minut, a następnie infuzji utrzymującej z szybkością 0,6 μg/kg/godz. Szybkość infuzji utrzymującej badanym lekiem była dopasowywana w zakresie od 0,2 do 1 μg/kg/godz. Liczba pacjentów, którzy osiągnęli celowy poziom uspokojenia (skala oceny czujności i uspokojenia, Observer’s Assessment of Alertness/Sedation Scale – OAA/S ≤ 4), bez konieczności podania rezerwowego środka uspokajającego midazolamu, wyniosła 54 % w grupie otrzymującej 1 μg/kg dekspemedetomidyny Kalcekс i 40 % w grupie otrzymującej 0,5 μg/kg dekspemedetomidyny Kalcekс, w porównaniu z 3 % w grupie placebo.
Różnica ryzyka liczby pacjentów zrandomizowanych do grupy dekspemedetomidyny Kalcekс 1 μg/kg i grupy dekspemedetomidyny Kalcekс 0,5 μg/kg, którzy nie wymagali podania rezerwowego środka uspokajającego midazolamu, wyniosła odpowiednio 48 % (95 % PU: 37–57 %) i 40 % (95 % PU: 28–48 %) w porównaniu z placebo. Średnia dawka rezerwowego środka uspokajającego midazolamu wyniosła 1,5 (0,5–7,0) mg w grupie dekspemedetomidyny Kalcekс 1,0 μg/kg, 2 (0,5–8,0) mg w grupie dekspemedetomidyny Kalcekс 0,5 μg/kg i 4,0 (0,5–14,0) mg w grupie placebo. Różnica średnich wartości dawki rezerwowej midazolamu w grupie dekspemedetomidyny Kalcekс 1 μg/kg i grupie dekspemedetomidyny Kalcekс 0,5 μg/kg w porównaniu z placebo wyniosła odpowiednio 3,1 mg (95 % PU: 3,8–2,5) i 2,7 mg (95 % PU: 3,3–2,1) na korzyść dekspemedetomidyny Kalcekс. Średni czas do podania pierwszej dawki rezerwowego midazolamu wyniósł 114 minut w grupie dekspemedetomidyny Kalcekс 1,0 μg/kg, 40 minut w grupie dekspemedetomidyny Kalcekс 0,5 μg/kg i 20 minut w grupie placebo.
Badanie 2. W badaniu 2 pacjenci poddawani fiberoptycznej intubacji tchawicy przy zachowanej przytomności pod znieczynieniem miejscowym zostali zrandomizowani do otrzymywania infuzji wstępnej dekspemedetomidyny Kalcekс w dawce 1 μg/kg (n=55) lub placebo (roztwór fizjologiczny) (n=50) przez ponad 10 minut, a następnie stałej infuzji utrzymującej z szybkością 0,7 μg/kg/godz. Pacjentom umożliwiono w razie potrzeby stosowanie terapii rezerwowej midazolamem w celu osiągnięcia i/lub utrzymania poziomu uspokojenia ≥ 2 według skali uspokojenia Ramsaya (Ramsay Sedation Scale – RSS).
Wyniki skuteczności wykazały, że dekspemedetomidyna Kalcekс jest bardziej skuteczna w porównaniu z placebo w uspokajaniu pacjentów nieintubowanych. Liczba pacjentów otrzymujących dekspemedetomidynę Kalcekс, którzy nie wymagali terapii rezerwowej midazolamem, wyniosła 53 % w porównaniu z 14 % w grupie placebo.
Farmakokinetyka.
Farmakokinetykę dekspemedetomidyny Kalcekс badano u zdrowych ochotników po krótkotrwałym wstrzykiwaniu dożylnej oraz u pacjentów w oddziale intensywnej terapii po długotrwałym wlewie dożylnym leku.
Rozkład. Farmakokinetyka dekspemedetomidyny Kalcekс opisana jest modelem dwukomorowym. U zdrowych ochotników obserwowano szybką fazę rozkładu ze średniem okresem półwylucania (t1/2α) około 6 minut. Średni okres półwylucania w fazie terminalnej (t1/2) wynosi około 1,9–2,5 godziny (wartość minimalna 1,35 godziny, maksymalna – 3,68 godziny), a średni wolumetryczny stan równowagi (Vss) – około 1,16–2,16 l/kg (90–151 litrów). Średnie oczyszczanie osocza (Cl) wynosi 0,46–0,73 l/godz/kg (35,7–51,1 l/godz). Średnia masa ciała pacjentów, na podstawie której obliczono parametry Vss i Cl, wynosiła 69 kg.
Farmakokinetyka osoczowa dekspemedetomidyny Kalcekс u pacjentów w OIT po podawaniu leku w postaci wlewu trwającego ponad 24 godziny była podobna. Obliczone parametry farmakokinetyczne wynosiły: t1/2 – około 1,5 godziny, Vss – około 93 litry i Cl – około 43 l/godz. W zakresie dawek od 0,2 do 1,4 μg/kg/godz farmakokinetyka dekspemedetomidyny Kalcekс jest liniowa, nie kumuluje się przy leczeniu trwającym do 14 dni. 94 % dekspemedetomidyny Kalcekс wiąże się z białkami osocza krwi. Stopień wiązania z białkami osocza krwi jest stały w zakresie stężeń od 0,85 do 85 ng/ml. Dekspemedetomidyna Kalcekс wiąże się zarówno z ludzkim albuminem surowicy, jak i z α1-glikoproteiną kwasową, głównie z ludzkim albuminem surowicy.
Biopreparacja i eliminacja. Dekspemedetomidyna Kalcekс jest całkowicie metabolizowana w wątrobie. Początkowy metabolizm zachodzi drogami trzech szlaków metabolicznych: bezpośrednia N-glukuronidacja, bezpośrednie N-metylowanie oraz pośrednie utlenianie katalizowane przez cytochrom P450. Głównymi metabolitami dekspemedetomidyny Kalcekс we krwi są dwa izomeryczne N-glukuronidy. Metabolit H-l (N-metylo-3-hydroksymetylo-dexmedetomidyny O-glukuronid) jest również głównym krążącym produktem biotransformacji dekspemedetomidyny Kalcekс. Cytochrom P450 katalizuje powstawanie dwóch wtórnych krążących metabolitów: 3-hydroksymetylo-dexmedetomidyna powstaje w wyniku hydroksylacji grupy 3-metylowej dekspemedetomidyny, a H-3 powstaje w wyniku utleniania pierścienia imidazolowego. Według dostępnych informacji powstawanie utlenionych metabolitów zachodzi przy udziale szeregu izoenzymów cytochromu P450 (CYP2A6, CYP1A2, CYP2E1, CYP2D6 i CYP2C19). Metabolity te nie wykazują istotnej aktywności farmakologicznej.
Po dożylnej podaniu radioaktywnie znaczonej dekspemedetomidyny Kalcekс w ciągu 9 dni około 95 % radioaktywności wykryto w moczu i 4 % w kale. Głównymi metabolitami w moczu są dwa izomeryczne N-glukuronidy, które stanowią 34 % podanej dawki, oraz N-metylo-3-hydroksymetylo-dexmedetomidyny O-glukuronid, który stanowi 14,51 % dawki. Metabolity wtórne: kwas dekspemedetomidynowy, 3-hydroksymetylo-dexmedetomidyna i jej O-glukuronid stanowią 1,11–7,66 % dawki. Mniej niż 1 % niezmienionej dekspemedetomidyny Kalcekс wykryto w moczu. Około 28 % metabolitów w moczu stanowi niezidentyfikowane metabolity wtórne.
Osobliwe grupy pacjentów.
Istotnych różnic w farmakokinetyce w zależności od wieku i płci nie zaobserwowano.
W porównaniu ze zdrowymi ochotnikami u osób z zaburzeniami funkcji wątroby obniża się stopień wiązania dekspemedetomidyny Kalcekс z białkami osocza krwi. Średnia część frakcji niezwiązanej dekspemedetomidyny Kalcekс wahala się od 8,5 % u zdrowych ochotników do 17,9 % u osób z ciężkimi zaburzeniami funkcji wątroby. U pacjentów z różnym stopniem niewydolności wątroby (klasy A, B i C według skali Childa-Pugh) obniżał się wątrobowy klirens dekspemedetomidyny Kalcekс i wydłużał się okres półwylucania z osocza (t1/2). Średnie wartości klirensu osoczowego niezwiązanej dekspemedetomidyny Kalcekс u osób z łagodną, umiarkowaną i ciężką niewydolnością wątroby wynosiły odpowiednio 59 %, 51 % i 32 % wartości obserwowanych u zdrowych ochotników. Średni t1/2 u osób z łagodną, umiarkowaną i ciężką niewydolnością wątroby wydłużał się odpowiednio do 3,9, 5,4 i 7,4 godziny. Mimo że doboru dawki dekspemedetomidyny Kalcekс dokonuje się na podstawie stopnia efektu uspokajającego, u pacjentów z zaburzeniami funkcji wątroby należy rozważyć możliwość zmniejszenia dawki początkowej lub utrzymującej w zależności od stopnia zaburzenia lub odpowiedzi klinicznej.
W porównaniu ze zdrowymi ochotnikami u pacjentów z ciężkimi zaburzeniami funkcji nerek (klirens kreatyniny <30 ml/min) farmakokinetyka dekspemedetomidyny Kalcekс nie ulega zmianie.
Dzieci. Dane dotyczące stosowania leku u dzieci, od noworodków (28–44 tygodnie ciąży) do dzieci w wieku do 17 lat, są ograniczone. Okres t1/2 dekspemedetomidyny Kalcekс u dzieci (od 1 miesiąca do 17 lat) odpowiada temu obserwowanemu u dorosłych, natomiast u noworodków (do 1 miesiąca życia) obserwuje się dłuższy t1/2. W grupach wiekowych od 1 miesiąca do 6 lat obserwowano dłuższy okres klirensu osoczowego skorygowany o masę ciała, natomiast u dzieci starszego wieku okres ten był krótszy. U noworodków (do 1 miesiąca życia) okres klirensu osoczowego skorygowany o masę ciała okazał się krótszy (0,9 l/godz/kg) niż u dzieci w starszych grupach z powodu niedojrzałości. Dostępne dane przedstawiono w tabeli 1.
Tabela 1
| Średnia wartość (95 % CI) |
|||
| Wiek |
N |
Cl (l/h/kg) |
T1/2 (godziny) |
| do 1 miesiąca |
28 |
0,93 (0,76; 1,14) |
4,47 (3,81; 5,25) |
| 1 do <6 miesięcy |
14 |
1,21 (0,99; 1,48) |
2,05 (1,59; 2,65) |
| 6 do <12 miesięcy |
15 |
1,11 (0,94; 1,31) |
2,01 (1,81; 2,22) |
| 12 do <24 miesięcy |
13 |
1,06 (0,87; 1,29) |
1,97 (1,62; 2,39) |
| 2 do <6 lat |
26 |
1,11 (1,00; 1,23) |
1,75 (1,57; 1,96) |
| 6 do <17 lat |
28 |
0,80 (0,69; 0,92) |
2,03 (1,78; 2,31) |
Właściwości kliniczne.
Wskazania.
Sedacja u dorosłych pacjentów przebywających na intensywnej terapii (IT), u których wymagana głębokość sedacji nie przekracza poziomu pobudzenia do odpowiedzi na bodźce głosowe (odpowiada zakresowi od 0 do -3 punktów według skali pobudzenia-sedacji Richmond (RASS)).
Sedacja u nieintubowanych dorosłych pacjentów przed i/lub podczas wykonywania zabiegów diagnostycznych lub chirurgicznych, tj. sedacja podczas udzielania pomocy anestezjologicznej/sedacja w świadomości.
Przeciwwskazania.
Podwyższona wrażliwość na dekspemedetomidynę lub którykolwiek z substancji pomocniczych preparatu.
Blokada przedsionkowo-komorowa II–III stopnia (przy braku sztucznego stymulatora serca).
Niekontrolowana hipotensja tętnicza.
Ostra patologia mózgowo-naczyniowa.
Współdziałanie z innymi lekami i inne rodzaje interakcji.
Badania interakcji leków prowadzono wyłącznie u dorosłych.
Jednoczesne stosowanie dekspemedetomidyny z anestetykami, lekami uspokajającymi, snodziejnymi oraz opioidami prowadzi do nasilenia ich efektów, takich jak sedacja, znieczulenie oraz działanie kardiorespiratoryjne. Założenie to potwierdzono w badaniach z izofluranem, propofolem, alfentanilem i midazolamem.
Nie stwierdzono farmakokinetycznych interakcji między dekspemedetomidyną a izofluranem, propofolem, alfentanilem i midazolamem. Jednak ze względu na możliwe interakcje farmakodynamiczne przy jednoczesnym stosowaniu tych leków z dekspemedetomidyną może być konieczne zmniejszenie dawki dekspemedetomidyny lub jednoczesnie stosowanych anestetyków, środków uspokajających, snodziejnych lub opioidów.
W badaniach in vitro na mikrosomach wątroby ludzkiej oceniano zdolność dekspemedetomidyny do hamowania cytochromu P450, w tym izoenzymu CYP2B6. Zgodnie z badaniami in vitro istnieje potencjalna możliwość interakcji między dekspemedetomidyną a substratami (głównie izoenzymu CYP2B6) in vivo.
Indukcja izoenzymów CYP1A2, CYP2B6, CYP2C8, CYP2C9 i CYP3A4 przez dekspemedetomidynę obserwowana była in vitro, dlatego nie można wykluczyć takiej interakcji in vivo. Kliniczne znaczenie jest nieznane.
U pacjentów przyjmujących leki powodujące obniżenie ciśnienia tętniczego i bradykardię, np. beta-adrenoblokery, należy wziąć pod uwagę możliwość nasilenia tych efektów (jednak dodatkowe efekty w badaniu interakcji z esmololem były umiarkowane).
Szczególne wskazania dotyczące stosowania.
Monitorowanie
Dekspemedetomidyna Kalcekс przeznaczona jest do stosowania w warunkach intensywnej terapii, w sali operacyjnej oraz podczas wykonywania zabiegów diagnostycznych; jej stosowanie w innych warunkach nie jest zalecane. Podczas infuzji leku należy prowadzić ciągłe monitorowanie czynności serca. U pacjentów nieintubowanych należy monitorować oddychanie ze względu na ryzyko抑ja oddechowego i, w niektórych przypadkach, wystąpienia apnei (patrz dział „Działania niepożądane”).
Czas odzyskania świadomości po zastosowaniu dekspemedetomidyny wynosi około jednej godziny. W przypadku stosowania ambulatoryjnego należy kontynuować dokładne monitorowanie przez co najmniej jedną godzinę (lub dłużej, w zależności od stanu pacjenta), a opiekę medyczną należy kontynuować przez kolejną godzinę, aby zapewnić bezpieczeństwo pacjenta.
Ogólne ostrzeżenia
Nie należy podawać dekspemedetomidyny w bolusie; nie zaleca się również stosowania dawki ładującej w warunkach OIT. Użytkownicy powinni być przygotowani na zastosowanie alternatywnego środka uspokajającego w celu szybkiego kontrolowania pobudzenia, szczególnie w pierwszych godzinach leczenia lub podczas wykonywania zabiegów medycznych. Podczas uspokojenia w trakcie anestezji można stosować niewielkie dawki bolusowe innego środka uspokajającego w celu szybkiego osiągnięcia pożądanego poziomu uspokojenia.
U niektórych pacjentów otrzymujących dekspemedetomidynę obserwowano lekkie przebudzenie i szybkie odzyskiwanie świadomości po stymulacji. W przypadku braku innych objawów klinicznych, ten objaw nie powinien być rozpatrywany izolacyjnie jako nieskuteczność leku.
Dekspemedetomidyna zazwyczaj nie powoduje głębokiego uspokojenia, dzięki czemu pacjentów można łatwo obudzić. Z tego powodu dekspemedetomidyna nie jest stosowana u pacjentów wymagających głębokiego uspokojenia.
Dekspemedetomidyna nie powinna być stosowana jako ogólny środek znieczulający podczas intubacji ani w celu zapewnienia uspokojenia podczas stosowania leków rozkurczających mięśnie.
Dekspemedetomidyna nie hamuje aktywności drgawkowej, dlatego nie powinna być stosowana jako monoterapia w stanie drgawkowym.
Należy zachować ostrożność przy jednoczesnym stosowaniu dekspemedetomidyny z lekami o działaniu uspokajającym lub wpływającymi na układ sercowo-naczyniowy, ze względu na możliwość efektu addytywnego.
Nie zaleca się stosowania dekspemedetomidyny w celu samokontrolowanego uspokojenia przez pacjenta. Brak odpowiednich danych.
Przy stosowaniu dekspemedetomidyny w warunkach ambulatoryjnych wypisywanie pacjentów jest możliwe pod nadzorem osób trzecich. Pacjentom należy zalecić powstrzymanie się od prowadzenia pojazdów lub wykonywania innych potencjalnie niebezpiecznych czynności oraz, jeśli to możliwe, unikanie stosowania innych środków, które mogą wywoływać działanie uspokajające (np. benzodiazepiny, opioidy, alkohol), przez odpowiedni czas, w zależności od obserwowanego działania dekspemedetomidyny, rodzaju procedury, stosowanych leków, wieku i stanu pacjenta.
Należy zachować ostrożność przy stosowaniu dekspemedetomidyny u pacjentów w podeszłym wieku. Pacjenci w wieku ≥65 lat mogą być bardziej narażeni na hipotensję tętniczą, szczególnie przy podawaniu dawki ładującej podczas procedur z zastosowaniem dekspemedetomidyny. Należy rozważyć możliwość zmniejszenia dawki (patrz dział „Sposób stosowania i dawki”).
Śmiertelność u pacjentów oddziałów intensywnej terapii w wieku ≤65 lat
W pragmatycznym, randomizowanym, kontrolowanym badaniu SPICE III, w którym wzięło udział 3904 ciężko chorych dorosłych pacjentów na oddziale intensywnej terapii, dekspemedetomidyna była stosowana jako główny środek uspokajający i porównywana z terapią standardową. Nie zaobserwowano ogólnych różnic w śmiertelności 90-dniowej między grupą otrzymującą dekspemedetomidynę a grupą otrzymującą terapię standardową (śmiertelność w obu grupach wynosiła 29,1%), ale zaobserwowano heterogeniczność wpływu wieku na śmiertelność. Dekspemedetomidyna była związana ze zwiększonym ryzykiem śmiertelności u pacjentów w wieku ≤65 lat (stosunek ryzyka 1,26; 95% przedział ufności od 1,02 do 1,56) w porównaniu z alternatywnymi środkami uspokajającymi. Choć mechanizm nie jest znany, ta heterogeniczność wpływu wieku na śmiertelność była najbardziej widoczna u pacjentów hospitalizowanych z powodów innych niż opieka pooperacyjna, nasilała się wraz ze wzrostem wyniku w skali APACHE II i malejącym wiekiem. Wyniki te należy ocenić z uwzględnieniem oczekiwanej korzyści klinicznej z zastosowania dekspemedetomidyny w porównaniu z alternatywnymi środkami uspokajającymi u młodszych pacjentów.
Wpływ na serce i naczynia; ostrzeżenia
Dekspemedetomidyna obniża częstość skurczów serca i ciśnienie tętnicze (działanie centralne sympatolityczne), ale w wyższych stężeniach powoduje skurcz naczyń obwodowych, prowadzący do podwyższenia ciśnienia tętniczego (patrz dział „Farmakodynamika”). Z tego powodu Dekspemedetomidyna Kalcekс nie powinna być stosowana u pacjentów z ciężką niestabilnością hemodynamiczną.
Należy zachować ostrożność przy podawaniu dekspemedetomidyny pacjentom z współistniejącą bradykardią. Dane dotyczące wpływu leku na pacjentów z częstością skurczów serca <60 są ograniczone, dlatego tacy pacjenci wymagają szczególnej kontroli i obserwacji. Bradykardia zazwyczaj nie wymaga leczenia i dobrze koryguje się podaniem blokerów m-cholinolitycznych lub zmniejszeniem dawki leku. Pacjenci uprawiający sport i o niskiej częstości skurczów serca mogą być szczególnie wrażliwi na negatywny chronotropowy efekt agonistów receptorów α2-adrenergicznych; opisano przypadki zatrzymania węzła zatokowego. Opisano również przypadki zatrzymania serca poprzedzone bradykardią lub blokadą przedsionkowo-komorową (patrz dział „Działania niepożądane”).
U pacjentów z współistniejącą hipotensją tętniczą (szczególnie oporną na wazokonstryktory), w tym przewlekłą, hipowolemię lub zmniejszony rezerw funkcjonalnych, takich jak pacjenci z ciężką dysfunkcją komorową i pacjenci w podeszłym wieku, efekt hipotensyjny dekspemedetomidyny może być bardziej wyrażony, co wymaga szczególnej uwagi (patrz dział „Przeciwwskazania”). Obniżenie ciśnienia tętniczego zazwyczaj nie wymaga szczególnych interwencji, ale w razie potrzeby należy być gotowym na zmniejszenie dawki, podanie środków uzupełniających objętość krwi obiegowej i/lub wazokonstryktorów.
U pacjentów z uszkodzeniem układu autonomicznego (np. w wyniku urazu rdzenia kręgowego) efekty hemodynamiczne po podaniu dekspemedetomidyny mogą być bardziej nasilone i wymagać szczególnej kontroli.
Przejściowa hipertensja tętnicza obserwowana była głównie podczas podawania dawki ładującej, z powodu obwodowego działania wazokonstrykcyjnego dekspemedetomidyny, dlatego podawanie dawki ładującej podczas uspokojenia w OIT nie jest zalecane. Leczenie podwyższonego ciśnienia tętniczego zazwyczaj nie jest konieczne, jednak należy rozważyć możliwość zmniejszenia szybkości podania leku.
Miejscowa wazokonstrykcja przy wysokim stężeniu może mieć większe znaczenie dla pacjentów z chorobą niedokrwienną serca lub ciężkimi chorobami mózgowymi; tych pacjentów należy dokładnie monitorować. W przypadku pojawienia się objawów niedokrwienia mięśnia sercowego lub mózgu należy zmniejszyć dawkę leku lub przerwać jego podawanie.
Zaleca się ostrożne stosowanie dekspemedetomidyny w połączeniu z znieczuleniem podpajęczynówkowym lub naczyniowym z uwagi na możliwe zwiększenie ryzyka hipotensji tętniczej i bradykardii.
Pacjenci z zaburzeniami funkcji wątroby
Należy zachować ostrożność u pacjentów z ciężką niewydolnością wątroby, ponieważ obniżenie klirensu dekspemedetomidyny przy nadmiernym podaniu leku u tych pacjentów może prowadzić do zwiększonego ryzyka działań niepożądanych, nadmiernej sedyacji i przedłużenia działania.
Pacjenci z zaburzeniami neurologicznymi
Doświadczenie stosowania dekspemedetomidyny w przypadku takich ciężkich stanów neurologicznych, jak uraz głowy czy okres pooperacyjny po zabiegach neurochirurgicznych, jest ograniczone, dlatego lek powinien być stosowany z ostrożnością w tych stanach, szczególnie gdy wymagane jest głębokie uspokojenie. Przy wyborze terapii należy wziąć pod uwagę, że dekspemedetomidyna może obniżać przepływ krwi mózgowej i ciśnienie wewnątrzczaszkowe.
Inne ostrzeżenia
Po nagłym odstawieniu agonistów receptorów α2-adrenergicznych po długotrwałym stosowaniu, w pojedynczych przypadkach wystąpił zespół odstawienia. W przypadku rozwoju agitacji i podwyższenia ciśnienia tętniczego bezpośrednio po odstawieniu dekspemedetomidyny należy wziąć pod uwagę możliwość wystąpienia tego stanu.
Dekspemedetomidyna może powodować hipertermię, która nie odpowiada na leczenie tradycyjnymi metodami chłodzenia. W przypadku wystąpienia trwającej, niejasnej gorączki należy przerwać stosowanie dekspemedetomidyny. Nie zaleca się jej stosowania u osób narażonych na złośliwą hipertermię.
Opisano przypadki rozwoju mocznicowego cukrzycy w związku z leczeniem dekspemedetomidyną. W przypadku wystąpienia poliurii zaleca się przerwanie stosowania dekspemedetomidyny oraz sprawdzenie stężenia sodu w surowicy i osmolalności moczu.
Dekspemedetomidyna Kalcekс zawiera mniej niż 1 mmol sodu (23 mg) na mililitr, czyli praktycznie nie zawiera sodu.
Stosowanie w czasie ciąży lub karmienia piersią.
Ciąża.
Brak danych lub są one ograniczone dotyczące stosowania dekspemedetomidyny u kobiet w ciąży. W badaniach na zwierzętach zaobserwowano toksyczność rozrodczą. Dekspemedetomidyna nie powinna być stosowana w czasie ciąży, z wyjątkiem przypadków, gdy stan kliniczny kobiety wymaga leczenia dekspemedetomidyną.
Karmienie piersią.
Dekspemedetomidyna wydzielana jest z mlekiem matki, jednak jej stężenia są poniżej granicy wykrywalności po 24 godzinach od przerwania podania leku. Ryzyko dla noworodka nie może być wykluczone. Decyzja o przerwaniu karmienia piersią lub o przerwaniu leczenia dekspemedetomidyną powinna być podjęta z uwzględnieniem korzyści z karmienia piersią dla noworodka oraz korzyści z leczenia dekspemedetomidyną dla matki.
Plodność.
W badaniu dotyczącym płodności przeprowadzonym na szczurach dekspemedetomidyna nie wpływała na funkcję rozrodczą samców ani samic. Brak danych dotyczących wpływu na funkcję rozrodczą u ludzi.
Wpływ na zdolność reagowania podczas prowadzenia pojazdów lub innych urządzeń.
Pacjentom zaleca się powstrzymanie się od prowadzenia pojazdów lub wykonywania innych niebezpiecznych zadań przez odpowiedni okres czasu po podaniu dekspemedetomidyny w celu uspokojenia podczas anestezji.
Sposób stosowania i dawki
Sedacja u dorosłych pacjentów w OIOM-ie, u których wymagana głębokość sedacji nie przekracza wybudzenia na bodziec głosowy (odpowiada zakresowi od 0 do -3 punktów według skali pobudzenia-sedacji Richmond (RASS))
Tylko dla warunków szpitalnych. Lek ten powinien być stosowany przez specjalistów z doświadczeniem w leczeniu pacjentów w warunkach intensywnej terapii.
Dawki dla dorosłych
Pacjentów, którzy są już intubowani i pozostają w stanie sedacji, można przechodzić na doksmedetomidynę z początkową prędkością infuzji 0,7 μg/kg/godz., którą można stopniowo dostosować w zakresie dawek od 0,2 do 1,4 μg/kg/godz. w celu osiągnięcia pożądanego poziomu sedacji, w zależności od reakcji pacjenta. U osłabionych pacjentów należy rozważyć niższą początkową prędkość infuzji. Doksmedetomidyna jest silnym środkiem, dlatego szybkość jej podawania podaje się na godzinę. Po dostosowaniu dawki osiągnięcie docelowej głębokości sedacji może trwać do jednej godziny.
Maksymalna dawka
Nie należy przekraczać maksymalnej dawki leku wynoszącej 1,4 μg/kg/godz. Pacjentów, którzy nie osiągnęli odpowiedniego poziomu sedacji przy maksymalnej dawce doksmedetomidyny, należy przełożyć na alternatywny lek uspokajający.
Podawanie dawki nasycenia doksmedetomidyny w OIOM-ie nie jest zalecane, ponieważ zwiększa częstość występowania niepożądanych reakcji lekowych. W razie potrzeby mogą być stosowane propofol lub midazolam do osiągnięcia efektu klinicznego doksmedetomidyny.
Czas trwania
Brak doświadczenia w stosowaniu doksmedetomidyny przez ponad 14 dni. W przypadku stosowania leku dłużej niż 14 dni należy regularnie oceniać stan pacjenta.
Sedacja u nieintubowanych dorosłych pacjentów przed i/lub podczas wykonywania zabiegów diagnostycznych lub chirurgicznych, czyli sedacja podczas anestezji/świadoma sedacja.
Doksmedetomidynę należy stosować wyłącznie przez specjalistów z doświadczeniem w prowadzeniu anestezji u pacjentów w sali operacyjnej lub podczas wykonywania zabiegów terapeutycznych lub diagnostycznych. Podczas stosowania doksmedetomidyny w celu świadomej sedacji pacjenci powinni być stale monitorowani przez osoby uczestniczące w zabiegu diagnostycznym lub chirurgicznym. Konieczne jest ciągłe monitorowanie pacjentów w celu wczesnego wykrywania objawów takich jak nadciśnienie tętnicze, hipotensja tętnicza, bradykardia, ucisk oddechowy, apneę, duszność i/lub spadek nasycenia.
Należy zapewnić dostępność terapii tlenowej, która powinna być natychmiast zastosowana w razie potrzeby. Nasycenie tlenu należy monitorować metodą pulsoksymetrii.
Podawanie doksmedetomidyny rozpoczyna się od dawki obciążającej, po której następuje infuzja utrzymująca. W zależności od rodzaju zabiegu może być wymagana odpowiednia anestezja miejscowa/rejonowa lub analgezja w celu osiągnięcia pożądanego efektu klinicznego. Zaleca się stosowanie dodatkowej analgezji lub środków uspokajających (np. opioidy, midazolam, propofol) w przypadku zabiegów bolesnych lub gdy wymagany jest głębszy poziom sedacji. Okres półtrwania farmakokinetyczny doksmedetomidyny szacuje się na około 6 minut. Należy go uwzględnić wraz z efektami innych stosowanych leków przy ocenie czasu niezbędnego do dozowania w celu osiągnięcia pożądanego efektu klinicznego doksmedetomidyny.
Rozpoczęcie sedacji podczas anestezji:
- Dawka obciążająca w postaci infuzji 1 μg/kg przez 10 minut. W mniej inwazyjnych zabiegach, np. operacjach okulistycznych, może być stosowana dawka obciążająca 0,5 μg/kg przez 10 minut.
Utrzymanie sedacji podczas anestezji:
- Infuzję utrzymującą zazwyczaj rozpoczyna się od dawki 0,6–0,7 μg/kg/godz. i dozuje się do osiągnięcia pożądanego efektu klinicznego w zakresie dawek od 0,2 do 1 μg/kg/godz. Szybkość infuzji utrzymującej należy dostosować do osiągnięcia docelowego poziomu sedacji.
Grupy specjalne pacjentów
Pacjenci w podeszłym wieku. Dostosowanie dawki zazwyczaj nie jest konieczne. Pacjenci w podeszłym wieku mogą mieć zwiększone ryzyko hipotensji tętniczej, jednak ograniczone dane dotyczące sedacji podczas anestezji nie wskazują na wyraźną zależność dawkową tego ryzyka.
Zaburzenia funkcji nerek. U pacjentów z zaburzeniami funkcji nerek dostosowanie dawki zazwyczaj nie jest konieczne.
Zaburzenia funkcji wątroby. Doksmedetomidyna jest metabolizowana w wątrobie, dlatego należy stosować ją z ostrożnością u pacjentów z zaburzeniami funkcji wątroby. Należy rozważyć możliwość zastosowania zmniejszonej dawki utrzymującej (patrz rozdziały „Farmakokinetyka” i „Szczególne ostrzeżenia i środki ostrożności”).
Sposób stosowania
Lek należy stosować wyłącznie po rozcieńczeniu w postaci dożylnego wlewu za pomocą kontrolowanego urządzenia infuzyjnego.
Jedna ampułka leku przeznaczona jest wyłącznie dla jednego pacjenta.
Przygotowanie roztworu
Przed zastosowaniem Dekspemedetomidyna Kalcekс można rozcieńczać w 5 % roztworze glukozy, roztworze Ringera, roztworze Ringera z laktem, manitolu lub w 0,9 % roztworze sodu chlorku w celu uzyskania wymaganej stężenia 4 μg/ml lub 8 μg/ml. W poniższych tabelach przedstawiono objętości niezbędne do przygotowania wlewu.
Tabela 2
W celu uzyskania stężenia 4 μg/ml:
| Objętość leku Dekspemedetomidyna Kalcekс, substancja do sporządzenia roztworu do infuzji, 100 μg/ml |
Objętość rozpuszczalnika, ml |
Całkowita objętość infuzji, ml |
| 2 ml |
48 |
50 |
| 4 ml |
96 |
100 |
| 10 ml |
240 |
250 |
| 20 ml |
480 |
500 |
Tabela 3
W celu osiągnięcia stężenia 8 μg/ml:
| Objętość leku Dekspemedetomidyna Kalcekс, substancja do sporządzenia roztworu do infuzji, 100 μg/ml |
Objętość rozpuszczalnika, ml |
Całkowita objętość infuzji, ml |
| 4 ml |
46 |
50 |
| 8 ml |
92 |
100 |
| 20 ml |
230 |
250 |
| 40 ml |
460 |
500 |
Ostrożnie potrząsnąć, aby dobrze wymieszać roztwór.
Przed podaniem roztwór należy wizualnie sprawdzić pod kątem obecności zanieczyszczeń lub zmiany barwy.
Dekspemedetomidyna Kalcekс jest farmaceutycznie zgodna z następującymi płynami do wlewu dożylnego i lekami: roztwór Ringer’a z laktem, 5 % roztwór glukozy, 0,9 % roztwór chlorku sodu, 20 % roztwór manitolu, tiopentalu sodu, etomidatu, wekuroniu bromku, pankuroniu bromku, sukcynylcholiny, atrakuriumu bezylianu, mivakuriumu chlorku, rokuroniui bromku, glikopiryoniu bromku, fenyloefryny hydrochloroku, atropiny siarczanu, dopaminy, noradrenaliny, dobutaminy, midazolamu, morfiny siarczanu, cytrynianu fentanilu, środki plazmazastępcze.
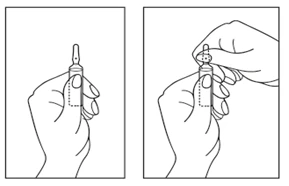
Jak otworzyć ampułkę:
- Obrócić ampułkę kolorowym punktem do siebie. Delikatnie stuknąć palcem w górną część ampułki, aby roztwór opadł do dolnej części ampułki.
- Użyć obu rąk, aby otworzyć ampułkę: trzymając dolną część ampułki w jednej ręce, drugą ręką nacisnąć na górną część ampułki w kierunku przeciwnym do kolorowego punktu (patrz rysunek poniżej).
Nieużywany lek należy zniszczyć zgodnie z lokalnymi przepisami.
Dzieci.
Bezpieczeństwo i skuteczność stosowania dekspemedetomidyny u dzieci w wieku od 0 do 18 lat nie zostały ustalone. Dane dotyczące stosowania leku u tej grupy pacjentów przedstawiono w rozdziałach „Działania niepożądane”, „Właściwości farmakologiczne”, jednakże nie można podać zaleceń dotyczących dawkowania.
Przedawkowanie.
Objawy.
W trakcie badań klinicznych i po rejestracji leku zgłaszano kilka przypadków przedawkowania dekspemedetomidyny. Według dostępnych danych szybkość podania w tych przypadkach wynosiła 60 µg/kg/godz. przez 36 minut oraz 30 µg/kg/godz. przez 15 minut u dziecka w wieku 20 miesięcy i dorosłego odpowiednio. Najczęstsze niepożądane reakcje lekowe spowodowane przedawkowaniem to bradykardia, hipotensja tętnicza, nadciśnienie tętnicze, nadmierne uspokojenie, depresja oddychania i zatrzymanie akcji serca.
Leczenie.
W przypadku przedawkowania z objawami klinicznymi należy zmniejszyć lub przerwać podawanie dekspemedetomidyny. Oczekiwane efekty są głównie kardiowaskularne i powinny być leczone zgodnie z wskazaniami klinicznymi (patrz rozdział „Szczególne ostrzeżenia i środki ostrożności podczas stosowania”). Przy wysokich stężeniach wzrost ciśnienia tętniczego może przeważać nad jego obniżeniem. W badaniach klinicznych zatrzymanie węzła zatokowego ustępowało samoistnie lub w odpowiedzi na podanie atropiny lub glikopirylolu. W pojedynczych przypadkach ciężkiego przedawkowania towarzyszącego zatrzymaniu akcji serca konieczne było przeprowadzenie zabiegów resuscytacyjnych.
Efekty niepożądane.
Podsumowanie profilu bezpieczeństwa
Sedacja u dorosłych pacjentów w OIOT
Efektami niepożądanymi, o których najczęściej raportowano podczas stosowania dekspemedetomidyny w warunkach OIOT, są hipotensja tętnicza, nadciśnienie tętnicze i bradykardia, występujące odpowiednio u około 25 %, 15 % i 13 % pacjentów. Hipotensja tętnicza i bradykardia były również najczęstszymi ciężkimi efektami niepożądanymi związanymi z dekspemedetomidyną, które wystąpiły odpowiednio u 1,7 % i 0,9 % pacjentów losowanych do OIOT.
Sedacja podczas anestezji / sedacja przy zachowaniu przytomności
Efekty niepożądane, o których najczęściej raportowano podczas stosowania dekspemedetomidyny w celu sedacji podczas anestezji, wymienione są poniżej:
- hipotensja tętnicza (55 % w grupie dekspemedetomidyny w porównaniu z 30 % w grupie placebo);
- osłabienie oddychania (38 % w grupie dekspemedetomidyny w porównaniu z 35 % w grupie placebo);
- bradykardia (14 % w grupie dekspemedetomidyny w porównaniu z 4 % w grupie placebo).
Niepożądane reakcje wymienione poniżej pochodzą z połączonych danych z badań klinicznych u pacjentów OIOT.
Częstotliwość występowania efektów niepożądanych sklasyfikowano następująco: bardzo często (≥ 1/10), często (≥ 1/100, < 1/10), rzadko (≥ 1/1000, < 1/100), bardzo rzadko (≥ 1/10000, < 1/1000), nieznana (niemożliwe do ustalenia na podstawie dostępnych danych).
Ze strony układu endokrynnego: nieznana – cukrzyca pozadiabetyczna.
Ze strony metabolizmu i odżywiania: często – hiperglikemia, hipoglikemia; rzadko – kwasica metaboliczna, hipoproteinemia.
Zaburzenia psychiczne: często – niepokój; rzadko – halucynacje.
Ze strony serca: bardzo często – bradykardia1,2; często – niedokrwienie lub zawał mięśnia sercowego, tachykardia; rzadko – blok przedsionkowo-komorowy1, zmniejszenie rzutu serca, zatrzymanie serca1.
Ze strony układu naczyniowego: bardzo często – hipotensja tętnicza1,2, nadciśnienie tętnicze1,2.
Ze strony układu oddechowego, klatki piersiowej i jamy międzyprzestrzeniowej: bardzo często – osłabienie oddychania2,3; rzadko – duszność, apnea.
Ze strony przewodu pokarmowego: często – nudności2, wymioty, suchość w ustach2; rzadko – wzdęcia brzucha.
Zespół ogólny i reakcje w miejscu podania: często – zespół odstawienia, hipertermia; rzadko – nieskuteczność leku, pragnienie.
1 Zob. poniżej opis poszczególnych niepożądanych reakcji.
2 Efekt niepożądany obserwowany również w badaniach sedacji podczas anestezji.
3 Częstotliwość „często” w badaniu sedacji w warunkach OIOT.
Opis poszczególnych efektów niepożądanych.
Klinicznie istotne obniżenie ciśnienia tętniczego i bradykardia powinny być leczone zgodnie z zaleceniami zawartymi w sekcji „Szczególne ostrzeżenia i środki ostrożności”.
U stosunkowo zdrowych ochotników, którzy nie przebywali w OIOT, podawanie dekspemedetomidyny powodowało czasem zatrzymanie węzła zatokowego lub pauzę zatokową. Objawy te ustępowały po uniesieniu nóg oraz stosowaniu leków antycholinergicznych, takich jak atropina lub glikopirinat. U pojedynczych pacjentów z wcześniejszą bradykardią dochodziło do epizodów asystolii. Opisywano również przypadki zatrzymania serca, którym poprzedzała bradykardia lub blok przedsionkowo-komorowy.
Nadciśnienie tętnicze było związane z podaniem dawki ładunkowej. Reakcję tę można zmniejszyć, unikając podania dawki ładunkowej lub zmniejszając szybkość infuzji lub dawkę ładunkową.
Dzieci.
Oceniano leczenie dzieci w wieku od 1 miesiąca, głównie po operacji, przebywających w OIOT przez okres do 24 godzin; wykazano profil bezpieczeństwa porównywalny z profilem u dorosłych. Dane u noworodków (28–44 tygodnie ciąży) są bardzo ograniczone, dawki ograniczone są do dawek utrzymujących ≤ 0,2 μg/kg/godz. W publikacjach opisano pojedynczy przypadek hipotermicznej bradykardii u noworodka.
Okres ważności. 5 lat.
Nie stosować po upływie terminu ważności podanego na opakowaniu.
Warunki przechowywania.
Nie wymaga specjalnych warunków przechowywania.
Przechowywać w miejscu niedostępnym dla dzieci.
Po rozcieńczeniu.
Po rozcieńczeniu stabilność chemiczna i fizyczna w warunkach stosowania została wykazana przez 36 godzin w temperaturze 25 °C.
Z mikrobiologicznego punktu widzenia roztwór należy stosować natychmiast. Jeżeli nie jest stosowany natychmiast, użytkownik ponosi odpowiedzialność za czas i warunki przechowywania przed zastosowaniem, który zazwyczaj nie powinien przekraczać 24 godzin w temperaturze od 2 do 8 °C, chyba że rozcieńczenie przeprowadzono w dobrze kontrolowanych i zwalidowanych warunkach aseptycznych.
Niezgodność.
Niniejszego leku nie wolno mieszać z innymi lekami, z wyjątkiem tych wymienionych w sekcji „Sposób stosowania i dawki”.
Istnieje możliwość adsorpcji dekspemedetomidyny do niektórych typów naturalnego kauczuku. Pomimo że dekspemedetomidyna stosowana jest do osiągnięcia efektu klinicznego, zaleca się stosowanie materiałów z powłoką z kauczuku syntetycznego lub naturalnego.
Opakowanie.
Po 2 ml w ampułce z bezbarwnego szkła klasy I hydroliotycznej z pierścieniami znacznikowymi i punktem złamania.
Po 5 ampułek w opakowaniu konturowym z folii poliwinylchlorowej.
Po 1 lub 5 opakowań konturowych w pudełku z tektury.
Kategoria wydania.
Na receptę.
Producent.
Producent odpowiedzialny za wydanie serii:
AT „Kalcekx”.
Adres siedziby producenta i miejsce prowadzenia działalności.
ul. Krustpils 71E, Ryga, LV-1057, Łotwa.
Wniosek.
AT „Kalcekx”.
Adres siedziby wnioskodawcy i/lub przedstawiciela wnioskodawcy.
ul. Krustpils 71E, Ryga, LV-1057, Łotwa.