Dexmedetomidina Kaleceks
UcraniaContenido
INSTRUCCIONES PARA USO MÉDICO DEL MEDICAMENTO DEXMEDETOMIDINA KALCEKS (DEXMEDETOMIDINE KALCEKS)
Composición:
Principio activo: dexmedetomidina;
1 ml de solución contiene dexmedetomidina 100 µg (en forma de clorhidrato de dexmedetomidina 118 µg);
Excipientes: cloruro de sodio, agua para inyección.
Forma farmacéutica. Concentrado para solución para perfusión.
Propiedades físico-químicas principales: solución transparente incolora o ligeramente amarillenta.
Grupo farmacoterapéutico. Psicolépticos. Otros sedantes e hipnóticos. Dexmedetomidina. Código ATC N05CM18.
Propiedades farmacodinámicas.
Farmacodinámica.
Dexmedetomidina es un agonista selectivo de los receptores α2-adrenérgicos con un amplio espectro de propiedades farmacológicas. Tiene un efecto simpaticolítico debido a la reducción de la liberación de noradrenalina desde las terminaciones nerviosas simpáticas. El efecto sedante se debe a la disminución de la excitación en la sustancia gris central del tallo cerebral (núcleo con predominio de neuronas noradrenérgicas).
Dexmedetomidina posee efectos analgésicos y conservadores de anestésicos-analgésicos. Los efectos cardiovasculares son dependientes de la dosis. Con velocidades bajas de infusión prevalecen los efectos centrales, lo que conduce a una reducción de la frecuencia cardíaca y de la presión arterial. Al administrar dosis más altas, predomina la vasoconstricción periférica, lo que provoca un aumento de la resistencia vascular sistémica y de la presión arterial, así como un mayor agravamiento de la bradicardia. Dexmedetomidina apenas tiene efecto depresor sobre el sistema respiratorio cuando se administra como monoterapia a voluntarios sanos.
Indicación 1. Sedación de pacientes adultos en la unidad de cuidados intensivos (UCI).
En estudios controlados con placebo en pacientes en la UCI posoperatoria, previamente intubados y sedados con midazolam o propofol, dexmedetomidina redujo significativamente la necesidad de sedación adicional (midazolam o propofol) y opioides durante 24 horas. La mayoría de los pacientes que recibieron dexmedetomidina no requirieron sedación adicional. Los pacientes pudieron ser extubados con éxito sin interrumpir la infusión de dexmedetomidina. Estudios realizados fuera de la UCI confirmaron que dexmedetomidina puede administrarse de forma segura a pacientes no intubados siempre que existan condiciones adecuadas de monitorización.
Dexmedetomidina fue comparable a midazolam (relación de riesgos 1,07; intervalo de confianza del 95 % (IC) 0,971; 1,176) y a propofol (relación de riesgos 1,00; IC del 95 % 0,922; 1,075) en cuanto al tiempo dentro del rango objetivo de sedación, principalmente en pacientes de la UCI que necesitaban sedación prolongada de intensidad leve a moderada (de 0 a -3 puntos en la escala de evaluación de agitación-sedación de Richmond (RASS)) durante hasta 14 días; redujo la duración de la ventilación mecánica en comparación con midazolam y acortó el tiempo hasta la extubación traqueal en comparación con midazolam y propofol. Los pacientes que recibieron dexmedetomidina despertaron más fácilmente, colaboraron mejor con el personal y comunicaron mejor la intensidad del dolor en comparación con los pacientes que recibieron midazolam o propofol.
En los pacientes que recibieron dexmedetomidina, se produjeron con mayor frecuencia hipotensión arterial y bradicardia (menos frecuentemente taquicardia) en comparación con los pacientes que recibieron midazolam, y se produjo taquicardia con mayor frecuencia, pero la frecuencia de hipotensión arterial fue similar en comparación con los pacientes que recibieron propofol.
En comparación con el grupo que recibió propofol, la frecuencia de taquicardia en los pacientes que recibieron dexmedetomidina fue más alta, mientras que la frecuencia de hipotensión arterial fue aproximadamente la misma. La evaluación mediante la escala CAM-ICU mostró que la frecuencia de delirio en los pacientes que recibieron dexmedetomidina fue menor en comparación con midazolam, y los eventos adversos relacionados con el delirio ocurrieron con menos frecuencia en el grupo de dexmedetomidina en comparación con propofol. Los pacientes en los que se interrumpió el tratamiento con dexmedetomidina debido a una profundidad insuficiente de sedación fueron cambiados a propofol o midazolam. El riesgo de nivel insuficiente de sedación fue mayor en los pacientes que habían sido difíciles de sedar con métodos estándar inmediatamente antes del cambio a otro método sedante.
La evidencia de eficacia en la población pediátrica se obtuvo en un estudio controlado por dosis en una gran población posoperatoria en la UCI de 1 mes a 17 años de edad. Aproximadamente el 50 % de los pacientes que recibieron dexmedetomidina no requirieron sedación adicional con midazolam durante un período de tratamiento medio de 20,3 horas, pero no más de 24 horas. No existen datos sobre el tratamiento con el medicamento durante más de 24 horas en niños. La información sobre la administración del medicamento a recién nacidos (después de 28-44 semanas de gestación) es muy limitada y se refiere únicamente a dosis bajas (≤ 0,2 mcg/kg/hora). Los recién nacidos pueden ser especialmente sensibles al efecto bradicárdico de dexmedetomidina en presencia de hipotermia y en estados en los que el gasto cardíaco depende de la frecuencia cardíaca.
En estudios controlados doble ciego en la UCI, la frecuencia de supresión de cortisol en pacientes que recibieron dexmedetomidina (n=778) fue del 0,5 % en comparación con el 0 % en pacientes que recibieron midazolam (n=338) o propofol (n=275). Este efecto se observó como leve en un caso y de intensidad media en tres casos.
Indicación 2. Sedación procedimental con conservación de la conciencia.
La seguridad y eficacia de dexmedetomidina para la sedación de pacientes no intubados antes y/o durante intervenciones quirúrgicas y diagnósticas se evaluaron en dos estudios aleatorizados, doble ciego, controlados con placebo y multicéntricos.
Estudio 1. En el estudio 1, se aleatorizaron pacientes que habían sido sometidos a ciertas operaciones/procedimientos bajo anestesia controlada y anestesia local/regional, y que recibieron una dosis de carga de dexmedetomidina de 1 mcg/kg (n=129) o 0,5 mcg/kg (n=134), o placebo (solución fisiológica) (n=63) durante más de 10 minutos, seguida de una infusión de mantenimiento a una velocidad de 0,6 mcg/kg/hora. La velocidad de infusión de mantenimiento del medicamento investigado se tituló entre 0,2 y 1 mcg/kg/hora. La cantidad de pacientes que alcanzaron el nivel objetivo de sedación (escala de evaluación de alerta y sedación, Observer’s Assessment of Alertness/Sedation Scale – OAA/S ≤ 4), sin necesidad de administrar midazolam como sedante de rescate, fue del 54 % en el grupo que recibió 1 mcg/kg de dexmedetomidina y del 40 % en el grupo que recibió 0,5 mcg/kg de dexmedetomidina, en comparación con el 3 % en el grupo placebo.
La diferencia de riesgo en la cantidad de pacientes aleatorizados al grupo de dexmedetomidina de 1 mcg/kg y al grupo de dexmedetomidina de 0,5 mcg/kg que no requirieron midazolam como sedante de rescate fue del 48 % (IC del 95 %: 37-57 %) y del 40 % (IC del 95 %: 28-48 %), respectivamente, en comparación con placebo. La dosis media de midazolam como sedante de rescate fue de 1,5 (0,5-7,0) mg en el grupo de dexmedetomidina de 1,0 mcg/kg, 2 (0,5-8,0) mg en el grupo de dexmedetomidina de 0,5 mcg/kg y 4,0 (0,5-14,0) mg en el grupo placebo. La diferencia en las dosis medias de rescate de midazolam en los grupos de dexmedetomidina de 1 mcg/kg y 0,5 mcg/kg en comparación con placebo fue de 3,1 mg (IC del 95 %: 3,8 - 2,5) y 2,7 mg (IC del 95 %: 3,3 - 2,1), respectivamente, a favor de dexmedetomidina. El tiempo medio hasta la primera dosis de midazolam de rescate fue de 114 minutos en el grupo de dexmedetomidina de 1,0 mcg/kg, 40 minutos en el grupo de dexmedetomidina de 0,5 mcg/kg y 20 minutos en el grupo placebo.
Estudio 2. En el estudio 2, se aleatorizaron pacientes que se sometieron a intubación traqueal con fibroscopio con conciencia conservada bajo anestesia local, para recibir una infusión de carga de dexmedetomidina a una dosis de 1 mcg/kg (n=55) o placebo (solución fisiológica) (n=50) durante más de 10 minutos, seguida de una infusión de mantenimiento constante a una velocidad de 0,7 mcg/kg/hora. Se permitió a los pacientes el uso de midazolam como terapia de rescate si era necesario para alcanzar y/o mantener un nivel de sedación ≥ 2 en la escala de sedación de Ramsay (Ramsay Sedation Scale – RSS).
Los resultados de eficacia mostraron que dexmedetomidina es más eficaz que el placebo en la sedación de pacientes no intubados. La cantidad de pacientes que recibieron dexmedetomidina y no requirieron terapia de rescate con midazolam fue del 53 % en comparación con el 14 % en el grupo placebo.
Farmacocinética.
La farmacocinética de dexmedetomidina se estudió en voluntarios sanos tras administración intravenosa a corto plazo y en pacientes de la unidad de cuidados intensivos tras administración intravenosa prolongada.
Distribución. La farmacocinética de dexmedetomidina se describe mediante un modelo de dos compartimentos. En voluntarios sanos se observa una fase rápida de distribución con un período medio de semidistribución (t1/2α) de aproximadamente 6 minutos. El período medio de semivida en la fase terminal (t1/2) es de aproximadamente 1,9–2,5 horas (mínimo 1,35 horas, máximo 3,68 horas), y el volumen medio de distribución en estado de equilibrio (Vss) es de aproximadamente 1,16–2,16 l/kg (90–151 litros). El aclaramiento plasmático medio (Cl) es de 0,46–0,73 l/h/kg (35,7–51,1 l/h). La masa corporal media de los pacientes utilizada para calcular los parámetros Vss y Cl fue de 69 kg.
La farmacocinética plasmática de dexmedetomidina en pacientes de la UCI tras administración por infusión durante más de 24 horas fue similar. Los parámetros farmacocinéticos calculados fueron: t1/2 de aproximadamente 1,5 horas, Vss de aproximadamente 93 litros y Cl de aproximadamente 43 l/hora. En el rango de dosis de 0,2 a 1,4 mcg/kg/hora, la farmacocinética de dexmedetomidina es lineal, y no se acumula con tratamientos de hasta 14 días. El 94 % de dexmedetomidina se une a las proteínas plasmáticas. El grado de unión a proteínas plasmáticas es constante en un rango de concentraciones de 0,85 a 85 ng/ml. Dexmedetomidina se une tanto a la albúmina sérica humana como al glucoproteína ácida alfa1, principalmente a la albúmina sérica.
Biocatálisis y eliminación. Dexmedetomidina se metaboliza completamente en el hígado. El metabolismo inicial ocurre a través de tres vías metabólicas: glucuronidación directa N-, metilación directa N- y oxidación mediada por citocromo P450. Los metabolitos principales de dexmedetomidina en la sangre son dos N-glucurónidos isoméricos. El metabolito H-l (N-metil-3-hidroximetildexmedetomidina O-glucurónido) también es el principal producto circulante de la biotransformación de dexmedetomidina. El citocromo P450 cataliza la formación de dos metabolitos circulantes secundarios: 3-hidroximetildexmedetomidina, formado por hidroxilación del grupo metilo en la posición 3 de dexmedetomidina, y H-3, formado por oxidación del anillo imidazólico. Según la información disponible, la formación de metabolitos oxidados ocurre con participación de varias isoformas del citocromo P450 (CYP2A6, CYP1A2, CYP2E1, CYP2D6 y CYP2C19). Estos metabolitos no tienen actividad farmacológica significativa.
Después de la administración intravenosa de dexmedetomidina marcada radiactivamente, aproximadamente el 95 % de la radiactividad se excretó en la orina y el 4 % en las heces durante 9 días. Los metabolitos principales en la orina son dos N-glucurónidos isoméricos, que representan el 34 % de la dosis administrada, y el O-glucurónido de N-metil-3-hidroximetildexmedetomidina, que representa el 14,51 % de la dosis. Los metabolitos secundarios son: ácido dexmedetomidina carbónico, 3-hidroximetildexmedetomidina y su O-glucurónido, que representan entre el 1,11 % y el 7,66 % de la dosis. Menos del 1 % de dexmedetomidina sin cambios se detectó en la orina. Aproximadamente el 28 % de los metabolitos en la orina son metabolitos secundarios no identificados.
Grupos de pacientes especiales.
No se observaron diferencias significativas en la farmacocinética según edad o sexo.
En comparación con voluntarios sanos, en personas con alteraciones de la función hepática se observó una reducción en el grado de unión de dexmedetomidina a las proteínas plasmáticas. La fracción media no unida de dexmedetomidina varió desde el 8,5 % en voluntarios sanos hasta el 17,9 % en personas con alteraciones hepáticas graves. En pacientes con diferentes grados de insuficiencia hepática (clases A, B y C según la escala Child-Pugh), se redujo el aclaramiento hepático de dexmedetomidina y se prolongó el período de semivida plasmática (t1/2). Los valores medios del aclaramiento plasmático de la fracción no unida de dexmedetomidina en personas con insuficiencia hepática leve, moderada y grave fueron del 59 %, 51 % y 32 %, respectivamente, en comparación con los observados en voluntarios sanos. El t1/2 medio en personas con insuficiencia hepática leve, moderada y grave se prolongó hasta 3,9, 5,4 y 7,4 horas, respectivamente. A pesar de que la selección de la dosis de dexmedetomidina se realiza según el grado de efecto sedante, en pacientes con alteraciones de la función hepática, según la gravedad de la alteración o la respuesta clínica, se debe considerar la posibilidad de reducir la dosis inicial o de mantenimiento del medicamento.
En comparación con voluntarios sanos, en pacientes con alteraciones graves de la función renal (aclaramiento de creatinina <30 ml/min), la farmacocinética de dexmedetomidina no cambia.
Pacientes pediátricos. Los datos sobre la administración del medicamento en niños, desde recién nacidos (28-44 semanas de gestación) hasta niños de hasta 17 años, son limitados. El t1/2 de dexmedetomidina en niños (de 1 mes a 17 años) es similar al observado en adultos, pero en recién nacidos (menores de 1 mes) se observa un t1/2 más prolongado. En los grupos de edad de 1 mes a 6 años se observó un aclaramiento plasmático más prolongado ajustado por peso corporal, pero en niños mayores esta duración fue más corta. En recién nacidos (menores de 1 mes), el aclaramiento plasmático ajustado por peso corporal fue más corto (0,9 l/h/kg) que en grupos pediátricos mayores debido a la inmadurez. Los datos disponibles se presentan en la tabla 1.
Tabla 1
| Valor medio (95 % IC) |
|||
| Edad |
N |
Cl (l/h/kg) |
T1/2 (horas) |
| hasta 1 mes |
28 |
0,93 (0,76; 1,14) |
4,47 (3,81; 5,25) |
| 1 a <6 meses |
14 |
1,21 (0,99; 1,48) |
2,05 (1,59; 2,65) |
| 6 a <12 meses |
15 |
1,11 (0,94; 1,31) |
2,01 (1,81; 2,22) |
| 12 a <24 meses |
13 |
1,06 (0,87; 1,29) |
1,97 (1,62; 2,39) |
| 2 a <6 años |
26 |
1,11 (1,00; 1,23) |
1,75 (1,57; 1,96) |
| 6 a <17 años |
28 |
0,80 (0,69; 0,92) |
2,03 (1,78; 2,31) |
Características clínicas.
Indicaciones.
Sedación en adultos hospitalizados en la UCI (unidad de cuidados intensivos), cuya profundidad de sedación no exceda la respuesta a estímulo verbal (corresponde al rango de 0 a -3 puntos en la escala de agitación-sedación de Richmond [RASS]).
Sedación en pacientes adultos no intubados antes y/o durante la realización de procedimientos diagnósticos o quirúrgicos, es decir, sedación durante la asistencia anestésica/sedación en estado de conciencia.
Contraindicaciones.
Hipersensibilidad al dexmedetomidina o a cualquiera de los excipientes del medicamento.
Bloqueo auriculoventricular de grado II-III (en ausencia de marcapasos artificial).
Hipotensión arterial no controlada.
Patología cerebrovascular aguda.
Interacción con otros medicamentos y otras formas de interacción.
Los estudios de interacción medicamentosa se han realizado únicamente en adultos.
La administración concomitante de dexmedetomidina con anestésicos, sedantes, hipnóticos y opioides provoca una potenciación de sus efectos, tales como sedación, anestesia y efectos cardiorespiratorios. Esta suposición ha sido confirmada en estudios con isoflurano, propofol, alfentanilo y midazolam.
No se han detectado interacciones farmacocinéticas entre la dexmedetomidina y el isoflurano, propofol, alfentanilo y midazolam. Sin embargo, debido a posibles interacciones farmacodinámicas al administrarlos conjuntamente con dexmedetomidina, puede ser necesario reducir la dosis de dexmedetomidina o de los anestésicos, sedantes, hipnóticos u opioides administrados simultáneamente.
En estudios realizados con microsomas hepáticos humanos, se evaluó la capacidad de la dexmedetomidina para inhibir el citocromo P450, incluyendo el isoenzima CYP2B6. Según estudios in vitro, existe una posible interacción entre la dexmedetomidina y los sustratos (principalmente del isoenzima CYP2B6) in vivo.
Se observó in vitro la inducción por dexmedetomidina de los isoenzimas CYP1A2, CYP2B6, CYP2C8, CYP2C9 y CYP3A4, por lo que no puede descartarse la posibilidad de dicha interacción in vivo. La relevancia clínica es desconocida.
En pacientes que toman medicamentos que provocan disminución de la presión arterial y bradicardia, como los betabloqueantes, se debe considerar la posibilidad de potenciación de estos efectos (sin embargo, los efectos adicionales observados en un estudio de interacción con esmolol fueron moderados).
Características de uso.
Monitorización
Dexmedetomidina Calceks está indicado para su uso en unidades de cuidados intensivos, en quirófanos y durante procedimientos diagnósticos; su uso en otras condiciones no se recomienda. Durante la infusión del medicamento debe realizarse un monitoreo continuo de la actividad cardíaca. En pacientes no intubados, debe monitorearse la función respiratoria debido al riesgo de depresión respiratoria y, en algunos casos, desarrollo de apnea (ver sección «Reacciones adversas»).
El tiempo necesario para la recuperación tras la administración de dexmedetomidina es de aproximadamente una hora. Cuando se utilice en condiciones ambulatorias, debe continuarse un monitoreo cuidadoso durante al menos una hora (o más tiempo, según el estado del paciente), y la supervisión médica debe continuar durante una hora adicional para asegurar la seguridad del paciente.
Precauciones generales
No se debe administrar dexmedetomidina en forma de bolo, y tampoco se recomienda el uso de una dosis de carga en unidades de terapia intensiva (UTI). Los usuarios deben estar preparados para utilizar un agente sedante alternativo para el control agudo de la agitación, especialmente durante las primeras horas de tratamiento o durante procedimientos médicos. Durante la sedación con anestesia asistida, pueden administrarse pequeñas dosis en bolo de otro agente sedante para lograr rápidamente el nivel deseado de sedación.
En algunos pacientes que recibieron dexmedetomidina se observó una ligera reactivación, y recuperaron rápidamente la conciencia tras la estimulación. En ausencia de otros síntomas clínicos, este signo aislado no debe considerarse como falta de eficacia del medicamento.
Normalmente, dexmedetomidina no provoca sedación profunda, por lo que los pacientes pueden despertarse fácilmente. Por este motivo, dexmedetomidina no debe administrarse a pacientes que requieran sedación profunda.
Dexmedetomidina no debe usarse como anestésico general durante la intubación ni para proporcionar sedación cuando se emplean relajantes musculares.
Dexmedetomidina no suprime la actividad convulsiva, por lo que no debe usarse como monoterapia en el estado epiléptico.
Debe tenerse precaución al administrar dexmedetomidina junto con medicamentos que produzcan efectos sedantes o que afecten al sistema cardiovascular, debido al posible efecto aditivo.
No se recomienda el uso de dexmedetomidina para sedación controlada por el paciente. No existen datos adecuados al respecto.
Cuando se administre dexmedetomidina en condiciones ambulatorias, el alta del paciente puede realizarse bajo supervisión de terceros. Se debe aconsejar a los pacientes abstenerse de conducir vehículos o realizar otras actividades potencialmente peligrosas, y, si es posible, evitar el uso de otros agentes que puedan producir efectos sedantes (por ejemplo, benzodiazepinas, opioides, alcohol) durante un tiempo suficiente, según los efectos observados de dexmedetomidina, dependiendo del procedimiento, medicamentos utilizados, edad y estado del paciente.
Debe tenerse precaución al administrar dexmedetomidina a pacientes de edad avanzada. Los pacientes de 65 años o más pueden ser más propensos a la hipotensión arterial, especialmente cuando se administra una dosis de carga durante procedimientos con dexmedetomidina. Debe considerarse la posibilidad de reducir la dosis (ver sección «Instrucciones de uso y dosis»).
Mortalidad en pacientes de unidades de cuidados intensivos de edad ≤65 años
En un estudio controlado aleatorizado pragmático SPICE III con 3904 pacientes adultos críticamente enfermos en unidades de cuidados intensivos, dexmedetomidina se utilizó como agente sedante principal y se comparó con la terapia habitual. No hubo diferencias generales en la mortalidad a los 90 días entre el grupo que recibió dexmedetomidina y el grupo que recibió terapia habitual (mortalidad del 29,1% en ambos grupos), pero se observó heterogeneidad en el efecto según la edad sobre la mortalidad. Dexmedetomidina se asoció con un aumento de la mortalidad en el grupo de edad ≤65 años (relación de riesgos 1,26; intervalo de confianza del 95%: 1,02 a 1,56) en comparación con otros agentes sedantes. Aunque el mecanismo no es claro, esta heterogeneidad del efecto de la edad sobre la mortalidad fue más evidente en pacientes hospitalizados por causas no relacionadas con el cuidado posoperatorio, y aumentó con el mayor puntaje en la escala APACHE II y con la disminución de la edad. Estos resultados deben evaluarse considerando el beneficio clínico esperado de dexmedetomidina en comparación con otros agentes sedantes cuando se utiliza en pacientes más jóvenes.
Efectos cardiovasculares; advertencias
Dexmedetomidina disminuye la frecuencia cardíaca y la presión arterial (efecto simpaticolítico central), pero en concentraciones más altas provoca vasoconstricción periférica, lo que conduce a un aumento de la presión arterial (ver sección «Farmacodinámica»). Por ello, Dexmedetomidina Calceks no debe administrarse a pacientes con inestabilidad hemodinámica grave.
Debe tenerse precaución al administrar dexmedetomidina a pacientes con bradicardia asociada. Los datos sobre el efecto del medicamento en pacientes con frecuencia cardíaca <60 son limitados, por lo que estos pacientes requieren control y observación especiales. La bradicardia generalmente no requiere tratamiento, pero si es necesario, responde bien a la administración de anticolinérgicos de tipo M o a la reducción de la dosis del medicamento. Los pacientes deportistas con frecuencia cardíaca baja pueden ser especialmente sensibles al efecto cronotrópico negativo de los agonistas de los receptores alfa-2 adrenérgicos; se han descrito casos de paro del nódulo sinusal. También se han notificado casos de paro cardíaco precedidos por bradicardia o bloqueo auriculoventricular (ver sección «Reacciones adversas»).
En pacientes con hipotensión arterial asociada (especialmente refractaria a vasopresores), incluyendo hipotensión crónica, hipovolemia o reserva funcional reducida, como pacientes con disfunción ventricular grave y pacientes de edad avanzada, el efecto hipotensor de dexmedetomidina puede ser más pronunciado, lo que requiere especial atención a estos pacientes (ver sección «Contraindicaciones»). La disminución de la presión arterial generalmente no requiere medidas especiales, pero si es necesario, debe estar preparado para reducir la dosis, administrar fluidos para reponer el volumen sanguíneo circulante y/o vasopresores.
En pacientes con afectación del sistema autónomo (por ejemplo, debido a trauma medular), los efectos hemodinámicos tras la administración de dexmedetomidina pueden ser más intensos y requerir un control especial.
Se ha observado hipertensión arterial transitoria principalmente durante la administración de la dosis de carga, debido al efecto vasoconstrictor periférico de dexmedetomidina; por ello, no se recomienda la administración de una dosis de carga durante la sedación en UTI. El tratamiento de la presión arterial elevada generalmente no es necesario, pero debe considerarse la posibilidad de reducir la velocidad de infusión del medicamento.
La vasoconstricción local en concentraciones altas puede tener mayor relevancia en pacientes con enfermedad isquémica cardíaca o enfermedades cerebrovasculares graves; estos pacientes deben ser vigilados estrechamente. Si aparecen signos de isquemia miocárdica o cerebral, debe reducirse la dosis o suspenderse la administración del medicamento.
Se recomienda precaución al usar dexmedetomidina en combinación con anestesia espinal o epidural debido al posible aumento del riesgo de hipotensión arterial y bradicardia.
Pacientes con alteraciones de la función hepática
Debe tenerse precaución en pacientes con insuficiencia hepática grave, ya que la reducción del aclaramiento de dexmedetomidina con una administración excesiva del medicamento en estos pacientes puede aumentar el riesgo de reacciones adversas, sedación excesiva y prolongación de los efectos.
Pacientes con trastornos neurológicos
La experiencia con el uso de dexmedetomidina en estados neurológicos graves, como traumatismo craneoencefálico y período posoperatorio tras cirugías neuroquirúrgicas, es limitada; por lo tanto, el medicamento debe usarse con precaución en tales condiciones, especialmente si se requiere sedación profunda. Al elegir el tratamiento, debe considerarse que dexmedetomidina puede reducir el flujo sanguíneo cerebral y la presión intracraneal.
Otras advertencias
Tras la interrupción repentina de agonistas de los receptores alfa-2 adrenérgicos tras su uso prolongado, en algunos casos se ha descrito un síndrome de abstinencia. Si aparecen agitación y aumento de la presión arterial inmediatamente tras la suspensión de dexmedetomidina, debe considerarse la posibilidad de este síndrome.
Dexmedetomidina puede provocar hipertermia que no responde a los métodos tradicionales de enfriamiento. Ante la aparición de fiebre persistente de causa desconocida, debe suspenderse la administración de dexmedetomidina. Su uso no se recomienda en personas predispuestas a la hipertermia maligna.
Se han notificado casos de diabetes insípida relacionados con el tratamiento con dexmedetomidina. Ante la aparición de poliuria, se recomienda suspender el uso de dexmedetomidina y verificar los niveles de sodio en suero y la osmolaridad urinaria.
Dexmedetomidina Calceks contiene menos de 1 mmol de sodio (23 mg) por mililitro, es decir, prácticamente carece de sodio.
Uso durante el embarazo o la lactancia.
Embarazo.
No existen o son limitados los datos sobre el uso de dexmedetomidina en mujeres embarazadas. En estudios en animales se ha observado toxicidad reproductiva. Dexmedetomidina no debe administrarse durante el embarazo, salvo que el estado clínico de la mujer requiera su tratamiento con este medicamento.
Lactancia.
Dexmedetomidina se excreta en la leche materna humana, pero sus niveles se encuentran por debajo del límite de detección a las 24 horas tras la suspensión de la administración. No puede descartarse el riesgo para el lactante. La decisión de suspender la lactancia o el tratamiento con dexmedetomidina debe tomarse considerando los beneficios de la lactancia para el lactante y los beneficios del tratamiento con dexmedetomidina para la madre.
Fertilidad.
En un estudio de fertilidad en ratas, dexmedetomidina no afectó la función reproductiva de machos ni hembras. No existen datos sobre su efecto en la función reproductiva humana.
Capacidad para afectar la velocidad de reacción al conducir vehículos o manejar maquinaria.
Se recomienda a los pacientes abstenerse de conducir vehículos o realizar otras tareas peligrosas durante un período adecuado tras la administración de dexmedetomidina para sedación durante asistencia anestésica.
Vía de administración y dosis.
Sedación en pacientes adultos en UCI cuya profundidad de sedación no exceda la necesidad de despertar ante una estimulación verbal (corresponde al rango de 0 a -3 puntos en la escala de sedación-agitación de Richmond (RASS))
Solo para uso hospitalario. Este medicamento debe ser administrado por profesionales con experiencia en el tratamiento de pacientes en unidades de cuidados intensivos.
Dosis para adultos
A los pacientes ya intubados y que se encuentran en estado de sedación se les puede iniciar la administración de dexmedetomidina con una velocidad inicial de infusión de 0,7 mcg/kg/hora, que puede ajustarse progresivamente dentro del rango de dosis de 0,2 a 1,4 mcg/kg/hora para alcanzar el nivel deseado de sedación, según la respuesta del paciente. En pacientes debilitados debe considerarse una velocidad inicial de infusión más baja. Dexmedetomidina es un fármaco potente, por lo que su velocidad de administración se indica por hora. Tras un ajuste de dosis, puede llevar hasta una hora alcanzar la profundidad objetivo de sedación.
Dosis máxima
No debe superarse la dosis máxima del medicamento de 1,4 mcg/kg/hora. Los pacientes que no alcancen un nivel adecuado de sedación con la dosis máxima de dexmedetomidina deben cambiarse a un fármaco sedante alternativo.
No se recomienda la administración de una dosis de carga de dexmedetomidina en UCI, ya que aumenta la frecuencia de reacciones adversas. Si fuera necesario, pueden utilizarse propofol o midazolam hasta alcanzar el efecto clínico deseado con dexmedetomidina.
Duración
No existe experiencia con el uso de dexmedetomidina por más de 14 días. Si se administra el medicamento durante más de 14 días, debe evaluarse regularmente el estado del paciente.
Sedación en pacientes adultos no intubados antes y/o durante procedimientos diagnósticos o quirúrgicos, es decir, sedación durante asistencia anestésica/sedación en conciencia.
Dexmedetomidina debe administrarse únicamente por profesionales con experiencia en asistencia anestésica, ya sea en quirófano o durante procedimientos diagnósticos o terapéuticos. Durante la administración de dexmedetomidina para sedación en conciencia, los pacientes deben estar bajo supervisión continua del personal que participa en el procedimiento diagnóstico o quirúrgico. Es necesario realizar un monitoreo constante para detectar signos precoces de hipotensión arterial, hipertensión arterial, bradicardia, depresión respiratoria, obstrucción de las vías respiratorias, apnea, disnea y/o caída de la saturación.
Debe garantizarse la disponibilidad de oxigenoterapia, que debe aplicarse inmediatamente si así lo requiere el caso. La saturación de oxígeno debe monitorizarse mediante oximetría de pulso.
La administración de dexmedetomidina comienza con una dosis de carga seguida de una infusión de mantenimiento. Dependiendo del tipo de procedimiento, puede ser necesaria una anestesia local o regional o analgesia adicional para lograr el efecto clínico deseado. Se recomienda el uso de analgésicos o sedantes adicionales (por ejemplo, opioides, midazolam, propofol) en procedimientos dolorosos o cuando se requiera un nivel más profundo de sedación. La vida media farmacocinética de distribución de dexmedetomidina se estima aproximadamente en 6 minutos. Este parámetro debe considerarse junto con los efectos de otros medicamentos utilizados para evaluar el tiempo necesario para la titulación y alcanzar el efecto clínico deseado con dexmedetomidina.
Inicio de la sedación durante asistencia anestésica:
- Dosis de carga mediante infusión de 1 mcg/kg durante 10 minutos. En procedimientos menos invasivos, por ejemplo cirugía oftalmológica, puede utilizarse una dosis de carga de 0,5 mcg/kg durante 10 minutos.
Mantenimiento de la sedación durante asistencia anestésica:
- La infusión de mantenimiento generalmente comienza con una dosis de 0,6–0,7 mcg/kg/hora y se titula para alcanzar el efecto clínico deseado en un rango de dosis de 0,2 a 1 mcg/kg/hora. La velocidad de la infusión de mantenimiento debe ajustarse hasta alcanzar el nivel objetivo de sedación.
Grupos especiales de pacientes
Pacientes de edad avanzada. Generalmente no se requiere ajuste de dosis. Los pacientes de edad avanzada pueden tener un mayor riesgo de hipotensión arterial, aunque los datos limitados disponibles sobre sedación durante asistencia anestésica no indican claramente una relación dosis-dependiente de este riesgo.
Alteración de la función renal. En pacientes con alteración de la función renal generalmente no se requiere ajuste de dosis.
Alteración de la función hepática. Dexmedetomidina se metaboliza en el hígado, por lo que debe administrarse con precaución en pacientes con alteración de la función hepática. Debe considerarse la conveniencia de utilizar una dosis de mantenimiento reducida (ver secciones «Farmacocinética» y «Precauciones de uso»).
Vía de administración
El medicamento debe administrarse únicamente después de dilución, mediante infusión intravenosa utilizando un dispositivo de infusión controlado.
Un vial del medicamento está destinado exclusivamente para un solo paciente.
Preparación de la solución
Antes de su uso, Dexmedetomidina Kalceks puede diluirse en solución glucosada al 5 %, solución de Ringer, solución de Ringer con lactato, manitol o solución de cloruro de sodio al 0,9 %, para alcanzar la concentración deseada de 4 mcg/ml o 8 mcg/ml. En las tablas siguientes se indican los volúmenes necesarios para la preparación de la infusión.
Tabla 2
Para alcanzar una concentración de 4 mcg/ml:
| Volumen del medicamento Dexmedetomidina Kalceks, concentrado para solución para perfusión, 100 mcg/ml |
Volumen del disolvente, ml |
Volumen total de la perfusión, ml |
| 2 ml |
48 |
50 |
| 4 ml |
96 |
100 |
| 10 ml |
240 |
250 |
| 20 ml |
480 |
500 |
Tabla 3
Para alcanzar una concentración de 8 µg/ml:
| Volumen del medicamento Dexmedetomidina Kalceks, concentrado para solución para perfusión, 100 mcg/ml |
Volúmen del diluyente, ml |
Volúmen total de la perfusión, ml |
| 4 ml |
46 |
50 |
| 8 ml |
92 |
100 |
| 20 ml |
230 |
250 |
| 40 ml |
460 |
500 |
Agitar cuidadosamente para mezclar bien la solución.
Antes de la administración, la solución debe inspeccionarse visualmente en busca de partículas extrañas o cambios de color.
Dexmedetomidina Calceks es farmacéuticamente compatible con los siguientes líquidos y medicamentos intravenosos: solución de Ringer con lactato, solución de glucosa al 5 %, solución de cloruro de sodio al 0,9 %, manitol al 20 %, tiopental sódico, etomidato, bromuro de vecuronio, bromuro de pancuronio, succinilcolina, besilato de atracurio, cloruro de mivacurio, bromuro de rocuronio, bromuro de glicopirrolato, clorhidrato de fenilefrina, sulfato de atropina, dopamina, noradrenalina, dobutamina, midazolam, sulfato de morfina, citrato de fentanilo, agentes plasmáticos.
Cómo abrir la ampolla:
- Gire la ampolla con el punto de color hacia usted. Toque ligeramente con el dedo la parte superior de la ampolla para que la solución baje a la parte inferior.
- Utilice ambas manos para abrir la ampolla: sujetando la parte inferior de la ampolla con una mano, con la otra mano presione la parte superior de la ampolla en dirección opuesta al punto de color (véase la figura inferior).
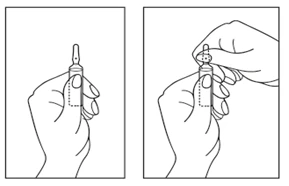
El medicamento no utilizado debe eliminarse de acuerdo con los requisitos locales.
Niños.
La seguridad y eficacia del uso de dexmedetomidina en niños de 0 a 18 años de edad no han sido establecidas. Los datos sobre el uso del medicamento en esta categoría de pacientes se proporcionan en las secciones «Reacciones adversas» y «Propiedades farmacológicas», pero no pueden darse recomendaciones sobre la dosificación.
Sobredosis.
Síntomas.
Durante los estudios clínicos y el uso poscomercialización se han notificado varios casos de sobredosis de dexmedetomidina. Según los datos disponibles, la velocidad de administración en tales casos alcanzó 60 mcg/kg/hora durante 36 minutos y 30 mcg/kg/hora durante 15 minutos en un niño de 20 meses y un adulto, respectivamente. Las reacciones adversas más frecuentes debidas a la sobredosis son bradicardia, hipotensión arterial, hipertensión arterial, sedación excesiva, depresión respiratoria y paro cardíaco.
Tratamiento.
En caso de sobredosis con síntomas clínicos, se debe reducir o interrumpir la administración de dexmedetomidina. Los efectos esperados son principalmente cardiovasculares y deben tratarse según las indicaciones clínicas (véase la sección «Precauciones de uso»). Con concentraciones elevadas, el aumento de la presión arterial puede predominar sobre su disminución. En estudios clínicos, la parada del nódulo sinusal se permitió que resolviera espontáneamente o en respuesta a la administración de atropina o glicopirrolato. En casos aislados de sobredosis grave acompañada de paro cardíaco, fue necesario realizar maniobras de reanimación.
Reacciones adversas.
Resumen según el perfil de seguridad
Sedación en pacientes adultos en UCI
Las reacciones adversas más frecuentemente notificadas con el uso de dexmedetomidina en pacientes en UCI son hipotensión arterial, hipertensión arterial y bradicardia, que ocurren aproximadamente en el 25 %, 15 % y 13 % de los pacientes, respectivamente. La hipotensión arterial y la bradicardia también fueron las reacciones adversas graves más frecuentes atribuibles a dexmedetomidina, que ocurrieron en el 1,7 % y 0,9 % de los pacientes aleatorizados en UCI, respectivamente.
Sedación durante la asistencia anestésica/sedación en pacientes conscientes
Las reacciones adversas más frecuentemente notificadas con el uso de dexmedetomidina durante la sedación durante la asistencia anestésica son las siguientes:
- hipotensión arterial (55 % en el grupo de dexmedetomidina frente al 30 % en el grupo placebo);
- depresión respiratoria (38 % en el grupo de dexmedetomidina frente al 35 % en el grupo placebo);
- bradicardia (14 % en el grupo de dexmedetomidina frente al 4 % en el grupo placebo).
Las reacciones adversas que se indican a continuación se obtuvieron a partir de datos combinados de estudios clínicos en pacientes en UCI.
La frecuencia de aparición de reacciones adversas se clasifica de la siguiente manera: muy frecuente (≥ 1/10), frecuente (≥ 1/100, < 1/10), poco frecuente (≥ 1/1000, < 1/100), rara (≥ 1/10000, < 1/1000), muy rara (< 1/10000), frecuencia no conocida (no puede determinarse con los datos disponibles).
Del sistema endocrino: frecuencia no conocida – diabetes insípida.
Del metabolismo y la nutrición: frecuente – hiperglucemia, hipoglucemia; poco frecuente – acidosis metabólica, hipoalbuminemia.
Alteraciones psiquiátricas: frecuente – agitación; poco frecuente – alucinaciones.
Del corazón: muy frecuente – bradicardia1,2; frecuente – isquemia o infarto de miocardio, taquicardia; poco frecuente – bloqueo auriculoventricular1, disminución del gasto cardíaco, paro cardíaco1.
Del sistema vascular: muy frecuente – hipotensión arterial1,2, hipertensión arterial1,2.
Del sistema respiratorio, órganos torácicos y mediastino: muy frecuente – depresión respiratoria2,3; poco frecuente – disnea, apnea.
Del tracto gastrointestinal: frecuente – náuseas2, vómitos, sequedad de boca2; poco frecuente – distensión abdominal.
Alteraciones generales y reacciones en el sitio de administración: frecuente – síndrome de abstinencia, hipertermia; poco frecuente – ineficacia del medicamento, sed.
1Ver descripción de reacciones adversas individuales a continuación.
2Reacción adversa también observada en estudios de sedación durante la asistencia anestésica.
3Frecuencia "frecuente" en el estudio de sedación en UCI.
Descripción de reacciones adversas individuales.
Las reducciones clínicamente significativas de la presión arterial y la bradicardia deben tratarse como se indica en la sección «Precauciones de uso».
En voluntarios sanos relativamente estables que no estaban en UCI, la administración de dexmedetomidina provocó en ocasiones bradicardia que condujo a la parada del nódulo sinusal o a pausa sinusal. Los síntomas se resolvieron mediante elevación de las piernas y administración de agentes anticolinérgicos como atropina o glicopirrolato. En casos aislados, en pacientes con bradicardia preexistente, esta progresó a episodios de asistolía. También se han notificado casos de paro cardíaco precedidos por bradicardia o bloqueo auriculoventricular.
La hipertensión arterial estuvo asociada con la administración de la dosis de carga. Esta reacción puede reducirse evitando la dosis de carga o disminuyendo la velocidad de infusión o la dosis de carga.
Pacientes pediátricos.
Se evaluó el tratamiento en niños a partir de 1 mes de edad, principalmente tras cirugía, que estuvieron en UCI durante hasta 24 horas; se demostró un perfil de seguridad comparable al de adultos. Los datos en recién nacidos (28-44 semanas de gestación) son muy limitados, y las dosis se limitan a dosis de mantenimiento ≤ 0,2 mcg/kg/hora. En publicaciones se ha descrito un caso aislado de bradicardia hipotérmica en un recién nacido.
Periodo de validez. 5 años.
No utilizar después de la fecha de caducidad indicada en el envase.
Condiciones de conservación.
No requiere condiciones especiales de conservación.
Conservar en lugar fuera del alcance de los niños.
Después de la dilución.
Después de la dilución, se ha demostrado estabilidad química y física durante 36 horas a 25 °C.
Desde el punto de vista microbiológico, la solución debe usarse inmediatamente. Si no se utiliza inmediatamente, el tiempo y las condiciones de almacenamiento son responsabilidad del usuario, generalmente no debe exceder las 24 horas a una temperatura de entre 2 y 8 °C, a menos que la dilución se haya realizado en condiciones asépticas bien controladas y validadas.
Incompatibilidades.
Este medicamento no debe mezclarse con otros medicamentos, excepto los indicados en la sección «Instrucciones de uso y dosis».
Existe la posibilidad de adsorción de dexmedetomidina con ciertos tipos de caucho natural. Aunque dexmedetomidina se utiliza hasta alcanzar el efecto clínico deseado, se recomienda emplear materiales con revestimiento de caucho sintético o natural.
Envase.
2 ml en ampolla de vidrio incoloro de clase I hidrolítica con anillos de identificación y punto de fractura.
5 ampollas en envase blíster de lámina de cloruro de polivinilo.
1 o 5 envases blíster en estuche de cartón.
Categoría de dispensación.
Medicamento sujeto a prescripción médica.
Fabricante.
Fabricante responsable del lanzamiento del lote:
S.A. «Kalceks».
Dirección del fabricante y lugar de actividad.
Calle Krustpils, 71E, Riga, LV-1057, Letonia.
Titular del medicamento.
S.A. «Kalceks».
Dirección del titular y/o representante del titular.
Calle Krustpils, 71E, Riga, LV-1057, Letonia.