Arixtra®
Ukraina
Spis treści
INSTRUKCJA dotyczÄ cza stosowania leku Arixtra®
SkÅ ad:
substancja czynna: fondaparynux sodu;
1 strzykawka (0,4 ml) zawiera 5 mg fondaparynuxu sodu;
1 strzykawka (0,6 ml) zawiera 7,5 mg fondaparynuxu sodu;
1 strzykawka (0,8 ml) zawiera 10 mg fondaparynuxu sodu;
substancje pomocnicze: chlorek sodu, woda do wstrzykiwań, wodorotlenek sodu lub kwas chlorowodorowy.
PostaÄ c leku. Roztwór do wstrzykiwaÅ„.
Główne cechy fizykochemiczne: wstÄ™pnie wypeÅ‚niona szklana strzykawka zawierajÄ ca przezroczystÄ lub prawie przezroczystÄ ciecz o barwie od bezbarwnej do Å‚agodnie żółtej, praktycznie bez widocznych czÄ…stek.
Grupa farmakoterapeutyczna. Leki przeciwcieniżdżowe. Kod ATC B01AX05.
Właściwości farmakologiczne.
Farmakodynamika.
Fondaparynux jest syntetycznym, selektywnym inhibitorem aktywowanego czynnika X (Xa). Działanie przeciwzakrzepowe fondaparynuksu wynika ze selektywnej inhibicji czynnika Xa, pośredniczonej przez antytrombinę III (AT III). Fondaparynux wiąże się selektywnie z AT III, co potęguje (około 300-krotnie) początkową neutralizację czynnika Xa przez antytrombinę III. Neutralizacja czynnika Xa przerywa łańcuch krzepnięcia krwi i hamuje zarówno powstawanie trombiny, jak i tworzenie się skrzeplin. Lek nie inaktywuje trombiny (aktywowanego czynnika IIa) i nie wpływa na płytki krwi.
W zalecanych dawkach terapeutycznych fondaparynux nie wywiera klinicznie istotnego wpływu na wyniki standardowych testów krzepnięcia, takich jak czas częściowego tromboplastynowego (APTT), czas krzepnięcia aktywowanego (ACT) czy czas protrombinowy (PT)/międzynarodowe znormalizowane stosunki (INR) we krwi, a także nie zmienia czasu krwawienia ani aktywności fibrynolitycznej. Jednak rzadko pojawiały się spontaniczne doniesienia o przedłużeniu APTT. W wyższych dawkach możliwe są umiarkowane zmiany APTT. W dawce 10 mg, stosowanej w badaniach interakcji, fondaparynux nie wykazywał istotnego wpływu na działanie przeciwkrzepne (INR) warfaryny.
Fondaparynux nie wywołuje reakcji krzyżowych z surowicą chorych z heparynową trombocytopenią indukowaną.
Farmakokinetyka.
Parametry farmakokinetyczne sodu fondaparynuksu ustalono na podstawie stężeń fondaparynuksu w osoczu krwi, ilościowo określonych według aktywności przeciwko czynnikowi Xa. Do kalibracji analizy przeciw-Xa można używać wyłącznie fondaparynuksu (międzynarodowe standardy heparyny lub heparyny o niskiej masie cząsteczkowej nie są do tego celu odpowiednie). Stężenia fondaparynuksu wyrażane są w miligramach (mg).
Wchłanianie.
Po podaniu podskórnej fondaparynux jest szybko i całkowicie wchłaniany (biologiczna dostępność absolutna – 100%). Po jednorazowym podaniu podskórnym 2,5 mg fondaparynuksu młodym zdrowym ochotnikom maksymalne stężenie w osoczu krwi (średnie C\max = 0,34 mg/l) osiągane było po 2 godzinach od podania dawki. Stężenie w osoczu równe połowie powyższego maksymalnego stężenia osiągane było po 25 minutach od podania dawki.
U zdrowych ochotników w podeszłym wieku farmakokinetyka fondaparynuksu jest liniowa w zakresie dawek 2–8 mg podawanych podskórnie. Po podawaniu raz na dobę stężenie równowagowe w osoczu krwi osiągane jest po 3–4 dniach, przy wzroście wartości C\max i AUC (pola pod krzywą) o 1,3-krotność.
Średnie (współczynnik zmienności – CV, %) parametry farmakokinetyczne fondaparynuksu w stanie równowagi u chorych po operacjach stawu biodrowego, stosujących lek w dawce 2,5 mg raz na dobę, były następujące: C\max – 0,39 mg/l (31%), T\max – 2,8 godz. (18%) i C\min – 0,14 mg/l (56%). U chorych w podeszłym wieku po operacjach związanych z złamaniami kości udowej stężenia równowagowe fondaparynuksu wynosiły: C\max – 0,50 mg/l (32%), C\min – 0,19 mg/l (58%).
W leczeniu ostrego zakrzepicy żył głębokich i zakrzembowego zatoru tętnicy płucnej u chorych otrzymujących Arixtra® w dawkach 5 mg (masa ciała < 50 kg), 7,5 mg (masa ciała 50–100 kg włącznie) oraz 10 mg (masa ciała > 100 kg) raz na dobę, średnie parametry farmakoterapeutyczne dostosowane do masy ciała były podobne we wszystkich kategoriach pacjentów. Obliczone średnie wartości (CV %) parametrów farmakokinetycznych fondaparynuksu w stanie równowagi u pacjentów z zakrzepicą żył głębokich (DVT), którym lek podawano w zalecanej dawce raz na dobę, były następujące: C\max (mg/l) – 1,41 (23%), T\max (godz.) – 2,4 (8%) i C\min (mg/l) – 0,52 (45%). Odpowiednie wartości percentyli 5. i 95. wynosiły 0,97 i 1,92 dla C\max (mg/l) oraz 0,24 i 0,95 dla C\min (mg/l).
Rozkład.
Objętość rozkładu jest ograniczona i wynosi 7–11 l. In vitro fondaparynux wiąże się w znacznym stopniu i specyficznie z białkiem AT III, a stopień wiązania zależy od stężenia leku w osoczu krwi (od 98,6 do 97,0% w zakresie stężeń od 0,5 do 2 mg/l). Wiązanie fondaparynuksu z innymi białkami osocza krwi, w tym z czynnikiem płytkowym IV, jest niewielkie.
Ponieważ fondaparynux nie wiąże się w znaczącym stopniu z innymi białkami osocza krwi poza antytrombiną, nie należy oczekiwać interakcji z innymi lekami poprzez wypieranie z miejsc wiązania z białkami.
Metabolizm.
Chociaż pełna ocena nie została przeprowadzona, nie stwierdzono oznak metabolizmu fondaparynuksu, a w szczególności powstawania metabolitów aktywnych.
Fondaparynux nie hamuje enzymów układu cytochromu CYP450 (CYP1A2, CYP2A6, CYP2C9, CYP2C19, CYP2D6, CYP2E1 ani CYP3A4) in vitro. Dlatego nie należy oczekiwać interakcji fondaparynuksu z innymi lekami na poziomie hamowania metabolizmu pośredniczonego przez układ CYP in vivo.
Wydalanie.
Fondaparynux jest wydalany głównie przez nerki w niezmienionej postaci – u zdrowych ochotników w 64–77%. Okres półtrwania (T1/2) wynosi około 17 godzin u młodych zdrowych ochotników i około 21 godzin u zdrowych ochotników w podeszłym wieku.
Grupy specjalne pacjentów.
Zaburzenia funkcji nerek.
W porównaniu z pacjentami o normalnej funkcji nerek (klirens kreatyniny > 80 ml/min), klirens osoczowy jest niższy o 1,2–1,4 razy u pacjentów z łagodnymi zaburzeniami funkcji nerek (klirens kreatyniny od 50 do 80 ml/min) i średnio 2-krotnie niższy u pacjentów z umiarkowanymi zaburzeniami funkcji nerek (klirens kreatyniny od 30 do 50 ml/min). W przypadku ciężkich zaburzeń funkcji nerek (klirens kreatyniny < 30 ml/min) klirens osoczowy jest około 5-krotnie niższy niż przy normalnej funkcji nerek. Odpowiednie końcowe okresy półtrwania wynosiły 29 godzin przy umiarkowanym i 72 godziny przy ciężkim stopniu niewydolności nerek. Analogiczna zależność między klirensiem fondaparynuksu a nasileniem niewydolności nerek obserwowana była u pacjentów leczonych z powodu zakrzepicy żył głębokich i zakrzembowego zatoru tętnicy płucnej.
Zaburzenia funkcji wątroby.
Zgodnie z danymi farmakokinetycznymi oczekuje się, że stężenie niezwiązanego fondaparynuksu pozostanie niezmienione u pacjentów z niewydolnością wątroby o lekkim i umiarkowanym stopniu, dlatego nie ma potrzeby zmiany dawki. Po jednorazowym podaniu podskórnym fondaparynuksu u chorych z umiarkowaną niewydolnością wątroby (skala Childa-Pugh, klasa B) C\max i AUC ogólnego (związanego i niezwiązanego) fondaparynuksu były niższe odpowiednio o 22% i 39% w porównaniu z pacjentami o normalnej funkcji wątroby. Niższe stężenie fondaparynuksu w osoczu wyjaśnia się zmniejszonym wiązaniem z AT III, ponieważ u pacjentów z niewydolnością wątroby stężenie AT III w osoczu krwi jest niższe. W rezultacie obserwuje się zwiększenie klirensu nerkowego fondaparynuksu.
Farmakokinetyka fondaparynuksu nie była badana u pacjentów z ciężką niewydolnością wątroby (patrz sekcje ↔Sposób stosowania i dawki» i ↔Szczególne wskazania»).
Dzieci. Stosowanie fondaparynuksu u dzieci nie było badane.
Pacjenci w podeszłym wieku.
Funkcja nerek może się obniżać z wiekiem, dlatego wydalanie fondaparynuksu u pacjentów powyżej 75. roku życia może być zaburzone. Po operacji ortopedycznej w badaniu, przy stosowaniu leku w dawce 2,5 mg raz na dobę, ogólny klirens fondaparynuksu był niższy o 1,2–1,4 razy u pacjentów powyżej 75. roku życia w porównaniu z pacjentami do 65. roku życia. Analogiczna zależność między klirensiem fondaparynuksu a wiekiem obserwowana była u pacjentów leczonych z powodu zakrzepicy żył głębokich i zakrzembowego zatoru tętnicy płucnej.
Płeć.
Po skorygowaniu dawki względem masy ciała nie stwierdzono różnic w farmakokinetyce u mężczyzn i kobiet.
Rasa.
Nie przeprowadzono planowanych badań różnic farmakokinetycznych. Jednak badania z udziałem zdrowych ochotników rasy mongolskiej nie wykazały różnic w profilu farmakokinetycznym w porównaniu z ochotnikami rasy europejskiej. Nie zaobserwowano różnic w klirensie osoczowym leku między pacjentami rasy europejskiej i negroidalnej po operacjach ortopedycznych.
Masa ciała.
Klirens fondaparynuksu z osocza krwi wzrasta wraz ze wzrostem masy ciała (o 9% na każde 10 kg masy ciała).
Właściwości kliniczne.
Wskazania.
Leczenie ostrego zakrzepicy żył głębokich, leczenie ostrej zakrzepicy tętnicy płucnej, z wyjątkiem pacjentów hemodynamicznie niestabilnych lub pacjentów wymagających trombolizy lub embolektomii płucnej.
Przeciwwskazania.
Stwierdzona alergia na substancję czynną lub którąkolwiek z substancji pomocniczych leku. Nasilone, klinicznie istotne krwawienie. Ostry bakteryjny zapalenie wsierdzia. Ciężka niewydolność nerek (klirens kreatyniny < 30 ml/min).
Współdziałanie z innymi lekami i inne rodzaje interakcji.
Leków, które mogą zwiększać ryzyko krwawienia, nie należy stosować jednocześnie z Arixtra®, z wyjątkiem antagonistów witaminy K stosowanych w leczeniu żylakowatości tętnic płucnych (patrz sekcja ↔Szczególne środki ostrożności przy stosowaniu»). Jeżeli konieczne jest takie wspólne stosowanie, należy je prowadzić pod ścisłym nadzorem.
W wyniku badań klinicznych fondaparynuksu wykazano, że wspólne stosowanie fondaparynuksu z doustnymi lekami przeciwzakrzepowymi (warfaryna), lekami przeciwpłytkowymi (kwas acetylosalicylowy), niesteroidowymi lekami przeciwzapalnymi (piroksykam) i glikozydami nasierdziowymi (digoxygen) nie wpływa istotnie na farmakokinetykę fondaparynuksu. Ponadto, lek w dawce 10 mg, stosowany w badaniach interakcji, nie wpływał ani na działanie przeciwzakrzepowe (według międzynarodowego znormalizowanego stosunku – INR) warfaryny, ani na czas krwawienia podczas leczenia kwasem acetylosalicylowym lub piroksykamem, ani na farmakokinetykę digoxyny w stanie równowagi.
Szczególne wskazania dotyczące stosowania.
Arixtra® przeznaczona jest wyłącznie do podania podskórnie. Nie należy stosować wstrzykiwań domięśniowych.
Doświadczenie w stosowaniu fondaparynyku su w przypadku pacjentów hemodynamicznie niestabilnych jest ograniczone, a doświadczenie w stosowaniu u pacjentów wymagających trombolizy, embolektomii lub wszczepienia filtru do żyły głównej dolnej – nie istnieje.
Okrwawienia.
Arixtra® należy stosować ostrożnie u chorych z podwyższonym ryzykiem krwawienia, w szczególności w przypadku wrodzonych lub nabytych zaburzeń krzepliwości krwi (np. liczba płytek krwi < 50 000/mm³), aktywnej choroby wrzodowej żołądka i jelit, niedawnego krwotoku śródczaszkowego, niedawnego zabiegu chirurgicznego mózgu lub rdzenia kręgowego lub operacji oczu, a także u innych grup pacjentów opisanych poniżej.
Tak jak w przypadku innych leków przeciwkrzepliwych, Arixtra® należy stosować ostrożnie u chorych, którzy niedawno przeszli zabieg chirurgiczny (< 3 doby), i to wyłącznie po osiągnięciu hemostazy.
Nie należy stosować jednocześnie z Arixtra® leków, które mogą zwiększać ryzyko krwawienia. Należą do nich substancje takie jak dezirudyna, leki fibrynolityczne, antagoniści receptorów GP IIb/IIIa, heparyna, heparynoidy lub heparyny o niskiej masie cząsteczkowej. W leczeniu zakrzepicy żył głębokich (ZŻG), jeśli konieczne jest jednoczesne podawanie leku wraz z terapią antagonistami witaminy K, należy wziąć pod uwagę informacje zamieszczone w sekcji „Interakcje z innymi lekami”. Inne leki przeciwpłytkowe (kwas acetylosalicylowy, dipyrydometol, sulfinpirazon, tyklopidyna lub klopidogrel) oraz niesteroidowe leki przeciwzapalne należy stosować ostrożnie. Jeśli terapia kombinowana jest absolutnie konieczna, należy ją prowadzić pod ścisłym nadzorem.
Znieczulenie przewodowe/naprawa lędźwiowa.
W przypadku stosowania Arixtry® w leczeniu zakrzepicy żył głębokich, a nie w celu jej zapobiegania, nie należy przeprowadzać znieczulenia przewodowego/naprawy lędźwiowej w przypadku konieczności zabiegu operacyjnego.
Pacjenci w podeszłym wieku.
Ryzyko wystąpienia krwawienia u pacjentów w podeszłym wieku jest wyższe niż u innych chorych. Ponieważ czynność nerek zazwyczaj zmniejsza się z wiekiem, u pacjentów w podeszłym wieku wydalanie fondaparynyku może być obniżone, a tym samym ekspozycja na lek zwiększa się (patrz sekcja „Właściwości farmakologiczne. Farmakokinetyka”). Częstość wystąpienia krwawień przy stosowaniu leku według zalecanej dawki w leczeniu zakrzepicy żył głębokich lub zakrzepicy płucnej u pacjentów w wieku odpowiednio < 65 lat, 65–75 lat i > 75 lat wynosiła 3,0 %, 4,5 % i 6,5 %. Odpowiednie wartości częstości zdarzeń u pacjentów otrzymujących enoksaparynę według zalecanej dawki w leczeniu zakrzepicy żył głębokich wynosiły 2,5 %, 3,6 % i 8,3 %, natomiast u pacjentów otrzymujących heparynę nieryzynową według zalecanej dawki w leczeniu zakrzepicy płucnej – odpowiednio 5,5 %, 6,6 % i 7,4 %. Dlatego Arixtra® należy stosować ostrożnie u pacjentów z tej grupy wiekowej (patrz sekcja „Sposób stosowania i dawki”).
Niska masa ciała.
Doświadczenie kliniczne w stosowaniu leku u pacjentów o masie ciała < 50 kg jest ograniczone. Pacjentom z tej grupy fondaparynyk należy stosować ostrożnie w dawce dobowej 5 mg (patrz sekcje „Sposób stosowania i dawki” oraz „Właściwości farmakologiczne. Farmakokinetyka”).
Niewydolność nerek.
Wraz ze wzrostem stopnia ciężkości niewydolności nerek wzrasta ryzyko krwawienia. Częstość krwawień przy stosowaniu leku według zalecanej dawki w leczeniu zakrzepicy żył głębokich lub zakrzepicy płucnej u pacjentów z prawidłową czynnością nerek, z lekkimi, umiarkowanymi i ciężkimi zaburzeniami czynności nerek wynosiła odpowiednio 3,0 % (34 z 1132 pacjentów), 4,4 % (32 z 733 pacjentów), 6,6 % (21 z 318 pacjentów) i 14,5 % (8 z 55 pacjentów). Częstość takich zdarzeń u pacjentów otrzymujących enoksaparynę według zalecanej dawki w leczeniu zakrzepicy żył głębokich wynosiła odpowiednio 2,3 % (13 z 559 pacjentów), 4,6 % (17 z 368 pacjentów), 9,7 % (14 z 145 pacjentów) i 11,1 % (2 z 18 pacjentów), natomiast u pacjentów otrzymujących heparynę nieryzynową według zalecanej dawki w leczeniu zakrzepicy płucnej – odpowiednio 6,9 % (36 z 523 pacjentów), 3,1 % (11 z 352 pacjentów), 11,1 % (18 z 162 pacjentów) i 10,7 % (3 z 28 pacjentów).
Arixtra® jest przeciwwskazana u chorych z ciężką niewydolnością nerek (klirens kreatyniny < 30 ml/min). Lek należy stosować ostrożnie w leczeniu pacjentów z umiarkowaną niewydolnością nerek (klirens kreatyniny 30–50 ml/min). Czas trwania leczenia nie powinien przekraczać czasu zastosowanego w badaniach klinicznych (średnio 7 dni) (patrz sekcje „Sposób stosowania i dawki”, „Przeciwwskazania” oraz „Właściwości farmakologiczne. Farmakokinetyka”).
Brak doświadczenia w stosowaniu leku u pacjentów o dużej masie ciała (> 100 kg), którzy jednocześnie mają umiarkowane zaburzenia czynności nerek (klirens kreatyniny 30–50 ml/min). Takim pacjentom fondaparynyk należy stosować ostrożnie. Po podaniu początkowej dawki dobowej 10 mg można rozważyć zmniejszenie dawki dobowej do 7,5 mg, opierając się na danych modelowania farmakokinetycznego (patrz sekcja „Sposób stosowania i dawki”).
Niewydolność wątroby ciężkiego stopnia.
Arixtra® należy stosować ostrożnie ze względu na zwiększone ryzyko krwawienia związanego z niedoborem czynników krzepliwości u pacjentów z niewydolnością wątroby ciężkiego stopnia (patrz sekcja „Sposób stosowania i dawki”).
Indukowana heparyną trombocytopenia.
Fondaparynyk nie wiąże się z czynnikiem IV płytek krwi i nie reaguje krzyżowo z surowicą pacjentów z indukowaną heparyną trombocytopenią typu II. Arixtra® należy stosować ostrożnie w leczeniu pacjentów z wywiadem indukowanej heparyną trombocytopenii. Skuteczność i bezpieczeństwo stosowania Arixtry® w leczeniu pacjentów z indukowaną heparyną trombocytopenią typu II nie zostały zbadane. Otrzymano pojedyncze spontaniczne doniesienia o rozwoju indukowanej heparyną trombocytopenii u pacjentów leczonych fondaparynykiem. Obecnie nie stwierdzono związku między leczeniem Arixtra® a wystąpieniem indukowanej heparyną trombocytopenii.
Alergia na lateks
Osłonka ochronna igły w prezentowanym strzykawce zawiera gumę z wysuszonego naturalnego lateksu, co może powodować reakcje alergiczne u osób wrażliwych na lateks.
Stosowanie w czasie ciąży lub karmienia piersią.
Ciąża.
Obecnie doświadczenie kliniczne w stosowaniu leku u kobiet w ciąży jest ograniczone. Badania na zwierzętach nie są wystarczające, aby ocenić wpływ na przebieg ciąży, rozwój embrionalny i płodowy, poród oraz rozwój poporodowy ze względu na ograniczoną ekspozycję. Dlatego Arixtra® nie powinna być stosowana u kobiet w ciąży, z wyjątkiem przypadków, gdy oczekiwana korzyść dla matki przewyższa potencjalne ryzyko dla płodu.
Karmienie piersią.
Arixtra® wydzielana jest z mlekiem szczurów, ale nie wiadomo, czy lek przenika do mleka kobiet. Dlatego karmienie piersią podczas leczenia tym lekiem nie jest zalecane. Jednakże wchłanianie doustne leku przez organizm dziecka jest mało prawdopodobne.
Niepłodność.
Brak danych dotyczących wpływu fondaparynyku na płodność człowieka. W badaniach na zwierzętach nie stwierdzono wpływu na płodność.
Możliwość wpływu na szybkość reakcji przy prowadzeniu samochodu lub innych maszyn.
Nie przeprowadzono badań wpływu leku na zdolność prowadzenia pojazdów i wykonywania prac wymagających zwiększonej uwagi, należy jednak wziąć pod uwagę możliwość wystąpienia działań niepożądanych ze strony układu nerwowego.
Sposób stosowania i dawki.
Leczenie ostrego zakrzepowego zapalenia żył głębokich i ostrej zakrzepowej embolii tętnicy płucnej.
Zalecaną dawką Arixtry® do podania podskórnie jest:
- 5 mg – dla pacjentów o masie ciała mniejszej niż 50 kg;
- 7,5 mg – dla pacjentów o masie ciała od 50 do 100 kg;
- 10 mg – dla pacjentów o masie ciała powyżej 100 kg.
Iniekcję podaje się raz na dobę. Leczenie powinno trwać co najmniej 5 dni i nie należy go kończyć wcześniej niż w momencie, gdy będzie można rozpocząć odpowiednią terapię doustnymi lekami przeciwzakrzepowymi (wartość międzynarodowego współczynnika znormalizowanego INR od 2 do 3). Terapię doustnymi lekami przeciwzakrzepowymi należy rozpocząć jak najszybciej, zazwyczaj w ciągu 72 godzin. Średnia długość stosowania leku w badaniach klinicznych wynosiła 7 dni; doświadczenie kliniczne dotyczące stosowania leku przez ponad 10 dni jest ograniczone.
Sposób stosowania.
Arixtrę® należy stosować w postaci głębokiej iniekcji podskórnej, pacjent powinien znajdować się w pozycji leżącej. Miejsca wstrzykiwania powinny być naprzemiennie lewą i prawą przednią boczną lub lewą i prawą tylną boczną ścianą brzucha. Aby uniknąć utraty leku, nie należy usuwać pęcherzyka powietrza z wstępnie napełnionego strzykawki przed iniekcją. Igłę należy wstrzyknąć na pełną długość pod kątem prostym do fałdu skóry zaciskanego między kciukiem a palcem wskazującym; fałd skóry należy trzymać zaciskany przez cały czas wstrzykiwania.
Arixtrę® należy stosować wyłącznie pod kontrolą lekarza.
Iniekcję podkórną należy wykonywać tak samo, jak w przypadku stosowania klasycznej strzykawki.
Przed zastosowaniem należy wizualnie ocenić roztwór pod kątem braku widocznych cząstek oraz zmiany zabarwienia.
Wstępnie napełnione strzykawki Arixtry® zostały opracowane z wykorzystaniem automatycznego systemu ochrony igły w celu zapobiegania urazom po wstrzyknięciu leku.
Instrukcja krok po kroku dotycząca stosowania leku Arixtra®
- Dokładnie umyj ręce wodą z mydłem i osusz ręcznikiem.
- Wyjmij strzykawkę z opakowania i upewnij się, że:
- data ważności leku nie upłynęła,
- roztwór jest przezroczysty i bezbarwny oraz nie zawiera cząstek,
- strzykawka nie była otwierana ani uszkodzona.
Wybierz miejsce w dolnej części okolicy brzusznej (brzucha), co najmniej 5 cm poniżej pępka (rysunek A). Kolejne zastrzyki wykonuj naprzemiennie po lewej i prawej stronie dolnej części brzucha. Zapobiegnie to nieprzyjemnym odczuciom w miejscu wstrzyknięcia. Jeśli nie można wykonać zastrzyku w dolną część okolicy brzusznej, skontaktuj się o pomoc z pielęgniarką lub lekarzem. |
|
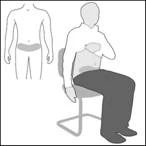 Rysunek A
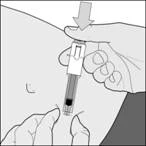
Rysunek A- Przetrzyj miejsce zastrzyku wacikiem nasączonym alkoholem.
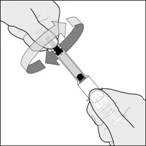
Wyrzuć nakrywkę igły. Ważna uwaga Nie dotykaj igły i nie pozwalaj, aby igła dotykała jakichkolwiek powierzchni przed wstrzyknięciem. Normalne jest, jeśli widzisz drobne pęcherzyki powietrza w tej strzykawce. Nie próbuj usuwać tych pęcherzyków powietrza przed wstrzyknięciem – możesz stracić część leku, jeśli to zrobisz. |
Rysunek B1 Rysunek B2 |
|
Rysunek C |
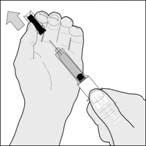
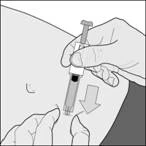
Wprowadź igłę na pełną długość pod kątem prostym do fałdu skóry. (rysunek D). |
Rysunek D |
|
|
| Rysunek E |
|
|
|
| Rysunek F |
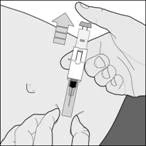
Nie wyrzucaj używanego strzykawki do odpadów komunalnych. Utylizuj ją zgodnie z instrukcją udzieloną przez lekarza lub farmaceutę.
Grupy pacjentów szczególnej uwagi.
Dzieci
Nie ustalono bezpieczeństwa i skuteczności stosowania Arixtry® u dzieci.
Pacjenci w wieku powyżej 75 lat
Nie ma potrzeby dostosowywania dawki u pacjentów w starszym wieku. Arixtrę® należy stosować z ostrożnością u pacjentów w starszym wieku, ponieważ z wiekiem zmniejsza się funkcja nerek (patrz sekcja „Szczególne ostrzeżenia i środki ostrożności dotyczące stosowania”).
Niewydolność nerek
Arixtrę® należy stosować z ostrożnością u pacjentów z umiarkowaną niewydolnością nerek (patrz sekcja „Szczególne ostrzeżenia i środki ostrożności dotyczące stosowania”). Brak doświadczenia w stosowaniu Arixtry® w leczeniu pacjentów o masie ciała powyżej 100 kg i z umiarkowaną niewydolnością nerek (klirens kreatyniny – 30–50 ml/min). U tej grupy pacjentów po podaniu dawki początkowej 10 mg na dobę może być konieczne zmniejszenie dawki do 7,5 mg na dobę, co wynika z właściwości farmakokinetycznych leku (patrz sekcja „Szczególne ostrzeżenia i środki ostrożności dotyczące stosowania”).
Arixtry® jest przeciwwskazana u pacjentów z ciężkim upośledzeniem czynności nerek (klirens kreatyniny < 30 ml/min) (patrz sekcja „Przeciwwskazania”).
Niewydolność wątroby
Nie ma potrzeby dostosowywania dawki u pacjentów z łagodnym i umiarkowanym upośledzeniem czynności wątroby. Arixtrę® należy stosować z ostrożnością u pacjentów z ciężkim upośledzeniem czynności wątroby, ponieważ nie prowadzono badań w tej grupie pacjentów (patrz sekcje „Szczególne ostrzeżenia i środki ostrożności dotyczące stosowania” oraz „Właściwości farmakologiczne. Farmakokinetyka”).
Dzieci.
Nie ustalono bezpieczeństwa i skuteczności stosowania Arixtry® u dzieci.
Przedawkowanie.
Przekroczenie zalecanych dawek Arixtry® może prowadzić do zwiększonego ryzyka wystąpienia krwawień. Nie istnieje znany antydot na fondaparynuksu.
W przypadku przedawkowania towarzyszącego powikłaniom krwotocznym należy przerwać leczenie i ustalić podstawową przyczynę krwawienia. Należy rozważyć wdrożenie odpowiedniej terapii, takiej jak hemostaza chirurgiczna, uzupełnienie utraty krwi, przetaczanie świeżej osocza krwi lub plazmafereza.
Niepożądane działania.
Najczęściej odnotowywanymi poważnymi niepożądanymi reakcjami podczas stosowania fondaparynuksu są powikłania krwotoczne (w różnych lokalizacjach, w tym rzadkie przypadki krwotoku wewnątrzczaszkowego/wewnątrzmózgowego oraz krwotoku do przestrzeni zaotrzewnowej). Fondaparynuks należy stosować z ostrożnością u pacjentów z podwyższonym ryzykiem wystąpienia krwotoku (patrz rozdział «Szczególne ostrzeżenia i środki ostrożności podczas stosowania»).
Bezpieczeństwo fondaparynuksu oceniano u 2517 pacjentów leczonych z powodu zakrzepicy żył głębokich i embolii płucnych, którym stosowano fondaparynuks średnio przez 7 dni. Najczęstszymi niepożądanymi reakcjami były powikłania krwotoczne (patrz rozdział «Szczególne ostrzeżenia i środki ostrożności podczas stosowania»).
Poniżej wymienione niepożądane reakcje pogrupowano według narządów i układów oraz częstości występowania. Częstość występowania sklasyfikowano jako bardzo często (≥ 1/10), często (≥ 1/100, < 1/10), rzadko (≥ 1/1000, < 1/100), bardzo rzadko (≥ 1/10000, < 1/1000), niezwykle rzadko (< 1/10000); niepożądane reakcje wymieniono w kolejności zmniejszającej się ciężkości.
| Układ narządów |
Objawy niepożądane(1) |
| Infekcje i inwazje |
Pojedyncze: infekcje rany pooperacyjnej. |
| Krew i układ limfatyczny |
Częste: krwawienie (krwawienie przewodu pokarmowego, hematuria, guzki krwawe, krwawienie z nosa, krwawe plwociny, krwawienie maciczno−vaginalne, krwawienie do stawów, krwawienie do oka, purpura, siniaki). Nieczęste: anemia, trombocytopenia, pojawienie się nieprawidłowych płytek krwi, zaburzenia krzepnięcia krwi. Pojedyncze: inne krwawienia (wątrobowe, zaotrzewnowe, wewnątrzczaszkowe/wewnątrz mózgowe), trombocytoza. |
| Układ odpornościowy |
Pojedyncze: reakcje alergiczne (w tym bardzo rzadkie przypadki obrzęku naczynioruchowego, reakcji anafilaktoidealnych/anafilaktycznych). |
| Przemiana materii i zaburzenia odżywiania |
Pojedyncze: hipokaliemia, podwyższenie stężenia azotu nienależącego do białek (Νπν)(2). |
| Układ nerwowy |
Nieczęste: ból głowy. Pojedyncze: niepokój, senność, zawroty głowy, zawroty, dezorientacja. |
| Układ sercowo-naczyniowy |
Pojedyncze: hipotensja tętnicza. |
| Układ oddechowy i narządy klatki piersiowej |
Pojedyncze: duszność, kaszel. |
| Układ pokarmowy |
Nieczęste: nudności, wymioty. Pojedyncze: dyspepsja, ból brzucha. |
| Układ wątrobowo-żółciowy |
Nieczęste: podwyższenie poziomu enzymów wątrobowych, zaburzenia funkcjonalnych testów wątrobowych. |
| Skóra i tkanki podskórne |
Nieczęste: zaczerwienienie, świąd. |
| Zaburzenia ogólne i w miejscu podania |
Nieczęste: ból, obrzęk, obrzęk obwodowy, gorączka, wydzielina z rany. Pojedyncze: ból w klatce piersiowej, ból nóg, zaczerwienienie, obrzęk narządów płciowych, uczucie ciepła, reakcja w miejscu podania, zwiększona zmęczalność, utrata przytomności. |
(1) Pojedyncze niepożądane zdarzenia nie były brane pod uwagę, z wyjątkiem przypadków, gdy miały one znaczenie medyczne.
(2) Νπν – azot niemiałkowy, taki jak azot mocznika, kwasu moczowego, aminokwasów itp.
W okresie po rejestracji leku zgłaszano rzadkie przypadki zapalenia żołądka, zaparć, biegunki i pojawienia się bilirubiny we krwi.
Zgłaszanie podejrzewanych działań niepożądanych
Zgłaszanie podejrzewanych działań niepożądanych po rejestracji leku jest ważne. Umożliwia to ciągłe monitorowanie stosunku korzyści do ryzyka związanego z lekiem. Osoby pracujące w zawodach medycznych proszone są o zgłaszanie podejrzewanych działań niepożądanych.
Okres ważności. 3 lata.
Warunki przechowywania.
Przechowywać w temperaturze nie wyższej niż 25 °C. Przechowywać w miejscu niedostępnym dla dzieci.
Niezgodność.
Arixtra® nie należy mieszać z innymi lekami, ponieważ nie przeprowadzono badań dotyczących ich wzajemnej zgodności.
Opakowanie.
Po 10 wstępnie wypełnionych strzykawek z automatycznym systemem bezpieczeństwa w tekturowym pudełku.
Kategoria wydania. Na receptę.
Producent.
Aspen Noter Dame de Bondeville
Miejsce położenia producenta oraz jego adres siedziby.
1, rue de l’Abbaye, 76960 Noter Dame de Bondeville, Francja