Actemra®
Ukraina
Spis treści
INSTRUKCJA dot. stosowania leku Actemra® (Actemra®)
Skład:
substancja czynna: tocylizumab;
1 prezentuje wypełniony strzykawka zawiera 162 mg/0,9 ml tocylizumabu;
substancje pomocnicze: polisorbat 80, chlorowodorek L-argininy, L-metionina, L-histydyna, monohydrat chlorowodoru L-histydyny, woda do wstrzykiwań
lub
polisorbat 80, L-arginina, chlorowodorek L-argininy, L-metionina, L-histydyna, monohydrat chlorowodoru L-histydyny, woda do wstrzykiwań.
Postać farmaceutyczna. Roztwór do wstrzykiwań.
Główne właściwości fizykochemiczne: ciecz od przezroczystej bezbarwnej do żółtawej, silnie opalizująca, o stopniu opalizacji nie przekraczającym 30,0 jednostek turbidymetrycznych formazynowych, zabarwiona nie intensywniej niż wzorzec Y4.
Grupa farmakoterapeutyczna.
Leki immunosupresyjne, inhibitory interleukiny.
Kod ATC L04A C07.
Właściwości farmakologiczne.
Farmakodynamika.
Tocylizumab – rekombinowany humanizowany przeciwciało monoklonalne z podklasy immunoglobulin G1 (IgG1), działające na rozpuszczalne i błonowe receptory interleukiny-6 (IL-6) człowieka.
Mechanizm działania
Tocylizumab wiąże się specyficznie zarówno z rozpuszczalnymi, jak i błonowymi receptorami IL-6 (sIL-6R i mIL-6R). Wykazano, że tocylizumab hamuje sygnalizację pośredniczoną przez sIL-6R i mIL-6R. IL-6 jest wielofunkcyjnym cytokinem prozapalnym, produkowanym przez różne typy komórek, w tym limfocyty T i B, monocyty oraz fibroblasty. IL-6 uczestniczy w różnych procesach fizjologicznych, takich jak stymulacja sekrecji immunoglobulin, aktywacja limfocytów T, stymulacja produkcji białek ostrej fazy w wątrobie oraz stymulacja hematopoezy. IL-6 bierze udział w patogenezie różnych chorób, w tym chorób zapalnych, osteoporozy i nowotworów.
Właściwości farmakodynamiczne
W badaniach klinicznych leku Actemra® obserwowano szybkie zmniejszenie stężenia białka C-reaktywnego (CRP), szybkości osiadania erytrocytów (SOE), surowiczego amyloidu A (SAA) oraz fibrynogenu. Zgodnie z wpływem na reagenty ostrej fazy, leczenie lekiem Actemra® wiązało się ze zmniejszeniem liczby płytek krwi w granicach normy. Obserwowano wzrost stężenia hemoglobiny w wyniku działania leku Actemra®, co wiązało się z osłabieniem wpływu IL-6 na produkcję hepcydyny i, w konsekwencji, z zwiększeniem dostępności żelaza. U pacjentów leczonych lekiem Actemra® obniżenie stężenia CRP do wartości normy obserwowano już w drugim tygodniu leczenia i utrzymywało się ono przez cały okres terapii.
W badaniu klinicznym leczenia tętnicy żółtaczkowej (WA28119) obserwowano podobne szybkie obniżenie stężenia CRP i SOE oraz nieznaczny wzrost średniego stężenia hemoglobiny w krwi.
U zdrowych ochotników, którym podawano lek Actemra® w dawkach od 2 do 28 mg/kg dożylnie oraz od 81 do 162 mg podskórnie, bezwzględna liczba neutrofili spadała do najniższego poziomu w ciągu 2–5 dnia po podaniu. Następnie liczba neutrofili wracała do poziomu wyjściowego, co zależało od dawki.
Pacjenci po podaniu leku Actemra® wykazują, w porównaniu ze zdrowymi osobami, obniżoną bezwzględną liczbę neutrofili.
Skuteczność leku Actemra® po podaniu podskórnym w łagodzeniu objawów reumatoidalnego zapalenia stawów (RZS) oraz odpowiedzi radiologicznej oceniano w dwóch randomizowanych, podwójnie ślepych, kontrolowanych, wieloośrodkowych badaniach. Do badania I (PS-I) zakwalifikowano pacjentów w wieku powyżej 18 roku życia z umiarkowanym lub ciężkim aktywnym RZS, zdiagnozowanym zgodnie z kryteriami Amerykańskiego Kolegium Reumatologii (ACR), którzy na początku badania mieli co najmniej 4 stawy bolesne i 4 stawy z obrzękiem. Wszyscy pacjenci otrzymywali podstawową niobiologiczną terapię chorobomodyfikującymi lekami przeciwzapalnymi (LZP). Do badania II (PS-II) zakwalifikowano pacjentów w wieku powyżej 18 roku życia z umiarkowanym lub ciężkim aktywnym RZS, zdiagnozowanym zgodnie z kryteriami ACR, którzy na początku badania mieli co najmniej 8 stawów bolesnych i 6 stawów z obrzękiem.
Zmiana z dożylnego podawania 8 mg/kg co 4 tygodnie na podawanie podskórne 162 mg raz w tygodniu wpływa na ekspozycję na lek u pacjenta. Stopień zmiany zależy od masy ciała pacjenta (zwiększony u pacjentów o niskiej masie ciała i zmniejszony u pacjentów o dużej masie ciała), ale efekt kliniczny odpowiada temu obserwowanemu u pacjentów po podaniu dożylnym.
Skuteczność i bezpieczeństwo leku Actemra® po podaniu podskórnym w tętnicy żółtaczkowej (GCA) oceniano w randomizowanym, wieloośrodkowym, placebo-kontrolowanym badaniu klinicznym WA28119.
Wszyscy pacjenci otrzymywali podstawową terapię glikokortykosteroidową (prednizolonem).
Stwierdzono istotny statystycznie efekt leczenia lekiem Actemra® w porównaniu z placebo w osiągnięciu trwałej remisji bezsterydowej w 52. tygodniu leczenia lekiem Actemra® w połączeniu z stopniowym zmniejszaniem dawki prednizolonu w ciągu 26 tygodni, w porównaniu z placebo w połączeniu ze stopniowym zmniejszaniem dawki prednizolonu w ciągu 26 tygodni oraz z placebo ze stopniowym zmniejszaniem dawki prednizolonu w ciągu 52 tygodni.
Farmakokinetyka.
Farmakokinetyka leku Actemra® charakteryzuje się nieliniową eliminacją, która stanowi połączenie eliminacji liniowej i eliminacji według kinetyki Michaelisa-Mentena. Nieliniowa część eliminacji leku Actemra® prowadzi do wzrostu ekspozycji większego niż proporcjonalny do dawki. Parametry farmakokinetyczne leku Actemra® nie zmieniają się w czasie. Ze względu na zależność całkowitego klirensu od stężenia leku Actemra® w surowicy, okres półtrwania leku również zależy od stężenia i zmienia się w zależności od poziomu stężenia w surowicy. Analiza farmakokinetyczna populacyjna w każdej populacji pacjentów, przetestowana do tej pory, wykazuje brak związku między pozornym klirensiem a obecnością przeciwciał przeciwko lekowi.
Reumatoidalne zapalenie stawów (RZS)
Podanie dożylne
Parametry farmakokinetyczne leku Actemra® oceniano za pomocą analizy populacyjnej farmakokinetyki na podstawie danych 3552 pacjentów z reumatoidalnym zapaleniem stawów, którzy otrzymywali lek Actemra® w dawce 4 lub 8 mg/kg w postaci jednogodzinnej infuzji co 4 tygodnie przez 24 tygodnie lub 162 mg podskórnie raz w tygodniu lub raz na 2 tygodnie przez 24 tygodnie.
Parametry (przewidywana średnia ± odchylenie standardowe (SD)), które oszacowano dla dawki leku Actemra® 8 mg/kg podawanej co 4 tygodnie: pole pod krzywą „stężenie–czas” w stanie stacjonarnym (AUC) wynosiło 38 000 ± 13 000 godz•μg/ml, minimalne stężenie (Cmin) wynosiło 15,9 ± 13,1 μg/ml, a maksymalne (Cmax) – 182 ± 50,4 μg/ml. Współczynniki akumulacji dla AUC i Cmax były niewielkie – odpowiednio 1,32 i 1,09. Współczynnik akumulacji był większy dla Cmin (2,49), co było oczekiwane z powodu wpływu nieliniowego klirensu przy niższych stężeniach. Stan stacjonarny dla Cmax osiągano po pierwszym podaniu, a dla AUC i Cmin odpowiednio po 8 i 20 tygodniach leczenia. AUC, Cmin i Cmax leku Actemra® wzrastały wraz ze wzrostem masy ciała pacjenta. Przy masie ciała ≥ 100 kg przewidywane średnie (± SD) AUC, Cmin i Cmax leku Actemra® w stanie stacjonarnym wynosiły odpowiednio 50 000 ± 16 800 μg•godz/ml, 24,4 ± 17,5 μg/ml i 226 ± 50,3 μg/ml, co przekracza średnie wartości ekspozycji w populacji pacjentów (tj. pacjentów o dowolnej masie ciała) podane powyżej. Krzywa „dawka–odpowiedź” dla tocylizumabu wypłaszcza się przy wyższej ekspozycji, co prowadzi do mniejszego przyrostu skuteczności przy każdym wzroście stężenia leku Actemra®, w ten sposób, że u pacjentów otrzymujących lek Actemra® w dawce > 800 mg nie stwierdzono klinicznie istotnego wzrostu skuteczności. Dlatego nie zaleca się stosowania dawki leku Actemra® przekraczającej 800 mg do infuzji (patrz sekcja „Sposób stosowania i dawki”).
Rozkład
U pacjentów z RZS objętość rozkładu centralna wynosiła 3,72 l, objętość rozkładu obwodowego – 3,35 l, w konsekwencji objętość rozkładu w stanie stacjonarnym – 7,07 l.
Eliminacja
Po podaniu dożylnej lek Actemra® jest usuwany z krążenia ogólnoustrojowego w dwóch etapach. Całkowity klirens leku Actemra® zależy od stężenia i stanowi sumę klirensu liniowego i nieliniowego. Klirens liniowy został oszacowany jako parametr w analizie populacyjnej farmakokinetyki i wynosił 9,5 ml/godz. Nieliniowy klirens, zależny od stężenia, ma największe znaczenie przy niskich stężeniach leku Actemra®. Przy wyższych stężeniach leku Actemra® dominuje klirens liniowy z powodu nasycenia ścieżki nieliniowego klirensu.
Okres półtrwania (t1/2) zależy od stężenia. W stanie stacjonarnym po podaniu dawki leku 8 mg/kg co 4 tygodnie efektywny okres półtrwania t1/2 zmniejszał się równolegle do spadku stężenia w przedziale od 18 do 6 dni.
Liniowość
Parametry farmakokinetyczne leku Actemra® nie zmieniają się w czasie. Obserwowano wzrost AUC i Cmin większy niż proporcjonalny do dawki dla dawek 4 i 8 mg/kg co 4 tygodnie. Cmax wzrastało wprost proporcjonalnie do wzrostu dawki. W stanie stacjonarnym obliczone AUC i Cmin były odpowiednio 3,2-krotnie i 30-krotnie wyższe przy dawce 8 mg/kg w porównaniu z dawką 4 mg/kg.
Podanie podskórne
Parametry farmakokinetyczne leku Actemra® określono za pomocą analizy populacyjnej farmakokinetyki na podstawie danych 3552 pacjentów z RZS, którzy otrzymywali podskórnie lek Actemra® w dawce 162 mg raz w tygodniu, 162 mg raz na 2 tygodnie lub 4 lub 8 mg/kg dożylnie co 4 tygodnie przez 24 tygodnie.
Parametry farmakokinetyczne leku Actemra® nie zmieniają się w czasie. Dla leku Actemra® w dawce 162 mg raz w tygodniu przewidywana średnia (± SD) AUC1 tydzień, Cmin i Cmax w stanie stacjonarnym wynosiły odpowiednio 7970 ± 3432 μg•godz/ml, 43,0 ± 19,8 μg/ml i 49,8 ± 21,0 μg/ml. Współczynniki akumulacji dla AUC, Cmin i Cmax wynosiły odpowiednio 6,32, 6,30 i 5,27. Stan stacjonarny dla AUC, Cmax i Cmin osiągnięto po 12 tygodniach leczenia.
Dla leku Actemra® w dawce 162 mg raz na 2 tygodnie przewidywana średnia (± SD) AUC2 tydzień, Cmin i Cmax w stanie stacjonarnym wynosiły odpowiednio 3430 ± 2660 μg•godz/ml, 5,7 ± 6,8 μg/ml i 13,2 ± 8,8 μg/ml. Współczynniki akumulacji dla AUC, Cmin i Cmax wynosiły odpowiednio 2,67, 6,02 i 2,12. Stan stacjonarny dla AUC i Cmin osiągnięto po 12 tygodniach, a dla Cmax – po 10 tygodniach leczenia.
Absorpcja
U pacjentów z RZS po podaniu podskórnym czas do osiągnięcia maksymalnego stężenia w surowicy (tmax) wynosił 2,8 dnia. Biodostępność postaci leku do podania podskórnego wynosiła 79%.
Eliminacja
Po podaniu leku w formie iniekcji podskórnej efektywny okres półtrwania (t1/2) wynosi do 13 dni przy dawkowaniu 162 mg raz w tygodniu u pacjentów z RZS w stanie stacjonarnym.
Uogólnione zapalenie stawów młodzieńcze idiopatyczne (uZSMI)
Podanie podskórne
Farmakokinetykę leku Actemra® u pacjentów z uZSMI określono za pomocą analizy populacyjnej farmakokinetyki, która obejmowała 140 pacjentów otrzymujących leczenie lekiem w dawce 8 mg/kg dożylnie co 2 tygodnie (pacjenci z masą ciała ≥ 30 kg), 12 mg/kg dożylnie co 2 tygodnie (pacjenci z masą ciała < 30 kg), 162 mg podskórnie co tydzień (pacjenci z masą ciała ≥ 30 kg), 162 mg podskórnie co 10 dni lub co 2 tygodnie (pacjenci z masą ciała < 30 kg).
Dane dotyczące ekspozycji po podaniu podskórnym leku pacjentom z uZSMI w wieku do 2 lat i masą ciała mniejszą niż 10 kg są ograniczone.
Minimalna masa ciała przy leczeniu lekiem Actemra® do podania podskórnego powinna wynosić co najmniej 10 kg (patrz sekcja „Sposób stosowania i dawki”).
Tabela 1
Przewidywane parametry farmakokinetyczne (średnie ± SD) w stanie stacjonarnym po podaniu podskórnym pacjentom z uZSMI
| Farmakokinetyczny parametr leku Actemra® |
162 mg raz w tygodniu przy masie ciała ≥ 30 kg |
162 mg raz na 2 tygodnie przy masie ciała < 30 kg |
| Cmax (μg/ml) |
99,8 ± 46,2 |
134 ± 58,6 |
| Cmin (μg/ml) |
79,2 ± 35,6 |
65,9 ± 31,3 |
| Cmean (μg/ml) |
91,3 ± 40,4 |
101 ± 43,2 |
| Cmax akumulacji |
3,66 |
1,88 |
| Cmin akumulacji |
4,39 |
3,21 |
| Cmean akumulacji lub AUCτ* |
4,28 |
2,27 |
* τ – 1 tydzień lub 2 tygodnie dla dwóch trybów podania podskórnie.
Cmean – średnie stężenie.
Po podaniu podskórnym stan równowagi osiągany był u około 90 % pacjentów do 12 tygodnia leczenia w schematach dawkowania 162 mg raz w tygodniu oraz raz na 2 tygodnie.
Absorpcja
U pacjentów z sJIA okres półwchłaniania po podaniu podskórnym wynosił około 2 dni. Biologiczna dostępność postaci leku do podania podskórnego u pacjentów z sJIA wynosiła 95 %.
Rozkład
U dzieci z sJIA objętość rozkładu centralna wynosiła 1,87 l, objętość rozkładu obwodowego – 2,14 l, co dawało objętość rozkładu w stanie równowagi – 4,01 l.
Eliminacja
Całkowity klirens tocylizumabu zależy od stężenia i jest sumą klirensu liniowego oraz nieliniowego. Klirens liniowy został oszacowany jako parametr w analizie farmakokinetyki populacyjnej i wynosił 5,7 ml/godz u dzieci z sJIA. Po podaniu podskórnym efektywny t1/2 leku Actemra® u pacjentów z sJIA w stanie równowagi wynosił do 14 dni w obu schematach dawkowania: 162 mg raz w tygodniu oraz raz na 2 tygodnie.
Poliarticularny młodzieńczy idiopatyczny zapalenie stawów (pJIA)
Podanie podskórne
Farmakokinetyka leku Actemra® u pacjentów z pJIA była określana za pomocą analizy farmakokinetyki populacyjnej, która objęła 237 pacjentów leczonych lekiem w dawce 8 mg/kg dożylnie co 4 tygodnie (pacjenci o masie ciała ≥ 30 kg), 10 mg/kg dożylnie co 4 tygodnie (pacjenci o masie ciała < 30 kg), 162 mg podskórnie co 2 tygodnie (pacjenci o masie ciała ≥ 30 kg), 162 mg podskórnie co 3 tygodnie (pacjenci o masie ciała < 30 kg).
Tabela 2
Prognozowane parametry farmakokinetyczne (średnie ± SD) w stanie równowagi po podaniu podskórnym pacjentom z pJIA
| Farmakokinetyczny parametr leku Actemra® |
162 mg raz na 2 tygodnie przy masie ciała ≥ 30 kg |
162 mg raz na 3 tygodnie przy masie ciała < 30 kg |
| Cmax (μg/ml) |
29,4 ± 13,5 |
75,5 ± 24,1 |
| Cmin (μg/ml) |
11,8 ± 7,08 |
18,4 ± 12,9 |
| Cmean (μg/ml) |
21,7 ± 10,4 |
45,5 ± 19,8 |
| Stosunek Cmax w stanie stacjonarnym |
1,72 |
1,32 |
| Stosunek Cmin w stanie stacjonarnym |
3,58 |
2,08 |
| Stosunek Cmean w stanie stacjonarnym lub AUCτ* |
2,04 |
1,46 |
* τ – 2 lub 1 tydzień dla dwóch trybów podania podskórnie.
Po podaniu dożylnym stan równowagi osiągany był u około 90 % pacjentów do 12 tygodnia stosowania przy dawkowaniu 10 mg/kg (masa ciała < 30 kg) i do 16 tygodnia stosowania przy dawkowaniu 8 mg/kg (masa ciała ≥ 30 kg). Po podaniu podskórnym stan równowagi osiągany był u około 90 % pacjentów do 12 tygodnia stosowania przy obu schematach dawkowania: 162 mg co 2 tygodnie lub co 3 tygodnie.
Absorpcja
U pacjentów z pJIA okres półwchłaniania po podaniu podskórnym wynosił około 2 dni, a biodostępność – 96 %.
Rozkład
U dzieci z pJIA objętość rozkładu centralna wynosiła 1,97 l, objętość rozkładu obwodowa – 2,03 l, w konsekwencji objętość rozkładu w stanie równowagi – 4,0 l.
Eliminacja
Analiza populacyjna farmakokinetyki u pacjentów z pJIA wykazała wpływ masy ciała na klarans liniowy. W związku z tym należy uwzględnić zależność dawki leku od masy ciała (patrz tabela 2).
Po podaniu podskórnym pacjentom z pJIA efektywny okres półtrwania leku Actemra® wynosi do 10 dni, gdy masa ciała < 30 kg (162 mg podskórnie, raz na 3 tygodnie) oraz do 7 dni, gdy masa ciała ≥ 30 kg (162 mg podskórnie, raz na 2 tygodnie), w okresie między dawkowaniem w stanie równowagi. Po podaniu dożylnym eliminacja tocyliczumabu z krążenia odbywa się w dwóch fazach. Całkowity klarans tocyliczumabu zależy od stężenia i jest sumą klaransu liniowego oraz nieliniowego. Klarns liniowy określono w trakcie analizy populacyjnej farmakokinetyki i wynosił 6,25 ml/godz. Klarns nieliniowy, zależny od stężenia, odgrywa główną rolę przy niskich stężeniach tocyliczumabu. Przy wyższych stężeniach leku Actemra® przeważa klarans liniowy z powodu nasycenia drogi eliminacji nieliniowej.
Gigantocellularny zapalenie tętnic (GCA)
Podanie podskórne
Farmakokinetyka leku Actemra® u chorych z GCA została określona przy użyciu modelu populacyjnej farmakokinetyki oraz zbioru danych analizy 149 pacjentów z GCA, którzy otrzymywali 162 mg leku podskórnie co tydzień lub 162 mg podskórnie co dwa tygodnie. Opracowany model miał taką samą strukturę, jak wcześniej opracowany model populacyjnej farmakokinetyki oparty na danych pacjentów z RA (patrz tabela 3).
Tabela 3
Prognozowane parametry farmakokinetyczne – średnia ± odchylenie standardowe w stanie równowagi po podaniu podskórnym dawki przy GCA
| Farmakokinetyczny parametr tocylicumabu |
Podskórnie |
|
| 162 mg raz na 2 tygodnie |
162 mg co tydzień |
|
| Cmax (μg/ml) |
19,3 ± 12,8 |
73 ± 30,4 |
| Cmin (μg/ml) |
11,1 ± 10,3 |
68,1 ± 29,5 |
| Cmean (μg/ml) |
16,2 ± 11,8 |
71,3 ± 30,1 |
| Cmax kumulacji |
2,18 |
8,88 |
| Cmin kumulacji |
5,61 |
9,59 |
| Cmean kumulacji lub AUCτ* |
2,81 |
10,91 |
Stacjonarny profil po podawaniu leku Actemra® co tydzień był prawie stały, z bardzo niewielkimi wahaniem między wartościami minimalnymi i maksymalnymi, podczas gdy podawanie Actemra® co dwa tygodnie charakteryzowało się znacznymi wahaniem stężenia. Osiągnięto około 90 % stanu stacjonarnego (AUCτ) w 14. tygodniu przy podawaniu co dwa tygodnie i w 17. tygodniu w grupie otrzymującej lek co tydzień.
Na podstawie obecnej charakterystyki farmakokinetycznej, stężenie minimalne leku Actemra® w stanie stacjonarnym jest o 50 % wyższe w tej populacji w porównaniu ze średnią koncentracją w dużej grupie danych populacyjnych z reumatoidalnym zapaleniem stawów (RA). Przyczyna tych różnic jest nieznana. Różnice farmakokinetyczne nie są towarzyszone wyraźnymi różnicami w parametrach farmakodynamicznych, dlatego znaczenie kliniczne tych różnic jest nieznane.
U pacjentów z zapaleniem tętnic skroniowych (GCA) wyższy poziom ekspozycji obserwowano u chorych o niższej masie ciała. W przypadku zastosowania dawki 162 mg co tydzień, średnie stężenie w stanie stacjonarnym (Cavg) było o 51 % wyższe u pacjentów o masie ciała poniżej 60 kg w porównaniu z pacjentami o masie ciała od 60 do 100 kg. Dla schematu dawkowania 162 mg co dwa tygodnie, średnie stężenie w stanie stacjonarnym było o 129 % wyższe u pacjentów o masie ciała poniżej 60 kg w porównaniu z pacjentami o masie ciała od 60 do 100 kg. Dane dotyczące pacjentów o masie ciała powyżej 100 kg są ograniczone (n = 7).
Absorpcja
Po podaniu podskórnie u pacjentów z GCA okres półtrwania wchłaniania (t½) wynosił około 4 dni. Bioavailability postaci podskórnej leku wynosiła 0,8. Średnie wartości Tmax wynosiły 3 dni po tygodniowej dawce leku Actemra® i 4,5 dnia po podaniu tocylicumabu raz na dwa tygodnie.
Rozkład
U pacjentów z GCA objętość rozkładu centralna wynosiła 4,09 l, objętość rozkładu obwodowa – 3,37 l, co dawało objętość rozkładu w stanie stacjonarnym wynoszącą 7,46 l.
Eliminacja
Całkowity klirens leku Actemra® zależy od stężenia i jest sumą klirensu liniowego oraz nieliniowego. Klirens liniowy szacowano jako parametr w analizie farmakokinetyki populacyjnej i wynosił 6,7 ml/godz. u pacjentów z GCA.
U pacjentów z GCA efektywny okres półtrwania (t½) leku Actemra® w stanie stacjonarnym wynosił 18,3–18,9 dnia przy dawce 162 mg co tydzień oraz 4,2–7,9 dnia przy dawce 162 mg raz na dwa tygodnie. Przy wysokim stężeniu w osoczu, gdy całkowity klirens leku Actemra® dominował nad klirensiem liniowym, efektywny t½ wynosił około 32 dni, co oszacowano na podstawie parametrów populacyjnych.
Grupy specjalne
Pacjenci z zaburzeniem funkcji nerek. Oficjalnych badań farmakokinetyki leku Actemra® u pacjentów z zaburzeniem funkcji nerek nie przeprowadzono. Większość pacjentów z RA i GCA w badaniach analizy farmakokinetyki populacyjnej miała prawidłową funkcję nerek lub łagodne zaburzenie funkcji nerek (szacowany klirens kreatyniny według wzoru Cockcrofta-Gaulta), co nie wpływało na farmakokinetykę leku Actemra®.
W badaniu GCA około jedna trzecia pacjentów miała początkowo umiarkowane zaburzenie funkcji nerek (szacowany klirens kreatyniny 30–59 ml/min). U tych pacjentów nie zaobserwowano żadnego wpływu na ekspozycję na lek Actemra®.
Pacjenci z łagodnym lub umiarkowanym zaburzeniem funkcji nerek nie wymagają dostosowania dawki.
Pacjenci z zaburzeniem funkcji wątroby. Oficjalnych badań farmakokinetyki leku Actemra® u pacjentów z zaburzeniem funkcji wątroby nie przeprowadzono.
Płeć, rasa, wiek pacjenta. Analiza farmakokinetyki populacyjnej u pacjentów z RA i GCA wykazała, że wiek, płeć i rasa nie wpływają na farmakokinetykę leku Actemra®.
Wyniki analizy farmakokinetyki populacyjnej zastosowania leku u pacjentów z sJIA lub pJIA potwierdzają, że tylko masa ciała jest niezależną zmienną (kowariantą), która istotnie wpływa na farmakokinetykę leku Actemra®, w szczególności na jego eliminację i absorpcję. Dlatego należy uwzględnić zależność dawki leku od masy ciała.
Charakterystyki kliniczne.
Wskazania.
Rzutawe zapalenie stawów
Zastosowanie leku Actemra® w połączeniu z metotreksatem (MT) jest wskazane w leczeniu:
- ciężkiego, aktywnego i postępującego rzutawego zapalenia stawów (RZS) u dorosłych pacjentów, którzy wcześniej nie byli leczeni metotreksatem;
- rzutawego zapalenia stawów (RZS) o umiarkowanym i ciężkim nasileniu u dorosłych pacjentów, u których stwierdzono niewystarczającą odpowiedź lub nietolerancję wcześniejszej terapii jednym lub więcej lekami modyfikującymi przebieg choroby (LZPC) lub inhibitorem czynnika martwicy nowotworów (TNF).
W przypadku takich pacjentów lek Actemra® może być stosowany jako monoterapia, gdy metotreksat jest nietolerowany lub gdy kontynuowanie leczenia metotreksatem jest nieuzasadnione.
W połączeniu z metotreksatem lek Actemra® hamuje postępowanie destrukcyjnych zmian stawowych, potwierdzonych radiologicznie, oraz poprawia funkcję fizyczną.
Poliartryt młodzieńczy idiopatyczny
Lek Actemra® w połączeniu z metotreksatem (MT) jest wskazany w leczeniu poliartrytu młodzieńczego idiopatycznego (pMII; czynnik reumatoidalny – dodatni lub ujemny oraz rozszerzony oligoartryt) u pacjentów od 2. roku życia, u których stwierdzono niewystarczającą odpowiedź na wcześniejszą terapię metotreksatem. W przypadku nietolerancji metotreksatu lub gdy kontynuowanie leczenia metotreksatem jest nieuzasadnione, lek Actemra® może być stosowany jako monoterapia.
Zapalenie tętnicy posoczniczej (ZTP)
Lek Actemra® jest wskazany w leczeniu zapalenia tętnicy posocznicowej (ZTP) u dorosłych pacjentów.
Systemowy młodzieńczy idiopatyczny artryt
Lek Actemra® jest wskazany w leczeniu aktywnego systemowego młodzieńczego idiopatycznego zapalenia stawów (sMII) u pacjentów od 1. roku życia, u których stwierdzono niewystarczającą odpowiedź na wcześniejszą terapię niesteroidowymi lekami przeciwzapalnymi (NSAID) i kortykosteroidami doustnymi. Lek Actemra® może być stosowany zarówno jako monoterapia (w przypadku nietolerancji MT lub gdy kontynuowanie leczenia MT jest nieuzasadnione), jak i w połączeniu z MT.
Przeciwwskazania.
Nadwrażliwość na tocyliczumab lub którykolwiek inny składnik leku.
Aktywne, ciężkie infekcje.
Interakcje z innymi lekami i inne rodzaje interakcji.
Badania interakcji przeprowadzono wyłącznie na dorosłych pacjentach.
Jednoczesne jednorazowe podanie leku Actemra® w dawce 10 mg/kg oraz metotreksatu w dawce 10–25 mg raz w tygodniu nie wpływało istotnie na ekspozycję na metotreksat.
Analiza farmakokinetyki populacyjnej nie wykazała wpływu metotreksatu, niesteroidowych leków przeciwzapalnych (NSAID) ani kortykosteroidów na klirens leku Actemra® u pacjentów z RZS. U pacjentów z ZTP nie obserwowano żadnego wpływu kumulacji dawki kortykosteroidów na ekspozycję na lek Actemra®.
Ekspresja wątrobowych enzymów CYP450 jest hamowana przez cytokiny, takie jak IL-6, które stymulują przewlekłe zapalenie. W związku z tym podczas terapii silnymi lekami hamującymi działanie cytokin (np. lekiem Actemra®) ekspresja enzymów CYP450 może być zaburzona.
Badania in vitro prowadzone na kulturach hepatocytów ludzkich wykazały, że IL-6 powodował obniżenie ekspresji enzymów CYP1A2, CYP2C9, CYP2C19 i CYP3A4. Stosowanie leku Actemra® normalizuje ekspresję tych izoenzymów.
W badaniu przeprowadzonym na pacjentach z RZS stężenie symwataryny (substrat CYP3A4) u pacjentów z RZS tydzień po jednorazowym podaniu tocyliczumabu było o 57% niższe niż stężenie symwataryny u zdrowych ochotników lub nieco wyższe.
Na początku lub na końcu terapii tocyliczumabem należy dokładnie obserwować stan pacjentów otrzymujących leki metabolizowane przez izoenzymy CYP450 3A4, 1A2 lub 2C9 (w indywidualnie dobranych dawkach, np. metylprednizolon, dexametazon (z możliwością wystąpienia zespołu odstawienia glikokortykosteroidów doustnych), atorwastatyna, blokery kanałów wapniowych, teofilina, warfaryna, fenprokumon, fenytoina, cyklosporyna lub benzodiazepiny). Może być konieczne zwiększenie dawki tych leków w celu zapewnienia skuteczności terapii. Ze względu na długi okres półtrwania (t1/2) tocyliczumabu jego wpływ na aktywność enzymów CYP450 może utrzymywać się przez kilka tygodni po zakończeniu leczenia.
Szczególne wskazania dotyczące stosowania.
Leku Actemra® do podskórnej iniekcji nie należy stosować do wstrzykiwania dożylnego.
Lek Actemra® do podskórnej iniekcji nie jest wskazany do stosowania u dzieci z sJIA o masie ciała mniejszej niż 10 kg.
Monitorowanie stosowania
W celu poprawy śledzenia biologicznych leków, nazwa handlowa i numer serii zastosowanego leku powinny być wyraźnie wpisane w dokumentacji medycznej pacjenta.
Zakażenia
U pacjentów otrzymujących leki immunosupresyjne, w tym lek Actemra®, obserwowano przypadki ciężkich zakażeń (czasem zakończone śmiercią) (patrz dział „Efekty niepożądane”). Nie należy rozpoczynać leczenia lekiem Actemra® u pacjentów z aktywnymi chorobami zakaźnymi (patrz dział „Przeciwwskazania”). W przypadku rozwoju ciężkich zakażeń leczenie lekiem Actemra® należy przerwać do całkowitego ustąpienia zakażenia (patrz dział „Efekty niepożądane”). Osoby pracujące w ochronie zdrowia powinny ostrożnie przepisywać lek Actemra® pacjentom z nawracającymi lub przewlekłymi zakażeniami w wywiadzie, a także z chorobami współistniejącymi sprzyjającymi rozwojowi zakażeń (np. zeszostnienie, cukrzyca, choroba interpłucna).
Należy zachować szczególną ostrożność, aby w porę wykryć ciężkie choroby zakaźne u pacjentów otrzymujących leki immunomodulujące, takie jak Actemra®, ponieważ objawy ostrego stanu zapalnego mogą być stłumione z powodu hamowania reakcji ostrej fazy. Należy wziąć pod uwagę wpływ tocylicumabu na poziom C-reaktywnego białka, neutrofilów i objawy zakażenia podczas oceny możliwości rozwoju zakażenia u pacjenta. Pacjentów (w tym młodszych pacjentów z sJIA lub pJIA, którzy mogą mieć trudności w donoszeniu objawów) oraz rodziców/opiekunów pacjentów z sJIA lub pJIA należy poinformować o konieczności natychmiastowego skontaktowania się z lekarzem w przypadku wystąpienia jakichkolwiek objawów wskazujących na zakażenie, w celu szybkiej diagnostyki i wdrożenia odpowiedniego leczenia.
Gruźlica
Przed przepisaniem leku Actemra®, tak jak w przypadku innych leków biologicznych, należy przebadać wszystkich pacjentów pod kątem obecności utajonej gruźlicy. W przypadku wykrycia utajonej gruźlicy należy przeprowadzić standardowy kurs terapii antymikobakterialnej przed rozpoczęciem leczenia lekiem Actemra®. Lekarze powinni pamiętać o ryzyku fałszywie ujemnych wyników próby skórnego tuberkuliny i testu krwi na gamma-interferonową tuberkulinę, szczególnie u pacjentów ciężko chorych i pacjentów z niedoborem odporności.
Pacjentów oraz rodziców/opiekunów pacjentów z sJIA lub pJIA należy poinstruować, że w przypadku wystąpienia objawów, które mogą wskazywać na rozwój zakażenia gruźliczego (w tym przewlekłego kaszlu, wyczerpania/utraty masy ciała, subfebrilia) podczas lub po terapii lekiem Actemra®, należy skontaktować się z lekarzem.
Reaktywacja zakażeń wirusowych
Podczas stosowania leków biologicznych w leczeniu RA obserwowano reaktywację zakażeń wirusowych (np. wirusa zapalenia wątroby typu B). Pacjenci z dodatnim wynikiem badania przesiewowego na wirusa zapalenia wątroby byli wykluczeni z badań klinicznych leku Actemra®.
Powikłania zeszostnienia
U pacjentów leczonych lekiem Actemra® rzadko wystąpiły przypadki perforacji zeszostnienia jako powikłanie zeszostnienia (patrz dział „Efekty niepożądane”). Lek Actemra® należy stosować z ostrożnością u pacjentów z chorobą wrzodową przewodu pokarmowego lub zeszostnieniem w wywiadzie. Pacjentów z objawami wskazującymi na możliwe powikłane zeszostnienie (ból brzucha, krwawienie i/lub niejasne zmiany rytmu wypróżnień towarzyszone podwyższeniem temperatury ciała) należy natychmiast przebadać w celu wczesnego wykrycia zeszostnienia, które może być powiązane z perforacją przewodu pokarmowego.
Reakcje nadwrażliwości
Podczas stosowania leku Actemra® obserwowano ciężkie reakcje nadwrażliwości, w tym anafilaksję (patrz dział „Efekty niepożądane”). Takie reakcje mogą być cięższe lub śmiertelne u pacjentów, u których wcześniej wystąpiły reakcje nadwrażliwości podczas wcześniejszego leczenia lekiem Actemra®, nawet jeśli otrzymywali oni leki przedlekowe w postaci steroidów i leków przeciwhistaminowych. W przypadku wystąpienia reakcji anafilaktycznej lub innej ciężkiej reakcji nadwrażliwości należy natychmiast przerwać podawanie leku Actemra®, rozpocząć odpowiednie leczenie i bezpowrotnie odstawić leczenie tocylicumabem.
Aktywne choroby i zaburzenia funkcji wątroby
Leczenie lekiem Actemra®, szczególnie w połączeniu z metotreksatem, może być związane ze wzrostem aktywności transaminaz wątrobowych, dlatego należy ostrożnie przepisywać leczenie pacjentom z aktywnymi chorobami lub zaburzeniami funkcji wątroby (patrz działy „Sposób stosowania i dawki” oraz „Efekty niepożądane”).
Hepatotoksyczność
Podczas leczenia lekiem Actemra® często obserwowano przemijające lub okresowe, łagodne lub umiarkowane podwyższenie aktywności transaminaz wątrobowych (patrz dział „Efekty niepożądane”). Podczas stosowania potencjalnie hepatotoksycznych leków (np. metotreksatu) w połączeniu z lekiem Actemra® obserwowano zwiększenie częstości takiego wzrostu enzymów. W przypadku wskazań klinicznych należy rozważyć wykonanie innych badań funkcji wątroby, w tym oznaczenie poziomu bilirubiny.
Podczas stosowania leku Actemra® obserwowano ciężkie uszkodzenia wątroby wywołane przez lek, w tym ostre niewydolności wątroby, zapalenie wątroby i żółtaczkę (patrz dział „Efekty niepożądane”). Ciężkie uszkodzenia wątroby występowały po upływie czasu od 2 tygodni do ponad 5 lat od rozpoczęcia leczenia lekiem Actemra®. Zgłaszano przypadki niewydolności wątroby, które wymagały przeszczepienia wątroby. Pacjentom należy zalecić natychmiastowe skorzystanie z pomocy medycznej w przypadku wystąpienia objawów uszkodzenia wątroby.
Należy ostrożnie podejść do decyzji o rozpoczęciu leczenia lekiem Actemra® u pacjentów z poziomem alaninotransaminazy (ALT) lub asparaginianotransaminazy (AST) przekraczającym górny limit normy (GLN) o więcej niż 1,5 raza. Leczenie nie jest zalecane, gdy poziom ALT lub AST przekracza GLN więcej niż 5 razy.
U pacjentów z RA, GCA, młodzieńczym zapaleniem stawów wielostawowym i systematycznym młodzieńczym zapaleniem stawów idiopatycznym należy oznaczać poziomy ALT i AST co 4–8 tygodni przez pierwsze 6 miesięcy leczenia, a następnie kontrolować co 12 tygodni. Rekomendacje dotyczące dawkowania, w tym odstawienie leku Actemra® w zależności od aktywności transaminaz wątrobowych, podano w dziale „Sposób stosowania i dawki”. W przypadku wzrostu poziomu ALT lub AST powyżej 3–5 razy wyższego niż GLN należy przerwać leczenie lekiem.
U pacjentów z sJIA lub pJIA po drugiej dawce leku i dalej należy kontrolować poziomy ALT i AST zgodnie z właściwą praktyką kliniczną (patrz dział „Sposób stosowania i dawki”).
Zaburzenia krwi
Po leczeniu tocylicumabem w dawce 8 mg/kg w połączeniu z metotreksatem obserwowano zmniejszenie liczby neutrofili i płytek krwi (patrz dział „Efekty niepożądane”). U pacjentów wcześniej leczonych antagonistami czynnika martwicy nowotworu może występować zwiększony ryzyko rozwoju neutropenii.
U pacjentów z liczbą absolutną neutrofili (ANC) poniżej 2 × 10⁹/l, którzy wcześniej nie otrzymywali leczenia lekiem Actemra®, nie zaleca się rozpoczynania leczenia tym lekiem. Należy zachować ostrożność przy rozważaniu rozpoczęcia leczenia lekiem Actemra® u pacjentów z niskim poziomem płytek krwi (tj. przy liczbie płytek krwi poniżej 100 × 10³/μl). Nie zaleca się kontynuowania leczenia u pacjentów z ANC < 0,5 × 10⁹/l lub liczbą płytek krwi < 50 × 10³/μl.
Ciężka neutropenia może być związana ze zwiększonym ryzykiem wystąpienia ciężkich chorób zakaźnych, choć dane z badań klinicznych leku Actemra® nie wykazały jednoznacznego związku między obniżeniem liczby neutrofili a przypadkami rozwoju ciężkich chorób zakaźnych.
U pacjentów z RA i GCA liczbę neutrofili i płytek krwi należy kontrolować co 4–8 tygodni od momentu rozpoczęcia leczenia lekiem Actemra® i dalej zgodnie ze standardową praktyką kliniczną. Rekomendacje dotyczące dostosowania dawki w zależności od poziomu ANC i liczby płytek krwi znajdują się w dziale „Sposób stosowania i dawki”.
U pacjentów z sJIA lub pJIA po drugiej dawce leku i dalej należy kontrolować liczbę neutrofili i płytek krwi zgodnie z właściwą praktyką kliniczną (patrz dział „Sposób stosowania i dawki”).
Zmiany parametrów metabolizmu lipidów
Obserwowano wzrost parametrów metabolizmu lipidów (w tym całkowity cholesterol, lipoproteiny o niskiej gęstości (LDL), lipoproteiny o wysokiej gęstości (HDL), trójglicerydy) (patrz dział „Efekty niepożądane”) u pacjentów leczonych lekiem Actemra®. U większości pacjentów nie obserwowano wzrostu indeksu aterogennego, a podwyższone poziomy całkowitego cholesterolu odpowiadały na leczenie lekami obniżającymi poziom lipidów.
U wszystkich pacjentów należy ocenić parametry metabolizmu lipidów 4–8 tygodni po rozpoczęciu terapii lekiem Actemra®. W prowadzeniu pacjentów należy kierować się krajowymi wytycznymi dotyczącymi leczenia hiperlipidemii.
Zaburzenia neurologiczne
Lekarze powinni zachować szczególną czujność w celu wczesnego wykrycia objawów, które mogą wskazywać na rozwój chorób demielinizacyjnych układu nerwowego centralnego. Obecnie nieznana jest zdolność leku Actemra® do wywoływania chorób demielinizacyjnych układu nerwowego centralnego.
Nowotwory złośliwe
U pacjentów z reumatoidalnym zapaleniem stawów ryzyko wystąpienia nowotworów złośliwych jest zwiększone. Stosowanie leków immunomodulujących zwiększa ryzyko nowotworów złośliwych.
Szczepienia
Nie należy przeprowadzać szczepień żywymi i osłabionymi szczepionkami żywymi równocześnie z leczeniem lekiem Actemra®, ponieważ bezpieczeństwo takiego połączenia nie zostało ustalone.
W otwartym, randomizowanym badaniu u dorosłych pacjentów z RA, którzy otrzymywali lek Actemra® w połączeniu z metotreksatem, po szczepieniu 23-walentną szczepionką przeciwko pneumokokom i szczepionką przeciwgruźliczą, zaobserwowano skuteczną odpowiedź immunologiczną porównywalną z odpowiedzią u pacjentów przyjmujących tylko metotreksat. Zaleca się, aby przed rozpoczęciem leczenia lekiem Actemra® wszyscy pacjenci, szczególnie dzieci i pacjenci starsi, zostali zaszczepieni zgodnie z obowiązującym krajowym kalendarzem szczepień. Należy przestrzegać zalecanego odstępu czasowego (zgodnie z obowiązującymi wytycznymi dotyczącymi szczepień pacjentów leczonych lekami immunosupresyjnymi) między szczepieniem żywymi szczepionkami a rozpoczęciem terapii lekiem Actemra®.
Ryzyko zaburzeń układu sercowo-naczyniowego
Pacjenci z reumatoidalnym zapaleniem stawów z czynnikami ryzyka (np. nadciśnienie tętnicze, hiperlipidemia) mają zwiększone ryzyko zaburzeń układu sercowo-naczyniowego, które podlegają leczeniu w ramach standardowej terapii.
Stosowanie łączone z antagonistami czynnika martwicy nowotworu
Brak doświadczenia w jednoczesnym stosowaniu leku Actemra® i antagonistów czynnika martwicy nowotworu lub innych leków biologicznych w leczeniu pacjentów z reumatoidalnym zapaleniem stawów. Nie zaleca się jednoczesnego stosowania leku Actemra® z innymi lekami biologicznymi.
GCA
Monoterapii lekiem Actemra® nie należy stosować w leczeniu ostrych zaostrzeń, ponieważ skuteczność w tych warunkach nie została ustalona. Leki przeciwpadaczkowe należy przepisywać zgodnie z oceną medyczną i wytycznymi klinicznymi.
sJIA
Zespół aktywacji makrofagów (MAS) to poważny, zagrażający życiu stan, który może się rozwijać u pacjentów z sJIA. W badaniach klinicznych nie badano działania leku Actemra® u pacjentów w okresie wystąpienia MAS.
Stosowanie w okresie ciąży lub karmienia piersią.
Kobiety w wieku rozrodczym
Kobietom w wieku rozrodczym należy stosować skuteczne metody antykoncepcji podczas leczenia i przez 3 miesiące po jego zakończeniu.
Ciąża
Brak odpowiednich danych dotyczących stosowania leku Actemra® u kobiet w ciąży. Badania na zwierzętach wykazały zwiększone ryzyko samoistnych poronień/śmierci embrionu/pleta po podaniu leku w wysokich dawkach. Potencjalne ryzyko dla ludzi jest nieznane.
Nie należy stosować leku Actemra® w czasie ciąży, z wyjątkiem przypadków ekstremalnej konieczności.
Karmienie piersią
Nie wiadomo, czy tocylicumab przenika do ludzkiego mleka matki. Przenikanie leku Actemra® do mleka matki u zwierząt nie było badane. Decyzję o kontynuowaniu/przerwaniu karmienia piersią lub kontynuowaniu/przerwaniu leczenia lekiem Actemra® należy podjąć po ocenie korzyści karmienia piersią dla dziecka i korzyści z leczenia lekiem Actemra® dla kobiety.
Plodność
Dostępne dane przedkliniczne wskazują na brak wpływu na płodność podczas leczenia lekiem Actemra®.
Wpływ na zdolność prowadzenia pojazdów lub obsługi urządzeń.
Lek Actemra® ma nieznaczny wpływ na zdolność prowadzenia pojazdów lub obsługi innych urządzeń (patrz dział „Efekty niepożądane”, zawroty głowy).
Sposób stosowania i dawki.
Tokolizumab w postaci leku do stosowania podskórnie podaje się za pomocą wstępnie napełnionego strzykawki z zamontowanym urządzeniem zabezpieczającym igłę.
Leczenie należy rozpoczynać lekarzom posiadającym doświadczenie w rozpoznawaniu i leczeniu reumatoidalnego zapalenia stawów (RZS), poliarticularnego młodzieńczego zapalenia stawów idiopatycznego (mZSI), systemowego młodzieńczego zapalenia stawów idiopatycznego (sZSI) oraz/lub tętnicy posurowatej (GCA). Pierwszą iniekcję należy wykonać pod nadzorem wykwalifikowanego personelu medycznego. Pacjent lub rodzice/opiekun mogą samodzielnie wykonywać iniekcje leku Actemra®, tylko jeśli lekarz uzna to za stosowne, a pacjent lub rodzice/opiekun wyrażą zgodę na konieczność dalszego nadzoru medycznego oraz ukończą szkolenie z właściwej techniki iniekcji.
Pacjentom przechodzącym z leczenia dożylnej terapii tokolizumabem na podskórne podawanie, pierwszą dawkę podkórną należy podać zamiast następnej zaplanowanej dawki dożylnej, pod nadzorem wykwalifikowanego personelu medycznego.
Wszystkim pacjentom leczonym lekiem Actemra® należy dostarczyć ulotkę dla pacjenta. Należy ocenić możliwość stosowania leku przez pacjenta lub rodziców/opiekunów w warunkach domowych oraz poinstruować ich o konieczności informowania personelu medycznego w przypadku wystąpienia objawów reakcji alergicznych. W przypadku wystąpienia objawów ciężkich reakcji alergicznych pacjent powinien natychmiast skontaktować się z placówką medyczną (patrz sekcja „Szczególne wskazania stosowania”).
Leczenie reumatoidalnego zapalenia stawów
Zalecana dawka w leczeniu reumatoidalnego zapalenia stawów to 162 mg raz w tygodniu w postaci iniekcji podskórnej.
Dane dotyczące przejścia pacjenta z leku Actemra® w postaci do stosowania dożylnej na postać do stosowania podkórnie w dawce ustalonej są ograniczone. Należy przestrzegać przedziału stosowania co tydzień.
W przypadku przejścia pacjenta z dożylnej iniekcji na podkórną, pierwszą dawkę podkórną należy podać zamiast następnej dawki dożylnej, pod nadzorem wykwalifikowanego personelu medycznego.
Leczenie tętnicy posurowatej
Zalecana dawka w leczeniu tętnicy posurowatej to 162 mg raz w tygodniu podskórnie, w połączeniu ze skróconym cyklem leczenia glikokortykosteroidami. Lek Actemra® w trybie monoterapii można stosować po zakończeniu leczenia glikokortykosteroidami.
Monoterapii lekiem Actemra® nie należy stosować w leczeniu ostrych nawrotów (patrz sekcja „Szczególne wskazania stosowania”).
Z uwagi na przewlekły charakter GCA, przy długości leczenia przekraczającej 52 tygodnie, należy wziąć pod uwagę aktywność choroby, zalecenia lekarza oraz preferencje pacjenta.
Leczenie reumatoidalnego zapalenia stawów i tętnicy posurowatej
Korekta dawki w przypadku zmiany wyników badań laboratoryjnych (patrz sekcja „Szczególne wskazania stosowania” oraz tabele 4–6)
Podwyższenie aktywności enzymów wątrobowych
Tabela 4
| Wartość parametru |
Korekta leczenia |
| Przekroczenie górnej granicy normy (GGN) o > 1–3 razy |
W razie potrzeby należy skorygować dawkę współprowadzonych modyfikujących przebieg choroby leków przeciwapłaszczykowych (DMARD) lub środków immunomodulujących u pacjentów z GCA. Przy trwałym wzroście aktywności transaminaz w tym zakresie zmniejszyć częstotliwość dawkowania leku Actemra® do 1 raz na 2 tygodnie lub przerwać leczenie lekiem do czasu powrotu wartości alaninotransaminazy (ALT) lub asparaginianotransaminazy (AST) do normy. Wznowić leczenie lekiem w dawce 1 raz w tygodniu lub 1 raz na 2 tygodnie, zgodnie z potrzebą kliniczną. |
| Przekroczenie GGN o > 3–5 razy |
Przerwać leczenie lekiem Actemra® do czasu spadku wartości poniżej < 3 × GGN; następnie stosować się do zaleceń w przypadku przekroczenia GGN o > 1–3 razy (patrz wyżej). Przestać stosować lek Actemra® przy trwałym wzroście wartości przekraczającej GGN więcej niż 3 razy (potwierdzonym w ponownym badaniu, patrz sekcja „Szczególne wskazania dotyczące stosowania”). |
| Przekroczenie GGN więcej niż 5 razy |
Przestać stosować lek Actemra®. |
Obniżona bezwzględna liczba neutrofili (ANC)
Nie zaleca się rozpoczynania leczenia lekiem Actemra® u pacjentów, którzy wcześniej nie byli leczeni tym lekiem, przy ANC poniżej 2 × 10⁹/l.
Tabela 5
| Wartość parametru (liczba komórek × 109/l) |
Korekta leczenia |
| ANC > 1 |
Nie zmieniać dawki. |
| ANC 0,5–1 |
Przerwać leczenie lekiem Actemra®. W przypadku wzrostu wskaźnika > 1×109/l wznowić leczenie lekiem w dawce 1 raz na 2 tygodnie i zwiększyć dawkę do 1 raz w tygodniu zgodnie z kliniczną potrzebą. |
| ANC < 0,5 |
Przerwać leczenie lekiem Actemra®. |
Mała liczba płytek krwi
Tabela 6
| Wartość parametru (liczba komórek × 103/μl) |
Korekta leczenia |
| 50–100 |
Przerwać leczenie lekiem Actemra®. W przypadku wzrostu wartości > 100 × 103/μl wznowić leczenie lekiem Actemra® w dawce 1 raz na 2 tygodnie i zwiększyć dawkę do 1 raz w tygodniu zgodnie z potrzebą kliniczną. |
| < 50 |
Przestać leczyć lekiem Actemra®. |
Leczenie reumatoidalnego zapalenia stawów i tętnicy szyjnej
Pominięta dawka
Jeśli pacjent pominie dawkę leku Actemra® (w postaci podskórnej w dawce raz w tygodniu w ciągu 7 dni planowanego przyjęcia), powinien przyjąć pominiętą dawkę w następnym zaplanowanym dniu. Jeśli pacjent pominie dawkę leku Actemra® (w postaci podskórnej w dawce raz na 2 tygodnie w ciągu 7 dni planowanego przyjęcia), powinien natychmiast przyjąć pominiętą dawkę, a kolejną dawkę – w następnym zaplanowanym dniu.
Grupy specjalne
Pacjenci w podeszłym wieku
Dostosowanie dawki nie jest wymagane u pacjentów w podeszłym wieku (> 65 lat).
Pacjenci z zaburzeniami funkcji nerek
Dostosowanie dawki nie jest wymagane u pacjentów z łagodnym lub umiarkowanym zaburzeniem funkcji nerek. Stosowanie leku Actemra® u pacjentów z ciężkim zaburzeniem funkcji nerek nie było badane. U tych pacjentów należy starannie monitorować funkcję nerek.
Pacjenci z zaburzeniami funkcji wątroby
Stosowanie leku Actemra® u pacjentów z zaburzeniami funkcji wątroby nie było badane, dlatego nie można podać zaleceń dotyczących dawkowania.
Dzieci
Skuteczność i bezpieczeństwo stosowania leku Actemra® w formie do wstrzykiwania podskórnie u dzieci od urodzenia do 1 roku życia nie są ustalone. Brakuje danych.
Zmiana dawki leku powinna opierać się wyłącznie na trwałej zmianie masy ciała pacjenta w czasie. Lek Actemra® można stosować jako monoterapię lub w połączeniu z MT.
Pacjenci z sJIA
Zalecana dawka dla pacjentów od 1 roku życia to 162 mg raz w tygodniu podskórnie przy masie ciała ≥ 30 kg lub 162 mg raz na 2 tygodnie podskórnie przy masie ciała < 30 kg.
W trakcie leczenia lekiem Actemra® do wstrzykiwania podskórnie minimalna masa ciała pacjenta musi wynosić co najmniej 10 kg.
Pacjenci z pJIA
Zalecana dawka leku dla pacjentów od 2 roku życia to 162 mg podskórnie raz na 2 tygodnie, jeśli masa ciała pacjenta wynosi ≥ 30 kg, oraz 162 mg podskórnie raz na 3 tygodnie, jeśli masa ciała < 30 kg.
Dostosowanie dawki w przypadku zmiany wyników badań laboratoryjnych (sJIA i pJIA)
W razie potrzeby dawkę metotreksatu i/lub innych leków towarzyszących należy dostosować lub przerwać ich stosowanie, a także zawiesić stosowanie tocylizumabu do czasu zakończenia oceny stanu klinicznego. Ponieważ istnieje wiele chorób współistniejących, które mogą wpływać na wyniki badań laboratoryjnych pacjentów z sJIA lub pJIA, decyzja o przerwaniu stosowania tocylizumabu z powodu zmiany wyników badań laboratoryjnych powinna opierać się na indywidualnej ocenie lekarskiej dla każdego pacjenta (patrz tabele 7–9).
Podwyższenie aktywności enzymów wątrobowych
Tabela 7
| Wartość parametru |
Korekta leczenia |
| Przekroczenie górnej granicy normy (GGN) o > 1–3 razy |
W razie potrzeby skorygować dawkę współpodawanego metotreksatu. W przypadku trwałego wzrostu wartości w tym zakresie przerwać leczenie lekiem Actemra® do czasu normalizacji poziomu alaninotransaminazy (ALT)/asparaginianotransaminazy (AST). |
| Przekroczenie GGN o > 3–5 razy |
W razie potrzeby skorygować dawkę współpodawanego metotreksatu. Przerwać stosowanie leku Actemra® do czasu spadku wartości poniżej poziomu < 3 × GGN, następnie stosować się do zaleceń dotyczących przekroczenia GGN o > 1–3 razy (patrz wyżej). |
| Przekroczenie GGN o > 5 razy |
Przestać stosować lek Actemra®. Decyzja o zaprzestaniu leczenia lekiem Actemra® w przypadku sJIA lub pJIA ze względu na zmianę parametru laboratoryjnego powinna opierać się na indywidualnej ocenie lekarskiej dla każdego pacjenta. |
Obniżona liczba całkowita neutrofili (ANC)
Tabela 8
| Wartość parametru (liczba komórek × 109/l) |
Korekta leczenia |
| ANC > 1 |
Dawkę nie należy zmieniać. |
| ANC 0,5–1 |
Przerwać leczenie lekiem Actemra®. W przypadku wzrostu ANC do > 1 × 109/l wznowić leczenie lekiem Actemra®. |
| ANC < 0,5 |
Przestać stosować leczenie lekiem Actemra®. Decyzja o przerwaniu leczenia lekiem Actemra® w przypadku sJIA lub pJIA ze względu na zmianę parametru laboratoryjnego powinna opierać się na ocenie medycznej dla każdego pacjenta indywidualnie. |
Niskie liczba płytek krwi
Tabela 9
| Wartość parametru (liczba komórek × 103/μl) |
Korekta leczenia |
| 50–100 |
W razie potrzeby dostosować dawkę współużywanego metotreksatu. Przerwać leczenie lekiem Actemra®. W przypadku wzrostu wartości powyżej 100 × 103/μl wznowić leczenie lekiem Actemra®. |
| < 50 |
Przestać stosować lek Actemra®. Decyzja o przerwaniu leczenia lekiem Actemra® w przypadku sJIA lub pJIA ze względu na zmianę parametru laboratoryjnego powinna opierać się na ocenie medycznej dla każdego pacjenta indywidualnie. |
Nie prowadzono badań nad zmniejszeniem częstości stosowania tocylizumabu w wyniku zmiany parametrów laboratoryjnych u pacjentów z uJZS lub pJZS.
W przypadku dzieci z chorobami innymi niż uJZS lub pJZS, bezpieczeństwo i skuteczność leku Actemra® w formie do podania podskórnie nie zostały ustalone.
Dostępne dane dotyczące stosowania formy do podania dożylnego wskazują na poprawę kliniczną obserwowaną w ciągu 12 tygodni od rozpoczęcia leczenia lekiem Actemra®. Jeżeli u pacjenta nie stwierdza się poprawy w tym okresie czasu, należy dokładnie przeanalizować celowość kontynuowania terapii.
Pominięta dawka
Jeśli pacjent z uJZS nie otrzymał tygodniowej iniekcji podskórnej leku Actemra® w ciągu 7 dni od zaplanowanego dnia iniekcji, pominiętą dawkę należy podać w następnym zaplanowanym terminie. Jeśli pacjent z uJZS pominął dawkę leku Actemra® w formie iniekcji podskórnej w dawce 1 raz na 2 tygodnie w ciągu 7 dni od zaplanowanego dnia podania, pominiętą dawkę należy podać natychmiast, a kolejną dawkę – w zaplanowanym terminie.
W przypadku pominięcia przez pacjenta z pJZS iniekcji podskórnej leku Actemra® w ciągu 7 dni od zaplanowanego dnia iniekcji, iniekcję należy wykonać tak szybko, jak tylko pacjent o niej pamięta, a następną iniekcję – zgodnie z pierwotnym harmonogramem. Jeśli pacjent z pJZS pominął iniekcję podkutą lekiem Actemra® przez więcej niż 7 dni od zaplanowanego dnia iniekcji lub nie jest pewien, kiedy należy wykonać iniekcję lekiem Actemra®, należy skontaktować się z lekarzem lub farmaceutą.
Sposób podania
Lek Actemra® podaje się w postaci iniekcji podskórnej.
Po odpowiednim przeszkoleniu w technice podania iniekcji pacjenci mogą samodzielnie podawać lek Actemra®, jeśli ich lekarz uzna to za stosowne. Całą zawartość wstępnie napełnionego strzykawki (0,9 ml) należy podać w postaci iniekcji podskórnej. Zalecane miejsca iniekcji (brzuch, uda lub ramię) należy zmieniać i nigdy nie podawać leku w brodawki, blizny ani w bolące, zasinione, zaczerwienione, zesztywniałe lub uszkodzone obszary skóry.
Lek Actemra® dostarczany jest w prezentacji w postaci strzykawki jednorazowej wstępnie napełnionej z wbudowanym urządzeniem zabezpieczającym igłę.
Po wyjęciu strzykawki wstępnie napełnionej z lodówki należy doprowadzić ją do temperatury pokojowej (od 18 do 28 °C), oczekując od 25 do 30 minut przed wykonaniem iniekcji lekiem Actemra®. Nie wolno wstrząsać strzykawką wstępnie napełnioną. Po zdjęciu osłonki iniekcję należy rozpocząć w ciągu 5 minut, aby uniknąć odparowania leku i zablokowania igły.
Jeśli strzykawka wstępnie napełniona nie została użyta w ciągu 5 minut po zdjęciu osłonki, należy ją zutylizować w pojemniku odpornym na przebicie i wziąć nową strzykawkę wstępnie napełnioną. Jeśli po wkłuciu igły nie można nacisnąć tłoka, strzykawkę należy zutylizować w pojemniku odpornym na przebicie i zastosować nową strzykawkę wstępnie napełnioną.
Nie wolno stosować leku, jeśli roztwór jest nieprzezroczysty, zawiera cząstki lub jego kolor różni się od bezbarwnego do żółtawego, lub jeśli stwierdzono oznaki uszkodzenia jakiejkolwiek części strzykawki wstępnie napełnionej.
Niezaopatrzone leki lub odpady po ich stosowaniu należy zutylizować zgodnie z lokalnymi wymogami.
Szczegółowe informacje zawarte są w podrozdziale „Instrukcja stosowania strzykawki wstępnie napełnionej”.
Po wyjęciu ze lodówki strzykawkę wstępnie napełnioną można przechowywać do 2 tygodni w temperaturze nie wyższej niż 30 °C.
Instrukcja stosowania strzykawki wstępnie napełnionej
Krok 1. Weryfikacja strzykawki wizualna
- Wyjmij z lodówki opakowanie kartonowe zawierające strzykawkę i otwórz je. Nie dotykaj spustów na strzykawce, ponieważ może to spowodować uszkodzenie strzykawki.
- Wyjmij strzykawkę z opakowania kartonowego i sprawdź ją wizualnie, a także bezpośrednio lek wewnątrz strzykawki. Ważne jest, aby upewnić się, że strzykawka i lek są bezpieczne do stosowania.
- Sprawdź datę ważności na opakowaniu kartonowym i na strzykawce, aby upewnić się, że nie upłynęła. Nie należy stosować strzykawki, jeśli upłynął termin ważności. Ważne jest, aby upewnić się, że strzykawka i lek są bezpieczne do stosowania.
Zutylizuj strzykawkę i nie stosuj jej, jeśli:
- lek jest mętny;
- lek zawiera cząstki;
- kolor leku różni się od bezbarwnego do żółtawego;
- jakakolwiek część strzykawki wygląda na uszkodzoną.
Krok 2. Doprowadzenie strzykawki do temperatury pokojowej
- Nie zdejmuj osłonki igły ze strzykawki do kroku 5. Wczesne zdjęcie osłonki igły może prowadzić do wyschnięcia leku i zablokowania igły.
- Połóż strzykawkę na czystej, równej powierzchni i pozostaw ją do ogrzania do temperatury pokojowej (18–28 °C) przez około 25–30 minut. Jeśli temperatura strzykawki nie zostanie doprowadzona do temperatury pokojowej, może to spowodować dyskomfort podczas wstrzykiwania i trudności z naciskaniem tłoka.
- Nie należy ogrzewać strzykawki w żaden inny sposób.
Krok 3. Umycie rąk
- Umij ręce wodą z mydłem.
Krok 4. Wybór i przygotowanie miejsca iniekcji
- Zalecanymi obszarami do wstrzyknięcia są przednia lub środkowa część uda oraz dolna część brzucha poniżej pępka (pępkowego), z wyjątkiem pięciocentymetrowego obszaru bezpośrednio wokół pępka (rys. 1). Jeśli iniekcję wykonuje osoba opiekująca się pacjentem, do wstrzyknięcia leku można również użyć zewnętrznej części ramienia.
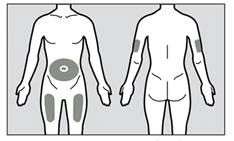
Rys. 1
- Należy za każdym razem używać innego miejsca do wstrzyknięcia leku podczas samodzielnego wykonywania iniekcji, zachowując co najmniej trzy centymetry od miejsca użytego do poprzedniej iniekcji.
- Nie należy wstrzykiwać leku w obszary podatne na podrażnienie przez pasek lub pas. Nie należy wstrzykiwać leku w znamiona, blizny, siniaki ani obszary, w których skóra jest delikatna, zaczerwieniona, zesztywniała lub uszkodzona.
- Oczyść wybrany obszar do iniekcji za pomocą chusteczki alkoholowej, aby zmniejszyć ryzyko infekcji.
- Pozwól skórze wyschnąć przez około 10 sekund.
- Nie dotykaj oczyszczonego obszaru przed wykonaniem iniekcji. Nie dmuchaj i nie dmuchaj na oczyszczony obszar.
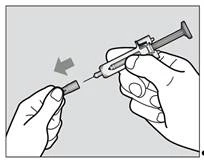
Krok 5. Zdjęcie osłonki igły
- Nie trzymaj strzykawki za tłok podczas zdjęcia osłonki igły.
- Trzymaj mocno osłonę igły strzykawki jedną ręką i zdjęcie osłonki igły drugą ręką (rys. 2). Jeśli nie możesz zdjąć osłonki igły, należy poprosić o pomoc osobę opiekującą się Tobą lub swojego lekarza.
Rys. 2
- Nie dotykaj igły i dbaj o to, aby igła nie dotykała żadnej powierzchni.
- Możesz zauważyć kropelkę cieczy na końcu igły. Jest to normalne.
- Wyrzuć osłonę igły do pojemnika odpornego na przebicie lub pojemnika na ostre przedmioty.
UWAGA: Po zdjęciu osłonki igły strzykawkę należy natychmiast użyć.
- Jeśli strzykawka nie została użyta w ciągu 5 minut po zdjęciu osłonki, należy ją zutylizować, używając pojemnika odpornego na przebicie lub pojemnika na ostre przedmioty, i należy użyć nowej strzykawki. Jeśli osłonka igły została zdjęta ponad 5 minut temu, może być trudniej wykonać iniekcję, ponieważ lek może wyschnąć i zablokować igłę.
- Nigdy nie zakładaj ponownie osłonki igły po jej zdjęciu.
Krok 6. Wykonanie iniekcji
- Trzymaj strzykawkę wygodnie w rękach.
- Aby upewnić się, że igła może być poprawnie wprowadzona pod skórę, zrób fałd skóry w oczyszczonym obszarze do iniekcji wolną ręką (rys. 3). Uformowanie fałdu skóry jest ważne, aby upewnić się, że lek jest wstrzykiwany pod skórę (do tkanki tłuszczowej), ale nie głębiej (do mięśnia). Iniekcja do mięśnia może powodować dyskomfort podczas wstrzykiwania.
- Nie trzymaj strzykawki za tłok i nie naciskaj tłoka podczas wkłuwania igły w skórę.
- Wprowadź igłę do końca w fałd skóry pod kątem od 45° do 90° szybkim, stanowczym ruchem.
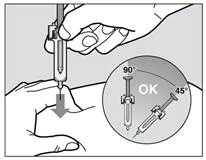
Rys. 3
Ważne jest wybranie odpowiedniego kąta, aby upewnić się, że lek jest wstrzykiwany pod skórę (do tkanki tłuszczowej), w przeciwnym razie iniekcja może być bolesna, a lek może nie zadziałać.
- Następnie, trzymając strzykawkę w tej samej pozycji, puść fałd skóry.
- Powoli podaj cały lek, delikatnie naciskając tłok do całkowitego opuszczenia go w dół (rys. 4). Należy naciskać tłok do całkowitego opuszczenia go w dół, aby upewnić się, że otrzymałeś pełną dawkę leku i aby upewnić się, że spusty są całkowicie odepchnięte. Jeśli tłok nie zostanie naciśnięty do oporu, osłona ochronna nie przykryje całkowicie igły po jej wyciągnięciu. Jeśli igła nie będzie całkowicie przykryta, należy zachować ostrożność i umieścić strzykawkę z igłą w pojemniku odpornym na przebicie, aby uniknąć urazu igłą.
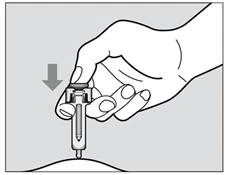
Rys. 4
- Gdy tłok jest całkowicie opuszczony, kontynuuj naciskanie w dół na tłok, aby upewnić się, że cały lek został podany przed wyciągnięciem igły ze skóry.
- Kontynuuj naciskanie na tłok podczas wyciągania igły ze skóry pod tym samym kątem, pod jakim została wprowadzona (rys. 5).
- Jeśli po wkłuciu igły nie możesz naciskać tłoka, należy wyrzucić strzykawkę wstępnie napełnioną do pojemnika odpornego na przebicie i użyć nowej strzykawki wstępnie napełnionej (należy ponownie rozpocząć od kroku 2). Jeśli nadal występują trudności, skonsultuj się z lekarzem.
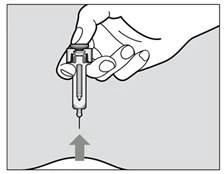
Rys. 5
- Po całkowitym wyciągnięciu igły ze skóry możesz zwolnić tłok, wówczas urządzenie ochronne igły schowa igłę (rys. 6).
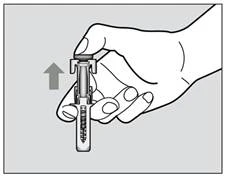
Rys. 6
- Jeśli zobaczysz krople krwi w miejscu wstrzyknięcia, możesz przycisnąć sterylną watę lub gazę do miejsca iniekcji na około 10 sekund.
- Nie należy tarmosić miejsca iniekcji.
Krok 7. Utylizacja strzykawki
- Nie próbuj ponownie zakładać osłonki na igłę używanej strzykawki.
- Wyrzuć używane strzykawki do pojemnika odpornego na przebicie lub pojemnika na ostre przedmioty. Poproś swojego lekarza lub farmaceutę o informacje, gdzie możesz uzyskać pojemnik na ostre przedmioty lub jakie inne typy pojemników odpornych na przebicie możesz użyć do bezpiecznej utylizacji używanych strzykawek, jeśli nie masz takiego pojemnika.
Skonsultuj się ze swoim lekarzem w celu uzyskania instrukcji dotyczących prawidłowego wyrzucania używanych strzykawek. Może istnieć lokalne lub państwowe prawo dotyczące utylizacji używanych strzykawek.
Nie wyrzucaj używanych strzykawek ani pojemników odpornych na przebicie do odpadów komunalnych i nie używaj ich ponownie.
- Utylizuj napełniony pojemnik zgodnie z zaleceniami swojego lekarza lub farmaceuty.
- Zawsze przechowuj pojemnik odporny na przebicie w miejscu niedostępnym dla dzieci.
Dzieci.
Skuteczność i bezpieczeństwo stosowania leku Actemra® w formie do podania podskórnie u dzieci od urodzenia do 1 roku życia nie zostały ustalone. Brak danych.
Przedawkowanie.
Dane dotyczące przedawkowania leku Actemra® są ograniczone. W jednym przypadkowym przypadku przedawkowania przy podaniu dożylnej dawki 40 mg/kg u pacjenta z szpiczakiem mnogim nie zaobserwowano żadnych niepożądanych reakcji.
Nie zaobserwowano również poważnych niepożądanych reakcji u zdrowych ochotników, którzy jednorazowo otrzymali lek Actemra® w dawce do 28 mg/kg, choć zaobserwowano ograniczającą dawkę neutropenię.
Efekty uboczne.
Krótki opis profilu bezpieczeństwa
Profil bezpieczeństwa oparto na danych z badań klinicznych u 4510 pacjentów otrzymujących lek Actemra®; większość z tych pacjentów uczestniczyła w badaniach nad reumatoidalnym zapaleniem stawów (RZS) u dorosłych (n = 4009), a reszta – w badaniach nad tętniczką z rumieniem guzowatym (GCA) (n = 149), młodzieńczym zapaleniem stawów wielopowstawowym (pJIA) (n = 240) oraz systemowym młodzieńczym zapaleniem stawów (sJIA) (n = 112). Profil bezpieczeństwa leku Actemra® w tych wskazaniach pozostaje podobny i niezróżnicowany.
Najczęściej zgłaszanymi efektami ubocznymi były: infekcje dróg oddechowych górnych, zapalenie nosa i gardła, ból głowy, nadciśnienie tętnicze oraz podwyższenie poziomu ALT.
Najpoważniejszymi efektami ubocznymi były: ciężkie infekcje, powikłania zapalenia zatok, reakcje nadwrażliwości.
Podsumowanie danych dotyczących efektów ubocznych
Poniżej przedstawiono zestawione dane dotyczące efektów ubocznych z badań klinicznych oraz/lub doświadczenia po rejestracji, spontanicznych zgłoszeń, literatury naukowej i badań nieinterwencyjnych, przy użyciu słownika medycznego do działalności regulacyjnej (MedDRA), pogrupowane według klas narządów i układów oraz częstości występowania: bardzo często (≥1/10), często (≥1/100 i <1/10), rzadko (≥1/1000 i <1/100), rzadko (≥1/10000 i <1/1000) lub bardzo rzadko (<1/10000). W każdej grupie według częstości występowania efekty uboczne są wymienione w kolejności malejącej ich powagi.
Zaburzenia układu krwi i układu chłonnego: często – leukopenia, neutropenia, hipofibrynogenemia.
Zaburzenia układu endokrynnego: rzadko – hipotyreozę.
Zaburzenia ze strony narządów wzroku: często – zapalenie spojówek.
Zaburzenia ze strony układu pokarmowego: często – owrzodzenie jamy ustnej, zapalenie żołądka, ból brzucha; rzadko – zapalenie jamy ustnej, owrzodzenie żołądka.
Zaburzenia ogólne i stan w miejscu podania: bardzo często – reakcje w miejscu wstrzyknięcia, często – obrzęki obwodowe, reakcje nadwrażliwości.
Zaburzenia ze strony układu wątrobowo-żółciowego: rzadko – uszkodzenie wątroby wywołane lekiem, zapalenie wątroby, żółtaczka; bardzo rzadko – niewydolność wątroby.
Zaburzenia ze strony układu odpornościowego: anafilaksja (śmiertelna)1,2,3.
Infekcje i inwazje: bardzo często – infekcje dróg oddechowych górnych; często – ropnie, zapalenie płuc, infekcje spowodowane przez Herpes simplex i Herpes zoster; rzadko – zapalenie zatok.
Badania: często – podwyższenie aktywności transaminaz wątrobowych, przyrost masy ciała, podwyższenie poziomu bilirubiny ogólnej*.
Zaburzenia przemiany materii i przemiany: bardzo często – hipercholesterolemia*; rzadko – hipertriglicerydemia.
Zaburzenia ze strony układu nerwowego: często – ból głowy, zawroty głowy.
Zaburzenia ze strony nerek i układu moczowego: rzadko – kamica nerkowa.
Zaburzenia ze strony układu oddechowego, klatki piersiowej i jamy piersiowej: często – kaszel, duszność.
Zaburzenia ze strony skóry i tkanki podskórnej: często – wysypka, świąd, pokrzywka; rzadko – zespół Stevensa-Johnsona3.
Zaburzenia ze strony układu naczyniowego: często – nadciśnienie tętnicze.
* Obejmuje podwyższenie w monitorowaniu laboratoryjnym (patrz niżej).
1 Zobacz sekcję „Przeciwwskazania”.
2 Zobacz sekcję „Szczególne ostrzeżenia i środki ostrożności”.
3 Ten efekt uboczny został wykryty podczas obserwacji po rejestracji, ale nie obserwowano go w kontrolowanych badaniach klinicznych.
Kategoria częstości została oszacowana jako górna granica 95% przedziału ufności, obliczonego na podstawie całkowitej liczby pacjentów, którzy otrzymywali tocylizumab w badaniach klinicznych.
Leczenie RZS
Podanie dożylnie
Bezpieczeństwo leku Actemra® badano w 5 podwójnie ślepych, kontrolowanych badaniach klinicznych fazy III oraz w okresach ich przedłużenia.
Wszystkie populacje kontrolne obejmowały wszystkich pacjentów z podwójnie ślepych okresów każdego badania podstawowego od momentu randomizacji do pierwszej zmiany dawki lub do osiągnięcia dwuletniego okresu uczestnictwa w badaniu. Okres kontrolny trwał 6 miesięcy w 4 z tych badań i do 2 lat w jednym badaniu. W podwójnie ślepych, placebo-kontrolowanych badaniach 774 pacjentów otrzymywało leczenie lekiem Actemra® w dawce 4 mg/kg masy ciała w połączeniu z metotreksatem, 1870 pacjentów otrzymywało tocylizumab w dawce 8 mg/kg masy ciała w połączeniu z metotreksatem/innymi modyfikującymi przebieg choroby lekami przeciwwątrobowymi (DMARD) oraz 288 pacjentów otrzymywało tocylizumab w dawce 8 mg/kg masy ciała jako monoterapię.
Populacja ogólna obejmowała wszystkich pacjentów, którzy otrzymali co najmniej jedną dawkę leku Actemra® w trakcie podwójnie ślepego, placebo-kontrolowanego okresu lub w trakcie otwartego, przedłużonego okresu badań. 3577 z 4009 pacjentów otrzymywało leczenie przez co najmniej 6 miesięcy, 3296 pacjentów – przez co najmniej 1 rok, 2806 pacjentów – przez co najmniej 2 lata oraz 1222 – przez 3 lata.
Opis poszczególnych efektów ubocznych
Infekcje
Na podstawie 6-miesięcznych badań kontrolnych częstość infekcji po podaniu leku Actemra® w dawce 8 mg/kg w połączeniu z DMARD wynosiła 127 przypadków na 100 pacjentów-rok w porównaniu do 112 przypadków na 100 pacjentów-rok w grupie pacjentów otrzymujących placebo w połączeniu z DMARD. W populacji z długotrwałym okresem ekspozycji ogólna częstość infekcji przy stosowaniu leku Actemra® wynosiła 108 na 100 pacjentów-rok ekspozycji.
Na podstawie 6-miesięcznych kontrolowanych badań klinicznych częstość ciężkich infekcji w grupie pacjentów otrzymujących lek Actemra® w dawce 8 mg/kg w połączeniu z DMARD wynosiła 5,3 przypadki na 100 pacjentów-rok ekspozycji w porównaniu do 3,9 przypadków na 100 pacjentów-rok ekspozycji w grupie pacjentów otrzymujących placebo w połączeniu z DMARD. W badaniu monoterapii częstość ciężkich infekcji wynosiła 3,6 przypadki na 100 pacjentów-rok ekspozycji w grupie leku Actemra® w porównaniu do grupy metotreksatu (1,5 przypadków na 100 pacjentów-rok ekspozycji).
W populacji wszystkich ekspozycji ogólna częstość ciężkich infekcji wynosiła 4,7 na 100 pacjentów-rok. Zarejestrowano ciężkie infekcje, niektóre z nich kończące się śmiercią, w tym zapalenie płuc, ropnie, ospy pęcherzykowe, zapalenia żołądka i jelit, zapalenie zatok, sepsę, bakteryjne zapalenia stawów. Zgłaszano przypadki wystąpienia infekcji oportunistycznych.
Choroba płuc międzybłonkowych
Zaburzenia funkcji płuc mogą zwiększać ryzyko rozwoju infekcji. Istnieją doniesienia po marketingu dotyczące choroby płuc międzybłonkowych (w tym zapalenia płuc i włóknienia płuc), niektóre z tych przypadków miały śmiertelny przebieg.
Przebicia przewodu pokarmowego
W trakcie 6-miesięcznych badań kontrolnych w grupie pacjentów otrzymujących leczenie lekiem Actemra® ogólna częstość przebicia przewodu pokarmowego wynosiła 0,26 na 100 pacjentów-rok. W populacji długotrwałej ekspozycji ogólna częstość przebicia przewodu pokarmowego wynosiła 0,28 na 100 pacjentów-rok. Ogólnie przypadki przebicia przewodu pokarmowego podczas stosowania leku Actemra® były powikłaniami zapalenia zatok i obejmowały rozlane ropne zapalenie otrzewnej, przebicie dolnych odcinków przewodu pokarmowego, przetoki i ropień.
Reakcje infuzyjne
W trakcie 6-miesięcznych badań kontrolnych reakcje niepożądane związane z podaniem leku (osobne reakcje występujące podczas podania leku lub w ciągu 24 godzin po jego podaniu) stwierdzono u 6,9% pacjentów otrzymujących tocylizumab w dawce 8 mg/kg w połączeniu z DMARD oraz u 5,1% pacjentów otrzymujących placebo wraz z DMARD. Niepożądane reakcje występujące podczas podania leku były głównie epizodami podwyższonego ciśnienia tętniczego. Efektami ubocznymi obserwowanymi w ciągu 24 godzin po zakończeniu podania leku były ból głowy oraz reakcje skórne (wysypka, pokrzywka). Te reakcje nie prowadziły do ograniczenia terapii.
Częstość reakcji anafilaktycznych (u 6 z 3778 pacjentów, 0,2%) była kilkakrotnie wyższa u pacjentów otrzymujących lek w dawce 4 mg/kg niż u pacjentów otrzymujących lek w dawce 8 mg/kg. W kontrolowanych i otwartych badaniach klinicznych klinicznie istotne reakcje nadwrażliwości spowodowane podaniem leku Actemra®, które wymagały przerwania leczenia, obserwowano u 13 z 3778 pacjentów (0,3%). Zazwyczaj te reakcje obserwowano między drugą a piątą infuzją tocylizumabu (patrz sekcja „Szczególne ostrzeżenia i środki ostrożności”). Anafilaksja z końcem śmiertelnym została zarejestrowana po rejestracji leku podczas leczenia lekiem Actemra® do podania dożylnego (patrz sekcja „Szczególne ostrzeżenia i środki ostrożności”).
Imunogenność
Ogółem 2876 pacjentów zostało przebadanych pod kątem obecności przeciwciał przeciwko lekowi Actemra® w 6-miesięcznych badaniach kontrolnych. U 6 (1,6%) z 46 pacjentów, u których wykryto przeciwciała przeciwko lekowi Actemra®, stwierdzono skojarzenie z klinicznie istotnymi reakcjami nadwrażliwości, które doprowadziły do całkowitego ostatecznego odstawienia leczenia u 5 pacjentów. U 30 pacjentów (1,1%) wykryto przeciwciała neutralizujące.
Zmiany laboratoryjne
Neutrofile
W 6-miesięcznych badaniach kontrolnych zmniejszenie liczby neutrofili poniżej 1 × 109/l stwierdzono u 3,4% pacjentów, którym lek Actemra® podawano w dawce 8 mg/kg w połączeniu z lekiem modyfikującym przebieg choroby, w porównaniu do mniej niż 0,1% pacjentów otrzymujących placebo w połączeniu z DMARD. Zmniejszenie ANC poniżej 1 × 109/l miało miejsce w ciągu 8 tygodni od rozpoczęcia leczenia u około połowy przypadków. Zmniejszenie liczby neutrofili poniżej 0,5 × 109/l stwierdzono u 0,3% pacjentów otrzymujących lek Actemra® w dawce 8 mg/kg w połączeniu z DMARD. Zgłaszano rozwój infekcji z towarzyszącą neutropenią.
W trakcie podwójnie ślepego, kontrolowanego okresu badań z długotrwałym okresem ekspozycji obraz i częstość zmniejszenia liczby neutrofili odpowiadały wynikom zarejestrowanym w 6-miesięcznych kontrolowanych badaniach klinicznych.
Trombocyty
W 6-miesięcznych badaniach kontrolnych zmniejszenie liczby trombocytów poniżej 100 × 103/μl obserwowano u 1,7% pacjentów otrzymujących lek Actemra® w dawce 8 mg/kg w połączeniu z lekiem modyfikującym przebieg choroby, w porównaniu do mniej niż 1% pacjentów otrzymujących placebo w połączeniu z DMARD. Te zmiany nie były towarzyszone wystąpieniem epizodów krwawień.
W trakcie podwójnie ślepego, kontrolowanego okresu badań z długotrwałym okresem ekspozycji obraz i częstość zmniejszenia liczby trombocytów odpowiadały wynikom zarejestrowanym w 6-miesięcznych kontrolowanych badaniach klinicznych.
Bardzo rzadko zgłaszano pancytopenię, która została zauważona w okresie po marketingu.
Podwyższenie aktywności transaminaz wątrobowych
W trakcie 6-miesięcznych kontrolowanych badań klinicznych przejściowe podwyższenie aktywności ALT/AST (przekroczenie GGN więcej niż 3-krotnie) obserwowano u 2,1% pacjentów otrzymujących lek Actemra® w dawce 8 mg/kg i u 4,9% pacjentów otrzymujących metotreksat. Te zmiany wystąpiły u 6,5% pacjentów otrzymujących lek Actemra® w dawce 8 mg/kg w połączeniu z DMARD i u 1,5% pacjentów otrzymujących placebo w połączeniu z DMARD.
Dodanie do monoterapii lekiem Actemra® leków potencjalnie hepatotoksycznych (np. metotreksatu) prowadziło do zwiększenia częstości przypadków podwyższenia aktywności transaminaz. Podwyższenie aktywności ALT/AST przekraczające GGN więcej niż 5-krotnie obserwowano u 0,7% pacjentów otrzymujących monoterapię tocylizumabem i u 1,4% pacjentów otrzymujących tocylizumab w połączeniu z DMARD. W większości przypadków terapię tocylizumabem trwale odstawiono. W trakcie podwójnie ślepego, kontrolowanego okresu przy rutynowym monitorowaniu laboratoryjnym częstość podwyższenia poziomu bilirubiny pośredniej powyżej górnej granicy normy, określonej jako rutynowy parametr laboratoryjny, u pacjentów otrzymujących lek Actemra® w dawce 8 mg/kg w połączeniu z DMARD wynosiła 6,2%. Ogólnie u 5,8% pacjentów obserwowano podwyższenie poziomu bilirubiny pośredniej od 1 do 2 razy powyżej górnej granicy normy i u 0,4% – więcej niż 2 razy powyżej GGN.
W trakcie podwójnie ślepego, kontrolowanego okresu badań z długotrwałym okresem ekspozycji charakter i częstość podwyższenia poziomu ALT/AST odpowiadały wynikom zarejestrowanym w 6-miesięcznych kontrolowanych badaniach klinicznych.
Zmiany parametrów metabolizmu lipidów
W trakcie rutynowego monitorowania laboratoryjnego w 6-miesięcznych badaniach kontrolnych często obserwowano podwyższenie parametrów metabolizmu lipidów (cholesterolu ogólnego, trójglicerydów, LDL i/lub HDL). Przy rutynowym monitorowaniu laboratoryjnym w badaniach klinicznych trwałe podwyższenie poziomu cholesterolu ogólnego ≥ 6,2 mmol/l obserwowano u 24% pacjentów i trwałe podwyższenie poziomu LDL ≥ 4,1 mmol/l – u 15% pacjentów otrzymujących lek Actemra®. Podwyższenie poziomu parametrów metabolizmu lipidów skutecznie korygowano lekami hipolipidemicznymi.
W trakcie podwójnie ślepego, kontrolowanego okresu badań z długotrwałym okresem ekspozycji charakter i częstość podwyższenia poziomu parametrów metabolizmu lipidów odpowiadały wynikom zarejestrowanym w 6-miesięcznych kontrolowanych badaniach klinicznych.
Złośliwe nowotwory
Dane kliniczne są niewystarczające do oceny możliwości rozwoju nowotworów złośliwych po zastosowaniu tocylizumabu. Trwa długoterminowa ocena bezpieczeństwa stosowania leku.
Reakcje skórne
O przypadkach zespołu Stevensa-Johnsona w okresie po marketingu zgłaszano rzadko.
Podanie podskórne
Leczenie RZS
Bezpieczeństwo stosowania leku Actemra® w postaci podskórnych wstrzyknięć u pacjentów z reumatoidalnym zapaleniem stawów badano w podwójnie ślepych, kontrolowanych, wieloośrodkowych badaniach SC-I. SC-I to badanie, w którym skuteczność i bezpieczeństwo stosowania leku Actemra® w dawce 162 mg raz w tygodniu porównywano z podaniem dożylnym w dawce 8 mg/kg u 1262 pacjentów z reumatoidalnym zapaleniem stawów. Wszyscy pacjenci otrzymywali terapię podstawową w postaci niobiologicznych leków modyfikujących przebieg choroby. Bezpieczeństwo i immunogenność leku Actemra® przy podaniu podskórnym odpowiadały znanemu profilowi bezpieczeństwa tocylizumabu przy podaniu dożylnym i nie zaobserwowano żadnych nowych lub nieoczekiwanych reakcji niepożądanych. Wyższa częstość reakcji w miejscu wstrzyknięcia obserwowana była w grupie z podaniem podskórnym niż w grupie z podaniem dożylnym, w której podawano placebo podskórnie.
Reakcje w miejscu wstrzyknięcia
W trakcie 6-miesięcznego kontrolowanego okresu badania SC-I częstość reakcji w miejscu wstrzyknięcia wynosiła 10,1% (64/631) i 2,4% (15/631) odpowiednio dla tocylizumabu w postaci podskórnego wstrzyknięcia w dawkowaniu raz w tygodniu i placebo w postaci podskórnego wstrzyknięcia (grupa z podaniem dożylnym). Te reakcje w miejscu wstrzyknięcia (w tym zaczerwienienie, świąd, ból i krwiak) miały od łagodnego do umiarkowanego stopnia nasilenia. Większość z nich była odwracalna bez żadnego leczenia i nie wymagała odstawienia leku.
Imunogenność
W badaniu klinicznym SC-I ogółem 625 pacjentów, którzy otrzymywali lek Actemra® w dawkowaniu 162 mg raz w tygodniu, zostało przebadanych w trakcie 6-miesięcznego kontrolowanego okresu pod kątem obecności przeciwciał przeciwko lekowi Actemra®. U pięciu pacjentów (0,8%) wykryto przeciwciała przeciwko lekowi Actemra®; u każdego z nich produkowane były przeciwciała neutralizujące przeciwko lekowi Actemra®. U jednego z pacjentów stwierdzono wynik dodatni na izotyp IgE (0,2%).
W badaniu klinicznym SC-II ogółem 434 pacjentów, którzy otrzymywali lek Actemra® w dawkowaniu 162 mg raz na 2 tygodnie, badano w trakcie 6-miesięcznego kontrolowanego okresu pod kątem obecności przeciwciał przeciwko lekowi Actemra®. U siedmiu pacjentów (1,6%) wykryto przeciwciała przeciwko lekowi Actemra®, u sześciu z nich (1,4%) produkowane były przeciwciała neutralizujące przeciwko lekowi Actemra®. U czterech pacjentów stwierdzono wynik dodatni na izotyp IgE (0,9%).
Nie zaobserwowano korelacji między produkcją przeciwciał a odpowiedzią kliniczną lub reakcjami niepożądanymi.
Zaburzenia hematologiczne
Neutrofile
W trakcie rutynowego monitorowania laboratoryjnego w 6-miesięcznym kontrolowanym badaniu tocylizumabu SC-I zmniejszenie liczby neutrofili poniżej 1 × 109/l obserwowano u 2,9% pacjentów, którym lek podawano podskórnie raz w tygodniu.
Nie zaobserwowano wyraźnej korelacji między zmniejszeniem liczby neutrofili poniżej 1 × 109/l a wystąpieniem ciężkich infekcji.
Trombocyty
W trakcie rutynowego monitorowania laboratoryjnego w 6-miesięcznym kontrolowanym badaniu SC-I leku Actemra® u żadnego z pacjentów, którym lek podawano podskórnie raz w tygodniu, nie zaobserwowano zmniejszenia liczby trombocytów ≤ 50 × 103/μl.
Podwyższenie aktywności transaminaz wątrobowych
W trakcie rutynowego monitorowania laboratoryjnego w 6-miesięcznym kontrolowanym badaniu leku Actemra® SC-I podwyższenie aktywności ALT lub AST ≥ 3 × GGN obserwowano odpowiednio u 6,5% i 1,4% pacjentów przy podaniu podskórnym leku raz w tygodniu.
Zmiany parametrów metabolizmu lipidów
W trakcie rutynowego monitorowania laboratoryjnego w 6-miesięcznym kontrolowanym badaniu leku Actemra® SC-I u 19% pacjentów obserwowano trwałe podwyższenie cholesterolu ogólnego > 6,2 mmol/l (240 mg/dl), a trwałe podwyższenie poziomu LDL do ≥ 4,1 mmol/l (160 mg/dl) – u 9% pacjentów, którym lek podawano podskórnie raz w tygodniu.
Podanie podskórne
Leczenie sJIA
Profil bezpieczeństwa leku Actemra® do podania podskórnego badano u 51 dzieci z sJIA (w wieku od 1 do 17 lat). Ogólnie u pacjentów z sJIA efekty uboczne pod względem typów były podobne do tych, które obserwowano u pacjentów z RZS (patrz powyższa sekcja „Efekty uboczne”).
Infekcje
Częstość infekcji u pacjentów z sJIA, którzy otrzymywali leczenie lekiem Actemra® do podania podskórnego, była porównywalna do częstości u pacjentów z sJIA, którzy otrzymywali leczenie lekiem Actemra® do podania dożylnego.
Reakcje w miejscu podania
W badaniu podania podskórnego leku Actemra® (WA28118) ogólnie u 41,2% (21/51) pacjentów z sJIA obserwowano reakcje w miejscu podania leku Actemra® podskórnego. Najczęstsze reakcje w miejscu podania to zaczerwienienie, świąd, ból i obrzęk w miejscu wstrzyknięcia. Większość zgłoszonych reakcji w miejscu podania była stopnia 1, wszystkie zgłoszone reakcje w miejscu podania były łagodne i żadna reakcja w miejscu podania nie doprowadziła do przerwania lub odstawienia leczenia.
Imunogenność
W badaniu podania podskórnego leku Actemra® (WA28118) 46 z 51 (90,2%) pacjentów zostało przebadanych pod kątem obecności przeciwciał przeciwko tocylizumabowi na poziomie wyjściowym i co najmniej raz podczas badania. U żadnego pacjenta nie rozwinęły się przeciwciała przeciwko tocylizumabowi po poziomie wyjściowym.
Odchylenia parametrów laboratoryjnych od normy
W 52-tygodniowym otwartym badaniu podania podskórnego (WA28118) zmniejszenie liczby neutrofili do poziomu poniżej 1 × 109/l obserwowano u 23,5% pacjentów otrzymujących leczenie lekiem Actemra® do podania podskórnego. Zmniejszenie liczby trombocytów do poziomu poniżej 100 × 103/μl obserwowano u 2% pacjentów otrzymujących leczenie lekiem Actemra® do podania podskórnego. Podwyższenie poziomu ALT lub AST do ≥ 3 × GGN obserwowano odpowiednio u 9,8% i 4,0% pacjentów otrzymujących leczenie lekiem Actemra® do podania podskórnego.
Zmiany parametrów metabolizmu lipidów
W 52-tygodniowym otwartym badaniu podania podskórnego leku (WA28118) u 23,4% i 35,4% pacjentów w dowolnym czasie w trakcie badania obserwowano podwyższenie poziomu cholesterolu LDL do ≥ 130 mg/dl i poziomu cholesterolu ogólnego do ≥ 200 mg/dl odpowiednio.
Podanie podskórne
Pacjenci z pJIA
Profil bezpieczeństwa podania podskórnego leku Actemra® oceniano również u 52 dzieci z pJIA. Ogólna ekspozycja leku Actemra® w ogólnej populacji ekspozycji pacjentów z pJIA wynosiła 184,4 pacjentów-rok dla tocylizumabu podawanego dożylnie i 50,4 pacjentów-rok dla tocylizumabu podawanego podskórnie. Ogólnie profil bezpieczeństwa obserwowany u pacjentów z pJIA odpowiadał znanemu profilowi bezpieczeństwa leku Actemra®, z wyjątkiem reakcji w miejscu podania. U pacjentów z pJIA reakcje w miejscu podania po podaniu podskórnym leku Actemra® występowały częściej niż u dorosłych pacjentów z RZS.
Infekcje
W badaniu podania podskórnego leku Actemra® częstość infekcji u pacjentów z pJIA, którzy otrzymywali leczenie lekiem Actemra® do podania podskórnego, była taka sama jak u pacjentów z pJIA, którzy otrzymywali leczenie lekiem Actemra® do podania dożylnego.
Reakcje w miejscu podania
Ogólnie u 28,8% (15/52) pacjentów z pJIA występowały reakcje w miejscu podania po podaniu podskórnym leku Actemra®. Te reakcje występowały u 44% pacjentów o masie ciała ≥ 30 kg w porównaniu do 14,8% pacjentów o masie ciała < 30 kg. Najczęstsze reakcje w miejscu podania to zaczerwienienie skóry, obrzęk, krwiak, ból i świąd. Wszystkie zgłoszone reakcje były łagodne – stopnia 1. Żadna z reakcji nie doprowadziła do przerwania lub odstawienia leczenia.
Imunogenność
W badaniu podania podskórnego u 5,8% (3 z 52) pacjentów obserwowano powstawanie przeciwciał neutralizujących przeciwko tocylizumabowi bez rozwoju ciężkich lub klinicznie istotnych reakcji nadwrażliwości. Jeden z tych 3 pacjentów później zakończył udział w badaniu. Nie zaobserwowano korelacji między powstawaniem przeciwciał a odpowiedzią kliniczną lub zjawiskami niepożądanymi.
Odchylenia parametrów laboratoryjnych od normy
Na podstawie standardowego monitorowania laboratoryjnego w ogólnej populacji zmniejszenie liczby neutrofili poniżej 1 × 109/l obserwowano u 15,4% pacjentów otrzymujących leczenie lekiem Actemra® przez podanie podskórne. Wzrost poziomu ALT lub AST ≥ 3 × GGN obserwowano odpowiednio u 9,6% i 3,8% pacjentów otrzymujących leczenie lekiem Actemra® przez podanie podskórne. U pacjentów otrzymujących leczenie lekiem Actemra® przez podanie podskórne nie obserwowano zmniejszenia liczby trombocytów ≤ 50 × 103/μl.
Parametry metabolizmu lipidów
W badaniu podania podskórnego leku Actemra® u 14,3% i 12,8% pacjentów w dowolnym czasie po rozpoczęciu badania obserwowano wzrost poziomu cholesterolu LDL do ≥ 130 mg/dl i poziomu cholesterolu ogólnego do ≥ 200 mg/dl odpowiednio.
Podanie podskórne
Leczenie GCA
Bezpieczeństwo podania podskórnego leku Actemra® badano w jednym badaniu fazy III (WA28119) u 251 pacjentów z GCA. Ogólna długość stosowania leku Actemra® wszystkim pacjentom wynosiła 138,5 pacjentów-rok w trakcie 12-miesięcznego podwójnie ślepego, placebo-kontrolowanego etapu badania. Ogólny profil bezpieczeństwa obserwowany u pacjentów z GCA, którym podawano lek Actemra®, odpowiadał znanemu profilowi bezpieczeństwa leku Actemra® (patrz „Podsumowanie danych dotyczących efektów ubocznych” powyżej).
Infekcje
Częstość infekcji/ciężkich infekcji była podobna w grupie tygodniowego podania leku Actemra® (200,2/9,7 przypadków na 100 pacjentów-rok), w grupie placebo z 26-tygodniowym zmniejszaniem dawki prednizolu (156,0/4,2 przypadki na 100 pacjentów-rok) i w grupie placebo z 52-tygodniowym zmniejszaniem dawki prednizolu (210,2/12,5 przypadków na 100 pacjentów-rok).
Reakcje w miejscu wstrzyknięcia
W grupie tygodniowego podania podskórnego leku Actemra® ogólnie 6% (6 z 100) pacjentów zgłosiło reakcję niepożądaną występującą w miejscu podania podskórnego. Nie zgłaszano ciężkich reakcji niepożądanych w miejscu podania ani takich, które wymagałyby przerwania leczenia.
Imunogenność
W grupie tygodniowego podania podskórnego leku Actemra® u jednego pacjenta (1,1%, 1 z 95) pojawiły się dodatnie przeciwciała neutralizujące przeciwko lekowi Actemra®, choć nie izotypu IgE. U tego pacjenta nie rozwinęła się reakcja nadwrażliwości ani reakcja w miejscu podania.
Zaburzenia hematologiczne
Neutrofile
Podczas planowego monitorowania laboratoryjnego w 12-miesięcznym kontrolowanym badaniu klinicznym leku Actemra® obserwowano zmniejszenie liczby neutrofili poniżej 1 × 109/l u 4% pacjentów w grupie tygodniowego podania podskórnego. Zjawisko to nie było obserwowane w żadnej z grup placebo przy zmniejszaniu dawki prednizolu.
Trombocyty
Podczas planowego monitorowania laboratoryjnego w 12-miesięcznym kontrolowanym badaniu klinicznym leku Actemra® jeden pacjent (1%, 1 z 100) w grupie tygodniowego podania podskórnego leku Actemra® miał pojedynczy przypadek tymczasowego zmniejszenia liczby trombocytów do < 100 × 103/μl bez powiązanych przypadków krwawienia. Zmniejszenie liczby trombocytów poniżej 100 × 103/μl nie było obserwowane w żadnej z grup placebo przy zmniejszaniu dawki prednizolu.
Podwyższenie poziomu transaminaz wątrobowych
Podczas planowego monitorowania laboratoryjnego w 12-miesięcznym kontrolowanym badaniu klinicznym leku Actemra® obserwowano podwyższenie poziomu ALT ≥ 3 × GGN u 3% pacjentów w grupie tygodniowego podania podskórnego leku Actemra® w porównaniu do 2% w grupie placebo przy 52-tygodniowym zmniejszaniu dawki prednizolu i 0% w grupie placebo przy 26-tygodniowym zmniejszaniu dawki prednizolu. Podwyższenie poziomu AST > 3 × GGN obserwowano u 1% pacjentów w grupie tygodniowego podania podskórnego leku Actemra® w porównaniu do 0% pacjentów w każdej z grup placebo przy zmniejszaniu dawki prednizolu.
Zmiany parametrów metabolizmu lipidów
Podczas planowego monitorowania laboratoryjnego w 12-miesięcznym kontrolowanym badaniu klinicznym leku Actemra® u 34% pacjentów obserwowano trwałe podwyższenie poziomu cholesterolu ogólnego > 6,2 mmol/l (240 mg/dl), u 15% obserwowano trwałe podwyższenie LDL do ≥ 4,1 mmol/l (160 mg/dl) w grupie tygodniowego podania podskórnego leku Actemra®.
Zgłaszanie efektów ubocznych po rejestracji leku ma duże znaczenie. Pozwala to na monitorowanie stosunku korzyści do ryzyka przy stosowaniu tego leku. Pracownicy medyczni i farmaceutyczni, a także pacjenci lub ich prawni przedstawiciele powinni zgłaszać wszystkie przypadki podejrzewanych efektów ubocznych i braku skuteczności leku poprzez zautomatyzowany system informacyjny nadzoru farmakologicznego pod adresem: https://aisf.dec.gov.ua»
Okres ważności.
36 miesięcy.
Warunki przechowywania.
Przechowywać w temperaturze od 2 do 8 °C w oryginalnym opakowaniu w celu ochrony przed światłem. Przechowywać w miejscu niedostępnym dla dzieci. Nie zamrażać.
Niezgodność.
W przypadku braku badań zgodności ten lek nie powinien być mieszany z innymi lekami.
Opakowanie.
Strzykawka wstępnie napełniona o pojemności 1 ml, cylinder wykonany z bezbarwnego szkła (klasa 1), nasmarowany olejem silikonowym, z przyczepioną adhezyjnie igłą ze stali nierdzewnej (26G 1/2), zamkniętą kapturkiem z poliizoprenu, oraz z tłokiem z butylorubberu, laminowanym polimerem fluorowym, wyposażonym w urządzenie bezpieczeństwa igły. Po 4 strzykawki wstępnie napełnione w pudełku kartonowym.
Kategoria wydania.
Na receptę.
Producent.
F. Hoffmann-La Roche Ltd
Miejsce zamieszkania producenta i adres miejsca prowadzenia działalności.
Wurmisweg, 4303 Kaiseraugst, Szwajcaria