GAMMAGARD
Włochy
Spis treści
- Ulotka: informacje dla użytkownika
- GAMMAGARD 50 mg/ml proszek i rozpuszczalnik do sporządzenia roztworu do wlewania
- 1. Co to jest GAMMAGARD i do czego służy
- 2. Co powinien wiedzieć przed podaniem GAMMAGARD
- 3. Jak ma być podawany lek GAMMAGARD
- 4. Możliwe działania niepożądane
- 5. Jak przechowywać GAMMAGARD
- 6. Skład opakowania i inne informacje
- 4. Trzymać fiolkę z rozpuszczalnikiem z założonym urządzeniem do transferu,
Ulotka: informacje dla użytkownika
GAMMAGARD 50 mg/ml proszek i rozpuszczalnik do sporządzenia roztworu do wlewania
immunoglobuliny ludzkie normalne
Przed zastosowaniem tego leku należy dokładnie przeczytać ulotkę,
gdyż zawiera ona ważne informacje dla Ciebie.
- Zachowaj tę ulotkę. Może się okazać konieczność ponownego jej przeczytania.
- W przypadku jakichkolwiek pytań skontaktuj się z lekarzem lub pielęgniarką.
- Jeśli wystąpią u Ciebie jakieś działania niepożądane, w tym te, których nie ma w niniejszej ulotce, skontaktuj się z lekarzem lub pielęgniarką. Zobacz punkt 4.
Zawartość tej ulotki:
- Co to jest GAMMAGARD i do czego służy
- Co należy wiedzieć przed podaniem GAMMAGARD
- Jak będzie Ci podawany GAMMAGARD
- Możliwe działania niepożądane
- Jak przechowywać GAMMAGARD
- Zawartość opakowania i inne informacje
1. Co to jest GAMMAGARD i do czego służy
GAMMAGARD zawiera ludzkie immunoglobuliny normalne do leczenia dożylnej.
Immunoglobuliny, zwane również przeciwciałami, to białka krążące we krwi i należące do układu odpornościowego, który umożliwia organizmowi obronę przed chorobami. Są one stosowane w leczeniu stanów, w których układ odpornościowy nie działa prawidłowo.
GAMMAGARD stosuje się u pacjentów w każdym wieku
Jako terapię uzupełniającą (terapię zastępczą) przeciwciał u pacjentów z niskim stężeniem przeciwciał krążących we krwi (hipogammaglobulinemia):
- Pierwotny zespół niedoboru odporności (PID), charakteryzujący się niedostateczną produkcją przeciwciał od urodzenia (patrz punkt „Ostrzeżenia i środki ostrożności”).
- Hipogammaglobulinemia i nawracające infekcje bakteryjne u pacjentów z przewlekłą białaczką limfocytyczną (nowotworem komórek krwi), którzy nie odpowiadają na leczenie antybiotykami.
- Hipogammaglobulinemia i nawracające infekcje bakteryjne u pacjentów z szpiczakiem plazmocytowym (nowotworem komórek szpiku kostnego), którzy nie odpowiadają na szczepienie przeciwko bakterii zwanej pneumokokiem (Streptococcus pneumoniae).
- Hipogammaglobulinemia wynikająca z przeszczepienia komórek macierzystych pochodzących od innego osobnika, które dadzą początek komórkom krwi (allogeniczny przeszczep komórek macierzystych krwiotwórczych).
- Wrodzony nabyte zespół niedoboru odporności (AIDS) z nawracającymi infekcjami bakteryjnymi.
Jako terapię regulującą działanie układu odpornościowego (immunomodulację) u pacjentów z:
- Pierwotną immunologiczną trombocytopenią (ITP), u pacjentów z wysokim ryzykiem krwawienia lub przed zabiegiem chirurgicznym w celu zwiększenia liczby płytek krwi – komórek krwi uczestniczących w procesie krzepnięcia (zatrzymanie krwawienia).
- Zespołem Guillaina-Barré, chorobą układu odpornościowego, która uszkadza nerwy i uniemożliwia ich prawidłowe działanie.
- Chorobą Kawasaki, chorobą dziecięcą, w której naczynia krwionośne (tętnice) się poszerzają.
Skontaktuj się z lekarzem, jeśli nie czujesz się lepiej lub jeśli czujesz się gorzej.
2. Co powinien wiedzieć przed podaniem GAMMAGARD
Nie będzie Ci podawany GAMMAGARD
- Jeśli jesteś uczulony na normalne ludzkie immunoglobuliny lub na którykolwiek z innych składników tego leku (wymienionych w punkcie 6).
- Jeśli posiadasz przeciwciała przeciwko immunoglobulinom A (IgA), białkom układu odpornościowego.
- Jeśli wcześniej miałeś reakcję alergiczną po podaniu GAMMAGARD.
Ostrzeżenia i środki ostrożności
Skonsultuj się z lekarzem lub pielęgniarką przed podaniem Ci GAMMAGARD.
W szczególności poinformuj lekarza, jeśli:
- wcześniej występowały u Ciebie ciężkie reakcje alergiczne
- miałeś zatorowość naczyń krwionośnych, taką jak zawał serca, udar mózgu, zakrzepica płucna lub zakrzepica żył
- cierpisz na cukrzycę
- cierpisz na nadciśnienie tętnicze
- cierpisz na trombofilię nabytą lub wrodzoną, czyli chorobę krzepnięcia krwi, która zwiększa ryzyko powstawania skrzeplin w naczyniach krwionośnych, które utrudniają lub uniemożliwiają normalny przepływ krwi
- masz ponad 65 lat
- jesteś otyły
- cierpiałeś wcześniej na choroby naczyń krwionośnych lub skrzepliny, które zwiększają ryzyko powstawania skrzeplin w naczyniach krwionośnych, utrudniających lub uniemożliwiających normalny przepływ krwi
- wcześniej przez dłuższy czas byłeś ograniczony w ruchu (po zabiegach chirurgicznych lub innych problemach zdrowotnych)
- wiesz, że cierpisz na choroby powodujące zwiększoną lepkość krwi
- cierpisz na chorobę nerek (niewydolność nerek)
- cierpisz na ciężkie zakażenie krwi (sepsę)
- cierpisz na paraproteinemię, chorobę charakteryzującą się obecnością w krwi białek podobnych do przeciwciał
- przyjmujesz leki, które mogą powodować uszkodzenia nerek
W przypadku rozwoju choroby nerek lekarz może zdecydować o przerwaniu terapii GAMMAGARD.
Badania laboratoryjne
Stosowanie GAMMAGARD może powodować fałszywie pozytywne wyniki niektórych badań krwi (np. Wirusowe zapalenie wątroby typu A, Wirusowe zapalenie wątroby typu B, odra, owsica, test antiglobulinowy bezpośredni DAT, bezpośredni test Coombsa).
Podawanie GAMMAGARD może prowadzić do fałszywie pozytywnych wyników testów diagnostyki infekcji grzybiczych opartych na wykrywaniu beta-D-glukanu; efekt ten może utrzymywać się przez kilka tygodni po wlewie produktu.
Przed poddaniem się badaniom krwi poinformuj lekarza lub personel laboratorium, że otrzymałeś GAMMAGARD.
Bezpieczeństwo wirusowe
GAMMAGARD to lek pochodzący z osocza ludzkiego (ciekłej części krwi).
Podczas wytwarzania leków z krwi lub osocza ludzkiego stosuje się szereg środków bezpieczeństwa, aby zapobiec przeniesieniu infekcji na pacjentów. Obejmują one staranne dobieranie dawców osocza i krwi, aby wykluczyć potencjalnych nosicieli infekcji, oraz analizę każdej darowizny i puli osocza w celu wykrycia obecności wirusów i infekcji.
Producenci tych leków stosują procedury przetwarzania krwi lub osocza, które inaktywują lub usuwają wirusy. Niemniej jednak, za każdym razem, gdy stosuje się leki przygotowane z krwi lub osocza ludzkiego, nie można całkowicie wykluczyć ryzyka przeniesienia infekcji. Dotyczy to również nieznanych lub nowo pojawiających się wirusów oraz innych rodzajów infekcji.
Środki podjęte przy produkcji GAMMAGARD są uznawane za skuteczne wobec wirusów z kapsydą lipidową, takich jak wirus HIV (ludzkiego wirusa niedoboru odporności), wirus WZW typu B (HBV) i wirus WZW typu C (HCV), a także wobec wirusów bez kapsydy lipidowej, takich jak wirus WZW typu A (HAV) i parwowirus B19.
Istnieje uspokajające doświadczenie kliniczne w zakresie braku przeniesienia WZW typu A lub parwowirusa B19 za pomocą immunoglobulin, a znaczna zawartość przeciwciał jest uważana za istotny czynnik bezpieczeństwa wirusowego.
Śledzenie produktu
W celu poprawy śledzenia produktów biologicznych, nazwa i numer serii leku podanego pacjentowi muszą być dokładnie zarejestrowane.
Inne leki i GAMMAGARD
Poinformuj lekarza, jeśli przyjmujesz, ostatnio przyjmowałeś lub możesz przyjmować inne leki.
W szczególności poinformuj lekarza, jeśli:
- planujesz szczepienie. GAMMAGARD bowiem może zmniejszyć skuteczność niektórych rodzajów szczepionek (szczepionek z wirusów osłabionych). W przypadku odry, świnki i ospy wietrznej należy odczekać do 3 miesięcy po podaniu GAMMAGARD przed szczepieniem. W przypadku odry należy odczekać do 1 roku.
Populacja pediatryczna
Nie przeprowadzono badań dotyczących interakcji GAMMAGARD z innymi lekami u dzieci.
Ciąża, karmienie piersią i płodność
Jeśli jesteś w ciąży, podejrzewasz ciążę lub planujesz zajść w ciążę, albo karmisz piersią, skonsultuj się z lekarzem przed podaniem tego leku.
Ciąża
GAMMAGARD zostanie Ci podany tylko w razie absolutnej konieczności podczas ciąży. Bezpieczeństwo stosowania GAMMAGARD w czasie ciąży nie zostało potwierdzone w kontrolowanych badaniach klinicznych, dlatego należy podawać go z ostrożnością kobietom w ciąży i karmiącym piersią.
Karmienie piersią
Immunoglobuliny przechodzą do mleka i mogą przyczynić się do ochrony noworodka.
Jednak GAMMAGARD zostanie Ci podany tylko w razie absolutnej konieczności podczas karmienia piersią.
Płodność
Doświadczenie kliniczne z immunoglobulinami sugeruje, że nie przewiduje się szkodliwego wpływu na płodność.
Kierowanie pojazdami i obsługa maszyn
Możliwość kierowania pojazdami lub obsługi maszyn może być zaburzona przez niektóre działania niepożądane związane z GAMMAGARD. Jeśli wystąpią u Ciebie działania niepożądane podczas leczenia GAMMAGARD, odczekaj do ich ustąpienia, zanim zaczniesz kierować pojazdami lub obsługiwać maszyny.
GAMMAGARD zawiera sód i glukozę
Ten lek zawiera około 668 mg sodu (główny składnik soli kuchennej) na fiolkę (10 g). Odpowiada to 34% maksymalnej zalecanej dziennej dawki sodu w diecie dla dorosłego.
Ilość sodu w maksymalnej dziennej dawce znacząco zwiększa zalecaną dzienne spożycie sodu u pacjentów stosujących dietę o ograniczonej zawartości sodu. U tych pacjentów ilość sodu pochodzącego z leku powinna być mierzona i uwzględniana przy określaniu dziennej dawki sodu z pożywienia. Roztwór GAMMAGARD o stężeniu 5% zawiera około 3340 mg/l sodu. Pacjent o masie 70 kg, któremu podano dawkę GAMMAGARD 1 g/kg (1,4 litra), otrzymuje 4676 mg sodu.
Roztwór GAMMAGARD o stężeniu 5% zawiera 20 mg glukozy na ml (400 mg/g IgG) jako substancję pomocniczą. Pacjent o masie 70 kg, któremu podano dawkę GAMMAGARD 1 g/kg, otrzymuje 28 g glukozy (112 kalorii). Należy to uwzględnić u pacjentów z cukrzycą utajoną (u których może wystąpić przejściowa glikozuria), u chorych na cukrzycę lub u pacjentów stosujących dietę o obniżonej zawartości węglowodanów.
3. Jak ma być podawany lek GAMMAGARD
Ten lek będzie podawany do żyły zawsze zgodnie z instrukcjami lekarza. W razie wątpliwości skonsultuj się z lekarzem lub pielęgniarką.
Dawkę zalecaną ustali lekarz i będzie ona zależeć od masy ciała oraz stanu zdrowia pacjenta.
Stosowanie u dzieci i młodzieży
Dzieciom i młodzieży (wiek: 0–18 lat) stosuje się takie same wskazania, dawkowanie oraz częstotliwość wlewu, jakie są stosowane u dorosłych.
4. Możliwe działania niepożądane
Tak jak wszystkie leki, ten lek może powodować działania niepożądane, choć nie u wszystkich osób one występują.
Poniżej wymieniono możliwe działania niepożądane według następującej częstości występowania:
częste (mogą dotyczyć do 1 osoby na 10)
- ból głowy
- uderzenia gorąca
- wymioty, nudności
- zmęczenie, dreszcze, gorączka
nieczęste (mogą dotyczyć do 1 osoby na 100)
- grypa
- zmniejszenie apetytu
- lęk, pobudzenie
- senność
- zamazane widzenie
- uczucie kołatania serca (palpitacje)
- częste zmiany ciśnienia krwi
- trudności w oddychaniu (dyspnę), krwawienie z nosa (epistaksja)
- biegunka, zapalenie wnętrza jamy ustnej (stomatyt), ból w górnej części brzucha, dolegliwości żołądkowe
- swędzące, zaczerwienione skóra (pokrzywka), swędzenie, zimny pot, nasilone potliwość
- ból w dolnej części pleców, skurcze mięśni, ból rąk i nóg
- ból w klatce piersiowej, ogólne niedobrze, ogólny ból, uczucie ucisku w klatce piersiowej, zaburzenia zmysłów, uczucie zimna lub gorąca, choroba przypominająca grypę, zaczerwienienie skóry w miejscu wlewu, wyciek roztworu poza żyłę, w której jest wlewany, ból w miejscu wlewu
- podwyższone ciśnienie krwi
nieznana (częstość nie może być określona na podstawie dostępnych danych)
- zapalenienie opon mózgowych, czyli błon otaczających mózg (zapalenie opon mózgowych nieswoiste)
- niszczenie czerwonych krwinek (hemoliza), zmniejszenie stężenia hemoglobiny, białka przenoszącego tlen we krwi (anemia), zmniejszenie liczby płytek krwi (trombocytopenia), powiększenie węzłów chłonnych (chłoniakowatość)
- reakcje alergiczne, również ciężkie (szok anafilaktyczny, reakcja anafilaktyczna lub anafilaksojadna, nadwrażliwość)
- niepokój
- niedokrwienie mózgu, czasem krótkotrwałe (udar mózgu, przejściowy atak niedokrwienny), niekontrolowane ruchy ciała (drżenie), bardzo silny ból głowy (migrena), zawroty głowy, mrowienie (parestezja), zaburzenia wrażliwości, wskutek których bodźce wywołują inne niż normalne reakcje (dyszestezja), omdlenie (zawał), drżenie
- zator naczynia krwionośnego oka spowodowany skrzepem (tromboza żyły siatkówki), zaburzenia wzroku, ból oczu, nadmierne wrażliwość na światło (fotofobia)
- atak serca (zawał mięśnia sercowego), skóra staje się niebieska z powodu niedotlenienia (cyjanóza), szybkie lub powolne bicie serca (tachykardia, bradykardia)
- zator naczynia krwionośnego spowodowany skrzepem (tromboza tętnicza, tromboza żyły głównej dolnej, głęboka zatorowość żył), zapalenie i zatorowość żył (tromboflebita), niskie lub wysokie ciśnienie krwi (hipotensja, nadciśnienie), bladość
- zator naczynia krwionośnego płuc (zatorowość płucna), płyn wokół płuc (obrzęk płuc), niedotlenienie (hipoksja), trudności w oddychaniu (bronchospazm), świsty podczas oddychania, zwiększenie częstości oddechów (hiperwentylacja), zamknięcie krtani, czyli narządu, w którym powstaje głos, co utrudnia oddychanie (laryngospazm), kaszel
- ból brzucha, trudności w trawieniu (dyspepsja)
- zapalenie wątroby (zapalenie wątroby nieswoiste)
- obrzęk skóry i twarzy (angioobrzęk), zapalenie skóry (dermatyt), zaczerwienienie skóry (rumień), wysypka skórna
- ból kości i mięśni (ból stawów, ból mięśni)
- choroba nerek (niewydolność nerek)
- reakcja w miejscu wlewu, osłabienie (astenia), obrzęk (edema)
- odchylenie wyniku badania krwi (dodatni bezpośredni test Coombsa)
Dodatkowe działania niepożądane u dzieci i nastolatków
U dzieci nie oczekuje się innych działań niepożądanych niż te, które występują u dorosłych.
Zgłaszanie działań niepożądanych
Jeśli wystąpi u Ciebie jakiekolwiek działanie niepożądane, w tym również takie, które nie są wymienione w niniejszym ulotce, skontaktuj się z lekarzem lub pielęgniarką. Możesz również zgłaszać działania niepożądane bezpośrednio za pośrednictwem systemu https://www.aifa.gov.it/content/segnalazioni-reazioni-avverse.
Zgłaszanie działań niepożądanych może pomóc w dostarczeniu dodatkowych informacji na temat bezpieczeństwa tego leku.
5. Jak przechowywać GAMMAGARD
Przechowywać w temperaturze poniżej 25°C.
Przechowywać w opakowaniu zewnętrznym w celu ochrony leku przed światłem.
Nie zamrażać, ponieważ naczynie z rozpuszczalnikiem może pęknąć.
Przechowywać ten lek w miejscu niedostępnym dla dzieci i poza ich zasięgiem wzroku.
Nie należy stosować tego leku po upływie daty ważności podanej na opakowaniu po napisie
WAŻNY DO. Data ważności odnosi się do ostatniego dnia danego miesiąca.
Nie należy stosować tego leku, jeśli przed odtworzeniem stwierdzi się, że proszek nie jest białym lub lekko żółtawym proszkiem/produktem, bez widocznych zanieczyszczeń.
Po odtworzeniu nie należy stosować tego leku, jeśli roztwór nie jest klarowny lub lekko opalizujący, bezbarwny lub lekko żółty.
Nie wyrzucać leków do kanalizacji ani do śmieci domowych. Zapytaj farmaceuty o sposób utylizacji leków, których już nie używasz. Pomogą w ten sposób chronić środowisko.
6. Skład opakowania i inne informacje
Co zawiera GAMMAGARD
- Substancją czynną w GAMMAGARD jest ludzka immunoglobulina normalna do stosowania dożylnego. Jeden ml zawiera 50 mg (roztwór odtworzony 5%) lub 100 mg (roztwór odtworzony 10%) białka, zawierającego co najmniej 90% ludzkiej immunoglobuliny typu G.
- Pozostałe składniki to: proszek: ludzka albumina (roztwór odtworzony 5%: 3 mg/ml; roztwór odtworzony 10%: 6 mg/ml), glicyna, sód chlorkowy, glukoza jednowodna (patrz punkt 2. GAMMAGARD zawiera sód i glukozę), polietylenoglikol. Roztwórnik: woda do wstrzykiwań.
Opis wyglądu GAMMAGARD i zawartości opakowania
GAMMAGARD to biały lub lekko żółtawy proszek, praktycznie pozbawiony widocznych niepożądanych cząstek, wraz z roztwornikiem do roztworu do infuzji (woda do wstrzykiwań).
Odtworzony roztwór jest zazwyczaj klarowny lub lekko mleczny, bezbarwny lub jasnożółty.
Dostępny jest w opakowaniach:
- 5 g/100 ml proszek i roztwornik do roztworu do infuzji – 1 fiolka z proszkiem + 1 fiolka z roztwornikiem o pojemności 96 ml + sterylny zestaw do przeniesienia + zestaw do infuzji
- 10 g/200 ml proszek i roztwornik do roztworu do infuzji – 1 fiolka z proszkiem + 1 fiolka z roztwornikiem o pojemności 192 ml + sterylny zestaw do przeniesienia + zestaw do infuzji
Może się zdarzyć, że nie wszystkie opakowania są wprowadzone do obrotu.
Właściciel pozwolenia na dopuszczenie do obrotu i producent
Właściciel pozwolenia na dopuszczenie do obrotu
Baxalta Innovations GmbH
Industriestrasse 67, A-1221 Wiedeń
Austria
Producent
Baxalta Belgium Manufacturing SA
Boulevard René Branquart 80
B-7860 Lessines
Belgia
Aby uzyskać dodatkowe informacje na temat tego leku, należy skontaktować się z lokalnym przedstawicielem właściciela pozwolenia na dopuszczenie do obrotu:
Takeda Italia S.p.A.
Tel. +39 06 502601
Następujące informacje są przeznaczone wyłącznie dla lekarzy lub personelu medycznego:
DAWKA, SPOSÓB I CZAS PODANIA
Terapię zastępczą należy rozpoczynać i monitorować pod nadzorem lekarza doświadczonych w leczeniu immunodeficytowości.
Dawka i schemat leczenia zależą od wskazań terapeutycznych.
W terapii zastępczej dawkę należy dostosować do indywidualnych potrzeb pacjenta, które są wynikiem farmakokinetyki i odpowiedzi klinicznej.
Poniższe schematy dawkowania podano jako wskazówki.
Terapia zastępcza w przypadku pierwotnych zespołów niedoboru odporności:
Schemat dawkowania powinien zapewnić minimalną stężenie IgG (mierzone przed kolejną infuzją) co najmniej 5–6 g/l. Aby osiągnąć stan równowagi, potrzeba od trzech do sześciu miesięcy od rozpoczęcia terapii. Zalecana dawka początkowa to 0,4–0,8 g/kg masy ciała podana jednorazowo, a następnie co najmniej 0,2 g/kg MC co trzy–cztery tygodnie.
Dawka potrzebna do osiągnięcia minimalnego stężenia 5–6 g/l wynosi około 0,2–0,8 g/kg MC/miesiąc. Odstęp między dawkami po osiągnięciu stanu równowagi waha się od 3 do 4 tygodni.
Należy monitorować minimalne stężenia oraz częstość występowania infekcji. Aby zmniejszyć częstość infekcji, może być konieczne zwiększenie dawki i osiągnięcie wyższych minimalnych stężeń.
Hipogammaglobulinemia i nawracające infekcje bakteryjne u chorych z przewlekłą białaczką limfocytarną, którzy nie odpowiadają na profilaktyczne leczenie antybiotykami; hipogammaglobulinemia i nawracające infekcje bakteryjne u chorych z postacią plateau szpiczaka mnogiego, którzy nie odpowiadają na szczepienie przeciwko pneumokokom; wrodzone AIDS z nawracającymi infekcjami bakteryjnymi.
Zalecana dawka to 0,2–0,4 g/kg co trzy–cztery tygodnie.
Hipogammaglobulinemia u pacjentów po allogenicznej transplantacji hematopoetycznych komórek macierzystych
Zalecana dawka to 0,2–0,4 g/kg co trzy–cztery tygodnie. Minimalne stężenia należy utrzymywać powyżej 5 g/l.
W leczeniu infekcji i profilaktyce choroby przeszczegodawcy przeciwko gospodarzowi dawkę należy dostosować indywidualnie do pacjenta.
Pierwotna immunologiczna trombocytopenia
Istnieją dwa alternatywne schematy leczenia:
- 0,8–1 g/kg podane w dniu 1; dawkę tę można powtórzyć raz w ciągu 3 dni;
- 0,4 g/kg codziennie przez dwa–pięć dni. Leczenie można powtórzyć w przypadku nawrotu.
Zespół Guillaina-Barré
0,4 g/kg/dzień przez 5 dni.
Zespół Kawasaki
Podaj 1,6–2,0 g/kg w dawkach podzielonych w ciągu dwóch–pięciu dni lub 2,0 g/kg w jednej dawce. Konieczne jest jednoczesne leczenie kwasem acetylosalicylowym.
Zalecane dawki podsumowano w poniższej tabeli:
| Wskazanie | Dawka | Częstotliwość infuzji |
| Leczenie zastępcze w zespole pierwotnej niedoborności odporności Leczenie zastępcze w zespole wtórnej niedoborności odporności Wrodzone AIDS |
| co 3–4 tygodnie w celu osiągnięcia minimalnego stężenia IgG co najmniej 5–6 g/l co 3–4 tygodnie w celu osiągnięcia minimalnego stężenia IgG co najmniej 5–6 g/l co 3–4 tygodnie |
| Hipogammaglobulinemia (< 4 g/l) u pacjentów po allogenicznym przeszczepie komórek macierzystych układu krwiotwórczego – leczenie zakażeń i profilaktyka choroby „przeszczep versus gospodarz” – trwała niewydolność produkcji przeciwciał | 0,2–0,4 g/kg | co 3–4 tygodnie w celu osiągnięcia minimalnego stężenia IgG powyżej 5 g/l co tydzień od dnia -7 przez okres do 3 miesięcy po przeszczepie co miesiąc aż do przywrócenia normalnych poziomów przeciwciał |
| Immunomodulacja: – Immune pierwotna trombocytopenia (Idiopatyczna plamica małopłytkowa) – Zespół Guillaina-Barrégo – Zespół Kawasaki | 0,8–1 g/kg lub 0,4 g/kg/dzień 0,4 g/kg/dzień 1,6–2 g/kg lub 2 g/kg | w dniu 1, z możliwością powtórzenia jednorazowo w ciągu 3 dni przez 2–5 dni przez 5 dni w dawkach podzielonych w ciągu 2–5 dni w połączeniu z kwasem acetylosalicylowym w jednorazowej dawce w połączeniu z kwasem acetylosalicylowym |
Populacja pediatryczna
Dawkowanie u dzieci i młodzieży (0–18 lat) nie różni się od dawkowania u dorosłych, ponieważ dawkowanie dla każdej wskazówki podawane jest według masy ciała i dostosowywane jest do wyników klinicznych powyższych stanów.
Sposób podania
Do użytku dożylnego.
Zaleca się podawanie, o ile to możliwe, roztworu GAMMAGARD o stężeniu 10% do żył przedramienia.
Może to zmniejszyć prawdopodobieństwo wystąpienia dyskomfortu w miejscu infuzji.
Przed wlewnym podaniem roztwór należy ogrzać do temperatury ciała lub temperatury pokojowej.
Ilość podawanego produktu wyrażana jest w ml na kg masy ciała/h.
GAMMAGARD 5% (50 mg/ml) należy podawać dożylnie z początkową szybkością 0,5 ml/kg/h. Jeśli jest dobrze tolerowane, szybkość podania można stopniowo zwiększać do maksymalnie 4 ml/kg/h.
Ogólnie zaleca się, aby pacjentów rozpoczynających leczenie GAMMAGARD lub przechodzących z innego produktu zawierającego IVIg początkowo wlewać z najniższą szybkością infuzji, a następnie stopniowo ją zwiększać do maksymalnej, jeśli dobrze tolerowali kilka infuzji z szybkością pośrednią.
Pacjentów, którzy dobrze tolerują GAMMAGARD w roztworze 5% przy szybkości 4 ml/kg/h, można wlewać roztworem odtworzonym do stężenia 10%, rozpoczynając od szybkości 0,5 ml/kg/h. W przypadku braku działań niepożądanych szybkość można stopniowo zwiększać do maksymalnie 8 ml/kg/h.
DZIAŁANIA NIEPOŻĄDANE
Niektóre natychmiastowe reakcje niepożądane po lekach, takie jak bóle głowy i zawroty głowy, ból brzucha, katar, nudności, skurcze oskrzeli, dreszcze, bóle mięśni, gorączka, mogą być związane ze szybkością wlewu.
Należy ściśle przestrzegać zalecanej szybkości wlewu podanej w punkcie „Dawka, sposób i czas podania”. Pacjentów należy dokładnie monitorować i obserwować pod kątem wystąpienia jakichkolwiek objawów w czasie trwania infuzji.
Niektóre działania niepożądane mogą występować częściej:
- przy wysokiej szybkości infuzji,
- u pacjentów przyjmujących po raz pierwszy ludzką immunoglobulinę normalną lub, rzadziej, gdy zmienia się produkt zawierający ludzką immunoglobulinę normalną lub gdy od poprzedniej infuzji upłynął długi okres czasu.
Często możliwe jest uniknięcie potencjalnych powikłań poprzez:
- upewnienie się, że pacjenci nie są wrażliwi na ludzką immunoglobulinę normalną, poprzez początkowe podanie produktu powoli z szybkością początkową 0,5 ml/kg/h,
- dokładne monitorowanie pacjentów pod kątem pojawienia się jakichkolwiek objawów przez cały czas trwania infuzji. W szczególności pacjentów leczonych po raz pierwszy ludzką immunoglobuliną normalną, pacjentów przechodzących z innego produktu IVIg lub u których od poprzedniej infuzji upłynął długi okres czasu, należy monitorować podczas pierwszej infuzji i przez pierwszą godzinę po jej zakończeniu w celu wykrycia potencjalnych działań niepożądanych. U wszystkich innych pacjentów należy prowadzić obserwację przez co najmniej 20 minut po podaniu.
W przypadku wystąpienia działań niepożądanych należy zmniejszyć szybkość podania lub przerwać infuzję. Zmniejszenie szybkości wlewu lub jego przerwanie zazwyczaj prowadzi do szybkiego ustąpienia objawów.
Infuzję można następnie wznowić z szybkością, która nie powoduje ponownego pojawienia się objawów.
Zalecane leczenie zależy od rodzaju i nasilenia działania niepożądanego. W przypadku wstrząsu należy rozpocząć standardowe leczenie wstrząsu.
U wszystkich pacjentów podawanie IVIg wymaga:
- odpowiedniego nawodnienia przed rozpoczęciem infuzji IVIg,
- monitorowania diurezy,
- monitorowania stężenia kreatyniny w surowicy,
- unikania jednoczesnego stosowania moczegonnych pętlowych.
Nadwrażliwość
Prawdziwe reakcje nadwrażliwości są rzadkie. Mogą wystąpić w rzadkich przypadkach niedoboru IgA z obecnością przeciwciał anty-IgA.
GAMMAGARD jest przeciwwskazany u pacjentów z izolowanym niedoborem IgA, w którym niedobór IgA jest jedynym składnikiem immunodeficytowości.
Rzadko ludzka immunoglobulina normalna może powodować gwałtowny spadek ciśnienia tętniczego z reakcją anafilaktyczną, nawet u pacjentów, którzy wcześniej dobrze tolerowali leczenie ludzką immunoglobuliną normalną.
Pacjenci z przeciwciałami anty-IgA lub z niedoborem IgA stanowiącym składnik pierwotnej immunodeficytowości, w której wskazane jest leczenie IVIg, mogą mieć zwiększone ryzyko wystąpienia reakcji anafilaktycznych.
Zarejestrowano przypadki anafilaksji przy użyciu GAMMAGARD, mimo że ten lek zawiera niskie stężenia IgA.
Pacjenci, którzy mieli ciężkie reakcje nadwrażliwości, powinni otrzymywać immunoglobuliny dożylne jedynie z największą ostrożnością i w miejscu, gdzie dostępna jest pomoc wspierająca leczenie reakcji zagrożonych życiem.
Zakrzepica i zatorowość
Istnieją dowody kliniczne wskazujące na związek między podawaniem IVIg a zdarzeniami trombotycznymi i zakrzepowo-zatorowymi, takimi jak zawał mięśnia sercowego, udar mózgu (w tym udar niedokrwienny), zatorowość płucna i głębokie zakrzepienie żył, które przypuszcza się, że są związane ze względnym wzrostem lepkości krwi w wyniku dużego przyjęcia immunoglobuliny u pacjentów z grupy ryzyka.
Należy zachować szczególną ostrożność przy przepisywaniu i wlewaniu IVIg u pacjentów otyłych lub z istniejącymi czynnikami ryzyka zdarzeń zakrzepowo-zatorowych (np. zaawansowany wiek, nadciśnienie tętnicze, cukrzyca, historia choroby naczyniowej lub epizodów trombotycznych, pacjenci z trombofilią nabytą lub wrodzoną, pacjenci długotrwałe unieruchomieni, pacjenci z ciężkim niedowodnieniem lub pacjenci z zaburzeniami powodującymi wzrost lepkości krwi).
Zapewnić odpowiednie nawodnienie pacjentów przed podaniem. Monitorować objawy i oznaki zakrzepicy oraz ocenić lepkość krwi u pacjentów z ryzykiem hipewiskozności.
U pacjentów z ryzykiem działań niepożądanych typu zakrzepowo-zatorowego GAMMAGARD należy podawać z najniższą możliwą szybkością i dawką.
Ostra niewydolność nerek
Zarejestrowano ciężkie przypadki ostrej niewydolności nerek (np. ostra niewydolność nerek, ostra martwica kanalików, choroba kanalików nerkowych bliższych, osmotyczna nefroza) u pacjentów leczonych IVIg, szczególnie produktami zawierającymi sacharozę (GAMMAGARD nie zawiera sacharozy).
W większości przypadków zidentyfikowano czynniki ryzyka, takie jak istniejąca uprzednio niewydolność nerek, cukrzyca, hipowolemia, nadwaga, jednoczesne stosowanie leków nefrotoksycznych, wiek powyżej 65 lat, sepsa lub paraproteinemia.
W przypadku zaburzeń czynności nerek należy rozważyć przerwanie terapii IVIg.
Chociaż przypadki zaburzeń czynności nerek i ostrej niewydolności nerek były związane z użyciem wielu produktów IVIg dopuszczonych do obrotu zawierających różne substancje pomocnicze, takie jak sacharoza, glukoza i maltoza, produkty zawierające sacharozę jako stabilizator stanowią bardzo wysoki procent ogólnej liczby przypadków. U pacjentów z grupy ryzyka należy rozważyć stosowanie produktów IVIg niezawierających sacharozy. GAMMAGARD nie zawiera sacharozy ani maltozy.
U pacjentów z ryzykiem ostrej niewydolności nerek GAMMAGARD należy podawać z najniższą możliwą szybkością i dawką.
Beziennicze zapalenie opon mózgowo-rdzeniowych (AMS)
Zarejestrowano wystąpienie bezienniczych zapaleń opon mózgowo-rdzeniowych (AMS) w związku z leczeniem IVIg (w tym GAMMAGARD). Przerwanie leczenia IVIg może prowadzić do ustąpienia AMS w ciągu kilku dni bez następstw. Zespół zazwyczaj pojawia się po upływie od kilku godzin do 2 dni od leczenia IVIg.
Badania płynu mózgowo-rdzeniowego często wykazują pozytywną plejocytozę do kilku tysięcy komórek/mm³, głównie granulocytów, oraz podwyższone stężenie białka do kilkuset mg/dl.
AMS może występować częściej przy jednoczesnym stosowaniu wysokich dawek IVIg (2 g/kg).
Dane z doniesień po wprowadzeniu na rynek nie wykazały jednoznacznie związku między dawką IVIg a wystąpieniem AMS, natomiast zaobserwowano wyższe występowanie AMS u pacjentek płci żeńskiej.
Anemia hemolityczna
GAMMAGARD zawiera przeciwciała przeciwko antygenom grupy krwi, które mogą działać jako hemolizyny i wywoływać in vivo przyłączanie immunoglobulin do czerwonych krwinek, powodując pozytywny bezpośredni test antiglobulinowy (test Coombsa) i rzadko hemolizę.
Anemia hemolityczna może się rozwinąć po terapii GAMMAGARD w wyniku zwiększonego uwięzienia czerwonych krwinek (GR); zarejestrowano ostry hemoliz, zgodny z hemolizą wewnątrznaczyniową.
Pacjentów otrzymujących IVIg należy poddawać monitorowaniu pod kątem objawów klinicznych i objawów hemolizy.
SZCZEGÓLNE OSTRZEŻENIA
Hiperproteinemia
U pacjentów otrzymujących leczenie IVIg może wystąpić hiperproteinemia i wzrost lepkości surowicy.
Zapewnić odpowiednie nawodnienie pacjentów przed podaniem. Monitorować objawy i oznaki zakrzepicy oraz ocenić lepkość krwi u pacjentów z ryzykiem hipewiskozności.
Ostra uszkodzenie płuc związane z przetaczaniem (TRALI)
Zarejestrowano przypadki niekardiogennego obrzęku płuc (Transfusion Related Acute Lung Injury, TRALI) u pacjentów, którym podano IVIg.
Śledzenie
W celu poprawy śledzenia produktów biologicznych nazwa i numer serii leku podanego muszą być wyraźnie zarejestrowane.
PRZEDAWKOWANIE
Przedawkowanie może spowodować przeciążenie objętościowe i hipewiskozność, szczególnie u pacjentów z grupy ryzyka, w tym u pacjentów starszych lub z niewydolnością serca lub nerek.
SKŁAD
Immunoglobuliny ludzkie normalne (IVIg).
GAMMAGARD może być odtworzony wodą do wstrzykiwań do roztworu o stężeniu 5% (50 mg/ml) lub 10% (100 mg/ml) białek zawierającego co najmniej 90% IgG. Poniższa tabela przedstawia objętości rozpuszczalnika do użycia w celu uzyskania obu stężeń.
Fiolka z proszkiem GAMMAGARD zawiera ilość całkowitej białek osocza, z których co najmniej 90% stanowi ludzka immunoglobulina normalna (IgG), odpowiednio 5 g i 10 g w zależności od opakowania.
| Opakowanie | Substancja czynna: całkowite białka osocza, z których co najmniej 90% stanowią normalne ludzkie immunoglobuliny | Stężenie całkowitych białek osocza, z których co najmniej 90% stanowią normalne ludzkie immunoglobuliny, po rekonstytucji za pomocą rozpuszczalnika dołączanego do opakowania | Objętość rozpuszczalnika do użycia (woda do wstrzykiwania) |
| Fiolka z 5 g proszku | 5 g | 50 mg/ml 100 mg/ml | 96 ml 48 ml |
| Fiolka z 10 g proszku | 10 g | 50 mg/ml 100 mg/ml | 192 ml 96 ml |
Rozkład podklas IgG: IgG1 > 56,9%
IgG2 > 16,0%
IgG3 > 3,3%
IgG4 > 0,3%
Maksymalna zawartość IgA: ≤ 3 mikrogramy/ml w roztworze 5%.
Produkt uzyskany z osocza ludzkich dawców.
Substancje pomocnicze to:
Proszek:
Albumina ludzka (roztwór odtworzony 5%: 3 mg/ml; roztwór odtworzony 10%: 6 mg/ml),
glicyna, chlorek sodu, glukoza monohydrat, polietylenoglikol.
Roztwórnik:
Woda do wstrzykiwań
INSTRUKCJE DOTYCZĄCE PRZYGOTOWANIA ROZTWORU
Produkt należy przed użyciem довести do temperatury pokojowej lub temperatury ciała.
Za każdym razem, gdy pozwala na to opakowanie, leki do stosowania parenteralnego należy sprawdzić wizualnie przed podaniem pod kątem obecności cząstek lub nietypowego zabarwienia.
Nie należy stosować roztworów o nietypowym zabarwieniu i/lub zawierających osad.
GAMMAGARD należy podawać wyłącznie drogą dożylną po odtworzeniu odpowiednią objętością wody do wstrzykiwań (rozcieńczalnik).
Po rozpuszczeniu za pomocą rozcieńczalnika dołączonego do opakowania, odtworzony roztwór 5% może być przechowywany przez 2 godziny w temperaturze poniżej 25 °C, jeśli odtworzenie przeprowadzono w warunkach bezpyłowych.
Nie chłodzić odtworzonego roztworu.
Z mikrobiologicznego punktu widzenia produkt należy stosować natychmiast po odtworzeniu. Jeśli nie jest stosowany natychmiast, warunki i czas przechowywania przed użyciem są odpowiedzialnością użytkownika.
Pozostałości nieużywanego roztworu należy usunąć ze względu na ryzyko zakażenia bakteryjnego. Odtworzony produkt należy sprawdzić wizualnie przed podaniem pod kątem obecności zawiesiny cząstek lub nietypowego zabarwienia. Nie należy stosować mętnych roztworów lub roztworów zawierających osad.
Odtworzenie – należy stosować technikę bezpyłową
Opakowania 5 i 10 g
Doprowadzić do temperatury pokojowej fiolki GAMMAGARD (lizofilizat) oraz wody do wstrzykiwań (rozcieńczalnik). Należy zachować tę temperaturę aż do całkowitego rozpuszczenia się produktu.
A) Odtworzenie produktu jako roztworu 5%:
- Usunąć korki z fiolek i zdezynfekować je roztworem bakteriobójczym.
- Usunąć korek z jednego z końców urządzenia do transferu. Nie dotykać końcówki.
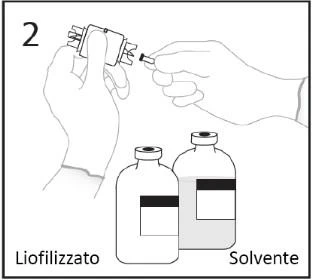
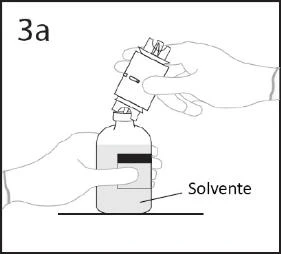
3a. Umieścić fiolkę z rozcieńczalnikiem na płaskiej powierzchni. Wprowadzić odsłoniętą końcówkę urządzenia do transferu w środek korka fiolki z rozcieńczalnikiem.
OSTRZEŻENIE: nieprawidłowe włożenie końcówki w środek korka może prowadzić do
wypchnięcia korka i utraty próżni.
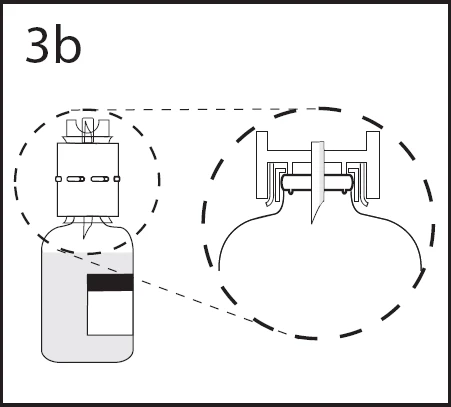
3b. Upewnić się, że urządzenie całkowicie wejdzie w szyjkę fiolki, mocno wciskając urządzenie do transferu.
Utrzymując urządzenie do transferu, usunąć osłonę z drugiej końcówki. Nie dotykać końcówki.
4. Trzymać fiolkę z rozpuszczalnikiem z założonym urządzeniem do transferu,
uważnie nachylona w stosunku do fiolki z liofilizatem, aby zapobiec wyciekowi
rozpuszczalnika. Uwaga: Nie trzymać fiolki z rozpuszczalnikiem do góry dnem, ponieważ może dojść do
wycieku rozpuszczalnika.
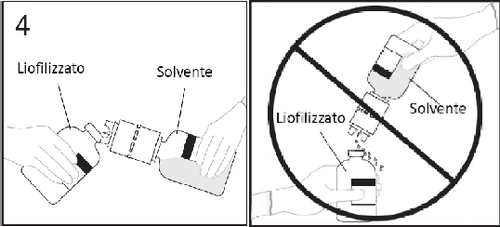
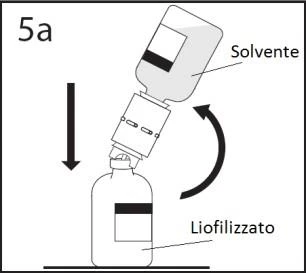
5a. Wprowadzić wolny koniec urządzenia do transferu w środek korka fiolki z liofilizatem i
szybko obrócić fiolkę z rozpuszczalnikiem do góry dnem, aby zapobiec wyciekowi rozpuszczalnika.
OSTRZEŻENIE: Nieprawidłowe umieszczenie końcówki w środku korka może prowadzić do
przesunięcia korka i utraty próżni.
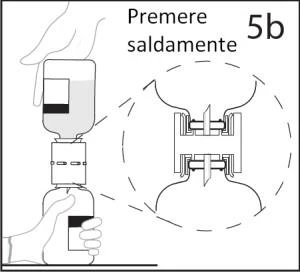
5b. Upewnić się, że urządzenie całkowicie wejdzie do szyjki fiolki, mocno dociskając fiolkę z rozpuszczalnikiem.
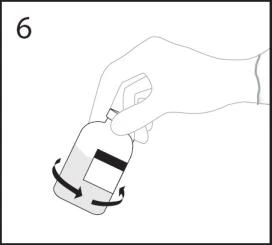
- Po zakończeniu transferu rozpuszczalnika usunąć urządzenie do transferu oraz pustą fiolkę z rozpuszczalnikiem. Natychmiast delikatnie wymieszać zawartość fiolki z liofilizatem, aby uzyskać jednolity roztwór.
OSTRZEŻENIE: Nie wstrząsać. Unikać powstawania piany.
Po jednym użyciu usunąć urządzenie do transferu.
B) Rekonstytucja produktu jako roztworu 10%:
- Usunąć korki z fiolki i zdezynfekować je roztworem germicydnym.
- Aby przygotować roztwór 10%, należy usunąć połowę objętości rozpuszczalnika. W tabeli 2 podano ilość rozpuszczalnika, którą należy usunąć z fiolki przed założeniem urządzenia do transferu w celu przygotowania stężenia 10%. Za pomocą techniki bezpyłowej, przy użyciu sterylnej strzykawki z igłą do wstrzykiwań, pobrać niepotrzebną ilość rozpuszczalnika. Usunąć napełnioną strzykawkę i igłę.
- Użyć pozostałego rozpuszczalnika znajdującego się w fiolce z rozpuszczalnikiem, postępując zgodnie z instrukcjami od kroku 2 do kroku 6, jak opisano wcześniej w punkcie A.
TABELA 2
Ilość rozpuszczalnika do usunięcia
Stężenie fiolka 5 g fiolka 10 g
5% Nie usuwać rozpuszczalnika przy rekonstytucji jako roztworu 5%
10% 48 mL 96 mL
Podawanie – stosować technikę bezpyłową
Infuzja (opakowania 5 i 10 g)
Podawać odtworzony roztwór za pomocą sterylnego zestawu do infuzji 15 dołączanego do każdego opakowania.
Usunąć wszelkie niewykorzystane pozostałości roztworu ze względu na ryzyko zakażenia bakteryjnego.
Nieużywany lek oraz odpady pochodzące z tego leku należy utylizować zgodnie z obowiązującymi
lokalnymi przepisami.