Dapagliflozyna Tecnigen
WłochySpis treści
- 1. NAZWA PRODUKTU LECZNICZEGO
- 2. SKŁAD JAKOŚCIOWY I ILOŚCIOWY
- 3. POSTAĆ LECZNICZA
- 4. INFORMACJE KLINICZNE
- 5. WŁAŚCIWOŚCI FARMACOLOGICZNE
- 6. INFORMACJE FARMACEUTYCZNE
- 7. WŁAŚCICIEL AUTORYZACJI DO WPUSZCZENIA NA RYNEK
- 8. NUMER WPROWADZENIA DO OBROTU
- 9. DATA PIERWSZEGO POZWOLENIA/ODNOWIENIA POZWOLENIA
- 10. DATA REWIZJI TEKSTU
STRESZCZENIE CHARAKTERYSTYKI PRODUKTU
1. NAZWA PRODUKTU LECZNICZEGO
Dapaglifozin Tecnigen 5 mg tabletki powlekane
Dapaglifozin Tecnigen 10 mg tabletki powlekane
2. SKŁAD JAKOŚCIOWY I ILOŚCIOWY
Dapaglifozin Tecnigen 5 mg tabletki powlekane
Każda tabletka zawiera dapagliflozyn propanediolo monohydryt odpowiadający 5 mg dapagliflozyny.
Dapaglifozin Tecnigen 10 mg tabletki powlekane
Każda tabletka zawiera dapagliflozyn propanediolo monohydryt odpowiadający 10 mg dapagliflozyny.
Aby uzyskać pełną listę substancji pomocniczych, zobacz punkt 6.1.
3. POSTAĆ LECZNICZA
Tabletka powlekana (tabletki).
Tabletki powlekane dapagliflozyny 5 mg to okrągłe, żółte tabletki powlekane o średnicy 7,0 mm ± 0,2 mm, z oznaczeniem „5” po jednej stronie.
Tabletki powlekane dapagliflozyny 10 mg to owalne, żółte tabletki powlekane o długości 11,5 mm ± 0,2 mm i szerokości 7,0 mm ± 0,2 mm.
4. INFORMACJE KLINICZNE
4.1 Wskazania terapeutyczne
Cukrzyca typu 2
Dapaglifozin Tecnigen jest wskazany u dorosłych i dzieci od 10. roku życia, u których nie osiągnięto odpowiedniej kontroli leczenia cukrzycy typu 2 w połączeniu z dietą i aktywnością fizyczną:
- w monoterapii, gdy stosowanie metformyny jest uznawane za niewłaściwe ze względu na nietolerancję.
- jako dodatek do innych leków stosowanych w leczeniu cukrzycy typu 2.
W celu uzyskania wyników badań dotyczących skojarzeń z innymi lekami, wpływu na kontrolę glikemiczną, zdarzeń kardiologicznych i nerkowych oraz badanych populacji, należy zapoznać się z punktami 4.4, 4.5 i 5.1.
Niewydolność serca
Dapaglifozin Tecnigen jest wskazany u dorosłych w leczeniu przewlekłej, objawowej niewydolności serca.
Przewlekła choroba nerek
Dapaglifozin Tecnigen jest wskazany u dorosłych w leczeniu przewlekłej choroby nerek.
4.2 Dawkowanie i sposób podania
Dawkowanie
Cukrzyca typu 2
Zalecana dawka to 10 mg dapagliflozyny raz dziennie.
Gdy dapagliflozyna jest stosowana w połączeniu z insuliną lub lekiem sekretagogiem insuliny, takim jak sulfonilomocznik, w celu zmniejszenia ryzyka hipoglikemii można rozważyć podanie niższej dawki insuliny lub leku sekretagogu insuliny (patrz punkty 4.5 i 4.8).
Niewydolność serca
Zalecana dawka to 10 mg dapagliflozyny raz dziennie.
Przewlekła choroba nerek
Zalecana dawka to 10 mg dapagliflozyny raz dziennie.
Osoby szczególne
Upośledzenie funkcji nerek
Nie jest wymagana dostosowanie dawki na podstawie czynności nerek.
U pacjentów z GFR < 25 mL/min, ze względu na ograniczone doświadczenie, nie zaleca się rozpoczynania leczenia dapagliflozyną.
U pacjentów z cukrzycą typu 2 skuteczność hipoglikemizująca dapagliflozyny jest zmniejszona, gdy szybkość filtracji kłębuszkowej (GFR) jest < 45 mL/min, a u pacjentów z ciężkim upośledzeniem czynności nerek jest prawdopodobnie nieobecna. Dlatego u pacjentów z cukrzycą typu 2, gdy GFR spadnie poniżej 45 mL/min, a konieczna jest dalsza kontrola glikemii, należy rozważyć dodatkowe leczenie hipoglikemizujące (patrz punkty 4.4, 4.8, 5.1 i 5.2).
Upośledzenie funkcji wątroby
Nie jest wymagane dostosowanie dawki u pacjentów z łagodnym lub umiarkowanym upośledzeniem czynności wątroby.
U pacjentów z ciężkim upośledzeniem czynności wątroby zaleca się dawkę początkową 5 mg. Jeśli jest dobrze tolerowana, dawkę można zwiększyć do 10 mg (patrz punkty 4.4 i 5.2).
Osoby starsze (≥ 65 lat)
Nie zaleca się dostosowania dawki w zależności od wieku.
Populacja pediatryczna
Nie jest wymagane dostosowanie dawki w leczeniu cukrzycy typu 2 u dzieci od 10. roku życia (patrz punkty 5.1 i 5.2). Brak danych dotyczących dzieci poniżej 10. roku życia.
Bezpieczeństwo i skuteczność dapagliflozyny w leczeniu niewydolności serca lub przewlekłej choroby nerek u dzieci < 18 lat nie zostały jeszcze ustalone. Brak dostępnych danych.
Sposób podania
Dapaglifozin Tecnigen można przyjmować doustnie raz dziennie niezależnie od posiłków, w dowolnym czasie dnia. Tabletki należy połykać całe.
4.3 Przeciwwskazania
Nadwrażliwość na substancję czynną lub którykolwiek z substancji pomocniczych wymienionych w punkcie 6.1.
4.4 Ostrzeżenia i środki ostrożności
Ogólne
Dapagliflozyna nie powinna być stosowana u pacjentów z cukrzycą typu 1 (patrz „Kwasica ketonowa cukrzycowa” w punkcie 4.4).
Upośledzenie funkcji nerek
U pacjentów z GFR < 25 mL/min, ze względu na ograniczone doświadczenie, nie zaleca się rozpoczynania leczenia dapagliflozyną.
Skuteczność hipoglikemizująca dapagliflozyny zależy od czynności nerek i jest zmniejszona u pacjentów z GFR < 45 mL/min, a u pacjentów z ciężkim upośledzeniem czynności nerek jest praktycznie nieobecna (patrz punkty 4.2, 5.1 i 5.2).
W badaniu przeprowadzonym u pacjentów z cukrzycą typu 2 i umiarkowanym upośledzeniem czynności nerek (GFR < 60 mL/min) większa liczba pacjentów leczonych dapagliflozyną doświadczyła działań niepożądanych, takich jak wzrost stężenia kreatyniny, fosforu, hormonu parathormonu (PTH) i hipotensji, w porównaniu do placebo.
Upośledzenie funkcji wątroby
Doświadczenie z badań klinicznych u pacjentów z upośledzeniem czynności wątroby jest ograniczone. Stężenie dapagliflozyny jest zwiększone u pacjentów z ciężkim upośledzeniem czynności wątroby (patrz punkty 4.2 i 5.2).
Stosowanie u pacjentów z ryzykiem wyczerpania objętości płynów i/lub hipotensji
Dzięki mechanizmowi działania dapagliflozyna zwiększa diurezę, co może prowadzić do umiarkowanego obniżenia ciśnienia krwi, obserwowanego w badaniach klinicznych (patrz punkt 5.1). Może to być bardziej wyrażone u pacjentów z bardzo wysokim stężeniem glukozy we krwi.
Należy zachować ostrożność u pacjentów, u których spadek ciśnienia krwi wywołany przez dapagliflozynę może stanowić ryzyko, np. u pacjentów leczonych lekami przeciwnadciśnieniowymi z historią hipotensji lub u osób starszych.
W przypadku stanów chorobowych prowadzących do wyczerpania objętości płynów (np. choroby przewodu pokarmowego) zaleca się staranne monitorowanie stanu wodno-elektrolitowego (np. badanie fizykalne, pomiar ciśnienia krwi, badania laboratoryjne w tym hematokryt i elektrolity). Zaleca się tymczasowe przerwanie leczenia dapagliflozyną u pacjentów, u których wystąpi wyczerpanie objętości, aż do jego skorygowania (patrz punkt 4.8).
Kwasica ketonowa cukrzycowa
Zarejestrowano rzadkie przypadki, w tym potencjalnie śmiertelne i śmiertelne, kwasicy ketonowej cukrzycowej (KDC) u pacjentów leczonych inhibitorami współprzewoźnika sodu-glukozy typu 2 (SGLT2), w tym dapagliflozyną. W wielu przypadkach stan kliniczny występował nietypowo, z tylko umiarkowanym wzrostem stężenia glukozy we krwi, poniżej 14 mmol/L (250 mg/dL).
Należy rozważyć ryzyko kwasicy ketonowej cukrzycowej w przypadku wystąpienia niespecyficznych objawów, takich jak nudności, wymioty, brak apetytu, ból brzucha, nadmierne pragnienie, trudności z oddychaniem, dezorientacja, nietypowa senność lub zmęczenie. W przypadku wystąpienia tych objawów pacjentów należy natychmiast ocenić pod kątem kwasicy, niezależnie od stężenia glukozy we krwi.
U pacjentów, u których podejrzewa się lub stwierdza KDC, leczenie dapagliflozyną należy natychmiast przerwać.
Leczenie należy przerwać u pacjentów hospitalizowanych do przeprowadzenia dużych zabiegów chirurgicznych lub z powodu ciężkich chorób w ostrym okresie. U tych pacjentów zaleca się monitorowanie poziomu ciał ketonowych. Pomiar stężenia ciał ketonowych we krwi jest preferowany nad pomiarem w moczu.
Leczenie dapagliflozyną można wznowić, gdy poziomy ciał ketonowych są normalne i stan pacjenta się ustabilizował.
Przed rozpoczęciem leczenia dapagliflozyną należy rozważyć czynniki w wywiadzie pacjenta, które mogą sprzyjać kwasicy ketonowej.
Pacjenci, którzy mogą być narażeni na większe ryzyko KDC, to osoby z niską rezerwą czynności beta-komórek (np. pacjenci z cukrzycą typu 2 z niskim peptydem C lub z ukrytą autoimmunologiczną cukrzycą dorosłych (LADA, latent autoimmune diabetes in adults)), pacjenci z warunkami prowadzącymi do zmniejszonego przyjmowania pokarmu lub ciężkiego odwodnienia, pacjenci, u których dawki insuliny są zmniejszone, oraz pacjenci z zwiększonym zapotrzebowaniem na insulinę z powodu choroby ostrej, zabiegu chirurgicznego lub nadużywania alkoholu. Inhibitory SGLT2 należy stosować z ostrożnością u tych pacjentów.
Wznowienie leczenia inhibitorami SGLT2 u pacjentów z wcześniejszą KDC nie jest zalecane, chyba że zidentyfikowano inny czynnik wyzwalający i został on wyeliminowany.
W badaniach nad cukrzycą typu 1 z dapagliflozyną KDC występowała często.
Dapagliflozyna nie powinna być stosowana w leczeniu pacjentów z cukrzycą typu 1.
Fascytyka nekrotyczna okolicy krocza (gangrena Fourniera)
Po wprowadzeniu na rynek zgłoszono przypadki fascytyki nekrotycznej okolicy krocza (znanej również jako gangrena Fourniera) u pacjentów płci żeńskiej i męskiej leczonych inhibitorami SGLT2 (patrz punkt 4.8). Jest to rzadkie, ale poważne i potencjalnie śmiertelne zdarzenie, wymagające natychmiastowego leczenia chirurgicznego i antybiotykoterapii.
Pacjentów należy zachęcać do kontaktu z lekarzem w przypadku wystąpienia kombinacji objawów bólu, bolesności, zaczerwienienia lub obrzęku w okolicy narządów płciowych lub krocza, w połączeniu z gorączką lub niedoborem samopoczucia. Należy pamiętać, że fascytyka nekrotyczna może poprzedzać infekcja dróg moczowych lub zgrzybienie okolicy krocza. W przypadku podejrzenia gangreny Fourniera należy przerwać leczenie Dapaglifozin Tecnigen i rozpocząć natychmiastowe leczenie (w tym antybiotyki i chirurgiczne usunięcie tkanek).
Zakażenia dróg moczowych
Wykrzepianie glukozy z moczem może być związane ze zwiększonym ryzykiem zakażeń dróg moczowych; dlatego należy rozważyć tymczasowe przerwanie leczenia dapagliflozyną podczas leczenia zapalenia nerek lub sepsy dróg moczowych.
Osoby starsze (≥ 65 lat)
Osoby starsze mogą być bardziej narażone na wyczerpanie objętości płynów i częściej przyjmują diuretyki.
Osoby starsze częściej mają upośledzoną czynność nerek i/lub przyjmują leki przeciwnadciśnieniowe, które mogą wpływać na czynność nerek, takie jak inhibitory enzymu konwertującego angiotensynę I (ACE, angiotensin converting enzyme) i blokery receptora angiotensyny II typu 1 (ARB, angiotensin receptor blockers). Te same zalecenia dotyczące czynności nerek dotyczą pacjentów starszych, jak i wszystkich pacjentów (patrz punkty 4.2, 4.4, 4.8 i 5.1).
Niewydolność serca
Doświadczenie z dapagliflozyną w klasach NYHA IV jest ograniczone.
Kardiomiopatia infiltracyjna
Pacjenci z kardiomiopatią infiltracyjną nie byli badani.
Przewlekła choroba nerek
Nie ma doświadczenia z dapagliflozyną w leczeniu przewlekłej choroby nerek u pacjentów bez cukrzycy, którzy nie mają albuminurii. Pacjenci z albuminurią mogą czerpać większy korzyść z leczenia dapagliflozyną.
Amputacje kończyn dolnych
Zauważono zwiększoną liczbę przypadków amputacji kończyn dolnych (głównie palców stóp) w długoterminowych badaniach klinicznych z cukrzycą typu 2 prowadzonych z inhibitorami SGLT2. Nie wiadomo, czy jest to efekt klasy. Ważne jest, aby doradzać pacjentom z cukrzycą regularną, zapobiegawczą opiekę nad stopami.
Badania moczu
Ze względu na mechanizm działania pacjenci przyjmujący Dapaglifozin Tecnigen będą wykazywali dodatni wynik testu glukozy w moczu.
4.5 Interakcje z innymi lekami i inne formy interakcji
Interakcje farmakodynamiczne
Diuretyki
Dapagliflozyna może nasilać działanie diuretyków tiazydowych i pętlowych oraz zwiększać ryzyko odwodnienia i hipotensji (patrz punkt 4.4).
Insulina i leki sekretagogi insuliny
Insulina i leki sekretagogi insuliny, takie jak sulfonilomoczniki, powodują hipoglikemię. Dlatego może być wymagana niższa dawka insuliny lub leku sekretagogu insuliny w celu zmniejszenia ryzyka hipoglikemii, gdy są stosowane w połączeniu z dapagliflozyną u pacjentów z cukrzycą typu 2 (patrz punkty 4.2 i 4.8).
Interakcje farmakokinetyczne
Dapagliflozyna jest metabolizowana głównie poprzez koniugację z glukuronidem katalizowaną przez UDP-glukuronosylotransferazę 1A9 (UGT1A9).
W badaniach in vitro dapagliflozyna nie hamowała ani nie indukowała cytochromu P450 (CYP) 1A2, CYP2A6, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, CYP3A4.
Dlatego nie oczekuje się, że dapagliflozyna będzie zmieniać klirens metaboliczny współpodawanych leków metabolizowanych przez te enzymy.
Wpływ innych leków na dapagliflozynę
Badania interakcji przeprowadzone u zdrowych osób, głównie w schemacie pojedynczej dawki, sugerują, że profil farmakokinetyczny dapagliflozyny nie jest zmieniany przez metformynę, pioglitazon, sitagliptynę, glimepiryd, voglibozę, hydrochlorotiazyd, bumetanid, walzartan ani simwastatynę.
Po współpodaniu dapagliflozyny z ryfampicyną (induktorem różnych aktywnych przenośników i enzymów metabolizujących leki) zaobserwowano zmniejszenie ekspozycji na dapagliflozynę (AUC) o 22%, bez istotnego klinicznie wpływu na wydalenie glukozy z moczem w ciągu 24 godzin. Nie zaleca się dostosowania dawki. Nie oczekuje się istotnego klinicznie wpływu z innymi induktorami (np. karbamazepiną, fenytoiną, fenobarbitalą).
Po współpodaniu dapagliflozyny z kwasem mefenamowym (inhibitorem UGT1A9) zaobserwowano wzrost ekspozycji na dapagliflozynę o 55%, bez istotnego klinicznie wpływu na wydalenie glukozy z moczem w ciągu 24 godzin. Nie zaleca się dostosowania dawki.
Wpływ dapagliflozyny na inne leki
Dapagliflozyna może zwiększać wydalanie litu z moczem, a stężenia litu we krwi mogą się zmniejszać. Po rozpoczęciu leczenia dapagliflozyną i zmianie dawki należy częściej monitorować stężenie litu w surowicy. Należy skierować pacjenta do lekarza, który przepisał lit, w celu monitorowania stężenia litu w surowicy.
W badaniach interakcji przeprowadzonych u zdrowych osób, głównie w schemacie pojedynczej dawki, dapagliflozyna nie zmieniała profili farmakokinetycznych metformyny, pioglitazonu, sitagliptyny, glimepirydu, hydrochlorotiazydu, bumetanidu, walzartanu, digoksyny (substrat glikoproteiny P, P-gp) ani warfaryny (S-warfaryna, substrat CYP2C9), ani efektów przeciwkrzepnych warfaryny mierzonych przez INR. Połączenie pojedynczej dawki dapagliflozyny 20 mg i simwastatyny (substrat CYP3A4) spowodowało wzrost AUC simwastatyny o 19% i wzrost AUC kwasu simwastatynowego o 31%. Wzrost ekspozycji na simwastatynę i kwas simwastatynowy nie jest uważany za klinicznie istotny.
Interferencja z analizą 1,5-anhydro-glucitolu (1,5 AG)
Monitorowanie kontroli glikemicznej za pomocą analizy 1,5 AG nie jest zalecane, ponieważ pomiary 1,5 AG nie są wiarygodne w ocenie kontroli glikemicznej u pacjentów przyjmujących inhibitory SGLT2. Zaleca się stosowanie alternatywnych metod monitorowania kontroli glikemicznej.
Populacja pediatryczna
Badania interakcji przeprowadzono wyłącznie u dorosłych.
4.6 Płodność, ciąża i karmienie piersią
Ciąża
Nie ma danych dotyczących stosowania dapagliflozyny u kobiet w ciąży. Badania na szczurach wykazały toksyczność w okresie rozwoju nerek odpowiadającym II i III trymestrowi ciąży u ludzi (patrz punkt 5.3). W związku z tym stosowanie dapagliflozyny nie jest zalecane w II i III trymestrze ciąży.
Po potwierdzeniu ciąży leczenie dapagliflozyną należy przerwać.
Karmienie piersią
Nie wiadomo, czy dapagliflozyna i/lub jej metabolity są wydzielane z mlekiem matki. Dane farmakodynamiczne/toksyczne dostępne u zwierząt wykazały wydzielanie dapagliflozyny/metabolitów z mlekiem oraz działanie farmakologiczne u potomstwa karmionego piersią (patrz punkt 5.3). Ryzyko dla noworodków/dzieci karmionych nie może być wykluczone. Dapagliflozyna nie powinna być stosowana podczas karmienia piersią.
Płodność
Wpływ dapagliflozyny na płodność nie był badany u ludzi. U szczurów męskich i żeńskich dapagliflozyna nie wykazała wpływu na płodność przy każdej badanej dawce.
4.7 Wpływ na zdolność prowadzenia pojazdów i obsługiwanie maszyn
Dapaglifozin Tecnigen nie wpływa lub wpływa w sposób pomijalny na zdolność prowadzenia pojazdów i obsługiwanie maszyn. Pacjentów należy uprzedzić o ryzyku hipoglikemii, gdy dapagliflozyna jest stosowana w połączeniu z sulfonilomocznikiem lub insuliną.
4.8 Działania niepożądane
Podsumowanie profilu bezpieczeństwa
Cukrzyca typu 2
W badaniach klinicznych dotyczących cukrzycy typu 2 leczono ponad 15 000 pacjentów dapagliflozyną.
Podstawową ocenę bezpieczeństwa i tolerancji przeprowadzono w analizie agregacyjnej 13 krótkoterminowych badań (do 24 tygodni) kontrolowanych placebo, w których uczestniczyło 2360 osób leczonych dapagliflozyną 10 mg i 2295 osób leczonych placebo.
W badaniu nad skutkami kardiologicznymi dapagliflozyny w cukrzycy typu 2 (badanie DECLARE, patrz punkt 5.1), 8574 pacjentów otrzymywało dapagliflozynę 10 mg, a 8569 otrzymywało placebo przez średni czas ekspozycji 48 miesięcy. Łącznie było 30 623 pacjentów-roku ekspozycji na dapagliflozynę.
Najczęściej zgłaszane działania niepożądane w badaniach klinicznych to infekcje narządów płciowych.
Niewydolność serca
W badaniu nad skutkami kardiologicznymi dapagliflozyny u pacjentów z niewydolnością serca i zmniejszoną frakcją wyrzutową (badanie DAPA-HF), 2368 pacjentów otrzymywało dapagliflozynę 10 mg, a 2368 pacjentów otrzymywało placebo przez medianę czasu ekspozycji 18 miesięcy. Grupa pacjentów obejmowała pacjentów z cukrzycą typu 2 i bez cukrzycy oraz pacjentów z eGFR ≥ 30 mL/min/1,73 m². W badaniu nad skutkami kardiologicznymi dapagliflozyny u pacjentów z niewydolnością serca i frakcją wyrzutową lewej komory > 40% (DELIVER), 3126 pacjentów otrzymywało dapagliflozynę 10 mg, a 3127 pacjentów otrzymywało placebo przez medianę czasu ekspozycji 27 miesięcy. Grupa pacjentów obejmowała pacjentów z cukrzycą typu 2 i bez cukrzycy oraz pacjentów z eGFR ≥ 25 mL/min/1,73 m².
Ogólny profil bezpieczeństwa dapagliflozyny u pacjentów z niewydolnością serca był zgodny z znanym profilem bezpieczeństwa dapagliflozyny.
Przewlekła choroba nerek
Wyniki badania nerkowego dapagliflozyny (DAPA-CKD), przeprowadzonego u pacjentów z przewlekłą chorobą nerek, 2149 pacjentów otrzymywało dapagliflozynę 10 mg, a 2149 pacjentów otrzymywało placebo przez medianę czasu ekspozycji 27 miesięcy. Grupa pacjentów obejmowała pacjentów z cukrzycą typu 2 i bez cukrzycy, z eGFR od ≥ 25 do ≤ 75 mL/min/1,73 m² i albuminurią (stosunek albuminy do kreatyniny w moczu [ACR] ≥ 200 i ≤ 5000 mg/g). Leczenie kontynuowano, jeśli eGFR spadło poniżej 25 mL/min/1,73 m².
U pacjentów z przewlekłą chorobą nerek ogólny profil bezpieczeństwa dapagliflozyny był zgodny z znanym profilem bezpieczeństwa dapagliflozyny.
Tabela działań niepożądanych
Następujące działania niepożądane zostały zidentyfikowane w kontrolowanych placebo badaniach klinicznych i nadzorze po wprowadzeniu na rynek. Żadne z nich nie było związane z dawką. Poniższe działania niepożądane sklasyfikowano według częstości i klasyfikacji według układów i narządów (SOC). Kategorie częstości zdefiniowano według następującej konwencji: bardzo często (≥ 1/10), często (≥ 1/100, < 1/10), nieczęsto (≥ 1/1000, < 1/100), rzadko (≥ 1/10000, < 1/1000), bardzo rzadko (< 1/10000), nieznana (częstość nie może być określona na podstawie dostępnych danych).
Tabela 1. Działania niepożądane zaobserwowane w kontrolowanych placebo badaniach klinicznych a i w doświadczeniu po wprowadzeniu na rynek
| Klasyfikacja wg narządów i układów | Bardzo często | Często* | Nieczęsto** | Rzadko | Bardzo rzadko |
| Infekcje i pasożyty | Zapalenie narządów płciowych, prącia i infekcje narządów rodnych związane z*,b,c Zakażenie dróg moczowych*,b,d | Zakażenie grzybicze** | Fasceit nekrotyczna okolicy krocza (gangrena Fourniera) b,i | ||
| Zaburzenia metabolizmu i odżywiania | hipoglikemia a (gdy stosowane z SU lub insulina) b | Ubytek objętości b,e Pragnienie** | Kwasicę ketonową cukrzycową (gdy stosowana w cukrzycy typu 2) b,i,k | ||
| Choroby układu nerwowego | Zawroty głowy | ||||
| Choroby układu pokarmowego | Wstyd** Sucha jamy ustnej** | ||||
| Choroby skóry i tkanki podskórnej | Obrzęk skórny j | Obrzęk naczyniowy | |||
| Choroby układu mięśniowo-szkieletowego i tkanki łącznej | Ból pleców* | ||||
| Choroby nerek i układu moczowego | Dysuria, poliuria*,f | Nikturia** | Nefryt naczyniowy | ||
| Choroby układu rozrodczego i piersi | Zapalenie narządów płciowych** Zapalenie narządów płciowych** | ||||
| Badania diagnostyczne | Zwiększony hematokryt g Zmniejszona clearance kreatyniny podczas wstępnego leczenia b Dyslipidemia h | Zwiększona kreatynina w surowicy podczas wstępnego leczenia**,b Zwiększony poziom mocznika w surowicy** Spadek masy ciała** |
Opis wybranych niepożądanych reakcji
Wulwowaginita, balanitis i powiązane infekcje narządów płciowych
W połączonym zbiorze danych z 13 badań bezpieczeństwa wulwowaginita, balanitis oraz powiązane infekcje narządów płciowych występowały u 5,5% i 0,6% pacjentów otrzymujących dapaglifyzynę 10 mg i placebo, odpowiednio. Większość infekcji była łagodna lub umiarkowana, a pacjenci odpowiadali na wstępną terapię, rzadko wymagającą przerwania leczenia dapaglifyzyną. Częstszym występowaniem tych infekcji charakteryzowały się kobiety (8,4% i 1,2% dla dapaglifyzyny i placebo, odpowiednio), a pacjenci z wywiadem infekcji mieli większe ryzyko ich nawrotu.
W badaniu DECLARE liczba pacjentów z ciężkimi niepożądanymi zdarzeniami infekcyjnymi narządów płciowych była niewielka i zrównoważona: 2 pacjenci w każdej z grup otrzymujących dapaglifyzynę i placebo.
W badaniu DAPA-HF żaden pacjent nie zgłosił ciężkich niepożądanych zdarzeń infekcyjnych narządów płciowych w grupie dapaglifyzyny, a jeden w grupie placebo. W grupie dapaglifyzyny odnotowano 7 (0,3%) pacjentów z niepożądanymi zdarzeniami prowadzącymi do przerwania leczenia z powodu infekcji narządów płciowych, natomiast w grupie placebo żadnego. W badaniu DELIVER jeden pacjent (<0,1%) w każdej z grup leczenia zgłosił ciężkie niepożądane zdarzenie infekcyjne narządów płciowych. W grupie dapaglifyzyny odnotowano 3 (0,1%) pacjentów z niepożądanymi zdarzeniami prowadzącymi do przerwania leczenia z powodu infekcji narządów płciowych, natomiast w grupie placebo żadnego.
W badaniu DAPA-CKD odnotowano 3 (0,1%) pacjentów z ciężkimi niepożądanymi zdarzeniami infekcyjnymi narządów płciowych w grupie dapaglifyzyny i żadnego w grupie placebo. W grupie dapaglifyzyny odnotowano 3 (0,1%) pacjentów z niepożądanymi zdarzeniami prowadzącymi do przerwania leczenia z powodu infekcji narządów płciowych, natomiast w grupie placebo żadnego. Nie odnotowano ciężkich niepożądanych zdarzeń infekcyjnych narządów płciowych ani niepożądanych zdarzeń prowadzących do przerwania leczenia z powodu infekcji narządów płciowych u żadnego pacjenta bez cukrzycy.
Zgłaszano przypadki fimosu/fimosu nabytej w przebiegu infekcji narządów płciowych, a w niektórych przypadkach konieczna była okoliczność.
Fascektomia nekrotyczna okolicy krocza (gangrena Fourniera)
Po wprowadzeniu leku na rynek zgłaszano przypadki gangreny Fourniera u pacjentów leczonych inhibitorem SGLT2, w tym dapaglifyzyną (patrz punkt 4.4).
W badaniu DECLARE obejmującym 17 160 pacjentów z cukrzycą typu 2 i średnim czasie ekspozycji 48 miesięcy, zgłoszono łącznie 6 przypadków gangreny Fourniera: jeden w grupie leczonej dapaglifyzyną i pięć w grupie placebo.
Hipoglikemia
Częstość występowania hipoglikemii zależała od rodzaju terapii podstawowej stosowanej w badaniach klinicznych u pacjentów z cukrzycą.
W badaniach z zastosowaniem dapaglifyzyny w monoterapii, jako terapii wspomagającej do metformyny lub jako terapii wspomagającej do sitagliptyny (z lub bez metformyny), częstość występowania lekkich epizodów hipoglikemii była podobna (< 5%) między grupami leczenia, w tym placebo, przez okres do 102 tygodni leczenia. We wszystkich badaniach ciężkie zdarzenia hipoglikemiczne występowały rzadziej i były porównywalne między grupami leczonymi dapaglifyzyną lub placebo. W badaniach z terapią wspomagającą zawierającą sulfonamidy i insulinę odnotowano wyższe stężenia hipoglikemii (patrz punkt 4.5).
W jednym z badań z terapią wspomagającą glimepirydą, lekkie epizody hipoglikemii zgłaszano częściej w grupie leczonej dapaglifyzyną 10 mg plus glimepiryda (6,0% i 7,9% odpowiednio w 24. i 48. tygodniu), w porównaniu z grupą placebo plus glimepiryda (2,1% i 2,1% odpowiednio).
W jednym z badań z terapią wspomagającą insuliną, ciężkie zdarzenia hipoglikemiczne zgłaszano u 0,5% i 1,0% pacjentów leczonych dapaglifyzyną 10 mg plus insulina odpowiednio w 24. i 104. tygodniu, oraz u 0,5% pacjentów z grupy placebo plus insulina w 24. i 104. tygodniu. Lekkie epizody hipoglikemii zgłaszano odpowiednio w 24. i 104. tygodniu u 40,3% i 53,1% pacjentów otrzymujących dapaglifyzynę 10 mg plus insulina oraz u 34,0% i 41,6% pacjentów otrzymujących placebo plus insulina.
W jednym z badań z terapią wspomagającą metformyną i sulfonamidem do 24 tygodnia nie odnotowano żadnych epizodów ciężkiej hipoglikemii. Lekkie epizody hipoglikemii zgłaszano u 12,8% pacjentów otrzymujących dapaglifyzynę 10 mg plus metformynę i sulfonamid oraz u 3,7% pacjentów otrzymujących placebo plus metformynę i sulfonamid.
W badaniu DECLARE nie zaobserwowano wzrostu ryzyka ciężkiej hipoglikemii przy leczeniu dapaglifyzyną w porównaniu z placebo. Ciężkie zdarzenia hipoglikemiczne zgłoszono u 58 (0,7%) pacjentów leczonych dapaglifyzyną i u 83 (1,0%) pacjentów leczonych placebo.
W badaniu DAPA-HF ciężkie zdarzenia hipoglikemiczne zgłoszono u 4 (0,2%) pacjentów w obu grupach leczenia dapaglifyzyną i placebo. W badaniu DELIVER ciężkie zdarzenia hipoglikemiczne zgłoszono u 6 (0,2%) pacjentów w grupie dapaglifyzyny i u 7 (0,2%) w grupie placebo. Ciężkie zdarzenia hipoglikemiczne występowały wyłącznie u pacjentów z cukrzycą typu 2.
W badaniu DAPA-CKD ciężkie zdarzenia hipoglikemiczne zaobserwowano u 14 (0,7%) pacjentów w grupie dapaglifyzyny i u 28 (1,3%) pacjentów w grupie placebo, wyłącznie u pacjentów z cukrzycą typu 2.
Wyczerpanie objętości krwi obwodowej
W połączonym zbiorze danych z 13 badań bezpieczeństwa reakcje wskazujące na wyczerpanie objętości (w tym przypadki odwodnienia, hipowolemii lub hipotensji) zgłaszano u 1,1% i 0,7% pacjentów leczonych odpowiednio dapaglifyzyną 10 mg i placebo. Ciężkie reakcje wystąpiły u < 0,2% pacjentów, zrównoważone między grupami dapaglifyzyny 10 mg i placebo (patrz punkt 4.4).
W badaniu DECLARE liczba pacjentów z zdarzeniami wskazującymi na wyczerpanie objętości była zrównoważona między grupami leczenia: 213 (2,5%) i 207 (2,4%) odpowiednio w grupach dapaglifyzyny i placebo. W grupie dapaglifyzyny i placebo zgłoszono odpowiednio 81 (0,9%) i 70 (0,8%) ciężkich niepożądanych zdarzeń. Zdarzenia te były ogólnie zrównoważone między grupami leczenia w różnych podgrupach wiekowych, stosujących diuretyki, z różnym ciśnieniem krwi oraz stosujących inhibitory enzymu konwertującego angiotensynę (ACE-I)/blokery receptora angiotensyny II typu 1 (ARB). U pacjentów z eGFR < 60 mL/min/1,73 m² na początku badania odnotowano 19 ciężkich niepożądanych zdarzeń wskazujących na wyczerpanie objętości w grupie dapaglifyzyny i 13 w grupie placebo.
W badaniu DAPA-HF liczba pacjentów z zdarzeniami wskazującymi na wyczerpanie objętości wyniosła 170 (7,2%) w grupie dapaglifyzyny i 153 (6,5%) w grupie placebo. Mniej pacjentów z ciężkimi objawami sugerującymi wyczerpanie objętości odnotowano w grupie dapaglifyzyny (23 [1,0%]) niż w grupie placebo (38 [1,6%]). Wyniki były podobne niezależnie od obecności cukrzycy i wartości eGFR na początku badania. W badaniu DELIVER liczba pacjentów z ciężkimi zdarzeniami z objawami sugerującymi wyczerpanie objętości wyniosła 35 (1,1%) w grupie dapaglifyzyny i 31 (1,0%) w grupie placebo.
W badaniu DAPA-CKD liczba pacjentów z zdarzeniami wskazującymi na wyczerpanie objętości wyniosła 120 (5,6%) w grupie dapaglifyzyny i 84 (3,9%) w grupie placebo. W grupie dapaglifyzyny odnotowano 16 (0,7%) pacjentów z ciężkimi objawami sugerującymi wyczerpanie objętości, a w grupie placebo 15 (0,7%) pacjentów.
Kwasicę ketonową w cukrzycy typu 2
W badaniu DECLARE, przy średnim czasie ekspozycji 48 miesięcy, zdarzenia CAD zgłoszono u 27 pacjentów w grupie z dapaglifyzyną 10 mg i u 12 pacjentów w grupie placebo. Zdarzenia te występowały równomiernie w trakcie badania. Spośród 27 pacjentów z zdarzeniami CAD, 22 otrzymywało terapię wspomagającą insuliną w momencie wystąpienia zdarzenia. Czynniki wywołujące CAD były zgodne z oczekiwaniami w populacji chorych na cukrzycę typu 2 (patrz sekcja 4.4).
W badaniu DAPA-HF zdarzenia CAD zgłoszono u 3 pacjentów z cukrzycą typu 2 w grupie dapaglifyzyny i żadnego w grupie placebo. W badaniu DELIVER zgłoszono zdarzenia DKA u 2 pacjentów z cukrzycą typu 2 w grupie dapaglifyzyny i żadnego w grupie placebo.
W badaniu DAPA-CKD nie zaobserwowano żadnych zdarzeń CAD u pacjentów w grupie dapaglifyzyny i u 2 pacjentów z cukrzycą typu 2 w grupie placebo.
Infekcje dróg moczowych
W połączonym zbiorze danych z 13 badań bezpieczeństwa infekcje dróg moczowych zgłaszano częściej dla dapaglifyzyny 10 mg w porównaniu do placebo (odpowiednio 4,7% vs 3,5%; patrz punkt 4.4). Większość infekcji była łagodna lub umiarkowana, a pacjenci odpowiadali na wstępną standardową terapię, rzadko prowadzącą do przerwania leczenia dapaglifyzyną. Takie infekcje zgłaszano częściej u kobiet, a pacjenci z wywiadem infekcji mieli większe ryzyko nawrotu.
W badaniu DECLARE ciężkie niepożądane zdarzenia infekcyjne dróg moczowych zgłaszano rzadziej dla dapaglifyzyny 10 mg w porównaniu do placebo: 79 (0,9%) zdarzeń kontra 109 (1,3%) zdarzeń, odpowiednio.
W badaniu DAPA-HF liczba pacjentów z ciężkimi niepożądanymi zdarzeniami infekcyjnymi dróg moczowych wyniosła 14 (0,6%) w grupie dapaglifyzyny i 17 (0,7%) w grupie placebo. W grupie dapaglifyzyny i placebo odnotowano po 5 (0,2%) pacjentów z niepożądanymi zdarzeniami prowadzącymi do przerwania leczenia z powodu infekcji dróg moczowych. W badaniu DELIVER liczba pacjentów z ciężkimi niepożądanymi zdarzeniami infekcyjnymi dróg moczowych wyniosła 41 (1,3%) w grupie dapaglifyzyny i 37 (1,2%) w grupie placebo. W grupie dapaglifyzyny odnotowano 13 (0,4%) pacjentów z niepożądanymi zdarzeniami prowadzącymi do przerwania leczenia z powodu infekcji dróg moczowych, a w grupie placebo 9 (0,3%).
W badaniu DAPA-CKD liczba pacjentów z ciężkimi niepożądanymi zdarzeniami infekcyjnymi dróg moczowych wyniosła 29 (1,3%) w grupie dapaglifyzyny i 18 (0,8%) w grupie placebo. W grupie dapaglifyzyny odnotowano 8 (0,4%) pacjentów z niepożądanymi zdarzeniami prowadzącymi do przerwania leczenia z powodu infekcji dróg moczowych, a w grupie placebo 3 (0,1%). Liczba pacjentów bez cukrzycy z ciężkimi niepożądanymi zdarzeniami infekcyjnymi dróg moczowych lub niepożądanymi zdarzeniami prowadzącymi do przerwania leczenia z powodu infekcji dróg moczowych była podobna między grupami leczenia (6 [0,9%] kontra 4 [0,6%] dla ciężkich niepożądanych zdarzeń i 1 [0,1%] kontra 0 dla niepożądanych zdarzeń prowadzących do przerwania, odpowiednio w grupach dapaglifyzyny i placebo).
Podwyższenie kreatyniny
Niepożądane reakcje związane ze wzrostem kreatyniny zostały pogrupowane (np. zmniejszona klirens kreatyniny, zaburzenia nerek, wzrost stężenia kreatyniny w osoczu i zmniejszenie szybkości filtracji kłębuszkowej). W połączonym zbiorze danych z 13 badań bezpieczeństwa ta grupa reakcji została zgłoszona u 3,2% i 1,8% pacjentów otrzymujących dapaglifyzynę 10 mg i placebo, odpowiednio. U pacjentów z prawidłową czynnością nerek lub umiarkowanym zaburzeniem czynności nerek (eGFR na początku badania ≥ 60 mL/min/1,73 m²) ta grupa reakcji została zgłoszona u 1,3% i 0,8% pacjentów otrzymujących dapaglifyzynę 10 mg i placebo, odpowiednio. Te reakcje występowały częściej u pacjentów z eGFR na początku badania ≥ 30 i < 60 mL/min/1,73 m² (18,5% u pacjentów leczonych dapaglifyzyną 10 mg i 9,3% u pacjentów leczonych placebo).
Dalsza analiza pacjentów, którzy doświadczyli niepożądanych reakcji nerkowych, wykazała, że większość z nich miała zmiany stężenia kreatyniny w surowicy ≤ 44 mikromole/L (≤ 0,5 mg/dL) od wartości wyjściowej. Wzrosty stężenia kreatyniny były zazwyczaj przejściowe podczas kontynuacji leczenia lub odwracalne po jego przerwaniu.
W badaniu DECLARE, obejmującym starszych pacjentów i pacjentów z zaburzeniem czynności nerek (eGFR < 60 mL/min/1,73 m²), eGFR zmniejszało się z czasem w obu grupach leczenia. Po 1 roku średnie eGFR było nieco niższe, a po 4 latach średnie eGFR było nieco wyższe w grupie dapaglifyzyny niż w grupie placebo.
W badaniach DAPA-HF i DELIVER eGFR zmniejszało się z czasem zarówno w grupie dapaglifyzyny, jak i w grupie placebo. W badaniu DAPA-HF początkowy spadek średniego eGFR wyniósł -4,3 mL/min/1,73 m² w grupie dapaglifyzyny i -1,1 mL/min/1,73 m² w grupie placebo. Po 20 miesiącach zmiana eGFR od wartości wyjściowej była podobna między grupami leczenia: -5,3 mL/min/1,73 m² dla dapaglifyzyny i -4,5 mL/min/1,73 m² dla placebo. W badaniu DELIVER spadek średniego eGFR po jednym miesiącu wyniósł -3,7 mL/min/1,73 m² w grupie dapaglifyzyny i -0,4 mL/min/1,73 m² w grupie placebo. Po 24 miesiącach zmiana eGFR od wartości wyjściowej była podobna między grupami leczenia:
-4,2 mL/min/1,73 m² w grupie dapaglifyzyny i -3,2 mL/min/1,73 m² w grupie placebo.
W badaniu DAPA-CKD eGFR zmniejszało się z czasem zarówno w grupie dapaglifyzyny, jak i w grupie placebo. Początkowy spadek (dzień 14) średniego eGFR wyniósł -4,0 mL/min/1,73 m² w grupie dapaglifyzyny i -0,8 mL/min/1,73 m² w grupie placebo. Po 28 miesiącach zmiana eGFR od wartości wyjściowej wyniosła -7,4 mL/min/1,73 m² w grupie dapaglifyzyny i -8,6 mL/min/1,73 m² w grupie placebo.
Populacja pediatryczna
Profil bezpieczeństwa dapaglifyzyny obserwowany w jednym badaniu klinicznym u dzieci od 10 roku życia z cukrzycą typu 2 (patrz punkt 5.1) był podobny do profilu obserwowanego w badaniach u dorosłych.
Zgłaszanie podejrzewanych niepożądanych reakcji
Zgłaszanie podejrzewanych niepożądanych reakcji po zatwierdzeniu leku ma istotne znaczenie, ponieważ pozwala na ciągłe monitorowanie stosunku korzyści do ryzyka. Operatorom medycznym zaleca się zgłaszanie wszelkich podejrzewanych niepożądanych reakcji za pośrednictwem krajowego systemu zgłaszania pod adresem https://www.aifa.gov.it/content/segnalazioni-reazioni-avverse.
4.9 Przedawkowanie
Dapaglifyzyna podana w pojedynczych dawkach doustnych do 500 mg (50-krotność maksymalnej zalecanej dawki u człowieka) nie wykazała żadnych objawów toksyczności u zdrowych osób. Te osoby wykazywały wykrywalne stężenie glukozy w moczu przez czas zależny od dawki (co najmniej 5 dni po dawce 500 mg), bez przypadków odwodnienia, hipotensji lub zaburzeń równowagi elektrolitowej oraz bez klinicznie istotnego wpływu na interwał QTc. Częstość występowania hipoglikemii była podobna do placebo. W badaniach klinicznych, w których podawano dobowe dawki do 100 mg (10-krotność maksymalnej zalecanej dawki u człowieka) przez 2 tygodnie u zdrowych osób i u pacjentów z cukrzycą typu 2, częstość hipoglikemii była nieco wyższa niż przy placebo i nie była zależna od dawki. Częstość występowania niepożądanych zdarzeń, w tym odwodnienia lub hipotensji, była podobna do placebo, a nie stwierdzono klinicznie istotnych zależnych od dawki zmian parametrów laboratoryjnych, w tym stężenia elektrolitów w surowicy i markerów czynności nerek.
W przypadku przedawkowania należy stosować odpowiednie leczenie wspomagające zgodnie z klinicznym stanem pacjenta. Nie przeprowadzono badań dotyczących usuwania dapaglifyzyny za pomocą hemodializy.
5. WŁAŚCIWOŚCI FARMACOLOGICZNE
5.1 Właściwości farmakodynamiczne
Kategoria farmakoterapeutyczna: Leki stosowane w cukrzycy, inhibitory współtransportera sodu-glukozy typu 2 (SGLT2), kod ATC: A10BK01
Mechanizm działania
Dapaglifyzyna jest bardzo silnym (Ki: 0,55 nM), selektywnym i odwracalnym inhibitorem SGLT2.
Inhibicja SGLT2 przez dapaglifyzynę zmniejsza resorpcję glukozy z filtratu kłębuszkowego w kanaliku nerkowym bliskim, co wiąże się z jednoczesnym zmniejszeniem resorpcji sodu, prowadzącym do wydalania glukozy z moczem i diurezy osmotycznej. Dapaglifyzyna zwiększa ponadto uwalnianie sodu w kanaliku dalszym, co zwiększa sprzężenie zwrotne tubulo-klubkowe i zmniejsza ciśnienie wewnątrzklubkowe. Wszystko to, w połączeniu z diurezą osmotyczną, prowadzi do zmniejszenia obciążenia objętościowego, obniżenia ciśnienia krwi, zmniejszenia obciążenia wstępnej i końcowej napełnienia serca, co może mieć korzystny wpływ na przebudowę serca i funkcję rozkurczową oraz chronić funkcję nerek. Korzyści sercowo-nerekowe dapaglifyzyny nie zależą wyłącznie od efektu obniżania glikemii i nie są ograniczone do pacjentów z cukrzycą, jak wykazano w badaniach DAPA-HF, DELIVER i DAPA-CKD. Inne efekty obejmują wzrost hematokrytu i redukcję masy ciała.
Dapaglifyzyna poprawia zarówno glikemię na czczo, jak i pożywienie, zmniejszając nerkową resorpcję glukozy, co prowadzi do jej wydalania z moczem. Takie wydalanie glukozy (efekt glikozuryczny) obserwuje się już po pierwszej dawce, jest ciągłe przez 24-godzinny okres podania i utrzymuje się przez cały czas leczenia. Ilość glukozy usuwanej z organizmu poprzez ten mechanizm zależy od stężenia glukozy we krwi i GFR. Dlatego u osób z normalnymi poziomami glukozy we krwi dapaglifyzyna ma niską skłonność do powodowania hipoglikemii. Dapaglifyzyna nie narusza normalnej endogennej produkcji glukozy w odpowiedzi na hipoglikemię. Działa niezależnie od wydzielania insuliny i działania insuliny. W badaniach klinicznych z użyciem dapaglifyzyny zaobserwowano poprawę w modelu oceny homeostazy funkcji komórek beta (HOMA beta-cell).
SGLT2 jest wyrażany selektywnie w nerkach. Dapaglifyzyna nie hamuje innych istotnych dla transportu glukozy w tkankach obwodowych transporterów glukozy i jest > 1400 razy bardziej selektywna dla SGLT2 niż dla SGLT1, głównego transportera w przewodzie pokarmowym odpowiedzialnego za wchłanianie glukozy.
Efekty farmakodynamiczne
Zaobserwowano wzrost ilości glukozy wydalonej z moczem u zdrowych osób i u osób z cukrzycą typu 2 po podaniu dapaglifyzyny. Oколо 70 g glukozy dziennie było wydawane z moczem (odpowiada to 280 kcal/dzień) przy dawce dapaglifyzyny 10 mg/dzień u osób z cukrzycą typu 2 przez 12 tygodni. U pacjentów z cukrzycą typu 2 otrzymujących dapaglifyzynę w dawce 10 mg/dzień przez okres do 2 lat zaobserwowano dowody na długotrwałe wydalanie glukozy z moczem.
To wydalanie glukozy z moczem wywołane przez dapaglifyzynę prowadzi również do diurezy osmotycznej i wzrostu objętości moczu u pacjentów z cukrzycą typu 2. Wzrost objętości moczu u pacjentów z cukrzycą typu 2 leczonych dapaglifyzyną w dawce 10 mg utrzymywał się do 12 tygodni i odpowiadał około 375 mL/dzień. Wzrost objętości moczu był związany z niewielkim i przejściowym wzrostem wydalania sodu z moczem, który nie był skojarzony ze zmianami stężenia sodu w surowicy.
Wydalanie kwasu moczowego z moczem wzrastało tymczasowo (przez 3–7 dni) i wiązało się z długotrwałym obniżeniem stężenia kwasu moczowego w surowicy. Po 24 tygodniach zmniejszenie stężenia kwasu moczowego w surowicy wahało się od –48,3 do –18,3 mikromol/L (od –0,87 do –0,33 mg/dL).
Skuteczność i bezpieczeństwo kliniczne
Cukrzyca typu 2
Poprawa kontroli glikemii oraz zmniejszenie współistniejącej choroby sercowo-nerekowej i śmiertelności są integralną częścią leczenia cukrzycy typu 2.
Przeprowadzono czternastu randomizowanych, podwójnie ślepych badań kontrolowanych u 7056 dorosłych pacjentów z cukrzycą typu 2 w celu oceny skuteczności glikemicznej i bezpieczeństwa Dapaglifyzyna Tecnigen; w tych badaniach 4737 pacjentów leczono dapaglifyzyną. Dwanaście badań obejmowało 24-tygodniowy okres leczenia, 8 badań miało długoterminowe fazy przedłużenia od 24 do 80 tygodni (do maksymalnego całkowitego czasu trwania badania wynoszącego 104 tygodnie), jedno badanie miało okres leczenia 28 tygodni, a jedno badanie trwało 52 tygodnie z długoterminowym przedłużeniem do 52 i 104 tygodni (całkowity czas trwania badania wyniósł 208 tygodni). Średni czas trwania cukrzycy wynosił od 1,4 do 16,9 roku. U 50% pacjentów stwierdzono łagodne zaburzenia czynności nerek, a u 11% – umiarkowane zaburzenia czynności nerek. 51% pacjentów stanowili mężczyźni, 84% – rasa biała, 8% – Azjaci, 4% – czarna rasa, 4% – inne grupy rasowe. 81% pacjentów miało wskaźnik masy ciała (BMI, body mass index) ≥ 27. Dodatkowo przeprowadzono dwa 12-tygodniowe, kontrolowane placebo badania u pacjentów z cukrzycą typu 2, u których nie osiągnięto odpowiedniej kontroli glikemii i u których stwierdzono nadciśnienie tętnicze.
Przeprowadzono badanie wyników sercowo-nerekowych (DECLARE) z użyciem dapaglifyzyny 10 mg w porównaniu do placebo u 17 160 pacjentów z cukrzycą typu 2 z obecną lub bez obecnej choroby sercowo-nerekowej w celu oceny wpływu na zdarzenia sercowo-nerekowe.
Kontrola glikemii
Monoterapia
Przeprowadzono jedno 24-tygodniowe, podwójnie ślepe, kontrolowane placebo badanie (z dodatkowym okresem przedłużenia) w celu oceny bezpieczeństwa i skuteczności monoterapii Dapaglifyzyna Tecnigen u pacjentów z cukrzycą typu 2, u których nie osiągnięto odpowiedniej kontroli glikemii. Leczenie dapaglifyzyną raz dziennie prowadziło do statystycznie istotnych zmniejszeń (p < 0,0001) hemoglobiny glikowanej (HbA1c) w porównaniu do placebo (Tabela 2).
W fazie przedłużenia zmniejszenia HbA1c utrzymywały się do 102. tygodnia (średnie skorygowane zmiany od punktu wyjściowego wynosiły odpowiednio –0,61% i –0,17% dla dapaglifyzyny 10 mg i placebo).
Tabela 2. Wyniki po 24 tygodniach (LOCFa) w badaniu klinicznym z dapaglifyzyną w monoterapii kontrolowanym placebo
Monoterapia
Dapaglifyzyna Placebo
10 mg
N 70 75
HbA1c (%)
Wyjściowy (średni) 8,01 7,79
Zmiana od wartości wyjściowejc –0,89 –0,23
Różnica w stosunku do placebo –0,66*
(IC 95%) (–0,96; –0,36)
Pacjenci (%) osiągający:
HbA1c < 7%
Skorygowane dla wartości wyjściowych 50,8§ 31,6
Masa ciała (kg)
Wyjściowa (średnia) 94,13 88,77
Zmiana od wartości wyjściowejc –3,16 –2,19
Różnica w stosunku do placebo –0,97
(IC 95%) (–2,20; 0,25)
Leczenie wspomagające (add-on)
W jednym badaniu ni-inferiorności z kontrolą aktywną, trwającym 52 tygodnie (z przedłużeniem do 52 i 104 tygodni), oceniano Dapaglifyzyna Tecnigen jako leczenie wspomagające do metforminy w porównaniu do sulfonilomocznika (glipizydu) jako leczenia wspomagającego do metforminy u pacjentów z nieadekwatną kontrolą glikemii (HbA1c > 6,5% i ≤ 10%). Wyniki wykazały podobne średnie zmniejszenie HbA1c od wartości wyjściowej do 52. tygodnia w porównaniu z glipizydem, co potwierdza ni-inferiorność leczenia (Tabela 3). Po 104 tygodniach zmiana od wartości wyjściowej średniej HbA1c wynosiła –0,32% dla dapaglifyzyny i –0,14% dla glipizydu. Po 208 tygodniach zmiana od wartości wyjściowej średniej HbA1c wynosiła –0,10% dla dapaglifyzyny i 0,20% dla glipizydu. Po 52, 104 i 208 tygodniach istotnie mniejszy odsetek pacjentów w grupie leczonej dapaglifyzyną (odpowiednio 3,5%, 4,3% i 5,0%) doświadczył co najmniej jednego zdarzenia hipoglikemicznego w porównaniu z grupą leczoną glipizydem (odpowiednio 40,8%, 47,0% i 50,0%). Odsetek pacjentów obecnych w badaniu po 104 i 208 tygodniach wynosił odpowiednio 56,2% i 39,7% w grupie leczonej dapaglifyzyną oraz 50,0% i 34,6% w grupie leczonej glipizydem.
Tabela 3. Wyniki po 52 tygodniach (LOCFa) w badaniu z kontrolą aktywną porównującym dapaglifyzynę z glipizydem jako leczenie wspomagające do metforminy
Dapaglifyzyna Glipizyd
+ metformina + metformina
Parametr
N 400 401
HbA1c (%)
Wyjściowy (średni) 7,69 7,74
Zmiana od wartości wyjściowej –0,52 –0,52
Różnica w stosunku do
glipizydu + metformina 0,00
(IC 95%) (–0,11; 0,11)
Masa ciała (kg)
Wyjściowa (średnia) 88,44 87,60
Zmiana od wartości wyjściowej –3,22 1,44
Różnica w stosunku do
glipizydu + metformina –4,65*
(IC 95%) (–5,14; –4,17)
Dapaglifyzyna dodana do metforminy, glimepirydu, metforminy i sulfonilomocznika, sitagliptyny (z lub bez metforminy) lub insuliny prowadziła do statystycznie istotnych zmniejszeń HbA1c po 24 tygodniach w porównaniu z pacjentami otrzymującymi placebo (p < 0,0001; Tabele 4, 5 i 6).
Zmniejszenia HbA1c obserwowane po 24 tygodniach utrzymywały się w badaniach leczenia wspomagającego (glimepiryd i insulina) na podstawie danych po 48 tygodniach (glimepiryd) i do 104 tygodnia (insulina). Po 48 tygodniach, gdy dodano do sitagliptyny (z lub bez metforminy), skorygowane średnie zmiany od wartości wyjściowej dla dapaglifyzyny 10 mg i placebo wynosiły odpowiednio –0,30% i 0,38%. Według badania leczenia wspomagającego do metforminy, zmniejszenia HbA1c utrzymywały się do 102. tygodnia (skorygowana średnia zmiana od wartości wyjściowej wynosiła odpowiednio –0,78% i 0,02% dla 10 mg i placebo). Po 104 tygodniach dla insuliny (z lub bez doustnych leków hipoglikemizujących) średnie zmniejszenia HbA1c od wartości wyjściowej wynosiły –0,71% i –0,06% odpowiednio dla dapaglifyzyny 10 mg i placebo. Po 48 i 104 tygodniach dawka insuliny pozostała stabilna w stosunku do wartości wyjściowej u pacjentów leczonych dapaglifyzyną 10 mg przy średniej dawce 76 J/dzień. W grupie placebo odnotowano średni wzrost od wartości wyjściowej odpowiednio o 10,5 J/dzień i 18,3 J/dzień (średnia dawka wynosiła 84 i 92 J/dzień) po 48 i 104 tygodniach. Odsetek pacjentów obecnych w badaniu po 104 tygodniach wynosił 72,4% w grupie leczonej dapaglifyzyną 10 mg i 54,8% w grupie placebo.
Tabela 4. Wyniki po 24 tygodniach (LOCFa) badań klinicznych z dapaglifyzyną kontrolowanych placebo w leczeniu wspomagającym z metforminą lub inhibitorami DPP-4 (sitagliptyna) (z lub bez metforminy)
Leczenie wspomagające
Metformina Inhibitory DPP-4
(sitagliptyna) ±
metformina
Dapaglifyzyna Placebo Dapaglifyzyna Placebo
10 mg 10 mg
N b 135 137 223 224
HbA1c (%)
Wyjściowy (średni) 7,92 8,11 7,90 7,97
Zmiana od wartości wyjściowej –0,84 –0,30 –0,45 0,04
Różnica w stosunku do placebo –0,54 –0,48
(IC 95%) (–0,74; –0,34) (–0,62; –0,34)
Pacjenci (%) osiągający:
HbA1c < 7%
Skorygowane dla wartości wyjściowych 40,6 25,9
Masa ciała (kg)
Wyjściowa (średnia) 86,28 87,74 91,02 89,23
Zmiana od wartości wyjściowej –2,86 –0,89 –2,14 –0,26
Różnica w stosunku do placebo –1,97 –1,89
(IC 95%) (–2,63; –1,31) (–2,37; –1,40)
Tabela 5. Wyniki po 24 tygodniach w badaniu klinicznym kontrolowanym placebo dotyczącego stosowania dapaglifyzyny jako leczenia wspomagającego do sulfonilomocznika (glimepirydu) lub metforminy i sulfonilomocznika
Leczenie wspomagające
Sulfonilomocznik (glimepiryd) Sulfonilomocznik + metformina2
Dapaglifyzyna Placebo Dapaglifyzyna Placebo
10 mg 10 mg
N 151 145 108 108
HbA1c (%)
Wyjściowy (średni) 8,07 8,15 8,08 8,24
Zmiana od wartości wyjściowej –0,82 –0,13 –0,86 –0,17
Różnica w stosunku do placebo –0,68* –0,69*
(IC 95%) (–0,86; –0,51) (–0,89; –0,49)
Pacjenci (%) osiągający:
HbA1c < 7%
(LOCF)
Skorygowane dla wartości wyjściowych 31,7* 13,0 31,8* 11,1
Masa ciała (kg)
(LOCF)
Wyjściowa (średnia) 80,56 80,94 88,57 90,07
Zmiana od wartości wyjściowej –2,26 –0,72 –2,65 –0,58
Różnica w stosunku do placebo –1,54* –2,07*
(IC 95%) (–2,17; –0,92) (–2,79; –1,35)
Tabela 6. Wyniki po 24 tygodniach (LOCFa) w badaniu klinicznym kontrolowanym placebo dotyczącego stosowania dapaglifyzyny w połączeniu z insulina (samej lub z doustnymi lekami hipoglikemizującymi)
Dapaglifyzyna 10 mg Placebo
+ insulina + insulina
± doustne leki hipoglikemizujące ± doustne leki hipoglikemizujące
Parametr
N 194 193
HbA1c (%)
Wyjściowy (średni) 8,58 8,46
Zmiana od wartości wyjściowej –0,90 –0,30
Różnica w stosunku do placebo –0,60*
(IC 95%) (–0,74; –0,45)
Masa ciała (kg)
Wyjściowa (średnia) 8,58 94,21
Zmiana od wartości wyjściowej –0,90 0,02
Różnica w stosunku do placebo –0,60*
(IC 95%) (–0,74; –0,45)
Średnia dawka dobową
insuliny (J) 77,96 73,96
Wyjściowa (średnia) –1,16 5,08
Zmiana od wartości wyjściowej
Różnica w stosunku do placebo –6,23*
(IC 95%) (–8,84; –3,63)
Pacjenci z redukcją
średniej dawki dobowej insuliny o co najmniej 10% (%) 19,7** 11,0
W połączeniu z metforminą u pacjentów leczonych po raz pierwszy
Łącznie 1236 pacjentów leczonych po raz pierwszy z cukrzycą typu 2, u których nie osiągnięto odpowiedniej kontroli glikemii (HbA1c ≥ 7,5% i ≤ 12%), wzięło udział w dwóch kontrolowanych badaniach aktywnych trwających 24 tygodnie w celu oceny skuteczności i bezpieczeństwa dapaglifyzyny (5 mg lub 10 mg) w połączeniu z metforminą u pacjentów leczonych po raz pierwszy w porównaniu z leczeniem pojedynczym składnikiem.
Leczenie dapaglifyzyną 10 mg w połączeniu z metforminą (do 2000 mg dziennie) zapewniło istotne poprawy HbA1c w porównaniu z pojedynczymi składnikami (Tabela 7) oraz prowadziło do większego zmniejszenia glikemii na czczo (FPG, fasting plasma glucose) (w porównaniu z pojedynczymi składnikami) i masy ciała (w porównaniu z metforminą).
Tabela 7. Wyniki po 24 tygodniach (LOCFa) w badaniu klinicznym kontrolowanym aktywnie dotyczącego terapii kombinowanej dapaglifyzyny i metforminy u pacjentów leczonych po raz pierwszy
Dapaglifyzyna 10 mg Dapaglifyzyna 10 mg Metformina
+
Parametr metformina
N 211 219 208
HbA1c (%)
Wyjściowy (średni) 9,10 9,03 9,03
Zmiana od wartości wyjściowej –1,98 –1,45 –1,44
Różnica w stosunku do dapaglifyzyny –0,53*
(IC 95%) (–0,74; –0,32)
Różnica w stosunku do metforminy –0,54* –0,01
(IC 95%) (–0,75; –0,33) (–0,22; 0,20)
Terapia kombinowana z egzenatydem o przedłużonym uwalnianiu
W jednym kontrolowanym, 28-tygodniowym, podwójnie ślepych badaniu z lekiem porównawczym, kombinację dapaglifyzyny i egzenatydu o przedłużonym uwalnianiu (agonisty receptora GLP-1) porównano z dapaglifyzyną samą i egzenatydem o przedłużonym uwalnianiu samym u pacjentów z nieadekwatną kontrolą glikemii przy samej metforminie (HbA1c ≥ 8% i ≤ 12%). Wszystkie grupy leczenia wykazały zmniejszenie HbA1c w porównaniu z wartością wyjściową.
Leczenie kombinowane dapaglifyzyną 10 mg i egzenatydem o przedłużonym uwalnianiu wykazało większe zmniejszenie HbA1c od wartości wyjściowej w porównaniu z dapaglifyzyną samą i egzenatydem o przedłużonym uwalnianiu samym (Tabela 8).
Tabela 8: Wyniki 28-tygodniowego badania z dapaglifyzyną i egzenatydem o przedłużonym uwalnianiu w porównaniu z dapaglifyzyną samą i egzenatydem o przedłużonym uwalnianiu samym, w połączeniu z metforminą (pacjenci „intencja do leczenia”)
Dapaglifyzyna 10 mg Dapaglifyzyna 10 mg Egzenatyd o przedłużonym
QD QD uwolnieniu 2 mg
-
- QW
egzenatyd o przedłużonym placebo QW placebo QD
uwolnieniu 2 mg +
Parametr QW
N 228 230 227
HbA1c (%)
Wyjściowy (średni) 9,29 9,25 9,26
Zmiana od wartości wyjściowej –1,98 –1,39 –1,60
Średnia różnica zmiany od wartości wyjściowej między kombinacją a pojedynczym lekiem –0,59* –0,38**
(IC 95%) (–0,84; –0,34) (–0,63; –0,13)
Pacjenci (%) osiągający 44,7 19,1 26,9
HbA1c < 7%
Masa ciała (kg)
Wyjściowa (średnia) 92,13 90,87 89,12
Zmiana od wartości wyjściowej –3,55 –2,22 –1,56
Średnia różnica zmiany od wartości wyjściowej między kombinacją a pojedynczym lekiem –1,33* –2,00*
(IC 95%) (–2,12; –0,55) (–2,79; –1,20)
- QW
Glikemia na czczo
Leczenie dapaglifyzyną 10 mg jako monoterapii lub jako leczenia wspomagającego do metforminy, glimepirydu, metforminy i sulfonilomocznika, sitagliptyny (z lub bez metforminy) lub insuliny prowadziło do statystycznie istotnych zmniejszeń FPG (od –1,90 do –1,20 mmol/L [od –34,2 do –21,7 mg/dL]) w porównaniu do placebo (od –0,33 do 0,21 mmol/L [od –6,0 do 3,8 mg/dL]). Ten efekt zaobserwowano już po 1 tygodniu leczenia i utrzymywał się w badaniach przedłużonych do 104. tygodnia.
Terapia kombinowana z dapaglifyzyną 10 mg i egzenatydem o przedłużonym uwalnianiu prowadziła do istotnie większych zmniejszeń FPG po 28 tygodniach: –3,66 mmol/L (–65,8 mg/dL) w porównaniu do –2,73 mmol/L (–49,2 mg/dL) dla dapaglifyzyny samej (p < 0,001) i –2,54 mmol/L (–45,8 mg/dL) dla egzenatydu samego (p < 0,001).
W jednym badaniu specjalistycznym u chorych cukrzycowych z eGFR od ≥ 45 do < 60 mL/min/1,73 m² leczenie dapaglifyzyną wykazało zmniejszenie FPG po 24 tygodniach: –1,19 mmol/L (–21,46 mg/dL) w porównaniu do –0,27 mmol/L (–4,87 mg/dL) dla placebo (p=0,001).
Glikemia pożywieniowa
Leczenie dapaglifyzyną 10 mg jako leczenie wspomagające do glimepirydu prowadziło po 24 tygodniach do statystycznie istotnych zmniejszeń glikemii pożywieniowej mierzonej po 2 godzinach, które utrzymywały się do 48. tygodnia.
Leczenie dapaglifyzyną 10 mg jako leczenie wspomagające do sitagliptyny (z lub bez metforminy) prowadziło po 24 tygodniach do zmniejszeń glikemii pożywieniowej mierzonej po 2 godzinach, które utrzymywały się do 48. tygodnia.
Terapia kombinowana z dapaglifyzyną 10 mg i egzenatydem o przedłużonym uwalnianiu prowadziła do istotnie większych zmniejszeń glikemii pożywieniowej po 2 godzinach po posiłku po 28 tygodniach w porównaniu z pojedynczym lekiem.
Masa ciała
Dapaglifyzyna 10 mg jako leczenie wspomagające do metforminy, glimepirydu, metforminy i sulfonilomocznika, sitagliptyny (z lub bez metforminy) lub insuliny prowadziła do statystycznie istotnego zmniejszenia masy ciała po 24 tygodniach (p < 0,0001, Tabele 4 i 5). Te efekty utrzymywały się w długoterminowych badaniach klinicznych. Po 48 tygodniach różnica dla dapaglifyzyny w połączeniu z sitagliptyną (z lub bez metforminy) w stosunku do placebo wynosiła –2,22 kg. Po 102 tygodniach różnica dla dapaglifyzyny w połączeniu z metforminą w stosunku do placebo lub w połączeniu z insulina w stosunku do placebo wynosiła odpowiednio –2,14 i –2,88 kg.
Jako leczenie wspomagające do metforminy w badaniu ni-inferiorności z kontrolą aktywną, dapaglifyzyna prowadziła do statystycznie istotnego zmniejszenia masy ciała w porównaniu z glipizydem o –4,65 kg po 52 tygodniach (p < 0,0001, Tabela 3), które utrzymywało się po 104 i 208 tygodniach (–5,06 kg i –4,38 kg odpowiednio).
Kombinacja dapaglifyzyny 10 mg i egzenatydu o przedłużonym uwalnianiu wykazała istotnie większe zmniejszenia masy ciała w porównaniu z pojedynczym lekiem (Tabela 8).
W jednym 24-tygodniowym badaniu klinicznym u 182 pacjentów cukrzycowych z zastosowaniem podwójnej absorpcyjności rentgenowskiej (DXA, X-ray absorptiometry) w celu oceny składu masy ciała zaobserwowano zmniejszenie masy ciała i masy tłuszczowej ciała przy dapaglifyzynie 10 mg w połączeniu z metforminą w porównaniu do placebo w połączeniu z metforminą, mierzone metodą DXA, a nie masy beztłuszczowej lub utraty płynów. Leczenie Dapaglifyzyna Tecnigen w połączeniu z metforminą prowadziło do zmniejszenia tkanki tłuszczowej wątrobowej w porównaniu do placebo w połączeniu z metforminą w badaniu podgrupowym z użyciem obrazowania rezonansu magnetycznego.
Ciśnienie krwi
W predefiniowanej analizie agregowanej 13 badań kontrolowanych placebo leczenie dapaglifyzyną 10 mg prowadziło do zmiany od wartości wyjściowej ciśnienia tętniczego skurczowego o –3,7 mmHg i ciśnienia tętniczego rozkurczowego o –1,8 mmHg w porównaniu do –0,5 mmHg (ciśnienie tętnicze skurczowe) i –0,5 mmHg (ciśnienie tętnicze rozkurczowe) dla grupy placebo po 24 tygodniach. Podobne zmniejszenia obserwowano do 104. tygodnia.
Terapia kombinowana z dapaglifyzyną 10 mg i egzenatydem o przedłużonym uwalnianiu prowadziła do istotnie większego zmniejszenia ciśnienia tętniczego skurczowego po 28 tygodniach (–4,3 mmHg) w porównaniu z dapaglifyzyną samą (–1,8 mmHg, p < 0,05) i egzenatydem o przedłużonym uwalnianiu samym (–1,2 mmHg, p < 0,01).
W dwóch 12-tygodniowych badaniach kontrolowanych placebo, łącznie 1062 pacjentów z cukrzycą typu 2, u których nie osiągnięto odpowiedniej kontroli i u których stwierdzono nadciśnienie tętnicze (mimo wcześniejszego stabilnego leczenia inhibitory ACE lub blokerami receptora angiotensyny w jednym badaniu i inhibitory ACE lub blokerami receptora angiotensyny plus inne leczenie przeciw nadciśnieniowe w drugim badaniu) leczono dapaglifyzyną 10 mg lub placebo. Po 12 tygodniach w obu badaniach dapaglifyzyna 10 mg w połączeniu z常规nym leczeniem przeciw cukrzycy prowadziła do poprawy HbA1c i zmniejszenia skorygowanego dla placebo ciśnienia tętniczego skurczowego średnio o 3,1 i 4,3 mmHg odpowiednio.
W jednym badaniu specjalistycznym u pacjentów cukrzycowych z eGFR od ≥ 45 do < 60 mL/min/1,73 m² leczenie dapaglifyzyną wykazało zmniejszenie ciśnienia tętniczego skurczowego po 24 tygodniach: –4,8 mmHg w porównaniu do –1,7 mmHg dla placebo (p < 0,05).
Kontrola glikemii u pacjentów z umiarkowanym zaburzeniem czynności nerek CKD 3A
(eGFR od ≥ 45 do < 60 mL/min/1,73 m²)
Skuteczność dapaglifyzyny oceniono w jednym badaniu specjalistycznym u pacjentów cukrzycowych z eGFR ≥ 45 do < 60 mL/min/1,73 m², u których nie osiągnięto odpowiedniej kontroli glikemii przy常规nym leczeniu. Leczenie dapaglifyzyną prowadziło do zmniejszenia HbA1c i masy ciała w porównaniu do placebo (Tabela 9).
Tabela 9. Wyniki po 24 tygodniach w badaniu z dapaglifyzyną kontrolowanym placebo u pacjentów cukrzycowych z eGFR ≥ 45 do < 60 mL/min/1,73 m²
Dapaglifyzyna Placebo
10 mg
N 159 161
HbA1c (%)
Wartość wyjściowa (średnia) 8,35 8,03
Zmiana od wartości wyjściowej –0,37 –0,03
Różnica w stosunku do placebo –0,34*
(IC 95%) (–0,53; –0,15)
Masa ciała (kg)
Wartość wyjściowa (średnia) 92,51 88,30
Procentowa zmiana od wartości wyjściowej –3,42 –2,02
Różnica procentowa w stosunku do placebo –1,43*
(IC 95%) (–2,15; –0,69)
Pacjenci z HbA1c na poziomie wyjściowym ≥ 9%
W predefiniowanej analizie pacjentów z HbA1c na poziomie wyjściowym ≥ 9,0% leczenie dapaglifyzyną 10 mg jako monoterapia prowadziło do statystycznie istotnych zmniejszeń HbA1c po 24 tygodniach (skorygowana średnia zmiana od wartości wyjściowej: –2,04% i 0,19% dla dapaglifyzyny 10 mg i placebo odpowiednio) oraz w leczeniu wspomagającym do metforminy (skorygowana średnia zmiana od wartości wyjściowej: –1,32% i –0,53% dla dapaglifyzyny i placebo odpowiednio).
Wyniki sercowo-nerekowe
Badanie wpływu dapaglifyzyny na zdarzenia sercowo-nerekowe (DECLARE) to międzynarodowe, wieloośrodkowe, randomizowane, podwójnie ślepe, kontrolowane placebo badanie mające na celu określenie wpływu dapaglifyzyny w porównaniu do placebo na wyniki sercowo-nerekowe, gdy jest dodawana do常规nego leczenia. Wszyscy pacjenci mieli cukrzycę typu 2 i co najmniej dwa dodatkowe czynniki ryzyka sercowo-nerekowego (wiek ≥ 55 lat u mężczyzn lub ≥ 60 lat u kobiet i co najmniej jedno z: dyslipidemia, nadciśnienie tętnicze lub palenie tytoniu) lub potwierdzoną chorobę sercowo-nerekową.
Spośród 17 160 pacjentów zakwalifikowanych do badania, 6974 (40,6%) miało potwierdzoną chorobę sercowo-nerekową, a 10 186 (59,4%) nie miało znanej choroby sercowo-nerekowej. 8582 pacjentów zostało losowo przydzielonych do grupy otrzymującej dapaglifyzynę 10 mg, a 8578 do grupy placebo i obserwowano ich przez średni czas 4,2 roku.
Średni wiek populacji badawczej wynosił 63,9 roku, kobiety stanowiły 37,4%. Łącznie 22,4% pacjentów miało rozpoznanie cukrzycy ≤ 5 lat, średni czas trwania cukrzycy wynosił 11,9 roku. Średnie HbA1c wynosiło 8,3%, a średnie BMI – 32,1 kg/m².
Na początku badania 10,0% pacjentów miało w wywiadzie niewydolność serca. Średnie eGFR wynosiło 85,2 mL/min/1,73 m², u 7,4% pacjentów eGFR < 60 mL/min/1,73 m², a u 30,3% pacjentów stwierdzono mikro- lub makroalbuminurię (ACR ≥ 30 do ≤ 300 mg/g lub > 300 mg/g odpowiednio).
Wielu pacjentów (98%) stosowało jeden lub więcej leków na cukrzycę na początku badania, w tym metforminę (82%), insulinę (41%) i sulfonilomoczniki (43%).
Pierwotnymi punktami końcowymi były czas do pierwszego zdarzenia w złożonym punkcie końcowym śmierci sercowo-nerekowej, zawału serca lub udaru niedokrwiennego (MACE) oraz czas do pierwszego zdarzenia hospitalizacji z powodu niewydolności serca lub śmierci sercowo-nerekowej. Punkty końcowe wtórne to złożony punkt końcowy nerek i śmiertelność z dowolnej przyczyny.
Zdarzenia sercowo-nerekowe
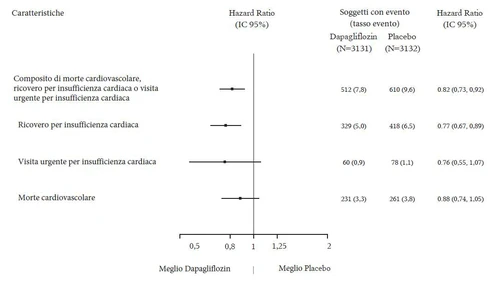
Dapaglifyzyna 10 mg wykazała ni-inferiorność w porównaniu do placebo dla złożonego punktu końcowego śmierci sercowo-nerekowej, zawału mięśnia sercowego i udaru niedokrwiennego (p jednostronnie < 0,001).
Niewydolność serca i śmierć sercowo-nerekowa
Dapaglifyzyna 10 mg wykazała przewagę nad placebo w zapobieganiu złożonemu punktowi końcowemu hospitalizacji z powodu niewydolności serca lub śmierci sercowo-nerekowej (Rysunek 1). Różnica w efekcie terapeutycznym była napędzana hospitalizacją z powodu niewydolności serca, bez różnicy w śmierci sercowo-nerekowej (Rysunek 2).
Korzyść z leczenia dapaglifyzyną w porównaniu do placebo obserwowano u pacjentów z potwierdzoną chorobą sercowo-nerekową i bez niej oraz z niewydolnością serca na początku badania i bez niej i była spójna w podgrupach, w tym wieku, płci, czynności nerek (eGFR) i regionie.
Rysunek 1: Czas do pierwszego zdarzenia hospitalizacji z powodu niewydolności serca lub śmierci sercowo-nerekowej.
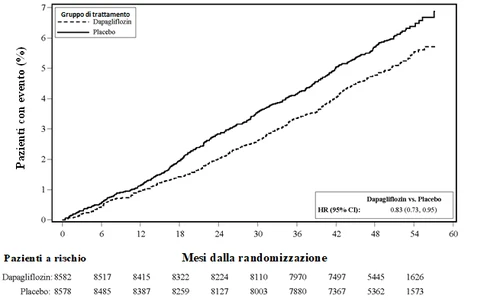
Pacjenci narażeni to liczba pacjentów narażonych na początku okresu.
HR = Hazard ratio IC = przedział ufności.
Wyniki punktów końcowych pierwotnych i wtórnych przedstawiono na Rysunku 2. Przewaga dapaglifyzyny nad placebo nie została wykazana dla MACE (p=0,172). Złożony punkt końcowy nerek i śmiertelność z dowolnej przyczyny nie zostały zatem testowane w ramach procedury potwierdzającej testowanie.
Rysunek 2: Efekty leczenia dla pierwotnych złożonych punktów końcowych i ich składowych oraz punktów końcowych wtórnych
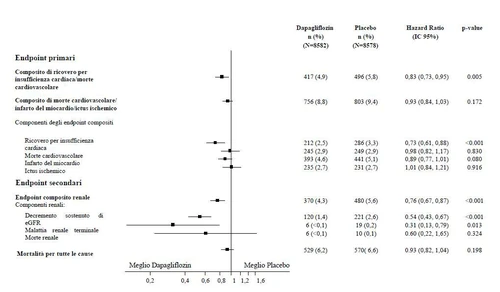
Złożony punkt końcowy nerek zdefiniowano jako: utrzymujące się i potwierdzone zmniejszenie eGFR ≥ 40%, eGFR <60 mL/min/1,73 m² i/lub końcowy etap choroby nerek (dializa ≥ 90 dni lub przeszczep nerki, utrzymujące się i potwierdzone eGFR < 15 mL/min/1,73 m²) i/lub śmierć nerek lub sercowo-nerekową.
Wartości p były dwustronne. Wartości p dla punktów końcowych wtórnych i pojedynczych składowych są nominalne. Czas do pierwszego zdarzenia analizowano w modelu proporcjonalnych ryzyków Coxa. Liczba pierwszych zdarzeń niepożądanych dla pojedynczych składowych to rzeczywista liczba pierwszych zdarzeń dla każdego składowego i nie sumuje się do liczby zdarzeń w złożonym punkcie końcowym.
IC = przedział ufności.
Nefropatia
Dapaglifyzyna zmniejszyła częstość występowania zdarzeń w złożonym punkcie końcowym: utrzymującego się i potwierdzonego zmniejszenia eGFR, końcowego stadium choroby nerek, śmierci nerek lub sercowo-nerekowej. Różnica między grupami była napędzana zmniejszeniem zdarzeń składowych nerek: utrzymującego się zmniejszenia eGFR, końcowego stadium choroby nerek i śmierci nerek (Rysunek 2).
Wskaźnik ryzyka (HR) dla czasu do nefropatii (utrzymujące się zmniejszenie eGFR, końcowe stadium choroby nerek i śmierć nerek) wynosił 0,53 (95% IC 0,43; 0,66) dla dapaglifyzyny w porównaniu do placebo.
Dodatkowo dapaglifyzyna zmniejszyła nowe wystąpienie utrzymującej się albuminurii (HR 0,79 [95% IC 0,72; 0,87]) i prowadziła do większej regresji makroalbuminurii (HR 1,82 [95% IC 1,51; 2,20]) w porównaniu z placebo.
Niewydolność serca
Badanie DAPA-HF: Niewydolność serca z frakcją wyrzutową ≤ 40% (LVEF ≤ 40%)
Dapaglifyzyna i zapobieganie niekorzystnym wynikom u pacjentów z niewydolnością serca (DAPA-HF) to międzynarodowe, wieloośrodkowe, randomizowane, podwójnie ślepe, kontrolowane placebo badanie u pacjentów z niewydolnością serca (New York Heart Association [NYHA] klasa czynnościowa II–IV) z obniżoną frakcją wyrzutową (frakcja wyrzutowa lewej komory [LVEF] ≤ 40%) mające na celu określenie wpływu dapaglifyzyny w porównaniu do placebo, gdy jest dodawana do常规nego leczenia, na częstość występowania śmierci sercowo-nerekowej i pogorszenia niewydolności serca.
Spośród 4744 pacjentów, 2373 zostało losowo przydzielonych do grupy otrzymującej dapaglifyzynę 10 mg, a 2371 do grupy placebo i obserwowano ich przez medianę 18 miesięcy. Średni wiek populacji badawczej wynosił 66 lat, 77% stanowili mężczyźni.
Na początku badania 67,5% pacjentów sklasyfikowano jako NYHA klasa II, 31,6% jako klasa III i 0,9% jako klasa IV, mediana LVEF wynosiła 32%, 56% niewydolności serca miało pochodzenie izemiczne, 36% miało pochodzenie nieizemiczne, a 8% miało nieznaną etiologię. W każdej grupie leczenia 42% pacjentów miało w wywiadie cukrzycę typu 2, a dodatkowe 3% pacjentów w każdej grupie sklasyfikowano jako mających cukrzycę typu 2 na podstawie HbA1c ≥ 6,5% zmierzonego zarówno przy zapisie, jak i randomizacji. Pacjenci byli leczeni zgodnie z standardem opieki nad niewydolnością serca; 94% pacjentów otrzymywało inhibitory ACE, ARB lub inhibitory receptora angiotensyny-neprylizyny (ARNI, 11%), 96% beta-blokery, 71% antagonistów receptora mineralokortykoidowego (MRA), 93% diuretyki, a 26% miało wszczepiony urządzenie (z funkcją defibrylatora).
Pacjentów z eGFR ≥ 30 mL/min/1,73 m² włączono do badania w momencie zapisu. Średnie eGFR wynosiło 66 mL/min/1,73 m², u 41% pacjentów eGFR < 60 mL/min/1,73 m², a u 15% eGFR < 45 mL/min/1,73 m².
Śmierć sercowo-nerekowa i pogorszenie niewydolności serca
Dapaglifyzyna była lepsza od placebo w zapobieganiu pierwotnemu złożonemu punktowi końcowemu śmierci sercowo-nerekowej, hospitalizacji z powodu niewydolności serca lub wizyty pilnej z powodu niewydolności serca (HR 0,74 [95% CI 0,65; 0,85], p < 0,0001). Efekt zaobserwowano wcześnie i utrzymywał się przez cały czas trwania badania (Rysunek 3).
Rysunek 3: Czas do pierwszego zdarzenia w pierwotnym złożonym punkcie końcowym śmierci sercowo-nerekowej, hospitalizacji z powodu niewydolności serca lub wizyty pilnej z powodu niewydolności serca
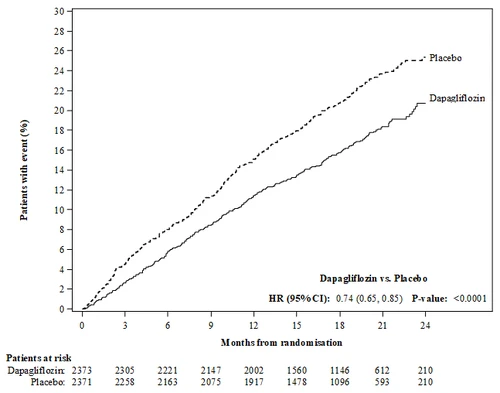
Pacjenci z zdarzeniem (%)
Miesiące od randomizacji
Wszystkie trzy składowe pierwotnego złożonego punktu końcowego indywidualnie przyczyniły się do efektu leczenia (Rysunek 4). Było niewiele wizyt pilnych z powodu niewydolności serca.
Rysunek 4: Efekty leczenia dla pierwotnego złożonego punktu końcowego, jego składowych i śmiertelności z dowolnej przyczyny
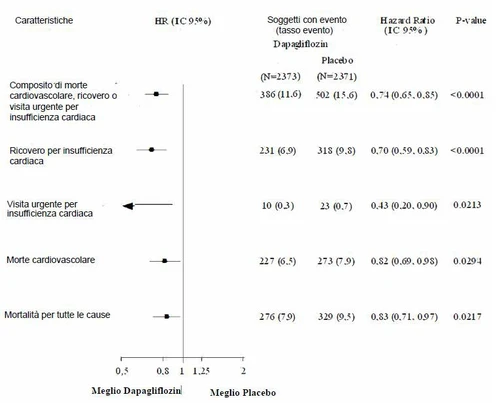
Dapaglifyzyna zmniejszyła również całkowitą liczbę zdarzeń hospitalizacji z powodu niewydolności serca (pierwszych i nawrotowych) i śmierci sercowo-nerekowej; było 567 zdarzeń w grupie dapaglifyzyny i 742 zdarzenia w grupie placebo (Rate Ratio 0,75 [95% CI 0,65; 0,88]; p=0,0002).
Korzyść z leczenia dapaglifyzyną była obserwowana u pacjentów z niewydolnością serca zarówno z cukrzycą typu 2, jak i bez niej. Dapaglifyzyna zmniejszyła pierwotny złożony punkt końcowy wystąpienia śmierci sercowo-nerekowej i pogorszenia niewydolności serca z HR 0,75 (95% CI 0,63; 0,90) u pacjentów z cukrzycą i 0,73 (95% CI 0,60; 0,88) u pacjentów bez cukrzycy.
Korzyść z leczenia dapaglifyzyną w stosunku do placebo dla pierwotnego punktu końcowego była spójna również w innych kluczowych podgrupach, w tym tych z jednoczesnym leczeniem niewydolności serca, czynnością nerek (eGFR), wiekiem, płcią i regionem.
Wynik zgłoszony przez pacjenta – objawy niewydolności serca
Efekt leczenia dapaglifyzyną na objawy niewydolności serca oceniano za pomocą całkowitego wyniku objawów z Kwestionariusza Kansas City Cardiomyopathy (KCCQ-TSS), który ilościowo określa częstość i nasilenie objawów niewydolności serca, w tym zmęczenia, obrzęku obwodowego, duszności i ortopnei. Wynik waha się od 0 do 100, przy czym wyższe wyniki oznaczają lepsze stany zdrowia.
Leczenie dapaglifyzyną prowadziło do statystycznie i klinicznie istotnej korzyści w porównaniu do placebo w objawach niewydolności serca, mierzonej zmianą od wartości wyjściowej do 8. miesiąca w KCCQ-TSS (Win Ratio 1,18 [95% CI 1,11; 1,26]; p < 0,0001).
Na wyniki wpłynęły zarówno częstość, jak i nasilenie objawów. Korzyść była widoczna zarówno w poprawie objawów niewydolności serca, jak i w zapobieganiu ich pogorszeniu.
W analizie pacjentów odpowiadających na leczenie, odsetek pacjentów z klinicznie istotną poprawą w KCCQ-TSS w stosunku do wartości wyjściowej po 8 miesiącach, zdefiniowaną jako wzrost o 5 punktów lub więcej, był wyższy w grupie leczonej dapaglifyzyną w porównaniu do placebo. Odsetek pacjentów z klinicznie istotnym pogorszeniem, zdefiniowanym jako spadek o 5 punktów lub więcej, był niższy w grupie leczonej dapaglifyzyną w porównaniu do placebo. Zaobserwowane korzyści z dapaglifyzyny utrzymywały się przy zastosowaniu bardziej konserwatywnych granic dla klinicznie istotniejszych zmian (Tabela 10).
Tabela 10. Liczba i odsetek pacjentów z klinicznie istotną poprawą i pogorszeniem w KCCQ-TSS po 8 miesiącach
| Zmiana względem wartości wyjściowej po 8 miesiącach: | Dapagliflozyna 10 mg n=2086 | Placebo n=2062 | ||
| Poprawa | n (%) poprawyb | n (%) poprawyb | Stosunek szansC (95% CI) | p-valuef |
| ≥ 5 punktów | 933 (44,7) | 794 (38,5) | 1,14 (1,06; 1,22) | 0,0002 |
| ≥ 10 punktów | 689 (33,0) | 579 (28,1) | 1,13 (1,05; 1,22) | 0,0018 |
| ≥ 15 punktów | 474 (22,7) | 406 (19,7) | 1,10 (1,01; 1,19) | 0,0300 |
| Pogorszenie | n (%) z pogorszeniemd | n (%) z pogorszeniemd | Stosunek szanse (95% CI) | p-valuef |
| ≥ 5 punktów | 537 (25,7) | 693 (33,6) | 0,84 (0,78; 0,89) | <0,0001 |
| ≥ 10 punktów | 395 (18,9) | 506 (24,5) | 0,85 (0,79; 0,92) | <0,0001 |
Nefropatia
Zarejestrowano niewiele zdarzeń w ramach złożonego punktu końcowego nerkowego (potwierdzony i utrzymujący się spadek eGFR ≥ 50%, ESKD lub śmierć wywołana niewydolnością nerek); częstość wyniosła 1,2% w grupie dapaglifyzyny i 1,6% w grupie placebo.
Badanie DELIVER: niewydolność serca z frakcją wyrzutową lewej komory > 40%
Badanie Dapagliflozin Evaluation to Improve the LIVEs of Patients with PReserved Ejection Fraction Heart Failure (DELIVER) było międzynarodowym, wieloośrodkowym, randomizowanym, podwójnie ślepym, kontrolowanym placebo badaniem przeprowadzonym u pacjentów w wieku ≥ 40 lat z niewydolnością serca (klasa NYHA II–IV) z LVEF > 40% oraz dowodami strukturalnej choroby serca, mającym na celu określenie wpływu dapaglifyzyny w porównaniu z placebo na częstość występowania zgonu z przyczyn sercowo-naczyniowych oraz nasilenia niewydolności serca. Spośród 6263 pacjentów, 3131 zostało losowo przydzielonych do grupy otrzymującej dapaglifyzynę, a 3132 do grupy placebo i obserwowanych przez medianę 28 miesięcy. Badanie uwzględniło 654 (10%) pacjentów z niewydolnością serca subostrą (określoną jako losowanie podczas hospitalizacji z powodu niewydolności serca lub w ciągu 30 dni od wypisu). Średni wiek populacji badawczej wynosił 72 lata, a mężczyźni stanowili 56%.
Na początku badania 75% pacjentów sklasyfikowano jako klasa NYHA II, 24% jako klasa III, a 0,3% jako klasa IV. Mediana LVEF wynosiła 54%, 34% pacjentów miało LVEF ≤ 49%, 36% miało LVEF 50–59%, a 30% miało LVEF ≥ 60%. W każdej grupie leczenia 45% pacjentów miało w wywiadzie cukrzycę typu 2. Standardowa terapia obejmowała ACEI/ARB/ARNI (77%), blokery beta (83%), diuretyki (98%) oraz MRA (43%).
Średnie eGFR wynosiło 61 mL/min/1,73 m², u 49% pacjentów eGFR było < 60 mL/min/1,73 m², u 23% eGFR < 45 mL/min/1,73 m², a u 3% eGFR < 30 mL/min/1,73 m².
Dapaglifyzyna okazała się lepsza niż placebo w redukcji częstości występowania głównego złożonego punktu końcowego, obejmującego zgon z przyczyn sercowo-naczyniowych, hospitalizację z powodu niewydolności serca lub wizytę w nagłej opiece z powodu niewydolności serca (HR 0,82 [95% CI 0,73, 0,92]; p=0,0008) (Rysunek 5).
Rysunek 5: Czas do pierwszego wystąpienia złożonego punktu końcowego obejmującego zgon z przyczyn sercowo-naczyniowych, hospitalizację z powodu niewydolności serca lub wizytę w nagłej opiece z powodu niewydolności serca
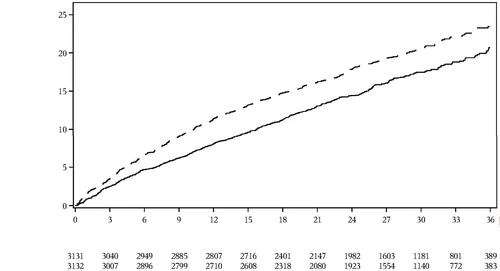

Wizytę w nagłej opiece z powodu niewydolności serca zdefiniowano jako nagłą, nieplanowaną ocenę przeprowadzoną przez lekarza, np. w izbie przyjęć, wymagającą leczenia z powodu
| Niewydolność serca (różne od prostego podania doustnych moczopędnych) | Miesiące od randomizacji |
| pogorszenie u pacjentów z ryzykiem Dapagliflozin: | nasilenie niewydolności serca (inne |
| ( |
Pilne wizyty z powodu niewydolności serca definiowano jako pilną i nieplanowaną ocenę przez lekarza, np. w izbie przyjęć, wymagającą leczenia z powodu nasilenia niewydolności serca (innych niż prosta korekta doustnych moczopędnych).
Pacjenci poddani ryzyku: liczba pacjentów poddanych ryzyku na początku okresu.
Rysunek 6 przedstawia wkład trzech składowych pierwotnego punktu końcowego złożonego na działanie leczenia.
Rysunek 6: Działanie leczenia na pierwotny punkt końcowy złożony i jego składowe
Pilne wizyty z powodu niewydolności serca definiowano jako pilną i nieplanowaną ocenę przez lekarza, np. w izbie przyjęć, wymagającą leczenia z powodu nasilenia niewydolności serca (innych niż prosta korekta doustnych moczopędnych).
Liczba pierwszych zdarzeń dla poszczególnych składowych to rzeczywista liczba pierwszych zdarzeń dla każdej składowej i nie sumuje się do liczby zdarzeń w punkcie końcowym złożonym.
Wskazniki zdarzeń przedstawiono jako liczbę przypadków na 100 pacjentów-rok obserwacji ( follow-up ). Śmiertelność sercowo-naczyniowa, przedstawiona tutaj jako składowa pierwotnego punktu końcowego, była testowana również pod formalną kontrolą błędu typu 1 jako wtórny punkt końcowy.
Dapaglifyzyna okazała się skuteczniejsza niż placebo w zmniejszaniu całkowitej liczby zdarzeń niewydolności serca (określonych jako hospitalizacja, pierwsze i nawrotowe przypadki hospitalizacji z powodu niewydolności serca lub pilne wizyty z powodu niewydolności serca) oraz śmiertelności sercowo-naczyniowej; wystąpiło 815 zdarzeń w grupie dapaglifyzyny w porównaniu do 1057 zdarzeń w grupie placebo (Rate Ratio 0,77 [95% CI 0,67, 0,89]; p=0,0003).
Korzyści z leczenia dapaglifyzyną w porównaniu do placebo w przypadku pierwotnego punktu końcowego obserwowano u podgrup pacjentów z LVEF ≤ 49%, 50-59% i ≥ 60%. Działania były spójne również w innych kluczowych podgrupach, takich jak wiek, płeć, klasa NYHA, poziom NT-proBNP, stan podostrej choroby i stan cukrzycy typu 2.
Wynik zgłoszony przez pacjenta – objawy niewydolności serca
Leczenie dapaglifyzyną przyniosło statystycznie istotne korzyści w porównaniu do placebo w zakresie objawów niewydolności serca, mierzonej jako zmiana w KCCQ-TSS w 8. miesiącu w porównaniu do stanu wyjściowego (Win Ratio 1,11 [95% CI 1,03, 1,21]; p=0,0086). Na wynik wpływała zarówno częstotliwość objawów, jak i ich nasilenie.
W analizach odpowiedzi, odsetek pacjentów, u których doszło do umiarkowanego (≥ 5 punktów) lub dużego (≥ 14 punktów) pogorszenia KCCQ-TSS w 8. miesiącu w porównaniu do stanu wyjściowego, był niższy w grupie leczonej dapaglifyzyną; 24,1% pacjentów leczonych dapaglifyzyną w porównaniu do 29,1% z placebo doświadczyło umiarkowanego pogorszenia (Odds Ratio 0,78 [95% CI 0,64, 0,95]) i 13,5% pacjentów leczonych dapaglifyzyną w porównaniu do 18,4% z placebo doświadczyło dużego pogorszenia (Odds Ratio 0,70 [95 %CI 0,55, 0,88]). Odsetek pacjentów z niewielką do umiarkowaną poprawą (≥ 13 punktów) lub dużą poprawą (≥ 17 punktów) nie różnił się między grupami leczenia.
Niewydolność serca w badaniach DAPA-HF i DELIVER
W analizie skumulowanej badań DAPA-HF i DELIVER, HR dla dapaglifyzyny w porównaniu do placebo w punkcie końcowym złożonym obejmującym śmiertelność sercowo-naczyniową, hospitalizację z powodu niewydolności serca lub pilną wizytę z powodu niewydolności serca wynosił 0,78 (95% CI 0,72, 0,85), p < 0,0001. Działanie leczenia było spójne w całym zakresie LVEF, bez osłabienia działania w zależności od LVEF.
W predefiniowanej analizie skumulowanej na poziomie pacjenta badań DAPA-HF i DELIVER, dapaglifyzyna w porównaniu do placebo zmniejszyła ryzyko śmiertelności sercowo-naczyniowej (HR 0,85 [95% CI 0,75, 0,96], p=0,0115). Oba badania przyczyniły się do efektu.
Przewlekła choroba nerek
Badanie mające na celu ocenę wpływu dapaglifyzyny na wyniki nerkowe i śmiertelność sercowo-naczyniową u pacjentów z przewlekłą chorobą nerek (DAPA-CKD) było międzynarodowym, wieloośrodkowym, randomizowanym, podwójnie ślepym, kontrolowanym placebo badaniem przeprowadzonym u pacjentów z przewlekłą chorobą nerek (CKD) z eGFR od ≥ 25 do 75 mL/min/1,73 m² i albuminurią (ACR ≥ 200 i ≤ 5000 mg/g), mającym na celu określenie w porównaniu do placebo wpływu dapaglifyzyny, dodanej do standardowego leczenia podstawowego, na wystąpienie złożonego punktu końcowego obejmującego trwałe obniżenie eGFR ≥ 50%, zaawansowaną chorobę nerek (ESKD) (określoną jako trwałe eGFR < 15 mL/min/1,73 m², przewlekła dializa lub przeszczep nerki), śmiertelność sercowo-naczyniową lub śmiertelność nerkową.
Spośród 4304 pacjentów, 2152 zostało zrandomizowanych do grupy dapaglifyzyny 10 mg, a 2152 do grupy placebo i obserwowanych przez medianę 28,5 miesiąca. Leczenie było kontynuowane, jeśli podczas badania eGFR spadło poniżej 25 mL/min/1,73 m² i mogło być kontynuowane w przypadkach wymagających dializy.
Średni wiek populacji badanej wynosił 61,8 roku, 66,9% stanowili mężczyźni. Na początku badania średnie eGFR wynosiło 43,1 mL/min/1,73 m², a medianę ACR 949,3 mg/g; 44,1% pacjentów miało eGFR od 30 do < 45 mL/min/1,73 m², a 14,5% miało eGFR < 30 mL/min/1,73 m². 67,5% pacjentów miało cukrzycę typu 2. Pacjenci otrzymywali standardowe leczenie (SOC); 97,0% pacjentów leczono inhibitorem konwertującego enzymu angiotensyny (ACEi) lub blokerem receptora angiotensyny (ARB).
Badanie zostało zakończone przedwcześnie z powodu skuteczności, przed zaplanowaną analizą, na podstawie rekomendacji Niezależnego Komitetu Monitorującego Dane. Dapaglifyzyna okazała się skuteczniejsza niż placebo w zapobieganiu pierwotnemu złożonemu punktowi końcowemu obejmującemu trwałe obniżenie eGFR ≥ 50%, osiągnięcie zaawansowanego stadium choroby nerek, śmiertelność sercowo-naczyniową lub śmiertelność nerkową. Na podstawie krzywej przeżycia Kaplana-Meiera, działanie leczenia było widoczne już od 4. miesiąca i utrzymywało się do końca badania (Rysunek 7).
Rysunek 7: Czas do osiągnięcia pierwotnego punktu końcowego złożonego, obejmującego trwałe obniżenie eGFR ≥ 50%, zaawansowaną chorobę nerek, śmiertelność sercowo-naczyniową lub śmiertelność nerkową
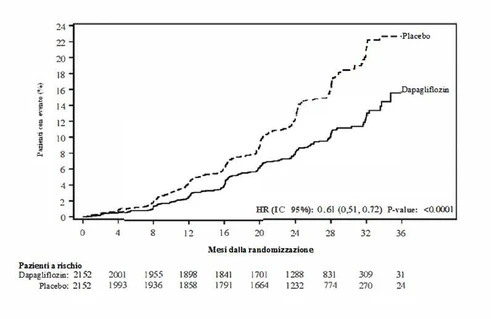
Wszystkie cztery składowe pierwotnego punktu końcowego złożonego indywidualnie przyczyniły się do działania leczenia. Dapaglifyzyna zmniejszyła również częstość występowania złożonego punktu końcowego obejmującego trwałe obniżenie eGFR ≥ 50%, zaawansowaną chorobę nerek lub śmiertelność nerkową oraz złożonego punktu końcowego obejmującego śmiertelność sercowo-naczyniową i hospitalizację z powodu niewydolności serca. U pacjentów z przewlekłą chorobą nerek leczenie dapaglifyzyną poprawiło całkowitą przeżywalność, z istotnym zmniejszeniem śmiertelności z powodu wszystkich przyczyn (Rysunek 8).
Rysunek 8: Działanie leczenia na pierwotne i wtórne punkty końcowe złożone, ich poszczególne składowe oraz śmiertelność z powodu wszystkich przyczyn
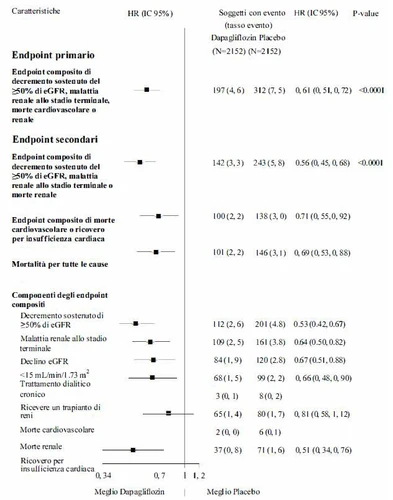
Korzyści z leczenia dapaglifyzyną były spójne u pacjentów z przewlekłą chorobą nerek z cukrzycą typu 2 i bez cukrzycy. Dapaglifyzyna zmniejszyła pierwotny punkt końcowy złożony obejmujący trwałe obniżenie eGFR ≥ 50%, zaawansowaną chorobę nerek, śmiertelność sercowo-naczyniową lub śmiertelność nerkową z HR 0,64 (95% CI 0,52; 0,79) u pacjentów z cukrzycą typu 2 i 0,50 (95% CI 0,35; 0,72) u pacjentów bez cukrzycy.
Korzyści z leczenia dapaglifyzyną w porównaniu do placebo w przypadku pierwotnego punktu końcowego były również spójne w innych kluczowych podgrupach, w tym w zależności od poziomu eGFR, wieku, płci i regionu.
Populacja pediatryczna
Cukrzyca typu 2
W badaniu klinicznym u dzieci i nastolatków w wieku od 10 do 24 lat z cukrzycą typu 2, 39 pacjentów zostało zrandomizowanych do grupy dapaglifyzyny 10 mg, a 33 do grupy placebo, w połączeniu z metforminą, insuliną lub kombinacją metforminy i insuliny. W chwili randomizacji 74% pacjentów miało poniżej 18 lat. Średnia skorygowana zmiana HbA1c dla dapaglifyzyny w porównaniu do placebo od stanu wyjściowego do 24. tygodnia wyniosła -0,75% (95% CI -1,65, 0,15).
W grupie wiekowej < 18 lat średnia skorygowana zmiana HbA1c dla dapaglifyzyny w porównaniu do placebo wyniosła -0,59% (95% CI -1,66, 0,48). W grupie wiekowej ≥ 18 lat średnia zmiana HbA1c od stanu wyjściowego wyniosła -1,52% w grupie leczonej dapaglifyzyną (n=9) i 0,17% w grupie leczonej placebo (n=6). Skuteczność i bezpieczeństwo były podobne do tych obserwowanych w populacji dorosłej leczonej dapaglifyzyną. Bezpieczeństwo i tolerancja zostały dodatkowo potwierdzone w 28-tygodniowym przedłużeniu badania bezpieczeństwa.
Niewydolność serca i przewlekła choroba nerek
Europejska Agencja Leków przewidziała zwolnienie z obowiązku przedstawiania wyników badań z dapaglifyzyną we wszystkich podgrupach populacji pediatrycznej w celu zapobiegania zdarzeniom sercowo-naczyniowym u pacjentów z przewlekłą niewydolnością serca oraz w leczeniu przewlekłej choroby nerek (patrz punkt 4.2 dla informacji na temat zastosowania u dzieci).
5.2 Właściwości farmakokinetyczne
Wchłanianie
Dapaglifyzyna jest szybko i skutecznie wchłaniana po doustnym podaniu. Maksymalne stężenia plazmatyczne (Cmax) dapaglifyzyny są osiągane zazwyczaj w ciągu 2 godzin po podaniu w warunkach głodówki. Średnia geometryczna wartości Cmax i AUCτ dapaglifyzyny w stanie stacjonarnym po podaniu jednorazowej dawki dziennie dapaglifyzyny 10 mg wynosi odpowiednio 158 ng/mL i 628 ng·h/mL. Absolutna biodostępność doustna dapaglifyzyny po podaniu dawki 10 mg wynosi 78%. Podawanie w połączeniu z posiłkiem o wysokiej zawartości tłuszczu zmniejszyło Cmax dapaglifyzyny do maksymalnie 50% i wydłużyło Tmax o około 1 godzinę, ale nie wpłynęło na AUC w porównaniu do warunków głodówki. Te zmiany nie są uważane za klinicznie istotne. Dlatego Dapaglifyzyna Technigen może być przyjmowana niezależnie od posiłków.
Rozkład
Dapaglifyzyna wiąże się z białkami osocza w około 91%. Wiązanie z białkami nie było zmieniane w obecności różnych stanów chorobowych (np. zaburzenia funkcji nerek lub wątroby). Średni objętość rozkładu dapaglifyzyny w stanie stacjonarnym wynosił 118 litrów.
Biodegradacja
Dapaglifyzyna jest szeroko metabolizowana, głównie do dapaglifyzyny 3-O-glukuronidu, który jest metabolitem nieaktywnym. Dapaglifyzyna 3-O-glukuronid ani inne metabolity nie przyczyniają się do efektów hipoglikemicznych. Tworzenie dapaglifyzyny 3-O-glukuronidu jest katalizowane przez UGT1A9, enzym występujący w wątrobie i nerkach, a proces metabolizmu katalizowany przez CYP stanowił u człowieka drogę wtórną eliminacji.
Eliminacja
Średnie końcowe półżycie plazmatyczne (t1/2) dapaglifyzyny wynosiło 12,9 godziny po podaniu jednorazowej doustnej dawki dapaglifyzyny 10 mg u zdrowych ochotników. Średnia całkowita klirens systemowy dapaglifyzyny podanej dożylnie wynosił 207 mL/min. Dapaglifyzyna i jej metabolity są usuwane głównie drogą nerkową, z mniej niż 2% w postaci niezmienionej. Po podaniu dawki [ 14 C]-dapaglifyzyny 50 mg odzyskano 96%, z czego 75% w moczu i 21% w kale. W kale około 15% dawki wydano w postaci leku pierwotnego.
Liniowość
Eksposycja na dapaglifyzynę wzrastała proporcjonalnie do dawki w zakresie 0,1-500 mg, a jej profil farmakokinetyczny nie zmieniał się w czasie po wielokrotnym podawaniu codziennym przez maksymalnie 24 tygodnie.
Grupy specjalne
Zaburzenia funkcji nerek
W stanie stacjonarnym (20 mg dapaglifyzyny podanej raz dziennie przez 7 dni), u pacjentów z cukrzycą typu 2 i łagodnym, umiarkowanym lub ciężkim zaburzeniem funkcji nerek (zgodnie z kliresem ioxynolowy) średnie ekspozycje systemowe na dapaglifyzynę były odpowiednio o 32%, 60% i ponad 87% wyższe niż u pacjentów z cukrzycą typu 2 i prawidłową funkcją nerek. Wydalenie glukozy z moczem w ciągu 24 godzin w stanie stacjonarnym było silnie zależne od funkcji nerek i u pacjentów z cukrzycą typu 2 z prawidłową funkcją nerek lub łagodnym, umiarkowanym lub ciężkim uszkodzeniem nerek wydano odpowiednio 85, 52, 18 i 11 g glukozy/dzień. Nieznany jest wpływ hemodializy na ekspozycję na dapaglifyzynę. Wpływ obniżonej czynności nerek na ekspozycję systemową oceniono w modelu farmakokinetycznym populacyjnym. Zgodnie z poprzednimi wynikami, AUC przewidywane przez model było wyższe u pacjentów z przewlekłą chorobą nerek niż u pacjentów z prawidłową czynnością nerek i nie różniło się istotnie u pacjentów z przewlekłą chorobą nerek z cukrzycą typu 2 i bez cukrzycy.
Zaburzenia funkcji wątroby
U pacjentów z łagodnym lub umiarkowanym uszkodzeniem wątroby (klasy Child-Pugh A i B), średnie wartości Cmax i AUC dapaglifyzyny były odpowiednio do 12% i 36% wyższe niż u zdrowych kontrolnych. Różnice te nie były uważane za klinicznie istotne. U pacjentów z ciężkim uszkodzeniem wątroby (klasa Child-Pugh C), średnie wartości Cmax i AUC dapaglifyzyny były odpowiednio o 40% i 67% wyższe niż u zdrowych kontrolnych.
Osoby starsze (≥ 65 lat)
Nie obserwuje się klinicznie istotnego wzrostu ekspozycji wyłącznie na podstawie wieku u osób do 70 roku życia. Jednakże można przewidywać wzrost ekspozycji związanego z wiekiem obniżeniem czynności nerek. Nie ma wystarczających danych, aby wyciągnąć wnioski dotyczące ekspozycji u pacjentów powyżej 70 roku życia.
Populacja pediatryczna
Profil farmakokinetyczny i farmakodynamiczny (glikozuria) u dzieci z cukrzycą typu 2 w wieku 10-17 lat był podobny do obserwowanego u dorosłych z cukrzycą typu 2.
Płeć
Średnie AUCss dapaglifyzyny u kobiet szacowane jest na około 22% wyższe niż u mężczyzn.
Etnia
Nie stwierdzono klinicznie istotnych różnic w ekspozycji systemowej między osobami białej, czarnej lub azjatyckiej etnii.
Masa ciała
Stwierdzono, że ekspozycja na dapaglifyzynę maleje wraz ze wzrostem masy ciała. W związku z tym pacjenci o niskiej masie ciała mogą czasem mieć zwiększoną ekspozycję, a osoby o wysokiej masie ciała mogą mieć zmniejszoną ekspozycję. Jednak różnice w ekspozycji nie były uważane za klinicznie istotne.
5.3 Dane przedkliniczne dotyczące bezpieczeństwa
Dane przedkliniczne nie wykazują szczególnych zagrożeń dla człowieka na podstawie konwencjonalnych badań farmakologii bezpieczeństwa, toksyczności przy wielokrotnym podawaniu, genotoksyczności, potencjału kancerogennego i płodności. Dapaglifyzyna nie powoduje guzów u myszy ani szczurów przy żadnej z dawek ocenianych w dwuletnich badaniach kancerogenności.
Toksykołogia rozrodcza i rozwojowa
Bezpośrednie podawanie dapaglifyzyny młodym szczurom zaraz po odstawieniu od mleka oraz pośrednie narażenie w ostatnim okresie ciąży (okresy odpowiadające drugiemu i trzeciemu trymestrowi ciąży w odniesieniu do rozwoju nerek u człowieka) i w czasie karmienia piersią były związane z zwiększeniem częstości i/lub nasilenia rozszerzeń kanalików i miedniczek nerkowych u potomstwa. W ramach badania toksyczności przeprowadzonego na młodych zwierzętach, gdy dapaglifyzyna była podawana bezpośrednio młodym szczurom od 21. do 90. dnia życia, stwierdzono rozszerzenia kanalików i miedniczek nerkowych przy wszystkich poziomach dawek; ekspozycja młodych przy najniższej dawce była ≥ 15-krotnie wyższa niż maksymalna zalecana dawka u człowieka. Te wyniki były związane ze wzrostem masy nerek zależnym od dawki i makroskopowym powiększeniem nerek obserwowanym przy wszystkich dawkach. Rozszerzenia miedniczek i kanalików nerkowych u młodych zwierząt nie zniknęły całkowicie w okresie rekonwalescencji trwającym około 1 miesiąca.
W jednym z badań rozwoju prenatalnego i postnatalnego niektóre matki szczurów były leczone od 6. dnia ciąży do 21. dnia po porodzie, podczas gdy potomstwo było narażone pośrednio in utero i przez cały okres karmienia piersią (przeprowadzono badanie satelitarne w celu oceny ekspozycji na dapaglifyzynę w mleku i potomstwie). Zaobserwowano zwiększenie częstości lub nasilenia rozszerzenia miedniczki nerkowej u dorosłego potomstwa leczonych matek, choć tylko przy najwyższej dawce testowanej (ekspozycja matek i potomstwa na dapaglifyzynę wynosiła odpowiednio 1415 i 137-krotnie wartości u człowieka przy maksymalnej zalecanej dawce). Dodatkowa toksyczność rozwojowa była ograniczona do dawkowo-zależnego zmniejszenia masy ciała potomstwa i obserwowana tylko przy dawkach ≥ 15 mg/kg/dzień (związanych z ekspozycją potomstwa ≥ 29-krotnie wartości u człowieka przy maksymalnej zalecanej dawce). Toksykołogia matek była widoczna tylko przy najwyższej dawce testowanej i ograniczała się do przejściowych zmniejszeń masy ciała i spożycia pokarmu po podaniu dawki. Poziom bez obserwowalnych szkodliwych efektów (NOAEL) dla toksyczności rozwojowej przy najniższej dawce testowanej był związany z wielokrotnością ekspozycji matki systematycznej około 19-krotnie wyższą niż wartość u człowieka przy maksymalnej zalecanej dawce u człowieka.
W dodatkowych badaniach rozwoju embrionalnego i płodowego przeprowadzonych na szczurach i królikach, dapaglifyzyna była podawana w odstępach odpowiadających najważniejszym fazom organogenezy w każdej z gatunków. U królików nie zaobserwowano żadnej toksyczności matek ani rozwoju przy żadnej dawce testowanej; maksymalna dawka testowana była związana z wielokrotnością ekspozycji systematycznej około 1191-krotnie wyższą niż maksymalna zalecana dawka u człowieka. U szczurów dapaglifyzyna nie była embriotoksyczna ani teratogenna przy ekspozycjach do 1441-krotnie wyższych niż maksymalna zalecana dawka u człowieka.
6. INFORMACJE FARMACEUTYCZNE
6.1 Wykaz substancji pomocniczych
Jądro tabletki
Celuloza mikrokryształowa silicowana (typ SMCC 50)
Crospowidon
Mannitol (typ M 100)
Talk
Stearynian magnezu
Powłoka filmowa
Alkohol poliwinylowy
Tlenek tytanu (E171)
Macrogol/polietylenoglikol
Talk
Tlenek żelaza żółty (E172).
6.2 Niekompatybilności
Nie dotyczy.
6.3 Okres ważności
2 lata
6.4 Szczególne środki ostrożności dotyczące przechowywania
Ten lek nie wymaga szczególnych warunków przechowywania.
6.5 Rodzaj i zawartość opakowania
Blister kompleksowy OPA szary matowy (poliamid 25 µm + aluminium 45 µm + PVC 60 µm)
zaplombowany folią aluminiową 20 µm.
Dapagliflozin Tecnigen 5 mg tabletki powlekane
Opakowania zawierające 14 i 28 tabletek powlekanych.
Dapagliflozin Tecnigen 10 mg tabletki powlekane
Opakowania zawierające 14 i 28 tabletek powlekanych.
Może być, że nie wszystkie opakowania są wprowadzone na rynek.
6.6 Szczególne środki ostrożności dotyczące usuwania
Nieużywany lek oraz odpady pochodzące z tego leku należy usuwać zgodnie z obowiązującymi przepisami lokalnymi.
7. WŁAŚCICIEL AUTORYZACJI DO WPUSZCZENIA NA RYNEK
Tecnigen S.r.l.
Via Galileo Galilei 40
Cinisello Balamo 20092 MI - Włochy
8. NUMER WPROWADZENIA DO OBROTU
051232015 - "tabletki powlekane 5 mg" 14 tabletek w blisterach OPA/AL/PVC
051232027 - "tabletki powlekane 5 mg" 28 tabletek w blisterach OPA/AL/PVC
051232039 - "tabletki powlekane 10 mg" 14 tabletek w blisterach OPA/AL/PVC
051232041 - "tabletki powlekane 10 mg" 28 tabletek w blisterach OPA/AL/PVC
9. DATA PIERWSZEGO POZWOLENIA/ODNOWIENIA POZWOLENIA
Data pierwszego pozwolenia: