Ozurdex®
Ucraina
Indice
ISTRUZIONI PER L'USO MEDICINALE DEL MEDICINALE OZURDEX® (OZURDEX®)
Composizione:
Principio attivo: desametasone;
1 impianto contiene 700 mcg di desametasone;
Eccipienti: copolimero acido lattico-acido glicolico 50:50 (Resomer® RG 502), copolimero acido lattico-acido glicolico 50:50 (Resomer® RG 502H).
Forma farmaceutica. Impianto per somministrazione intravitreale.
Principali caratteristiche fisico-chimiche: dispositivo monouso per iniezione contenente un impianto in forma di cilindro, non visibile. L’impianto ha un diametro di circa 0,46 mm e una lunghezza di 6 mm.
Categoria farmacoterapeutica. Preparati oftalmologici antinfiammatori.
Codice ATC S01B A01.
Proprietà farmacodinamiche.
È noto che il desametasone è un corticosteroide potente che esercita un marcato effetto antinfiammatorio e inibisce l'edema riducendo il deposito di fibrina, la permeabilità capillare e l'infiltrazione fagocitaria in risposta all'infiammazione. Il fattore di crescita dell'endotelio vascolare (VEGF) è una citochina prodotta in concentrazioni elevate in caso di regolazione dell'edema maculare. È un potente stimolatore della permeabilità vascolare. È stato dimostrato che i corticosteroidi inibiscono l'espressione del fattore di crescita dell'endotelio vascolare. Inoltre, i corticosteroidi impediscono il rilascio di prostaglandine, alcune delle quali sono state identificate come mediatori dell'edema maculare cistoide.
Edema maculare diabetico
L'efficacia dell'impianto Ozurdex® è stata valutata in due studi multicentrici, randomizzati, in doppio cieco, controllati con placebo, di 3 anni di durata, condotti secondo uno schema parallelo, che insieme hanno coinvolto 1048 pazienti (studi 206207-010 e 206207-011). In totale, 351 pazienti sono stati randomizzati al gruppo Ozurdex**®**, 347 al gruppo desametasone alla dose di 350 µg e 350 pazienti al gruppo placebo.
I pazienti dovevano sottoporsi a un nuovo impianto sulla base di uno spessore retinico > 175 micron, misurato mediante tomografia a coerenza ottica (OCT), oppure se, secondo l'investigatore, i risultati dell'OCT indicavano la presenza di un edema retinico residuo costituito da più cisti intraretiniche o da più aree di aumento dello spessore retinico all'interno o al di fuori del sottocampo centrale. Ai pazienti sono stati somministrati fino a 7 impianti con intervalli non più frequenti di ogni 6 mesi.
Era consentito l'uso di terapie alternative a discrezione dell'investigatore in qualsiasi momento dello studio, ma ciò comportava l'esclusione dallo studio stesso.
Nel complesso, il 36% dei pazienti trattati con Ozurdex**®** ha interrotto la partecipazione allo studio per qualsiasi motivo, rispetto al 57% dei pazienti del gruppo placebo. Il rapporto tra partecipanti che hanno interrotto lo studio a causa di reazioni avverse era molto simile sia nel gruppo Ozurdex**®** che nel gruppo placebo (13% contro 11%). Il rapporto tra partecipanti che hanno interrotto lo studio per mancanza di efficacia è risultato inferiore nel gruppo Ozurdex**®** rispetto al gruppo placebo (7% contro 24%).
I punti finali primari e secondari chiave negli studi 206207-010 e 011 sono riportati nella Tabella 1. Dopo un miglioramento della vista nei pazienti del gruppo desametasone (DEX 700), si è verificato un peggioramento dovuto alla formazione di cataratta. Il miglioramento della vista è stato rivalutato dopo l'asportazione della cataratta.
Tabella 1. Efficacia negli studi 206207-010 e 206207-011 (ITT - gruppo per protocollo di trattamento assegnato)
| Punto finale |
Studio 206207-010 |
Studio 206207-011 |
Dati cumulativi degli studi 206207-010 e 206207-011 |
|||
| DEX 700, numero di partecipanti – 163 |
Placebo, numero di partecipanti – 165 |
DEX 700, numero di partecipanti – 188 |
Placebo, numero di partecipanti – 185 |
DEX 700, numero di partecipanti – 351 |
Placebo, numero di partecipanti – 350 |
|
| Variazione media dei punteggi sulla scala BCVA* nel corso di 3 anni in base all'AUC** (lettere) |
4,1 |
1,9 |
2,9 |
2,0 |
3,5 |
2,0 |
| Valore p |
0,016 |
0,366 |
0,023 |
|||
| BCVA ≥ miglioramento di 15 lettere rispetto ai valori basali al 3° anno dopo l'iniezione/ultimo visita (%) |
22,1 |
13,3 |
22,3 |
10,8 |
22,2 |
12,0 |
| Valore p |
0,038 |
0,003 |
< 0,001 |
|||
| Variazione media dei punteggi sulla scala BCVA dal basale al 3° anno dopo l'iniezione/ultimo visita (lettere) |
4,1 |
0,8 |
1,3 |
|
2,6 |
0,4 |
| Valore p |
0,020 |
0,505 |
0,054 |
|||
| Variazione media dello spessore retinico nei parametri di tomografia a coerenza ottica (OCT) nella regione centrale nel corso di 3 anni in base all'AUC (µm) |
|
|
|
|
|
|
| Valore p |
<0,001 |
< 0,001 |
< 0,001 |
|||
* Acuità visiva corretta massima (BCVA)
** Area sotto la curva (AUC)
I punti finali primari e secondari chiave nell'analisi aggregata dei pazienti pseudofachici sono riportati nella Tabella 2.
Tabella 2. Efficacia nei pazienti pseudofachici (studi combinati 206207-010 e 206207-011)
| Punto finale |
DEX 700, numero di partecipanti – 86 |
Placebo, numero di partecipanti – 101 |
Valore p |
| Variazione media del punteggio BCVA nel corso di 3 anni in base all'AUC (lettere) |
6,5 |
1,7 |
< 0,001 |
| BCVA ≥ 15 lettere di miglioramento rispetto al valore basale al terzo anno dopo l'iniezione/ultimo controllo (%) |
23,3 |
10,9 |
0,024 |
| Variazione media del punteggio BCVA dal valore basale al terzo anno dopo l'iniezione/ultimo controllo |
6,1 |
1,1 |
0,004 |
| Variazione media dello spessore retinico nel campo centrale misurato con tomografia a coerenza ottica (OCT) nel corso di 3 anni in base all'AUC (µm) |
|
|
< 0,001 |
I punti finali primari e secondari chiave dell'analisi aggregata nei pazienti precedentemente trattati sono riportati nella Tabella 3.
Tabella 3. Efficacia nei pazienti precedentemente trattati (studi combinati 206207-010 e 206207-011)
| Endpoint |
DEX 700, numero di partecipanti – 247 |
Placebo, numero di partecipanti – 261 |
Valore P |
| Variazione media del punteggio BCVA nel corso di 3 anni in base all'AUC (lettere) |
3,2 |
1,5 |
0,024 |
| BCVA ≥ 15 lettere di miglioramento rispetto ai valori basali al terzo anno dopo l'iniezione/ultimo appuntamento (%) |
21,5 |
11,1 |
0,002 |
| Variazione media del punteggio BCVA rispetto ai valori basali al terzo anno dopo l'iniezione/ultimo appuntamento |
2,7 |
0,1 |
0,055 |
| Variazione media dello spessore retinico ai valori di tomografia a coerenza ottica (OCT) nel quadrante centrale nel corso di 3 anni in base all'AUC (µm) |
|
|
< 0,001 |
Occlusione della vena centrale della retina / occlusione di un ramo della vena della retina
L'efficacia dell'impianto Ozurdex® è stata valutata in due studi multicentrici, in doppio cieco, randomizzati, controllati con placebo, di tipo parallelo e identici, che hanno coinvolto 1267 pazienti, randomizzati e trattati con desametasone alle dosi di 350 mcg o 700 mcg oppure con placebo (studio 206207-008 e studio 206207-009). In totale, 427 pazienti sono stati randomizzati al gruppo del farmaco Ozurdex®, 414 al gruppo desametasone 350 mcg e 426 pazienti al gruppo placebo.
Sulla base dei risultati degli studi combinati, il trattamento con impianti Ozurdex® ha mostrato un numero statisticamente significativo maggiore di pazienti considerati come quelli che hanno ottenuto un miglioramento dell'acuità visiva corretta (BCVA) ≥ 15 caratteri rispetto ai valori basali a 90 giorni dall'iniezione di un singolo impianto, rispetto al gruppo placebo (p < 0,001).
La percentuale di pazienti che hanno ottenuto un miglioramento dell'efficacia primaria di ≥ 15 caratteri nell'acuità visiva corretta rispetto ai valori basali dopo l'iniezione di un singolo impianto è riportata nella Tabella 4.
L'effetto terapeutico è stato osservato già al primo punto di osservazione al giorno 30. L'effetto massimo del trattamento è stato osservato al giorno 60 e la differenza nel numero di pazienti considerati come quelli che hanno ottenuto un miglioramento è risultata statisticamente significativa nel gruppo del farmaco Ozurdex® rispetto al gruppo placebo per tutto il periodo fino al giorno 90 dopo l'iniezione. È persistita una tendenza al miglioramento dell'acuità visiva corretta ≥ 15 caratteri rispetto ai valori basali in una percentuale significativamente maggiore di soggetti trattati con l'impianto Ozurdex® rispetto al gruppo placebo anche al giorno 180.
Tabella 4. Rapporto di pazienti con miglioramento dell'acuità visiva corretta ≥ 15 caratteri rispetto ai valori basali nell'occhio in studio (risultati combinati del gruppo ITT)
| Giorno della visita |
Ozurdex® numero di pazienti – 427 |
Placebo numero di pazienti – 426 |
| Giorno 30 Giorno 60 Giorno 90 Giorno 180 |
21,3 % a 29,3 % a 21,8 % a 21,5 % |
7,5 % 11,3 % 13,1 % 17,6 % |
a Il coefficiente nel gruppo del farmaco Ozurdex® è significativamente più alto rispetto al gruppo placebo (p < 0,001).
Il valore medio di variazione dell'acuità visiva massima corretta rispetto ai valori basali è risultato significativamente maggiore nel gruppo Ozurdex® rispetto al gruppo placebo in tutte le osservazioni.
In ciascuno studio di fase III e nell'analisi combinata, il tempo necessario per raggiungere un miglioramento dell'acuità visiva massima corretta di almeno ≥ 15 lettere (3 linee) sulle curve cumulative di risposta differiva significativamente nel gruppo del farmaco Ozurdex® rispetto al gruppo placebo (p < 0,001); i pazienti trattati con l’impianto Ozurdex® raggiungevano prima il miglioramento dell'acuità visiva massima corretta di 3 linee rispetto ai pazienti del gruppo placebo.
Numericamente, l’impianto Ozurdex® si è dimostrato significativamente più efficace del placebo nel prevenire la perdita visiva, come evidenziato da una percentuale inferiore di pazienti che presentavano un peggioramento visivo ≥ 15 lettere nel gruppo Ozurdex® durante il periodo di valutazione di 6 mesi.
In ciascuno studio di fase III e nell'analisi combinata, lo spessore medio della retina era significativamente minore e la riduzione media rispetto ai valori basali era significativamente maggiore nel gruppo del farmaco Ozurdex® (–207,9 micron) rispetto al gruppo placebo (–95,0 micron) al giorno 90 (p < 0,001, dati combinati).
L'effetto terapeutico, confermato dalla valutazione dell'acuità visiva massima corretta al giorno 90, è stato supportato da questo reperto anatomico. Tuttavia, la riduzione media (–119,3 micron) dello spessore della retina al giorno 180, rispetto al gruppo placebo, non è risultata significativa.
I pazienti con un valore di acuità visiva massima corretta < 84 oppure uno spessore retinico > 250 micron misurato mediante tomografia a coerenza ottica, e che secondo l'opinione dello sperimentatore non erano esposti a rischio in caso di trattamento, potevano essere selezionati per il trattamento con l’impianto Ozurdex® in uno studio in aperto. Il 98% dei pazienti trattati nella fase in aperto dello studio ha ricevuto l’impianto Ozurdex® tra il quinto e il settimo mese dall'inizio del trattamento.
La massima risposta è stata osservata al giorno 60 dall'inizio del trattamento nella fase in aperto dello studio. I coefficienti cumulativi di risposta sono risultati più elevati durante la fase in aperto dello studio nei pazienti che hanno ricevuto due somministrazioni consecutive dell’impianto Ozurdex®, rispetto ai pazienti che non hanno ricevuto l’iniezione dell’impianto Ozurdex® nella fase iniziale.
La percentuale di rispondenti in ogni intervallo temporale è stata sempre maggiore dopo la seconda somministrazione rispetto alla prima. Tuttavia, un ritardo nella somministrazione di 6 mesi porta a una percentuale inferiore di rispondenti in tutti gli intervalli temporali, rispetto ai pazienti che hanno ricevuto la seconda iniezione di Ozurdex® nella fase in aperto dello studio.
Uveite
L'efficacia clinica dell’impianto Ozurdex® è stata valutata in uno studio multicentrico, randomizzato, in doppio cieco, per il trattamento dell'infiammazione non infettiva del segmento posteriore dell'occhio in pazienti con uveite.
Complessivamente, 229 pazienti sono stati randomizzati nei gruppi di trattamento con impianti di desametasone alle dosi di 350 mcg o 700 mcg oppure con placebo. Di questi, 77 pazienti hanno ricevuto l’impianto Ozurdex®, 76 la desametasone 350 mcg e 76 il placebo. Nel complesso, il 95% dei pazienti ha completato lo studio della durata di 26 settimane.
La percentuale di pazienti con opacità del corpo vitreo con punteggio 0 nell'occhio in studio alla settimana 8 dello studio (endpoint primario) è risultata 4 volte maggiore (46,8%) rispetto al gruppo placebo (11,8%), p < 0,001. Il vantaggio statistico si è mantenuto fino alla settimana 26 inclusa (p ≤ 0,014), come indicato nella tabella 5.
Le curve cumulative del coefficiente di risposta (tempo fino al raggiungimento di un punteggio 0 per l'opacità del corpo vitreo) differivano significativamente nei pazienti trattati con l’impianto Ozurdex® rispetto al gruppo placebo (p < 0,001). Il miglioramento si manifesta prima e l'efficacia terapeutica è maggiore.
La riduzione dell'opacità del corpo vitreo è stata accompagnata da un miglioramento dell'acuità visiva. Il rapporto tra pazienti con un miglioramento dell'acuità visiva massima corretta di almeno ≥ 15 lettere rispetto ai valori basali nell'occhio in studio alla settimana 8 è risultato 6 volte maggiore (42,9%) nel gruppo Ozurdex® (42,9%) rispetto al gruppo placebo (6,6%), p < 0,001. Il vantaggio statistico è stato raggiunto alla settimana 3 e si è mantenuto fino alla settimana 26 inclusa (p < 0,001), come indicato nella tabella 5.
La percentuale di pazienti che hanno richiesto un trattamento alternativo entro la settimana 8 è risultata 3 volte inferiore nel gruppo del farmaco Ozurdex® (7,8%) rispetto al gruppo placebo (22,4%), p = 0,012, rispetto ai valori basali.
Tabella 5. Rapporto tra pazienti con punteggio 0 per l'opacità del corpo vitreo e con miglioramento dell'acuità visiva massima corretta di ≥ 15 lettere rispetto ai valori basali nell'occhio in studio (gruppo ITT)
| Data di revisione |
Turbamento del corpo vitreo con indice pari a 0 |
Miglioramento dell'acuità visiva con correzione massima ≥ 15 caratteri rispetto al livello iniziale |
||
| Dexametasone 700 N. di pazienti – 77 |
Placebo N. di pazienti – 76 |
Dexametasone 700 N. di pazienti – 77 |
Placebo N. di pazienti – 76 |
|
| Settimana 3 |
23,4 % |
11,8 % |
32,5 %a |
3,9 % |
| Settimana 6 |
42,9 %a |
9,2 % |
41,6 %a |
7,9 % |
| Settimana 8 |
46,8 %a |
11,8 % |
42,9 %a |
6,6 % |
| Settimana 12 |
45,5 %a |
13,2 % |
41,6 %a |
13,2 % |
| Settimana 16 |
40,3 %b |
21,1 % |
39,0 %a |
13,2 % |
| Settimana 20 |
39,0 %c |
19,7 % |
40,3 %a |
13,2 % |
| Settimana 26 |
31,2 %d |
14,5 % |
37,7 %a |
13,2 % |
a p< 0,001; b p = 0,010; c p = 0,009; d p = 0,014
Pediatria
L’Agenzia europea per i medicinali ha rinunciato all’obbligo di presentare i risultati degli studi sull’uso dell’impianto Ozurdex**®** in tutte le sottopopolazioni pediatriche per quanto riguarda l’occlusione della vena retinica e l’edema maculare diabetico.
Farmacocinetica.
I dati sulla concentrazione plasmatica sono stati ottenuti da un sottogruppo di 21 pazienti durante due studi di efficacia di 6 mesi, prima della somministrazione e ai giorni 7, 30, 60 e 90 dopo l’iniezione dell’impianto intravitreale monouso contenente 350 mcg o 700 mcg di desametasone. Il 95% dei livelli plasmatici di desametasone dopo somministrazione di 350 mcg e l’86% dopo somministrazione di 700 mcg risultavano al di sotto del limite inferiore della valutazione quantitativa (0,05 ng/ml). La concentrazione plasmatica massima di 0,094 ng/ml è stata osservata in un paziente del gruppo trattato con 700 mcg. È emerso che la concentrazione plasmatica di desametasone non dipende dall’età, dal peso corporeo o dal sesso del paziente.
I dati sulla concentrazione nel plasma sono stati ottenuti da un sottogruppo di pazienti in due studi principali sull’edema maculare diabetico, prima della somministrazione e ai giorni 1, 7, 21 e a 1,5 e 3 mesi dopo l’iniezione intravitreale dell’impianto intravitreale monouso contenente 350 mcg o 700 mcg di desametasone. Il 100% dei livelli di desametasone nel plasma dopo somministrazione di 350 mcg e il 90% dopo somministrazione di 700 mcg risultavano al di sotto del limite inferiore della valutazione quantitativa (0,05 ng/ml). La concentrazione plasmatica massima di 0,102 ng/ml è stata osservata in un paziente del gruppo trattato con 700 mcg. È emerso che le concentrazioni di desametasone nel plasma non dipendono dall’età, dal peso corporeo o dal sesso dei pazienti.
In uno studio di 6 mesi condotto su scimmie, dopo una singola iniezione intravitreale dell’impianto Ozurdex**®**, la concentrazione massima (Cmax) di desametasone nel corpo vitreo è stata di 100 ng/ml al giorno 42 e di 5,57 ng/ml al giorno 91. Il desametasone è stato rilevato nel corpo vitreo anche dopo 6 mesi dall’iniezione. L’ordine di concentrazione del desametasone è risultato: retina > iride > corpo ciliare > corpo vitreo > umore acqueo > plasma.
In uno studio in vitro sul metabolismo, dopo 18 ore di incubazione di desametasone marcato con C14 con cornea umana, iride, corpo ciliare, coroide, retina, corpo vitreo e tessuti della sclera, non sono stati osservati metaboliti del farmaco. Questi risultati sono in accordo con gli studi sul metabolismo dei tessuti oculari negli animali.
Il desametasone viene metabolizzato in metaboliti finali idrosolubili e liposolubili, che vengono escreti con la bile e l’urina.
La matrice di Ozurdex**®** si degrada lentamente in acido lattico e acido glicolico attraverso un semplice processo di idrolisi, e successivamente in anidride carbonica e acqua.
Caratteristiche cliniche.
Indicazioni.
Trattamento di adulti:
- con edema maculare secondario ad occlusione della vena centrale della retina o ad occlusione di un ramo venoso della retina;
- con infiammazione del segmento posteriore dell’occhio sotto forma di uveite non infettiva;
- con perdita visiva secondaria a edema maculare diabetico, in cui l’occhio sia afachico o per i quali una terapia non steroidea non sia efficace o non sia accettabile.
Controindicazioni.
- Ipersensibilità alla sostanza attiva o a uno qualsiasi degli eccipienti del medicinale.
- Infezione oculare o perioculare acuta o potenziale, inclusi la maggior parte dei disturbi virali della cornea e della congiuntiva, in particolare cheratite erpetica epiteliale attiva (cheratite dendritica), malattia da vaccino, varicella, infezioni micobatteriche e malattie fungine.
- Glaucoma avanzato (con terapia farmacologica insufficientemente efficace).
- Afachia con rottura della capsula posteriore del cristallino.
- Presenza di lenti intraoculari anteriori impiantate, clip per lente intraoculare ad iris o lente intraoculare posteriore con fissazione transsclerale in presenza contemporanea di rottura della capsula posteriore del cristallino.
Precauzioni particolari di sicurezza.
Qualsiasi iniezione intravitreale (inclusa l’iniezione di Ozurdex®) può essere associata a endoftalmite, infiammazione intraoculare, aumento della pressione intraoculare e distacco della retina. Si deve sempre utilizzare un’adeguata tecnica asettica per l’iniezione. Inoltre, dopo l’iniezione, i pazienti devono essere sottoposti a controllo per garantire un trattamento precoce in caso di infezione o aumento della pressione intraoculare. Il controllo può includere la valutazione della perfusione del disco ottico immediatamente dopo l’iniezione, la misurazione della pressione arteriosa entro 30 minuti dall’iniezione e una biomicroscopia oculare tra il secondo e il settimo giorno successivo all’iniezione.
I pazienti devono essere istruiti a segnalare immediatamente la comparsa di qualsiasi sintomo che possa indicare endoftalmite o uno qualsiasi degli stati sopra menzionati, come dolore oculare o offuscamento della vista.
Tutti i pazienti con rottura della capsula posteriore del cristallino, in particolare pazienti con cristallino nella camera posteriore (ad esempio, a seguito di intervento chirurgico per cataratta) e/o pazienti con iride aperta nella cavità del corpo vitreo (ad esempio, a seguito di iridectomia), con o senza vitrectomia, sono a rischio di migrazione dell’impianto nella camera anteriore dell’occhio. La migrazione dell’impianto nella camera anteriore può causare edema corneale. In caso di edema corneale persistente e grave, potrebbe rendersi necessario un trapianto di cornea.
Ad altri gruppi di pazienti, oltre a quelli per i quali l’uso dell’impianto Ozurdex® è controindicato (vedi sezione «Controindicazioni»), l’impianto deve essere somministrato con cautela e solo dopo un’attenta valutazione del rapporto rischio-beneficio. Questi pazienti devono essere sottoposti a un monitoraggio attento per rilevare eventuali segni di migrazione dell’impianto.
L’uso di corticosteroidi, incluso il medicinale Ozurdex®, può portare allo sviluppo di cataratta (inclusa cataratta sottocapsulare posteriore), aumento della PIO, glaucoma steroideo e infezione secondaria dell’occhio.
Nei tre anni di studi clinici sull’edema maculare diabetico, il 59% dei pazienti fachici trattati con Ozurdex® è stato sottoposto a intervento chirurgico per cataratta nell’occhio trattato.
L’incidenza di cataratta dopo la prima iniezione è più alta nei pazienti con uveite non infettiva del segmento posteriore rispetto ai pazienti con occlusione di un ramo venoso della retina o occlusione della vena centrale della retina. Negli studi clinici su occlusione di un ramo venoso della retina/occlusione della vena centrale della retina, la cataratta è stata riportata più frequentemente nei pazienti fachici dopo la seconda iniezione (vedi sezione «Effetti indesiderati»).
Solo 1 paziente su 368 ha richiesto un intervento chirurgico per cataratta dopo la prima somministrazione e 3 pazienti su 302 dopo il secondo ciclo di trattamento. Nello studio sull’uveite non infettiva, 1 paziente su 62 pazienti fachici ha ricevuto un trattamento chirurgico per cataratta dopo una singola somministrazione.
L’emorragia congiuntivale è più frequente nei pazienti con uveite non infettiva del segmento posteriore rispetto ai pazienti con occlusione di un ramo venoso della retina o della vena centrale della retina e con edema maculare diabetico. Questo può essere direttamente correlato alla procedura di iniezione intravitreale o all’uso concomitante di corticosteroidi locali e/o sistemici o di farmaci antiinfiammatori non steroidei. Non è necessaria assistenza medica, poiché i sintomi si risolvono spontaneamente.
Come previsto, con l’uso di corticosteroidi oftalmici e iniezioni intravitreali, è possibile un aumento della pressione intraoculare (PIO). L’aumento della PIO può generalmente essere controllato con farmaci per la riduzione della PIO. Nei pazienti con aumento della PIO ≥ 10 mmHg rispetto al valore basale, il picco massimo di PIO è stato osservato tra il 45° e il 60° giorno dopo l’iniezione. Pertanto, è necessario effettuare un controllo regolare della pressione intraoculare e qualsiasi aumento deve essere trattato adeguatamente immediatamente dopo l’iniezione.
L’aumento della pressione intraoculare (PIO) si verifica più frequentemente nei pazienti di età inferiore a 45 anni con edema maculare secondario ad occlusione venosa della retina o con infiammazione del segmento posteriore dell’occhio sotto forma di uveite non infettiva.
Destinato esclusivamente all’uso monouso.
Ogni applicatore può essere utilizzato per il trattamento di un solo occhio.
Se la pellicola protettiva della confezione in foglio di alluminio contenente l’applicatore è danneggiata, non si deve utilizzare. Una volta aperta la confezione in foglio di alluminio, l’applicatore deve essere utilizzato immediatamente.
Qualsiasi farmaco medicinale non utilizzato o rifiuti devono essere smaltiti in conformità ai requisiti locali.
Disturbi visivi
Disturbi visivi possono verificarsi con l’uso di corticosteroidi sistemici o locali. Se un paziente sviluppa sintomi come peggioramento della vista o altri disturbi visivi, si deve effettuare una valutazione delle possibili cause, tra cui cataratta, glaucoma o malattie rare come la retinopatia sierosa centrale (RSC), che sono state osservate dopo l’uso di corticosteroidi sistemici o locali.
Interazioni con altri medicinali ed altre forme di interazione.
Non sono stati condotti studi specifici sulle interazioni del medicinale. Tuttavia, è noto che l’assorbimento sistemico è minimo e non sono previste interazioni.
Caratteristiche di impiego.
Non è stata studiata la sicurezza ed efficacia dell’applicazione contemporanea dell’impianto Ozurdex® in entrambi gli occhi. Pertanto, l’applicazione contemporanea del medicinale in entrambi gli occhi non è raccomandata.
I corticosteroidi devono essere utilizzati con cautela nei pazienti con anamnesi di infezioni oculari virali (herpes simplex) e non devono essere utilizzati in caso di herpes oculare attivo. L’uso dell’impianto Ozurdex® nel trattamento di pazienti con edema maculare secondario ad occlusione della vena retinica associata a significativa ischemia retinica non è stato studiato; pertanto, l’uso del medicinale non è raccomandato in questo gruppo di pazienti.
Negli studi di fase III è stata coinvolta un numero limitato di pazienti con diabete di tipo I, e la risposta all’applicazione di Ozurdex® in questi pazienti non differiva sostanzialmente da quella osservata nei pazienti con diabete di tipo II.
Nell’occlusione della vena retinica, una terapia anticoagulante è stata utilizzata nel 2% dei pazienti trattati con impianto Ozurdex®; non sono state riportate reazioni avverse emorragiche in questo gruppo di pazienti.
Nell’edema maculare diabetico, una terapia anticoagulante è stata utilizzata nell’8% dei pazienti. Tra i pazienti che assumevano farmaci anticoagulanti, la frequenza di reazioni avverse emorragiche è risultata simile tra il gruppo trattato con Ozurdex® e il gruppo placebo (29% contro 32%). Tra i pazienti che non assumevano farmaci anticoagulanti, le reazioni avverse emorragiche si sono verificate nel 27% dei pazienti trattati con Ozurdex®, rispetto al 20% nel gruppo placebo. L’emorragia nel corpo vitreo nei pazienti trattati con Ozurdex® si è verificata più frequentemente tra coloro che assumevano farmaci anticoagulanti (11%) rispetto a coloro che non li assumevano (6%).
Farmaci antiaggreganti come il clopidogrel sono stati utilizzati in alcune fasi durante gli studi clinici in oltre il 56% dei pazienti. Tra i pazienti che assumevano farmaci concomitanti insieme ad antiaggreganti, le reazioni avverse emorragiche si sono verificate più frequentemente nei pazienti trattati con Ozurdex® (fino al 29%) rispetto al gruppo placebo (fino al 23%), indipendentemente dall’indicazione o dal numero di iniezioni. La reazione avversa emorragica più comune osservata è stata l’emorragia congiuntivale (fino al 24%).
L’impianto Ozurdex® deve essere utilizzato con cautela nel trattamento di pazienti che assumono anticoagulanti o farmaci antiaggreganti.
Uso durante la gravidanza o l’allattamento.
Gli studi sugli animali hanno evidenziato un effetto teratogeno dopo l’applicazione oftalmica locale.
Non esistono dati adeguati sull’uso di iniezioni intravitreali di desametasone in donne in gravidanza. Il trattamento prolungato e sistematico con glucocorticoidi durante la gravidanza aumenta il rischio di ritardo della crescita intrauterina e di insufficienza surrenalica nei neonati. Pertanto, anche se si ritiene che l’effetto sistemico del desametasone sia molto basso dopo un trattamento intraoculare locale, l’uso dell’impianto Ozurdex® non è raccomandato durante la gravidanza, salvo nei casi in cui il beneficio atteso per la madre superi il potenziale rischio per il feto.
Il desametasone viene escreto nel latte materno. Considerando il metodo di somministrazione dell’impianto Ozurdex® e la sua bassa concentrazione sistemica, non ci si attende alcun effetto sul neonato. Tuttavia, l’uso dell’impianto Ozurdex® non è raccomandato durante l’allattamento, a meno che non sia strettamente necessario.
Non sono disponibili dati sull’effetto sulla fertilità femminile.
Capacità di influenzare la capacità di guidare veicoli a motore o di utilizzare macchinari.
Ozurdex® può influire in modo lieve sulla capacità di reazione durante la guida di veicoli a motore o l’utilizzo di macchinari.
Dopo la procedura di iniezione intravitreale dell’impianto Ozurdex®, i pazienti possono riscontrare un peggioramento temporaneo della vista (vedere sezione «Effetti indesiderati»). In tal caso, i pazienti devono astenersi dalla guida e dall’uso di macchinari fino al ripristino della funzione visiva.
Modalità di somministrazione e dosaggio
Ozurdex® deve essere somministrato esclusivamente da un medico oculista qualificato con esperienza nelle iniezioni intravitreali.
Dose raccomandata: un impianto di Ozurdex® deve essere somministrato per via intravitreale nell’occhio interessato. Non è raccomandato l’uso contemporaneo in entrambi gli occhi.
Edema maculare diabetico
Le somministrazioni ripetute devono essere effettuate quando il paziente presenta una risposta iniziale e, a giudizio del medico, un trattamento ripetuto determinerebbe un miglioramento dello stato clinico senza un rischio significativo per il paziente.
Nell’edema maculare diabetico, un’iniezione ripetuta può essere effettuata circa ogni 6 mesi, qualora si osservi un peggioramento della vista e/o un aumento dello spessore retinico in seguito a recidiva o progressione dell’edema maculare diabetico.
Non sono disponibili dati sull’efficacia e sulla sicurezza di un impiego superiore a 7 iniezioni nell’edema maculare diabetico.
Occlusione della vena retinica e uveite
È necessario un ripetuto trattamento se il paziente risponde al medicinale con un progressivo deterioramento dell’acuità visiva e se, a giudizio del medico, il beneficio dell’iniezione ripetuta supera il rischio significativo.
Nei pazienti in cui la vista è migliorata e si mantiene stabile, non è indicato un trattamento ripetuto. Nei pazienti in cui si osserva un peggioramento della vista che non si riduce dopo l’applicazione dell’impianto Ozurdex®, non è indicato un trattamento ripetuto.
Esistono informazioni limitate riguardo alla ripetuta somministrazione con intervalli inferiori ai 6 mesi. Per informazioni sull’esperienza relativa alla ripetuta somministrazione sicura di più di 2 impianti nell’uveite posteriore non infettiva o nell’occlusione della vena retinica, vedere la sezione «Effetti indesiderati».
I pazienti devono essere monitorati dopo l’iniezione per garantire un trattamento tempestivo in caso di insorgenza di infezione o aumento della pressione intraoculare.
Pazienti anziani (≥ 65 anni): non è necessaria alcuna modifica della dose nei pazienti anziani.
L’uso di Ozurdex® in pazienti con insufficienza renale o epatica non è stato studiato; tuttavia, non sono necessarie misure specifiche per questi pazienti.
L’impianto Ozurdex® per somministrazione intravitreale nell’applicatore monouso è destinato esclusivamente all’uso intravitreale.
Ogni applicatore deve essere utilizzato per il trattamento di un solo occhio.
La procedura di iniezione intravitreale deve essere eseguita esclusivamente in condizioni asettiche, che comprendono l’uso di guanti sterili, garze sterili e specillo per palpebre sterile (o equivalente).
Tre giorni prima e dopo la procedura, deve essere somministrato ogni giorno un agente antibatterico topico a spettro ampio. Prima dell’iniezione, è necessario disinfettare la pelle intorno all’occhio, le palpebre e la superficie oculare (come effettuato negli studi clinici con l’impianto Ozurdex®, ad esempio applicando gocce di soluzione al 5% di povidone-iodio sulla congiuntiva) e deve essere applicata un’adeguata anestesia locale. Rimuovere la busta in alluminio dalla confezione di cartone e verificarne l’integrità (vedere la sezione «Confezionamento»). Successivamente, in condizioni sterili, aprire la busta in alluminio ed estrarre con cautela l’applicatore, posizionandolo su un vassoio sterile. Rimuovere con attenzione il cappuccino protettivo dall’applicatore.
Utilizzare l’applicatore immediatamente dopo l’apertura della busta in alluminio.
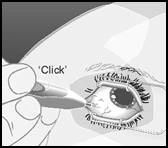
Tenere l’applicatore in una mano ed estrarre il dispositivo di sicurezza. Non ruotare né piegare il dispositivo di sicurezza. Avvicinare la punta dell’ago dell’applicatore direttamente alla sclera e inserire l’ago di circa 1 mm all’interno della sclera, quindi reindirizzare l’applicatore verso il centro dell’occhio, nella cavità del corpo vitreo, finché la guaina in silicone non si trovi allineata con la congiuntiva. Premere lentamente il pulsante di attivazione fino a sentire un caratteristico clic. Prima di rimuovere l’applicatore dall’occhio, assicurarsi che il pulsante di attivazione sia completamente premuto e che abbia bloccato il reflusso di liquido dalla superficie dell’applicatore. Rimuovere l’ago dal corpo vitreo seguendo il percorso inverso rispetto a quello utilizzato per l’inserimento.
Uso di Ozurdex®
| |
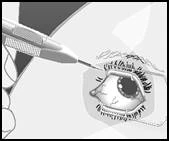
Mantenere l'asse longitudinale dell'applicatore parallelo al limbo. |
|
| |
Avvicinare l'applicatore direttamente alla sclera con un angolo acuto, orientando il taglio dell'ago verso l'alto, lontano dalla sclera. Inserire la punta dell'ago nella sclera per circa 1 mm, mantenendola parallela al limbo. |
|
| |
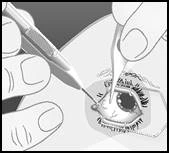
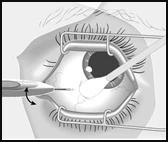
Ruotare l'applicatore verso il centro dell'occhio, entrando nella cavità del corpo vitreo. Questo creerà un ampio canale sclerale. Continuare a inserire l'ago fino a quando non si entra nella cavità del corpo vitreo. Interrompere l'inserimento dell'ago quando la ghiera dell'applicatore entra in contatto con la congiuntiva. |
|
| |
Premere lentamente il pulsante di attuazione fino a sentire un caratteristico clic. Prima di rimuovere l'applicatore dall'occhio, assicurarsi che il pulsante di attuazione sia completamente premuto e che abbia bloccato il deflusso di liquido dalla superficie dell'applicatore. |
|
| |
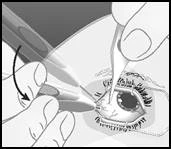
Estrarre l'applicatore dal corpo vitreo seguendo il percorso inverso rispetto a quello utilizzato per l'inserimento. |
|
| |
Smaltire in sicurezza l'applicatore immediatamente dopo l'uso. L'applicatore Ozurdex® è destinato all'uso monouso. |
Immediatamente dopo l'iniezione dell'impianto Ozurdex® effettuare una oftalmoscopia indiretta nel settore dell'iniezione per confermare l'avvenuto impianto. La visualizzazione è possibile nella maggior parte dei casi. Nel caso in cui l'impianto non sia visibile, utilizzare un bastoncino sterile di ovatta e premere leggermente sulla zona del sito di iniezione per rendere l'impianto visibile.
Dopo l'iniezione intravitreale, ai pazienti deve essere continuato il trattamento con un agente antibatterico a spettro ampio.
Popolazione pediatrica
Non vi sono dati adeguati sull'uso dell'impianto Ozurdex® nei bambini con
- edema maculare diabetico;
- edema maculare secondario ad occlusione della vena centrale della retina o ad occlusione di un ramo della vena della retina.
La sicurezza e l'efficacia dell'uso del medicinale Ozurdex® nell'uveite nei bambini non sono state stabilite. I dati sono mancanti.
Sovradosaggio
Non vi sono informazioni riguardo al sovradosaggio.
In caso di sovradosaggio, deve essere monitorata e normalizzata la pressione intraoculare, se il medico lo ritiene necessario.
Effetti indesiderati.
Riassunto del profilo di sicurezza
Gli effetti indesiderati più comunemente riportati sono quelli che si verificano frequentemente durante la terapia con corticosteroidi o sono associati alla procedura di iniezione intravitreale (aumento della pressione oculare, formazione di cataratta e sanguinamenti subcongiuntivali o intravitreali, rispettivamente).
Effetti indesiderati meno comuni ma più gravi includono: endoftalmite, retinite necrotizzante, distacco della cornea e lacerazione della cornea.
A parte cefalea e emicrania, non sono stati identificati effetti indesiderati sistemici con l'uso del medicinale Ozurdex®.
L'elenco degli effetti indesiderati è riportato nella Tabella 6.
Gli effetti indesiderati ritenuti correlati all'uso dell'impianto Ozurdex® in base ai risultati degli studi clinici di Fase III (edema maculare diabetico, occlusione della vena centrale della retina/occlusione del ramo della vena della retina e uveite) e alle segnalazioni spontanee sono elencati nella Tabella 6 secondo la classificazione per sistemi e organi MedDRA.
La frequenza degli effetti indesiderati è definita come segue:
molto frequente (≥ 1/10); frequente (da ≥1/100 a <1/10); non frequente (da ≥1/1000 a <1/100); raro (da ≥1/10000 a <1/1000); molto raro (<1/10000). All'interno di ciascun gruppo di frequenza, gli effetti indesiderati sono elencati in ordine decrescente di rilevanza clinica.
Tabella 6. Effetti indesiderati
| Sistemi degli organi |
Frequenza |
Reazione avversa |
| Patologie del sistema nervoso |
Comune |
Cefalea |
| Non comune |
Emicrania |
|
| Patologie dell'occhio |
Molto comune |
Aumento della pressione intraoculare**, cataratta**, emorragia congiuntivale* |
| Comune |
Oftalmopotensione, cataratta subcapsulare, emorragia nel corpo vitreo**, riduzione dell'acuità visiva*, peggioramento della vista, distacco del corpo vitreo*, "miodesopsie" (corpi mobili nel vitreo)*, opacizzazione del corpo vitreo*, blefarite, dolore all'occhio*, fotopsia*, edema congiuntivale*, iperemia congiuntivale* |
|
| Non comune |
Retinite necrotizzante, endoftalmite*, glaucoma, distacco della retina*, rottura della retina*, ipotensione oculare*, infiammazione della camera anteriore dell'occhio*, opalescenza cellulare della camera anteriore dell'occhio*, sensazione anomala all'occhio*, prurito delle palpebre, iperemia sclerale* |
|
| Disturbi generali e condizioni relative alla sede di somministrazione |
Non comune |
Dislocazione del prodotto* (migrazione dell'impianto) con o senza edema della cornea (vedere sezione "Precauzioni particolari di sicurezza"), complicazioni nell'applicazione del prodotto che portano a lesioni del tessuto oculare* (posizionamento errato dell'impianto) |
* Si ritiene che queste reazioni avverse siano associate alla procedura di iniezione intravitreale (la frequenza di queste reazioni avverse è proporzionale al numero di iniezioni somministrate).
** Durante uno studio osservazionale di 24 mesi condotto in condizioni reali per il trattamento dell'edema maculare conseguente all'occlusione della vena retinica e dell'uveite non infettiva del segmento posteriore dell'occhio, queste reazioni avverse si sono verificate più frequentemente nei pazienti che hanno ricevuto >2 iniezioni rispetto a quelli che hanno ricevuto ≤2 iniezioni: formazione di cataratta (24,7% vs 17,7%), sviluppo di cataratta (32,0% vs 13,1%), emorragia nel corpo vitreo (6,0% vs 2,0%) e aumento della PTIO (24,0% vs 16,6%).
Edema maculare diabetico
La sicurezza clinica dell'impianto Ozurdex**®** nei pazienti con edema maculare diabetico è stata valutata in due studi di fase III, randomizzati, in doppio cieco, controllati con placebo. Nel complesso, nei due studi hanno partecipato 347 pazienti assegnati al gruppo trattato con l'impianto Ozurdex**®** e 350 pazienti ai quali è stato somministrato il placebo.
Le reazioni avverse più comuni osservate negli studi clinici nei pazienti trattati con Ozurdex**®** sono state cataratta e aumento della PTIO (vedi sotto).
Nel corso di uno studio clinico triennale sull'edema maculare diabetico, all'inizio dello studio l'87% dei pazienti afachici trattati con l'impianto Ozurdex**®** presentava un certo grado di opacizzazione del cristallino/stadio iniziale di cataratta. La frequenza di comparsa di tali sintomi durante lo studio triennale, in tutti i pazienti esaminati con diversi tipi di cataratta (cataratta corticale, cataratta diabetica, cataratta nucleare, cataratta sottocapsulare, cataratta lenticolare, cataratta senile), è stata del 68% nei pazienti afachici trattati con Ozurdex**®**. Il 59% dei pazienti afachici trattati con l'impianto ha richiesto un intervento chirurgico per la rimozione della cataratta al momento dell'ultima visita dello studio triennale, e nella maggior parte dei casi l'intervento è stato eseguito durante il secondo e il terzo anno dello studio.
All'inizio dello studio, i valori medi della PTIO nell'occhio trattato erano simili nei due gruppi (15,3 mmHg). L'aumento medio della PTIO rispetto ai valori basali non ha superato i 3,2 mmHg in tutte le visite nel gruppo trattato con Ozurdex**®, con un picco della PTIO osservato a 1,5 mesi dall'iniezione, mentre la PTIO è tornata ai livelli basali entro il sesto mese dopo ciascuna iniezione. La frequenza e l'entità dell'aumento della PTIO dopo l'applicazione dell'impianto Ozurdex®** non sono aumentate con le somministrazioni ripetute.
Nel 28% dei pazienti trattati con Ozurdex**®** si è verificato un aumento della PTIO ≥10 mmHg rispetto ai valori basali durante una o più visite nel corso dello studio. All'inizio dello studio, il 3% dei pazienti richiedeva farmaci per ridurre la PTIO. Nel complesso, il 42% dei pazienti ha richiesto farmaci per ridurre la PTIO nell'occhio trattato in diversi momenti dello studio triennale, e la maggior parte di questi pazienti ha richiesto più di un farmaco aggiuntivo. La percentuale massima di farmaci per ridurre la PTIO (33%) è stata utilizzata nei primi 12 mesi e si è mantenuta a questo livello successivamente.
Complessivamente, 4 pazienti (1%) trattati con Ozurdex**®** hanno richiesto procedure chirurgiche per ridurre la PTIO nell'occhio trattato. Un paziente trattato con Ozurdex**®** ha richiesto un intervento chirurgico (trabeculectomia) per ridurre la PTIO indotta da steroidi; un paziente è stato sottoposto a trabeculectomia a causa di un blocco dell'afflusso dell'umore acqueo nella camera anteriore dell'occhio dovuto a fibrina, che ha causato un aumento della PTIO; un paziente è stato sottoposto a iridectomia per glaucoma ad angolo chiuso e un altro paziente è stato sottoposto a iridectomia in seguito a un intervento chirurgico per la cataratta. Nessun paziente ha richiesto la rimozione dell'impianto mediante vitrectomia per ridurre la PTIO.
Occlusione della vena centrale della retina/occlusione della ramificazione della vena retinica
La sicurezza clinica dell'impianto Ozurdex**®** è stata valutata in due studi randomizzati, in doppio cieco, controllati con placebo di fase III, condotti su pazienti con edema maculare causato da occlusione della vena centrale della retina o della ramificazione della vena retinica. Nel complesso, per i due studi di fase III sono stati randomizzati 427 pazienti trattati con Ozurdex**®** e 426 pazienti trattati con placebo. Nel complesso, 401 pazienti (94%) del gruppo trattato con Ozurdex**®** hanno completato il periodo iniziale di trattamento (fino al giorno 180).
Complessivamente, il 47,3% dei pazienti ha manifestato almeno una reazione avversa.
Le reazioni avverse più comuni riportate dopo la somministrazione dell'impianto Ozurdex**®** sono state l'aumento della pressione intraoculare (24%) e l'emorragia congiuntivale (14,7%).
Il profilo di reazioni avverse nei pazienti con occlusione della ramificazione della vena retinica è stato simile a quello osservato nei pazienti con occlusione della vena centrale della retina, sebbene nel complesso il numero di reazioni avverse nel sottogruppo di pazienti con occlusione della vena centrale della retina fosse più elevato.
L'aumento della pressione intraoculare (PTIO) dopo la somministrazione dell'impianto Ozurdex**®** è stato massimo al giorno 60 ed è tornato ai livelli basali al giorno 180. L'aumento della pressione intraoculare è stato gestito senza intervento terapeutico oppure con l'uso temporaneo di farmaci che riducono la PTIO. Durante il periodo iniziale di trattamento, lo 0,7% (3/421) dei pazienti trattati con Ozurdex**®** ha richiesto terapia laser o intervento chirurgico per ridurre l'aumento della PTIO, rispetto allo 0,2% nel gruppo placebo (1/423).
Il profilo di reazioni avverse dopo la seconda iniezione (341 pazienti) di Ozurdex**®** è stato simile a quello osservato dopo la prima iniezione. Nel 54% dei pazienti si è verificata almeno una reazione avversa. La frequenza di aumenti della PTIO (24,9%) è stata simile a quella osservata dopo la prima iniezione ed è tornata ai livelli basali al giorno 180. La frequenza complessiva di cataratta è risultata maggiore dopo un anno rispetto ai dati dei primi 6 mesi.
Uveite
La sicurezza clinica dell'impianto Ozurdex**®** nei pazienti con infiammazione del segmento posteriore dell'occhio sotto forma di uveite non infettiva è stata valutata in un singolo studio randomizzato, in cieco, multicentrico.
In totale, 77 pazienti sono stati randomizzati nel gruppo trattato con Ozurdex**®** e 76 nel gruppo placebo. 73 pazienti (95%) del gruppo trattato con l'impianto Ozurdex**®** hanno completato lo studio della durata di 26 settimane.
Le reazioni avverse più comuni osservate dopo la somministrazione dell'impianto Ozurdex**®** negli studi clinici sono state emorragia congiuntivale (30,3%), aumento della pressione intraoculare (25%) e cataratta (11,8%).
Segnalazione delle reazioni avverse sospette
La segnalazione delle reazioni avverse sospette è di fondamentale importanza poiché consente di continuare a monitorare il rapporto rischio/beneficio del medicinale. Si raccomanda agli operatori sanitari di segnalare qualsiasi reazione avversa sospetta.
Durata della validità.
3 anni.
Condizioni di conservazione.
Non richiede condizioni particolari di conservazione.
Conservare in un luogo inaccessibile ai bambini.
Incompatibilità.
Non sono noti casi di incompatibilità.
Confezione.
Ogni confezione contiene:
1 impianto sterile cilindrico contenente 700 mcg di desametasone, posizionato all'interno dell'ago (acciaio inossidabile) dell'applicatore.
L'applicatore è costituito da un cilindro con ago (acciaio inossidabile), all'interno del quale l'impianto è posizionato mediante un manicotto (silicone). Il cilindro è controllato da un dispositivo di sicurezza presente sull'applicatore. L'ago è protetto da un cappuccio e il dispositivo di sicurezza da un fermo.
L'applicatore contenente l'impianto, insieme a un pacchetto disidratante, è sigillato in un sacchetto in foglio di alluminio. Il sacchetto in foglio di alluminio è confezionato in una scatola di cartone.
Categoria di prescrizione.
Sotto prescrizione medica.
Produttore.
Allergan Pharmaceuticals Ireland.
Sede del produttore e indirizzo del luogo di produzione.
Castlebar Road, Westport, Co. Mayo, F28 AW83, Irlanda.