Filgrastim-Vista
Ucraina
Indice
ISTRUZIONE per l'uso medico del medicinale FILGRASTIM-VISTA (FILGRASTIM-VISTA)
Composizione:
Principio attivo: filgrastim;
1 siringa preriempita (0,5 ml di soluzione) contiene filgrastim 300 µg (30 milioni di UI);
1 siringa preriempita (0,8 ml di soluzione) contiene filgrastim 480 µg (48 milioni di UI);
Eccipienti: acido acetico glaciale, polisorbato 80, idrossido di sodio, sorbitolo, acqua per preparazioni iniettabili.
Forma farmaceutica. Soluzione iniettabile o per infusione.
Principali caratteristiche fisico-chimiche: soluzione incolore e limpida.
Gruppo farmacoterapeutico. Immunostimolanti. Fattori stimolanti le colonie. Filgrastim. Codice ATC L03A A02.
Proprietà farmacodinamiche
Il fattore stimolante le colonie di granulociti umani (G-CSF) è una glicoproteina che regola la produzione e il rilascio di neutrofili funzionali dal midollo osseo. Filgrastim-Vista, contenente r-metHuG-CSF (filgrastim), induce un marcato aumento del numero di neutrofili nel sangue periferico entro ventiquattro ore, con un lieve incremento del numero di monociti. In alcuni pazienti con sindrome da neutropenia cronica (SNC), il filgrastim può determinare anche un lieve aumento del numero di eosinofili e basofili circolanti rispetto ai livelli iniziali; alcuni di questi pazienti possono presentare eosinofilia o basofilia già prima del trattamento. L'aumento del numero di neutrofili è dipendente dalla dose alle dosi raccomandate. I neutrofili prodotti in risposta al filgrastim mostrano una funzionalità normale o potenziata, come dimostrato dai test di chemotassi e di attività fagocitaria. Dopo l'interruzione della terapia con filgrastim, il numero di neutrofili circolanti si riduce del 50% entro 1-2 giorni e ritorna ai livelli normali entro 1-7 giorni.
L'uso di filgrastim in pazienti sottoposti a chemioterapia citotossica determina una significativa riduzione della frequenza, gravità e durata della neutropenia e della neutropenia febbrile. Il trattamento con filgrastim riduce significativamente la durata della neutropenia febbrile, l'uso di antibiotici e la durata del ricovero ospedaliero dopo chemioterapia induttiva per leucemia mieloidi acuta o terapia mieloablativa seguita da trapianto di midollo osseo. Tuttavia, l'incidenza di febbre e il numero di infezioni documentate non sono risultati ridotti. La durata della febbre non si è ridotta nei pazienti sottoposti a terapia mieloablativa seguita da trapianto di midollo osseo.
L'uso di filgrastim, singolarmente o in seguito a chemioterapia, stimola il rilascio nel sangue periferico di cellule progenitrici ematopoietiche. Queste cellule progenitrici autologhe del sangue periferico possono essere raccolte ed infuse dopo terapia citotossica ad alta dose, in sostituzione o in aggiunta al trapianto di midollo osseo. L'infusione di cellule progenitrici del sangue periferico accelera il ripristino dell'emopoiesi, riducendo la durata del rischio di complicanze emorragiche e la necessità di trasfusioni di piastrine. Nei riceventi di cellule progenitrici allogeniche del sangue periferico mobilizzate con filgrastim, il recupero ematologico si è verificato significativamente più rapidamente, con una riduzione statisticamente significativa del tempo necessario al recupero indipendente delle piastrine rispetto al trapianto allogenico di midollo osseo.
Uno studio retrospettivo europeo che ha valutato l'uso di G-CSF dopo trapianto allogenico di midollo osseo in pazienti con leucemie acute ha mostrato un aumento del rischio di reazione di rigetto "trapianto contro ospite" (GvHD), della mortalità correlata al trattamento (TRM) e della mortalità generale nei pazienti trattati con G-CSF. In uno studio retrospettivo internazionale separato che includeva pazienti con leucemia mieloide acuta e cronica, non è stato osservato alcun effetto sul rischio di GvHD, TRM e mortalità. Una meta-analisi degli studi sul trapianto allogenico, compresi i risultati di nove studi randomizzati controllati, otto studi retrospettivi e uno studio controllato, non ha evidenziato alcun effetto sui rischi di GvHD acuta, GvHD cronica o mortalità precoce correlata al trattamento.
| Rischio relativo (IC 95% [intervallo di confidenza]) di malattia del trapianto contro l'ospite (GvHD) e mortalità associata al trattamento (TRM) Dopo il trattamento con G-CSF successivo al trapianto di midollo osseo |
|||||
| Pubblicazione |
Periodo dello studio |
Numero |
GvHD acuta di grado II-IV |
GvHD cronica |
TRM |
| Meta-analisi (2003) |
1986–2001a |
1198 |
1,08 (0,87, 1,33) |
1,02 (0,82, 1,26) |
0,70 (0,38, 1,31) |
| Studio retrospettivo europeo (2004) |
1992–2002b |
1789 |
1,33 (1,08, 1,64) |
1,29 (1,02, 1,61) |
1,73 (1,30, 2,32) |
| Studio retrospettivo internazionale (2006) |
1995–2000b |
2110 |
1,11 (0,86, 1,42) |
1,10 (0,86, 1,39) |
1,26 (0,95, 1,67) |
a L'analisi comprende studi relativi al trapianto di midollo osseo durante questo periodo; alcuni studi hanno utilizzato il G-CSF.
b L'analisi comprende pazienti sottoposti a trapianto di midollo osseo durante questo periodo.
Uso di filgrastim per la mobilizzazione delle cellule progenitrici del sangue periferico in donatori sani prima del trapianto allogenico di cellule progenitrici del sangue periferico. In donatori normali, una dose di 10 µg/kg/die somministrata per via sottocutanea per 4-5 giorni consecutivi consente di raccogliere ≥ 4 × 106 cellule CD34+ / kg di peso corporeo del ricevente nella maggior parte dei donatori dopo due leucaferesi.
L'uso di filgrastim in pazienti, bambini o adulti, con NCA (neutropenia congenita grave, neutropenia ciclica e neutropenia idiopatica) determina un aumento sostenuto del conteggio assoluto di neutrofili nel sangue periferico e riduce le infezioni e le complicanze ad esse correlate. L'uso di filgrastim in pazienti con infezione da HIV mantiene un conteggio normale di neutrofili, consentendo la somministrazione programmata di farmaci antivirali e/o di altri agenti mielosoppressivi. Non vi sono evidenze che nei pazienti con infezione da HIV trattati con filgrastim aumenti la replicazione dell'HIV.
Come altri fattori emopoietici di crescita, il G-CSF ha dimostrato proprietà stimolanti in vitro sulle cellule endoteliali umane.
Farmacocinetica
È stato dimostrato che l'eliminazione del filgrastim segue una cinetica di primo ordine sia dopo somministrazione sottocutanea che endovenosa. L'emivita sierica del filgrastim è di circa 3,5 ore, la clearance è di circa 0,6 ml/min/kg. L'infusione continua di filgrastim per un periodo fino a 28 giorni in pazienti in fase di recupero dopo trapianto autologo di midollo osseo non ha determinato un accumulo del farmaco e l'emivita è risultata comparabile. Esiste una correlazione lineare positiva tra dose e concentrazione sierica di filgrastim indipendentemente dalla via di somministrazione (endovenosa o sottocutanea). Dopo somministrazione sottocutanea delle dosi raccomandate, le concentrazioni sieriche sono state mantenute superiori a 10 ng/ml per 8-16 ore. Il volume di distribuzione nel sangue è di circa 150 ml/kg.
Caratteristiche cliniche
Indicazioni
- Per ridurre la durata della neutropenia e diminuire la frequenza della neutropenia febbrile in pazienti sottoposti a chemioterapia citotossica per neoplasie maligne (esclusa la leucemia mieloide cronica e il sindrome mielodisplastico).
- Per ridurre la durata della neutropenia in pazienti sottoposti a terapia mieloablativa seguita da trapianto di midollo osseo, con alto rischio di neutropenia grave prolungata.
- Per la mobilizzazione delle cellule staminali del sangue periferico (CBSC).
- Per una terapia a lungo termine al fine di aumentare il numero di neutrofili e ridurre la frequenza e la durata delle complicanze infettive in bambini e adulti con neutropenia congenita grave, periodica o idiopatica (numero assoluto di neutrofili ≤ 0,5×109/l) e con infezioni gravi o ricorrenti in anamnesi.
- Per ridurre il rischio di infezioni batteriche in caso di neutropenia persistente (numero assoluto di neutrofili ≤ 1,0×109/l) in pazienti con infezione da HIV in fase avanzata, quando altri trattamenti per il controllo della neutropenia si sono dimostrati inefficaci.
Controindicazioni
Ipersensibilità al principio attivo o a qualsiasi eccipiente.
Precauzioni particolari di sicurezza
Gli scarti del medicinale non utilizzato devono essere smaltiti secondo le normative vigenti.
Le siringhe con dispositivo di sicurezza devono essere smaltite secondo le normative vigenti.
Interazioni con altri medicinali ed altre forme di interazione
Non sono state stabilite la sicurezza e l'efficacia dell'uso contemporaneo di filgrastim e agenti citotossici mielosoppressivi nello stesso giorno. Poiché le cellule mieloidi in rapida divisione sono sensibili alla chemioterapia citotossica mielosoppressiva, non si raccomanda di somministrare filgrastim meno di 24 ore prima o meno di 24 ore dopo l'amministrazione di tali agenti. Dati preliminari ottenuti da un numero ridotto di pazienti che hanno ricevuto contemporaneamente filgrastim e 5-fluorouracile indicano un rischio di peggioramento della gravità della neutropenia. Possibili interazioni con altri fattori di crescita emopoietici e citochine non sono state studiate in studi clinici.
Considerando che il litio stimola la mobilizzazione dei neutrofili, un effetto potenziato di filgrastim può verificarsi in caso di somministrazione concomitante. Sebbene studi specifici su questa interazione non siano stati condotti, non esistono evidenze di effetti dannosi derivanti da tale interazione.
Caratteristiche particolari di impiego
Ipersensibilità
Sono state osservate reazioni di ipersensibilità, comprese reazioni anafilattiche, all'inizio o durante il proseguimento del trattamento con filgrastim. In caso di comparsa di reazioni di ipersensibilità clinicamente significative, l'uso di filgrastim deve essere interrotto e non deve essere ripreso successivamente. Filgrastim non deve essere somministrato ai pazienti con anamnesi di ipersensibilità al filgrastim o al pegfilgrastim.
Reazioni avverse a carico del polmone
Sono state riportate reazioni avverse a carico del polmone, in particolare malattia polmonare interstiziale, dopo somministrazione di G-CSF. I pazienti con infiltrati polmonari o pneumonite nella storia clinica recente hanno un rischio maggiore di sviluppare tali reazioni. L'insorgenza di sintomi respiratori, come tosse, febbre e dispnea, in combinazione con segni radiologici di infiltrati polmonari e peggioramento della funzionalità polmonare, possono rappresentare i primi segni di sindrome da distress respiratorio acuto (ARDS). È necessario interrompere il trattamento con filgrastim e iniziare un trattamento appropriato.
Glomerulonefrite
Sono stati riportati casi di glomerulonefrite in pazienti trattati con filgrastim e pegfilgrastim. Generalmente la glomerulonefrite regredisce dopo riduzione della dose o interruzione del trattamento con filgrastim e pegfilgrastim. Si raccomanda un regolare esame delle urine.
Sindrome da perdita capillare
Sono stati riportati casi di sindrome da perdita capillare dopo somministrazione di G-CSF. Questa condizione è caratterizzata da ipotensione, ipoalbuminemia, edema e ispessimento del sangue e può essere potenzialmente letale in assenza di trattamento tempestivo. I pazienti che sviluppano sintomi di sindrome da perdita capillare devono essere attentamente monitorati e trattati con terapia sintomatica standard e, se necessario, con terapia intensiva.
Splenomegalia e rottura della milza
Sono stati osservati casi generalmente asintomatici di splenomegalia e rari casi di rottura della milza in pazienti e donatori sani trattati con filgrastim. Alcuni casi di rottura della milza sono stati letali. Per questo motivo si raccomanda il monitoraggio delle dimensioni della milza (ad esempio esame clinico, ecografia) in tali pazienti. Si deve considerare la possibilità di rottura della milza in donatori e/o pazienti che riferiscono dolore nell'area superiore sinistra dell'addome o nella spalla sinistra. La riduzione della dose di filgrastim ha rallentato o arrestato l'ingrandimento della milza in pazienti con neutropenia cronica grave, e il 3% dei pazienti ha richiesto splenectomia.
Crescita di cellule maligne
Il G-CSF può indurre la crescita di cellule mieloidi in vitro. Effetti simili sono possibili anche per alcune cellule non mieloidi.
Sindrome mielodisplastica o leucemia mieloide cronica
La sicurezza e l'efficacia dell'impiego di filgrastim nei pazienti con sindrome mielodisplastica o leucemia mieloide cronica non sono state stabilite. Filgrastim non è raccomandato per l'uso in queste patologie. È necessaria particolare attenzione nella diagnosi differenziale tra crisi blastica nella leucemia mieloide cronica e leucemia mieloide acuta. Leucemia mieloide acuta
La sicurezza e l'efficacia dell'uso di filgrastim nei pazienti con leucemia mieloide acuta secondaria non sono state sufficientemente studiate; pertanto, il filgrastim deve essere somministrato con cautela. Non sono state stabilite la sicurezza e l'efficacia dell'uso di filgrastim nella leucemia mieloide acuta de novo in pazienti di età inferiore a 55 anni con fattori citogenetici prognostici favorevoli [t(8;21), t(15;17) e inv(16)].
Trombocitopenia
Sono stati riportati casi di trombocitopenia in pazienti trattati con filgrastim. È necessario monitorare attentamente il numero di piastrine, specialmente durante le prime settimane di trattamento con filgrastim. Se nei pazienti con neutropenia cronica grave si sviluppa trombocitopenia (numero di piastrine < 100×10⁹/l), si deve considerare l'interruzione temporanea o la riduzione della dose di filgrastim.
Leucocitosi
In meno del 5% dei pazienti oncologici trattati con filgrastim a dosi superiori a 0,3 milioni UI/kg/die (3 µg/kg/die), il numero di leucociti è aumentato a oltre 100×10⁹/l. Non sono state descritte reazioni avverse direttamente correlate a tale leucocitosi. Tuttavia, considerato il rischio associato a una marcata leucocitosi, durante il trattamento con filgrastim è necessario monitorare regolarmente il numero di leucociti. Se dopo il previsto calo del numero di leucociti questo supera 50×10⁹/l, il trattamento con filgrastim deve essere immediatamente interrotto. Tuttavia, se filgrastim è utilizzato per la mobilizzazione delle CSE, il trattamento deve essere sospeso o la dose ridotta quando il numero di leucociti supera 70×10⁹/l.
Immunogenicità
Come per tutti i farmaci proteici, esiste il rischio di reazioni immunogeniche. La frequenza di sviluppo di anticorpi contro filgrastim è generalmente bassa. La comparsa di anticorpi leganti è attesa, come per altri farmaci biologici; tuttavia, attualmente non ci sono dati sulla loro attività neutralizzante.
Aortite
Sono stati riportati casi di aortite dopo somministrazione di G-CSF in soggetti sani e in pazienti oncologici. I sintomi possono includere febbre, dolore addominale, malessere, dolore alla schiena e possono essere associati a un aumento dei marcatori infiammatori (ad esempio proteina C-reattiva, numero di leucociti). Nella maggior parte dei casi, l'aortite è stata diagnosticata mediante tomografia computerizzata ed è generalmente regredita dopo l'interruzione del G-CSF.
Caratteristiche particolari di impiego in presenza di patologie concomitanti
Anomalia a cellule falciformi e anemia falciforme
In pazienti con anomalia a cellule falciformi o anemia falciforme, durante il trattamento con filgrastim sono stati osservati episodi di crisi falciforme, talvolta con esito fatale. Pertanto, filgrastim deve essere somministrato con cautela ai pazienti con anomalia a cellule falciformi o anemia falciforme.
Osteoporosi
Ai pazienti con osteoporosi concomitante che ricevono terapia continua con filgrastim per oltre 6 mesi è raccomandato il monitoraggio della densità minerale ossea.
Avvertenze particolari per pazienti con patologie oncologiche
Filgrastim non deve essere somministrato per aumentare il dosaggio della chemioterapia citotossica oltre i limiti stabiliti.
Rischio associato alla chemioterapia ad alto dosaggio
Particolare cautela è richiesta nel trattamento di pazienti sottoposti a chemioterapia ad alto dosaggio, poiché in questi casi l'efficacia del trattamento non è stata stabilita, mentre dosi più elevate di agenti chemioterapici hanno mostrato una tossicità più marcata, causando reazioni cardiache, polmonari, neurologiche e dermatologiche (vedere il foglio illustrativo del corrispondente agente chemioterapico).
Effetto della chemioterapia su eritrociti e piastrine
La monoterapia con filgrastim non previene la trombocitopenia e l'anemia indotte da chemioterapia mielosoppressiva. A causa della possibilità di utilizzare dosi più elevate di agenti chemioterapici (ad esempio dosi complete secondo gli schemi), il paziente può essere esposto a un rischio maggiore di sviluppare trombocitopenia e anemia; pertanto si raccomanda di determinare regolarmente il numero di piastrine e l'ematocrito. Filgrastim deve essere somministrato con particolare cautela in combinazione con schemi chemioterapici monocomponente o combinati che possono causare trombocitopenia grave.
La somministrazione di CSE mobilizzate con filgrastim riduce l'intensità e la durata della trombocitopenia dopo chemioterapia mielosoppressiva o mieloablativa.
Sindrome mielodisplastica e leucemia mieloide acuta in pazienti con cancro al seno e al polmone
In uno studio osservazionale post-marketing, la sindrome mielodisplastica (MDS) e la leucemia mieloide acuta (AML) sono state associate all'uso di pegfilgrastim, un G-CSF alternativo, in combinazione con chemioterapia e/o radioterapia in pazienti con cancro al seno e al polmone. Tale associazione non è stata osservata con filgrastim. Tuttavia, i pazienti con cancro al seno e al polmone devono essere monitorati per segni e sintomi di MDS/AML.
Altre avvertenze particolari
L'effetto di filgrastim in pazienti con marcata riduzione delle cellule progenitrici mieloidi non è stato studiato. Filgrastim aumenta il numero di neutrofili agendo principalmente sulle cellule progenitrici dei neutrofili. Pertanto, in pazienti con un numero ridotto di cellule progenitrici (ad esempio, dopo radioterapia intensiva o chemioterapia, o in presenza di infiltrazione tumorale del midollo osseo), l'aumento del numero di neutrofili può essere limitato.
Occasionalmente, in pazienti sottoposti a chemioterapia ad alto dosaggio seguita da trapianto autologo di midollo osseo, sono stati osservati disturbi vascolari, in particolare malattia da occlusione venosa e alterazioni dell'equilibrio idrico.
Sono stati riportati casi di reazione di trapianto contro ospite con esito fatale in pazienti trattati con fattore stimolante le colonie granulocitarie dopo trapianto allogenico di midollo osseo.
Sono stati riportati aumenti transitori dell'attività emopoietica del midollo osseo in risposta al trattamento con fattore stimolante le colonie granulocitaria, manifestati da risultati positivi temporanei negli scintigrammi ossei. Tale aspetto deve essere considerato nell'interpretazione degli esami scintigrafici.
Avvertenze particolari per pazienti sottoposti a mobilizzazione delle CSE
Mobilizzazione
Non è stato effettuato un confronto prospettico randomizzato tra i due metodi raccomandati di mobilizzazione (solo filgrastim o filgrastim in combinazione con chemioterapia mielosoppressiva) nella stessa popolazione di pazienti. Le caratteristiche individuali dei pazienti in diversi studi e le differenze nei metodi di determinazione del numero di cellule CD34+ rendono difficile un confronto diretto dei risultati. Pertanto, è difficile raccomandare un metodo ottimale. La scelta del metodo di mobilizzazione deve essere basata sull'obiettivo terapeutico del paziente.
Prima della somministrazione di agenti citotossici
In pazienti precedentemente sottoposti a terapia mielosoppressiva intensiva, potrebbe non verificarsi una sufficiente attivazione delle CSE al livello minimo raccomandato (≥ 2,0×10⁶ cellule CD34+/kg) o un rapido recupero del numero di piastrine. Alcuni citostatici hanno una tossicità specifica per le cellule progenitrici emopoietiche e possono influire negativamente sulla loro mobilizzazione. Farmaci come melfalan, carmustina e carboplatino, se somministrati per un lungo periodo prima della mobilizzazione delle cellule staminali, possono ridurne l'efficacia. Tuttavia, l'uso combinato di melfalan, carboplatino o carmustina con filgrastim si è dimostrato efficace nell'attivazione delle cellule staminali. Se si prevede un trapianto di CSE, si raccomanda di pianificare la mobilizzazione delle cellule staminali in una fase precoce del trattamento. Particolare attenzione deve essere prestata al numero di cellule staminali mobilizzate in questi pazienti prima della chemioterapia ad alto dosaggio. Se i risultati della mobilizzazione non sono sufficienti secondo i criteri sopra indicati, si devono considerare alternative terapeutiche che non richiedano l'uso di cellule progenitrici.
Valutazione del numero di cellule staminali mobilizzate nel sangue periferico
Nella valutazione del numero di CSE mobilizzate con filgrastim, è necessario prestare particolare attenzione al metodo di quantificazione. I risultati dell'analisi citometrica del numero di cellule CD34+ variano a seconda della metodica utilizzata; pertanto, si deve procedere con cautela nell'applicare raccomandazioni basate su studi effettuati in altri laboratori. I risultati dell'analisi statistica della relazione tra il numero di cellule CD34+ infuse e la velocità di recupero del numero di piastrine dopo chemioterapia ad alto dosaggio indicano una relazione complessa ma costante. Il numero minimo raccomandato di CSE è ≥ 2,0×10⁶ cellule CD34+/kg, basato su dati pubblicati sull'esperienza di un adeguato recupero ematologico. Un numero superiore a questo valore sembra accelerare il recupero, mentre un numero inferiore determina un recupero più lento della composizione ematica.
Avvertenze particolari per donatori sani sottoposti a mobilizzazione delle CSE
La mobilizzazione delle CSE non offre un beneficio clinico diretto ai donatori sani e deve essere considerata esclusivamente ai fini di trapianto allogenico di cellule staminali.
La mobilizzazione delle CSE nei donatori può essere effettuata solo se soddisfatti i criteri clinici e di laboratorio abituali per la donazione di cellule staminali, con particolare attenzione agli indici ematologici e alla presenza di malattie infettive.
La sicurezza ed efficacia dell'uso di filgrastim nei donatori sani di età inferiore ai 16 anni o superiore ai 60 anni non è stata valutata.
Una trombocitopenia transitoria (numero di piastrine < 100×10⁹/l) dopo somministrazione di filgrastim e leucaferesi è stata osservata nel 35% dei soggetti studiati. Tra questi, 2 casi hanno mostrato trombocitopenia con numero di piastrine < 50×10⁹/l, correlata alla procedura di leucaferesi.
Nel caso di necessità di più di una leucaferesi, particolare attenzione deve essere prestata ai donatori con numero di piastrine inferiore a 100×10⁹/l prima della procedura; in generale, l'aferesi non deve essere effettuata se il numero di piastrine è inferiore a 75×10⁹/l. La leucaferesi non deve essere effettuata in donatori in trattamento con anticoagulanti o con alterazioni dell'omeostasi ematica.
I donatori che assumono G-CSF per la mobilizzazione delle CSE devono essere monitorati fino alla normalizzazione degli indici ematologici.
In donatori che ricevono G-CSF sono state osservate alterazioni citogenetiche transitorie. L'importanza clinica di tali cambiamenti è sconosciuta. Tuttavia, non può essere escluso il rischio di stimolazione di un clone mieloide maligno. Ai centri di aferesi si raccomanda di registrare e monitorare i donatori di cellule staminali per almeno 10 anni per garantire il monitoraggio della sicurezza a lungo termine.
Avvertenze particolari per i riceventi di CSE allogeniche mobilizzate con filgrastim
I dati disponibili indicano che l'interazione immunologica tra CSE allogeniche e ricevente comporta un rischio maggiore di sviluppare reazioni di trapianto contro ospite acuta e cronica rispetto al trapianto di midollo osseo.
Avvertenze particolari per pazienti con neutropenia cronica grave
Filgrastim non deve essere somministrato a pazienti con neutropenia congenita grave che sviluppano leucemia o mostrano segni di sviluppo leucemico.
Determinazione del numero di cellule ematiche
Possono verificarsi altri cambiamenti nel profilo ematico, in particolare anemia e aumento transitorio del numero di cellule progenitrici mieloidi, che richiedono un attento monitoraggio del numero di cellule.
Trasformazione in leucemia o sindrome mielodisplastica
Particolare cautela è richiesta nella diagnosi delle neutropenie croniche gravi. È necessario differenziarle da altre patologie ematologiche come anemia aplastica, mielodisplasia e leucemia mieloide. Prima dell'inizio del trattamento, si deve effettuare un esame ematico completo con formula leucocitaria e conta delle piastrine, nonché un esame morfologico del midollo osseo e cariotipo.
Negli studi clinici, solo una piccola percentuale (3%) di pazienti con neutropenia cronica grave trattati con filgrastim ha sviluppato sindrome mielodisplastica (MDS) o leucemia. Tale osservazione è stata fatta solo in pazienti con neutropenia congenita. MDS e leucemia sono complicanze comuni di questa malattia; il loro legame con il trattamento con filgrastim non è stato definito. In circa il 12% dei pazienti con citogenetica basale normale, nei controlli successivi sono state riscontrate anomalie, in particolare monosomia 7. Attualmente non è noto se il trattamento prolungato con filgrastim favorisca lo sviluppo di anomalie citogenetiche, MDS e leucemia nei pazienti con neutropenia cronica grave. In questi pazienti si raccomanda di effettuare esami morfologici e citogenetici del midollo osseo regolarmente (circa ogni 12 mesi).
Altre avvertenze particolari
È necessario escludere cause di neutropenia transitoria come infezioni virali.
In un numero ridotto di pazienti sono state osservate ematuria e/o proteinuria; per il loro monitoraggio si raccomanda un esame urinario regolare.
La sicurezza ed efficacia dell'uso di filgrastim nei neonati e nei pazienti con neutropenia autoimmune non sono state stabilite.
Avvertenze particolari per pazienti con infezione da HIV
Determinazione del numero di cellule ematiche
È necessario monitorare attentamente il numero di neutrofili, specialmente durante le prime settimane di trattamento con filgrastim. In alcuni pazienti, già dopo la prima iniezione, si osserva rapidamente un effetto terapeutico con marcato aumento del numero di neutrofili. Si raccomanda di controllare il numero di neutrofili giornalmente nei primi 2-3 giorni di trattamento con filgrastim, poi almeno due volte alla settimana durante le prime due settimane e almeno una volta alla settimana o ogni due settimane durante il trattamento di mantenimento. Se la dose di filgrastim di 30 milioni UI (300 µg) al giorno viene somministrata in modo non quotidiano, nel tempo possono verificarsi forti oscillazioni del numero di neutrofili. Per determinare la riduzione del numero di neutrofili o il livello minimo effettivo, si raccomanda di prelevare campioni di sangue immediatamente prima della somministrazione della dose successiva di filgrastim.
Rischio associato all'uso di dosi elevate di farmaci mielosoppressivi
La monoterapia con filgrastim non previene la trombocitopenia e l'anemia indotte da chemioterapia mielosoppressiva. A causa della possibilità di utilizzare dosi maggiori o più elevate di agenti chemioterapici insieme a filgrastim, il paziente può essere esposto a un rischio maggiore di sviluppare trombocitopenia e anemia; pertanto si raccomanda di determinare regolarmente il numero di cellule ematiche (vedere sopra).
Infezioni e neoplasie maligne che causano mielosoppressione
La neutropenia può essere causata dall'infiltrazione del midollo osseo da agenti infettivi opportunistici (come Mycobacterium avium) o da neoplasie (linfoma). A tali pazienti, oltre al filgrastim per il trattamento della neutropenia, deve essere somministrato un trattamento specifico per la patologia di base. L'effetto di filgrastim sulla neutropenia causata da agenti infettivi o neoplasie maligne del midollo osseo non è stato sufficientemente studiato.
Altre avvertenze
Tracciabilità. Per migliorare la tracciabilità dei fattori stimolanti le colonie granulocitarie, nella cartella clinica del paziente deve essere chiaramente indicato il nome commerciale del farmaco prescritto e il numero di lotto del prodotto somministrato.
Informazioni importanti sulle sostanze ausiliarie
Sorbitolo. Il medicinale contiene sorbitolo. È necessario considerare l'effetto additivo dell'uso contemporaneo di medicinali contenenti sorbitolo (o fruttosio) e l'assunzione di sorbitolo (o fruttosio) con l'alimentazione.
Per somministrazione endovenosa. Il filgrastim non deve essere somministrato a pazienti con intolleranza ereditaria al fruttosio (IEF), tranne nei casi di assoluta necessità. Nei neonati e nei bambini piccoli (fino a 2 anni) l'IEF può non essere ancora diagnosticata. I medicinali per somministrazione endovenosa contenenti sorbitolo/fruttosio possono essere letali; pertanto non devono essere somministrati a questa categoria di pazienti, salvo nei casi di assoluta necessità clinica acuta e in assenza di alternative. Prima di prescrivere questo medicinale, è necessario raccogliere un'anamnesi dettagliata sui sintomi di IEF in ogni paziente.
Sodio. Un'ampolla preriempita contiene meno di 1 mmol (23 mg) di sodio, cioè il medicinale è praticamente privo di sodio.
Uso durante la gravidanza o l'allattamento
Gravidanza. I dati sull'uso di filgrastim in donne in gravidanza sono limitati o assenti. Studi sugli animali hanno mostrato tossicità riproduttiva. È stata osservata una maggiore frequenza di perdita embrionale negli animali dopo dosi ripetute e alte dosi e in presenza di tossicità per la madre. Sono stati riportati casi di passaggio di filgrastim attraverso la barriera placentare. Filgrastim non è raccomandato durante la gravidanza.
Allattamento. Non è noto se filgrastim o i suoi metaboliti passino nel latte materno umano. Non può essere escluso un rischio per neonati/lattanti allattati al seno. La decisione di interrompere l'allattamento o di interrompere/sospendere la terapia con filgrastim deve essere presa considerando il beneficio dell'allattamento per il bambino e il beneficio del trattamento per la donna.
Fertilità. Negli studi sugli animali, è stato dimostrato che filgrastim non influisce sulla funzione riproduttiva e sulla fertilità.
Capacità di guidare veicoli o utilizzare macchinari
Filgrastim può influire in modo lieve sulla capacità di guidare veicoli o utilizzare macchinari. Dopo somministrazione di filgrastim può verificarsi capogiro (vedere il paragrafo «Effetti indesiderati»).
Modalità e posologia
Il trattamento deve essere effettuato in collaborazione con un centro oncologico dotato delle necessarie attrezzature diagnostiche e di personale esperto con adeguata esperienza nel campo dell'emmatologia e del trattamento con fattore di stimolazione delle colonie granulocitarie umano. Le procedure di mobilizzazione e di aferesi cellulare devono essere eseguite da specialisti con sufficiente esperienza in questo settore e con adeguato monitoraggio dei precursori emopoietici.
Chemioterapia citotossica abituale
Posologia
La dose raccomandata di filgrastim è di 0,5 milioni UI (5 µg)/kg di peso corporeo una volta al giorno. La prima dose deve essere somministrata non prima di 24 ore dopo il termine del ciclo di chemioterapia citotossica. Negli studi clinici randomizzati è stata somministrata per via sottocutanea una dose di 23 milioni UI (230 µg)/m²/die (4,0–8,4 µg/kg di peso corporeo al giorno).
Il medicinale deve essere somministrato ogni giorno fino al ripristino dei livelli normali di neutrofili dopo il calo atteso. Nei pazienti sottoposti a chemioterapia citotossica per tumori solidi, linfomi e linfoleucosi, la durata prevista del trattamento necessaria per soddisfare questi criteri è fino a 14 giorni. Dopo la terapia induttiva e di consolidamento per la leucemia mieloide acuta, la durata del trattamento può aumentare notevolmente (fino a 38 giorni), in base al tipo, alle dosi e allo schema di chemioterapia citotossica impiegati.
Solitamente si osserva un aumento temporaneo del numero di neutrofili entro 1-2 giorni dall'inizio del trattamento con filgrastim nei pazienti sottoposti a chemioterapia citotossica. Tuttavia, per ottenere un effetto terapeutico stabile, il trattamento con filgrastim non deve essere interrotto prima che il numero di neutrofili, dopo il calo massimo atteso, sia tornato ai livelli normali. Non è raccomandato interrompere prematuramente il trattamento, prima del calo massimo atteso del numero di neutrofili.
Modalità di somministrazione
Il medicinale può essere somministrato mediante iniezioni sottocutanee giornaliere o infusioni endovenose giornaliere brevi (di 30 minuti) in soluzione glucosata al 5%. La via sottocutanea è preferibile, poiché dati da studi clinici sull'uso singola dose indicano che la durata dell'effetto del filgrastim può ridursi con la somministrazione endovenosa. La rilevanza clinica di questi dati per l'uso ripetuto non è stata chiarita. La scelta della via di somministrazione deve essere effettuata in base alle caratteristiche di ciascun caso clinico.
Somministrazione sottocutanea
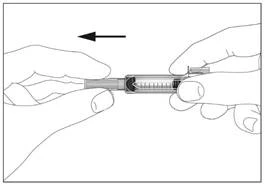
1
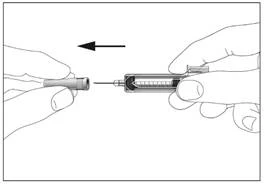
2
Siringa con dispositivo di sicurezza per la prevenzione di lesioni da ago o riutilizzo (fig. 1, 2).
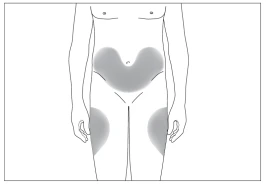
3
4
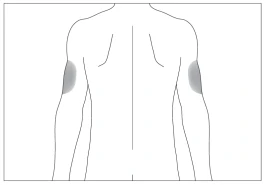
Aree corporee preferite per la somministrazione sottocutanea (fig. 3, 4).
5
6
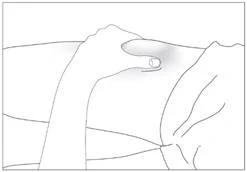
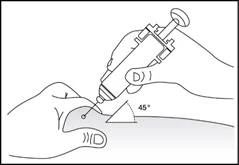
Somministrazione sottocutanea del medicinale (fig. 5–6).
- Disinfettare il sito di iniezione con una garza imbevuta di soluzione alcolica. Afferrare l'area della pelle tra pollice e indice, senza stringerla (fig. 5).
- Inserire l'ago per tutta la sua lunghezza nella pelle (fig. 6).
- Tirare leggermente il pistone della siringa verso di sé per verificare di non aver colpito un vaso sanguigno. Se nella siringa appare sangue, estrarre l'ago ed effettuare nuovamente l'iniezione in un'altra area della pelle.
Il filgrastim deve essere somministrato ogni giorno alla stessa ora.
Per evitare dolore, è preferibile cambiare ogni giorno il sito di iniezione.
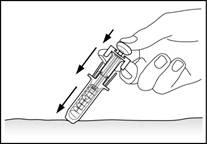
7
Siringhe con dispositivo di sicurezza
Il filgrastim deve essere somministrato lentamente e uniformemente, mantenendo la pelle afferrata tra le dita, fino al completo rilascio della dose e all'arresto del movimento del pistone della siringa.
È vietato esercitare una pressione eccessiva sul pistone.
Dopo la somministrazione del medicinale, estrarre l'ago continuando a premere con il dito sul pistone, quindi rilasciare la pelle (fig. 7).
Una volta rimosso il dito dal pistone, il dispositivo di protezione della siringa si chiuderà rapidamente sull'ago per coprirlo.
Per ogni iniezione successiva utilizzare una siringa separata. È vietato riutilizzare il filgrastim rimasto nella siringa.
Terapia mieloablativa seguita da trapianto di midollo osseo
Posologia
La dose iniziale raccomandata è di 1,0 milione UI (10 µg)/kg di peso corporeo al giorno. La prima dose di filgrastim deve essere somministrata non prima di 24 ore dopo la chemioterapia citotossica e non prima di 24 ore dopo il trapianto di midollo osseo.
Dopo il calo massimo del numero di neutrofili, la dose giornaliera viene aggiustata in base alle variazioni del numero di neutrofili (vedi tab. 1).
Tabella 1
| Numero di neutrofili |
Dosaggio di filgrastim |
| > 1,0×109/l per 3 giorni consecutivi |
Ridurre a 0,5 milioni UI (5 µg)/kg/giorno |
| Poi, se il numero assoluto di neutrofili rimane > 1,0×109/l per i successivi 3 giorni consecutivi |
Interrompere l'uso di filgrastim |
| Se durante il trattamento il numero assoluto di neutrofili diminuisce fino a livelli < 1,0×109/l, aumentare nuovamente la dose di filgrastim secondo lo schema sopra indicato. |
|
Modalità di somministrazione
Il medicinale può essere somministrato mediante infusione endovenosa della durata di 30 minuti o di 24 ore oppure mediante infusione sottocutanea continua della durata di 24 ore. Filgrastim deve essere diluito in 20 ml di soluzione glucosata al 5% per infusione.
Mobilizzazione delle cellule staminali del sangue periferico in pazienti sottoposti a terapia mielosoppressiva o mieloablative seguita da trapianto autologo di CSCH
Dosaggio
Quando filgrastim viene utilizzato come monoterapia per la mobilizzazione delle CSCH, il dosaggio raccomandato è di 1,0 milione UI (10 µg)/kg/die per 5-7 giorni consecutivi. Di norma sono sufficienti uno o due leucocitaferesi effettuati al quinto o al sesto giorno. Nel caso sia necessaria un’ulteriore leucocitaferesi, il trattamento con filgrastim alla stessa dose deve proseguire fino al completamento della procedura.
Per la mobilizzazione delle CSCH dopo chemioterapia mielosoppressiva, il dosaggio raccomandato è di 0,5 milioni UI (5 µg)/kg/die, iniziando dal primo giorno successivo al termine della chemioterapia e proseguendo finché il numero di neutrofili, dopo il previsto calo massimo, non si sia ripristinato a valori normali. La leucocitaferesi deve essere effettuata durante il periodo di aumento del conteggio assoluto dei neutrofili da meno di 0,5×109/l a oltre 5,0×109/l. In alcuni pazienti che non hanno ricevuto chemioterapia intensiva, a volte è sufficiente una singola leucocitaferesi. In altri casi si raccomandano leucocitaferesi aggiuntive.
Modalità di somministrazione
Quando filgrastim viene utilizzato come monoterapia per la mobilizzazione delle CSCH, può essere somministrato come infusione sottocutanea continua di 24 ore o mediante iniezione sottocutanea. Per l’infusione, il medicinale deve essere diluito in 20 ml di soluzione glucosata al 5% per infusione. Quando filgrastim viene utilizzato per la mobilizzazione delle CSCH dopo chemioterapia mielosoppressiva, deve essere somministrato mediante iniezione sottocutanea.
Mobilizzazione delle cellule staminali del sangue periferico in donatori sani per trapianto allogenico di CSCH
Dosaggio
Per la mobilizzazione delle CSCH in donatori sani, filgrastim deve essere somministrato alla dose di 1,0 milione UI (10 µg)/kg/die per 4-5 giorni consecutivi. La leucocitaferesi deve iniziare il quinto giorno di trattamento e, se necessario, proseguire fino al sesto giorno, al fine di ottenere 4×106 cellule CD34+/kg di peso corporeo del ricevente.
Modalità di somministrazione
Filgrastim deve essere somministrato mediante iniezione sottocutanea.
Neutropenia cronica grave
Dosaggio
Neutropenia congenita. Il dosaggio iniziale raccomandato è di 1,2 milioni UI (12 µg)/kg/die, somministrato in una o più somministrazioni.
Neutropenia idiopatica o periodica. Il dosaggio iniziale raccomandato è di 0,5 milioni UI (5 µg)/kg/die, somministrato in una o più somministrazioni.
Adeguamento del dosaggio. Filgrastim deve essere somministrato ogni giorno mediante iniezione sottocutanea fino al raggiungimento e al mantenimento stabile di un conteggio dei neutrofili superiore a 1,5×109/l. Dopo aver raggiunto l’effetto terapeutico, deve essere determinata la dose efficace minima necessaria per mantenere questo livello. Per mantenere il conteggio dei neutrofili richiesto, è necessaria la somministrazione quotidiana a lungo termine di filgrastim. Dopo 1 o 2 settimane di trattamento, la dose iniziale può essere raddoppiata o dimezzata, a seconda dell’effetto terapeutico. Successivamente, ogni 1-2 settimane deve essere effettuato un adeguamento individuale del dosaggio per mantenere il conteggio medio dei neutrofili compreso tra 1,5×109/l e 10×109/l. Nei pazienti con infezioni gravi può essere utilizzato uno schema con aumento più rapido della dose. Negli studi clinici, nel 97% dei pazienti che hanno risposto al trattamento, la risposta completa è stata osservata dopo la somministrazione di una dose ≤ 2,4 milioni UI (24 µg)/kg/die. La sicurezza dell’uso di filgrastim a lungo termine nei pazienti con neutropenia cronica grave con dosi superiori a 2,4 milioni UI (24 µg)/kg/die non è stata stabilita.
Modalità di somministrazione
Neutropenia congenita, idiopatica o periodica. Filgrastim deve essere somministrato mediante iniezione sottocutanea.
Pazienti con infezione da HIV
Dosaggio
Per la correzione della neutropenia. Il dosaggio iniziale raccomandato è di 0,1 milione UI (1 µg)/kg/die, con aumento della dose fino a un massimo di 0,4 milioni UI (4 µg)/kg/die, fino al raggiungimento di una normalizzazione stabile del numero di neutrofili (conteggio assoluto dei neutrofili > 2,0×109/l). Negli studi clinici, oltre il 90% dei pazienti ha risposto a questo dosaggio, raggiungendo il ripristino del numero di neutrofili in media entro 2 giorni.
Una piccola percentuale di pazienti (meno del 10%) ha richiesto una dose fino a 1 milione UI (10 µg)/kg/die per correggere la neutropenia.
Per il mantenimento di un numero normale di neutrofili. Dopo la correzione della neutropenia, deve essere determinata la dose efficace minima di filgrastim necessaria per mantenere un numero normale di neutrofili. Si raccomanda di iniziare con 30 milioni UI (300 µg)/die ogni due giorni. Successivamente potrebbe essere necessario un adeguamento individuale della dose in base al livello dei neutrofili del paziente, per mantenere un conteggio di neutrofili > 2,0×109/l. Negli studi clinici, la dose di 30 milioni UI (300 µg)/die dal 1° al 7° giorno della settimana si è dimostrata sufficiente per mantenere un conteggio assoluto dei neutrofili > 2,0×109/l, con una frequenza media di somministrazione di 3 volte alla settimana. Per mantenere un conteggio assoluto dei neutrofili > 2,0×109/l potrebbe essere necessario un uso prolungato di filgrastim.
Modalità di somministrazione
Correzione della neutropenia o mantenimento di un numero normale di neutrofili. Filgrastim deve essere somministrato mediante iniezione sottocutanea.
Categorie speciali di pazienti
Pazienti anziani
Gli studi clinici su filgrastim hanno incluso un numero limitato di pazienti anziani, ma non sono stati condotti studi specifici in questo gruppo. Pertanto, non possono essere formulate raccomandazioni specifiche sul dosaggio.
Pazienti con compromissione renale o epatica
I pazienti con grave compromissione della funzione renale o epatica non richiedono alcun aggiustamento del dosaggio, poiché gli studi hanno dimostrato che i loro parametri farmacocinetici e farmacodinamici sono simili a quelli dei volontari sani.
Bambini con neutropenia cronica grave e con malattie oncologiche
Il 65% dei pazienti arruolati nel programma di studi clinici sul trattamento della neutropenia cronica grave aveva meno di 18 anni. L’efficacia del trattamento è risultata evidente in questo gruppo d’età, che comprendeva la maggior parte dei pazienti con neutropenia congenita. Non sono state osservate differenze nei profili di sicurezza nei bambini trattati per neutropenia cronica grave. Gli studi clinici hanno dimostrato un’efficacia e una sicurezza simili di filgrastim in adulti e bambini sottoposti a chemioterapia citotossica.
Le raccomandazioni sul dosaggio nei bambini sono le stesse di quelle per gli adulti sottoposti a chemioterapia citotossica mielosoppressiva.
Il congelamento accidentale occasionale delle siringhe non influisce sulla stabilità di filgrastim.
Precauzioni per la manipolazione del medicinale
Il medicinale, se necessario, deve essere diluito con soluzione glucosata al 5% (50 mg/ml) per infusione. Non è raccomandata alcuna diluizione a una concentrazione finale inferiore a 0,2 milioni UI (2 µg)/ml. La soluzione deve essere ispezionata visivamente prima dell’uso. Deve essere utilizzata solo una soluzione limpida, priva di particelle visibili.
Se filgrastim viene diluito a una concentrazione inferiore a 1,5 milioni UI (15 µg)/ml, deve essere aggiunta albumina umana in quantità tale che la concentrazione finale sia di 2 mg/ml. Ad esempio, quando si diluisce una dose totale di filgrastim inferiore a 30 milioni UI (300 µg) fino a un volume finale di 20 ml, si deve aggiungere 0,2 ml di soluzione di albumina umana al 20% (200 mg/ml).
Il medicinale non contiene conservanti. Pertanto, a causa del rischio di contaminazione microbica, le siringhe di filgrastim sono destinate all’uso monouso. Filgrastim diluito con soluzione glucosata al 5% è compatibile con il vetro e con diversi tipi di plastica, tra cui il cloruro di polivinile (PVC), il poliolefine (copolimero di polipropilene e polietilene) e il polipropilene. La stabilità chimica e fisica della soluzione diluita per infusione è mantenuta per 24 ore a una temperatura compresa tra 2 e 8 °C.
Dal punto di vista microbiologico, il medicinale deve essere utilizzato immediatamente. Se filgrastim non viene somministrato immediatamente dopo la diluizione, il tempo e le condizioni di conservazione prima dell’uso sono di responsabilità dell’utilizzatore e in genere non devono superare le 24 ore a una temperatura compresa tra 2 e 8 °C, a meno che la diluizione non sia stata effettuata in condizioni asettiche controllate e validate.
Bambini
I dati degli studi clinici indicano che la sicurezza e l’efficacia del trattamento con filgrastim non differiscono tra adulti e bambini sottoposti a chemioterapia citotossica. Il dosaggio raccomandato per bambini e adulti sottoposti a chemioterapia citotossica mielosoppressiva è lo stesso.
Sovradosaggio
Sintomi. L’effetto di filgrastim in caso di sovradosaggio non è stato stabilito.
Trattamento. Dopo l’interruzione del medicinale, il numero di neutrofili circolanti di solito si riduce del 50% entro 1-2 giorni e successivamente ritorna alla normalità entro 1-7 giorni.
Reazioni avverse
Le reazioni avverse più gravi che possono verificarsi durante il trattamento con filgrastim includono: reazione anafilattica, gravi reazioni avverse a carico dei polmoni (in particolare pneumonite interstiziale e sindrome da distress respiratorio acuto), sindrome da leak capillare, grave splenomegalia/rottura del milza, trasformazione in sindrome mielodisplastica o leucemia in pazienti con neutropenia cronica grave, reazione di trapianto contro ospite in pazienti sottoposti a trapianto allogenico di midollo osseo o trapianto di cellule staminali del sangue periferico, e crisi a cellule falciformi in pazienti con anemia falciforme.
Le reazioni avverse più comunemente riportate sono: ipertermia, dolore muscoloscheletrico (inclusi dolore osseo, dolore alla schiena, artralgia, mialgia, dolore agli arti, dolore muscoloscheletrico, dolore muscoloscheletrico al torace, dolore al collo), anemia, vomito, nausea. Negli studi clinici condotti su pazienti oncologici, il dolore muscoloscheletrico è stato di lieve o moderata intensità nel 10% dei pazienti e grave nel 3% dei pazienti.
Le reazioni avverse osservate negli studi clinici e riportate in modo spontaneo sono elencate nella Tabella 2. All'interno di ciascuna sottocategoria per frequenza, le reazioni avverse sono elencate in ordine decrescente di gravità.
Tabella 2
| Sistemi corporei |
Reazioni avverse |
|||
| Molto frequenti (≥1/10) |
Frequenti (≥1/100 e <1/10) |
Non frequenti (≥1/1000 e <1/100) |
Rari (≥1/10000 e <1/1000) |
|
| Infezioni e infestazioni |
Setticemia Bronchite Infezioni delle vie respiratorie superiori Infezioni del tratto urinario |
|||
| Sangue e sistema linfatico |
Trombocitopenia Anemia e |
Splenomegalia a Diminuzione dell'emoglobina e |
Leucocitosi a |
Rottura del milza a Anemia falciforme con crisi, Emopoiesi extramidollare |
| Sistema immunitario |
Ipersensibilità Ipersensibilità ai farmaci a Reazione di trapianto contro l'ospite b |
Reazione anafilattica |
||
| Metabolismo e nutrizione |
Diminuzione dell'appetito e Aumento del livello ematico di lattato deidrogenasi |
Iperuricemia Aumento del livello ematico di acido urico |
Diminuzione del livello ematico di glucosio Pseudogotta a (condrocalcinosi) Disturbi dell'equilibrio idrico |
|
| Psichiatrico |
Insonnia |
|||
| Sistema nervoso |
Cefalea a |
Vertigini Ipoestesia Parestesia |
||
| Vasi sanguigni |
Ipertensione Ipotesione |
Malattia da occlusione venosa d |
Sindrome da perdita capillare a Aortite |
|
| Sistema respiratorio |
Emottisi Dispnea Tosse a Dolore orofaringeo a, e Epistassi |
Sindrome da distress respiratorio acuto a Insufficienza respiratoria a Edema polmonare a Emorragia polmonare Malattia interstiziale del polmone a Infiltrazione polmonare a Ipossia |
||
| Apparato gastrointestinale |
Diarrea a, e Vomito a, e Nausea a |
Dolore orale Costipazione e |
||
| Sistema epatobiliare |
Epatomegalia Aumento del livello ematico di fosfatasi alcalina |
Aumento del livello ematico di aspartato aminotransferasi Aumento del livello ematico di gamma-glutamil transferasi |
||
| Pelle e tessuto sottocutaneo |
Alopecia a |
Eruzione cutanea a Eritema |
Eruzioni maculopapulari |
Vasculite cutanea a Sindrome di Sweet (dermatosi neutrofila febbrile acuta) |
| Sistema muscoloscheletrico e tessuto connettivo |
Dolore muscoloscheletrico c |
Crampi muscolari |
Osteoporosi |
Diminuzione della densità ossea Riacutizzazione dell'artrite reumatoide |
| Renali e sistema urinario |
Disuria Ematuria |
Proteinuria |
Glomerulonefrite Anomalie nell'analisi delle urine |
|
| Condizioni generali e sede di somministrazione |
Stanchezza a Infiammazione delle mucose a Ipertermia |
Dolore al torace a Dolore a Astenia a Malessere e Edema periferico e |
Reazione nel sito di iniezione |
|
| Lesioni, avvelenamenti e complicanze da procedure |
Reazione da trasfusione e |
|||
a Vedere il paragrafo «Descrizione di reazioni avverse specifiche».
b Sono stati riportati casi di reazione «trapianto contro ospite» e decessi in pazienti dopo trapianto allogenico di midollo osseo (vedere il paragrafo «Descrizione di reazioni avverse specifiche» di seguito).
c Include dolore osseo, dolore alla schiena, artralgia, mialgia, dolore agli arti, dolore muscoloscheletrico, dolore muscoloscheletrico al torace, dolore al collo.
d I casi si sono verificati nel periodo post-commercializzazione in pazienti sottoposti a trapianto di midollo osseo o a mobilizzazione delle cellule staminali emopoietiche.
e Reazioni avverse che si verificano con maggiore frequenza nei pazienti in trattamento con filgrastim rispetto al gruppo placebo e che sono correlate alle conseguenze della patologia oncologica di base o della chemioterapia citotossica.
Descrizione di reazioni avverse specifiche
Ipersensibilità
Sono state osservate reazioni di ipersensibilità, in particolare anafilassi, eruzioni cutanee, orticaria, edema angioneurotico, dispnea e ipotensione, che si sono manifestate all'inizio o durante il successivo utilizzo di filgrastim negli studi clinici e nel periodo post-commercializzazione. In generale, tali reazioni sono state più frequenti dopo somministrazione endovenosa di filgrastim. Talvolta la ripresa del trattamento è stata accompagnata dalla ricomparsa dei sintomi, indicando un rapporto causale. In caso di reazioni allergiche gravi, l'uso di filgrastim deve essere interrotto e non deve essere ripreso successivamente.
Reazioni avverse a carico dell'apparato respiratorio
Negli studi clinici e nel periodo post-commercializzazione sono state riportate reazioni avverse a carico dei polmoni, comprese malattia polmonare interstiziale, edema polmonare, infiltrati polmonari, che talvolta hanno portato a insufficienza respiratoria o sindrome da distress respiratorio acuto, potenzialmente letali (vedere la sezione «Speciali avvertenze e precauzioni per l'uso»).
Splenomegalia e rottura del milza
Durante l'uso di filgrastim sono stati osservati casi di splenomegalia e rottura del milza. Alcuni casi di rottura del milza sono stati letali (vedere la sezione «Speciali avvertenze e precauzioni per l'uso»).
Sindrome da perdita capillare
Sono stati riportati casi di sindrome da perdita capillare durante l'uso di fattore stimolante le colonie di granulociti. Tali casi si verificano generalmente in pazienti con malattie maligne in evoluzione, sepsi, in concomitanza con l'uso di più farmaci chemioterapici o durante aferesi (vedere la sezione «Speciali avvertenze e precauzioni per l'uso»).
Vasculite cutanea
Sono stati riportati casi di vasculite cutanea in pazienti in trattamento con filgrastim. Il meccanismo della vasculite nei pazienti in trattamento con filgrastim non è chiaro. Con l'uso prolungato, la vasculite cutanea si è verificata nel 2% dei pazienti con neutropenia cronica grave.
Leucocitosi
La leucocitosi (conteggio dei leucociti > 50×109/l) è stata osservata nel 41% dei donatori sani; trombocitopenia transitoria (conteggio delle piastrine < 100×109/l) dopo somministrazione di filgrastim e leucaferesi è stata osservata nel 35% dei donatori (vedere la sezione «Speciali avvertenze e precauzioni per l'uso»).
Sindrome di Sweet
Nei pazienti in trattamento con filgrastim sono stati osservati casi di sindrome di Sweet (dermatosi neutrofila febbrile acuta).
Pseudogotta (condrocalcinosi)
È stata riportata pseudogotta (condrocalcinosi) in pazienti oncologici in trattamento con filgrastim.
Reazione «trapianto contro ospite»
Sono stati riportati casi di reazione «trapianto contro ospite» con esito fatale in pazienti trattati con G-CSF dopo trapianto allogenico di midollo osseo (vedere le sezioni «Farmacodinamica», «Speciali avvertenze e precauzioni per l'uso»).
Bambini
I dati degli studi clinici condotti nei bambini dimostrano un'efficacia e una sicurezza di filgrastim sovrapponibili a quelle osservate negli adulti sottoposti a chemioterapia citotossica, indicando l'assenza di differenze legate all'età nella farmacocinetica di filgrastim. L'unica reazione avversa riportata costantemente è il dolore muscoloscheletrico, simile a quanto osservato negli adulti. I dati disponibili sono insufficienti per una valutazione ulteriore dell'uso di filgrastim nei bambini.
Altre categorie speciali di pazienti
Pazienti anziani
Non sono state osservate differenze generali significative in termini di sicurezza ed efficacia nei pazienti di età superiore a 65 anni rispetto ai pazienti adulti più giovani (> 18 anni) sottoposti a chemioterapia citotossica; l'esperienza clinica non ha evidenziato differenze nella risposta terapeutica tra pazienti anziani e pazienti adulti più giovani. I dati disponibili sono insufficienti per valutare l'uso di filgrastim nei pazienti geriatrici per altre indicazioni approvate.
Pazienti pediatrici con neutropenia cronica grave
Sono stati riportati casi di riduzione della densità minerale ossea e osteoporosi in bambini con neutropenia cronica grave sottoposti a trattamento prolungato con filgrastim.
Segnalazione di reazioni avverse sospette
La segnalazione delle reazioni avverse sospette dopo l'autorizzazione del medicinale è di grande importanza. Permette un monitoraggio continuo del rapporto beneficio/rischio del medicinale. I professionisti sanitari e farmaceutici, nonché i pazienti o i loro rappresentanti legali, devono segnalare qualsiasi caso sospetto di reazione avversa o di mancata efficacia del medicinale attraverso il Sistema informatizzato automatizzato di farmacovigilanza al seguente indirizzo: https://aisf.dec.gov.ua
Durata della conservazione. 2,5 anni.
Condizioni di conservazione. Conservare nella confezione originale a una temperatura compresa tra 2 °C e 8 °C. Conservare in un luogo inaccessibile ai bambini.
Incompatibilità
Il medicinale non deve essere diluito con soluzioni di cloruro di sodio.
Il medicinale non deve essere mescolato con altri prodotti, eccetto quelli indicati nella sezione «Modalità di somministrazione e posologia».
Filgrastim, dopo la diluizione, può essere adsorbito dal vetro e da alcune plastiche; ciò non si verifica quando la diluizione viene effettuata nel modo indicato nella sezione «Modalità di somministrazione e posologia».
Confezionamento
0,5 ml (30 milioni UI) in siringa preriempita con dispositivo di sicurezza; confezione da 5 siringhe preriempite con dispositivo di sicurezza.
0,8 ml (48 milioni UI) in siringa preriempita con dispositivo di sicurezza; confezione da 5 siringhe preriempite con dispositivo di sicurezza.
Categoria di prescrivibilità. Sotto prescrizione medica.
Produttore. Teva Pharmaceutical Industries Ltd.
Sede del produttore e indirizzo del luogo di esercizio dell'attività. Industrial Zone, 18 Hurvitz Street, P.O. Box 353, Kfar Saba, 4410202, Israele.