Filgrastim-vista
Ukraine
Table of Contents
INSTRUCTIONS FOR MEDICAL USE OF THE MEDICINAL PRODUCT FILGRASTIM-VISTA (FILGRASTIM-VISTA)
Composition:
Active substance: filgrastim;
1 pre-filled syringe (0.5 mL solution) contains 300 mcg of filgrastim (30 million IU);
1 pre-filled syringe (0.8 mL solution) contains 480 mcg of filgrastim (48 million IU);
Excipients: glacial acetic acid, polysorbate 80, sodium hydroxide, sorbitol, water for injections.
Pharmaceutical form. Solution for injection or infusion.
Main physicochemical properties: colorless and clear solution.
Pharmacotherapeutic group. Immunostimulants. Colony-stimulating factors. Filgrastim. ATC code L03A A02.
Pharmacological Properties
Pharmacodynamics
Human granulocyte colony-stimulating factor (G-CSF) is a glycoprotein that regulates the production and release of functional neutrophils from the bone marrow. FILGRASTIM-VISTA, containing r-metHuG-CSF (filgrastim), induces a marked increase in the number of neutrophils in peripheral blood within twenty-four hours, with a slight increase in the number of monocytes. In some patients with chronic neutropenia syndrome (CNS), filgrastim may also cause a slight increase in the number of circulating eosinophils and basophils compared to baseline levels; some of these patients may have eosinophilia or basophilia even prior to treatment. The increase in neutrophil count is dose-dependent at recommended doses. Neutrophils produced in response to filgrastim demonstrate normal or enhanced function, as shown by tests of chemotactic and phagocytic activity. After discontinuation of filgrastim therapy, the number of circulating neutrophils decreases by 50% within 1–2 days and returns to normal levels within 1–7 days.
The use of filgrastim in patients undergoing cytotoxic chemotherapy significantly reduces the frequency, severity, and duration of neutropenia and febrile neutropenia. Filgrastim treatment significantly shortens the duration of febrile neutropenia, reduces antibiotic use, and shortens hospitalization following induction chemotherapy for acute myeloid leukemia or myeloablative therapy followed by bone marrow transplantation. However, the incidence of fever and the number of documented infection episodes were not reduced. The duration of fever was not reduced in patients undergoing myeloablative therapy followed by bone marrow transplantation.
The use of filgrastim, either alone or following chemotherapy, stimulates the mobilization of hematopoietic progenitor cells into peripheral blood. These autologous peripheral blood progenitor cells can be collected and reinfused after high-dose cytotoxic therapy as an alternative or adjunct to bone marrow transplantation. Infusion of peripheral blood progenitor cells accelerates hematopoietic recovery, reducing the duration of risk for hemorrhagic complications and the need for platelet transfusions. In recipients of allogeneic peripheral blood progenitor cells mobilized by filgrastim, hematologic recovery occurred significantly faster, resulting in a significantly shorter time to unassisted platelet recovery compared to allogeneic bone marrow transplantation.
One retrospective European study evaluating the use of G-CSF after allogeneic bone marrow transplantation in patients with acute leukemias showed an increased risk of graft-versus-host disease (GvHD), treatment-related mortality (TRM), and overall mortality with G-CSF administration. In a separate retrospective international study involving patients with acute and chronic myeloid leukemia, no effect was observed on the risk of GvHD, TRM, or mortality. A meta-analysis of allogeneic transplantation studies, including data from nine prospective randomized trials, eight retrospective studies, and one controlled study, found no impact on the risks of acute GvHD, chronic GvHD, or early treatment-related mortality.
| Relative risk (95% CI [confidence interval]) of graft-versus-host disease (GVHD) and treatment-related mortality (TRM) Following post-transplantation G-CSF treatment |
|||||
| Publication |
Study period |
Number |
Acute GVHD grade II–IV |
Chronic GVHD |
TRM |
| Meta-analysis (2003) |
1986–2001a |
1198 |
1.08 (0.87, 1.33) |
1.02 (0.82, 1.26) |
0.70 (0.38, 1.31) |
| European retrospective study (2004) |
1992–2002b |
1789 |
1.33 (1.08, 1.64) |
1.29 (1.02, 1.61) |
1.73 (1.30, 2.32) |
| International retrospective study (2006) |
1995–2000b |
2110 |
1.11 (0.86, 1.42) |
1.10 (0.86, 1.39) |
1.26 (0.95, 1.67) |
a The analysis includes studies involving bone marrow transplantation during this period; some studies used G-CSF.
b The analysis includes patients who received bone marrow transplantation during this period.
Use of filgrastim for mobilization of peripheral blood progenitor cells in healthy donors prior to allogeneic peripheral blood progenitor cell transplantation. In normal donors, a dose of 10 mcg/kg/day administered subcutaneously for 4–5 consecutive days allows collection of ≥ 4 × 106 CD34+ cells/kg recipient body weight in most donors after two leukapheresis procedures.
Administration of filgrastim to patients, children or adults, with SCN (severe congenital, cyclic, and idiopathic neutropenia) results in a sustained increase in the absolute neutrophil count in peripheral blood and reduction in infections and infection-related complications. Administration of filgrastim to HIV-infected patients maintains normal neutrophil counts, allowing scheduled administration of antiviral and/or other myelosuppressive agents. There is no evidence that HIV replication increases in HIV-infected patients treated with filgrastim.
Like other hematopoietic growth factors, G-CSF has demonstrated in vitro stimulatory effects on human endothelial cells.
Pharmacokinetics
Filgrastim clearance has been shown to follow first-order pharmacokinetics after both subcutaneous and intravenous administration. The serum half-life of filgrastim is approximately 3.5 hours, and clearance is approximately 0.6 mL/min/kg. Continuous infusion of filgrastim for periods up to 28 days in patients recovering from autologous bone marrow transplantation did not result in drug accumulation, and the half-life was comparable. There is a positive linear correlation between dose and serum filgrastim concentration regardless of whether it is administered intravenously or subcutaneously. After subcutaneous administration of recommended doses, serum concentrations were maintained above 10 ng/mL for 8–16 hours. The volume of distribution in blood is approximately 150 mL/kg.
Clinical characteristics
Indications
- To reduce the duration of neutropenia and the incidence of febrile neutropenia in patients receiving cytotoxic chemotherapy for malignant diseases (with the exception of chronic myeloid leukemia and myelodysplastic syndrome).
- To reduce the duration of neutropenia in patients undergoing myeloablative therapy followed by bone marrow transplantation, who are at high risk of prolonged severe neutropenia.
- For mobilization of peripheral blood progenitor cells (PBPC).
- For long-term therapy to increase neutrophil counts and reduce the frequency and duration of infectious complications in children and adults with severe congenital, cyclic, or idiopathic neutropenia (absolute neutrophil count ≤ 0.5×10⁹/L) and a history of severe or recurrent infections.
- To reduce the risk of bacterial infections in patients with persistent neutropenia (absolute neutrophil count ≤ 1.0×10⁹/L) and advanced HIV infection when other methods of controlling neutropenia have been ineffective.
Contraindications
Hypersensitivity to the active substance or to any of the excipients.
Safety precautions
Any unused medicinal product and waste materials should be disposed of in accordance with current requirements.
Syringes with safety devices should be disposed of in accordance with current requirements.
Interaction with other medicinal products and other forms of interaction
The safety and efficacy of administering filgrastim on the same day as myelosuppressive cytotoxic agents have not been established. Due to the sensitivity of rapidly dividing myeloid cells to myelosuppressive cytotoxic chemotherapy, filgrastim should not be administered less than 24 hours before or earlier than 24 hours after administration of these agents. Preliminary data from a small number of patients who received filgrastim and 5-fluorouracil concurrently suggest an increased risk of severe neutropenia. Interactions with other hematopoietic growth factors and cytokines have not been studied in clinical trials.
Given that lithium stimulates neutrophil release, combination with filgrastim may enhance its effect. Although studies on this interaction have not been conducted, there is no evidence of harmful effects from this interaction.
Special precautions for use
Hypersensitivity
Hypersensitivity reactions, including anaphylactic reactions, have been observed in patients at the start or during further treatment with filgrastim. If clinically significant hypersensitivity reactions occur, filgrastim should be discontinued and not resumed thereafter. Do not administer filgrastim to patients with a history of hypersensitivity to filgrastim or pegfilgrastim.
Pulmonary adverse reactions
Pulmonary adverse reactions, including interstitial lung disease, have been reported after administration of G-CSF. Patients with recent history of pulmonary infiltrates or pneumonia have a higher risk of such reactions. The onset of respiratory symptoms such as cough, fever, and dyspnea, in combination with radiological signs of pulmonary infiltrates and worsening pulmonary function, may be the first signs of acute respiratory distress syndrome (ARDS). Filgrastim administration should be discontinued and appropriate treatment initiated.
Glomerulonephritis
Cases of glomerulonephritis have been reported in patients receiving filgrastim and pegfilgrastim. Glomerulonephritis usually resolves after dose reduction or discontinuation of filgrastim and pegfilgrastim. Regular urinalysis is recommended.
Capillary leak syndrome
Cases of capillary leak syndrome have been reported after administration of G-CSF. This condition is characterized by hypotension, hypoalbuminemia, edema, and hemoconcentration, and may be life-threatening if not treated promptly. Patients who develop symptoms of capillary leak syndrome should be closely monitored and provided with standard symptomatic treatment and, if necessary, intensive care.
Splenomegaly and splenic rupture
Asymptomatic cases of splenomegaly and isolated cases of splenic rupture have been observed in patients and healthy donors receiving filgrastim. Several cases of splenic rupture were fatal. Therefore, monitoring of spleen size (e.g., clinical examination, ultrasound) is recommended in such patients. Splenic rupture should be considered in donors or patients reporting pain in the upper left quadrant of the abdomen or left shoulder. Dose reduction of filgrastim slowed or halted spleen enlargement in patients with severe chronic neutropenia, and 3% of patients required splenectomy.
Growth of malignant cells
G-CSF may promote the growth of myeloid cells in vitro. Similar effects may be possible for certain non-myeloid cells.
Myelodysplastic syndrome or chronic myeloid leukemia
The safety and efficacy of filgrastim in patients with myelodysplastic syndrome or chronic myeloid leukemia have not been established. Filgrastim is not recommended for use in these conditions. Particular attention should be paid to differential diagnosis between blast crisis in chronic myeloid leukemia and acute myeloid leukemia.
Acute myeloid leukemia
The safety and efficacy of filgrastim in patients with secondary acute myeloid leukemia have not been sufficiently studied; therefore, filgrastim should be used with caution in these patients. The safety and efficacy of filgrastim in de novo acute myeloid leukemia in patients under 55 years of age with favorable cytogenetic factors [t(8;21), t(15;17), and inv(16)] have not been established.
Thrombocytopenia
Cases of thrombocytopenia have been reported in patients receiving filgrastim. Platelet counts should be closely monitored, especially during the first few weeks of filgrastim treatment. If thrombocytopenia (platelet count < 100×10⁹/L) develops in patients with severe chronic neutropenia, temporary discontinuation or dose reduction of filgrastim should be considered.
Leukocytosis
In less than 5% of oncology patients receiving filgrastim at doses exceeding 0.3 million IU/kg/day (3 μg/kg/day), white blood cell counts increased to 100×10⁹/L or higher. No adverse reactions directly related to such leukocytosis have been described. However, due to the risk associated with high leukocytosis, white blood cell counts should be monitored regularly during filgrastim treatment. If white blood cell counts exceed 50×10⁹/L after the expected decline, filgrastim treatment should be discontinued immediately. However, if filgrastim is used for stem cell mobilization, it should be discontinued or the dose reduced when white blood cell counts exceed 70×10⁹/L.
Immunogenicity
As with all therapeutic proteins, there is a potential for immunogenicity. The rate of antibody formation against filgrastim is generally low. The appearance of binding antibodies is expected, as with other biological agents; however, data on their neutralizing activity are currently lacking.
Aortitis
Cases of aortitis have been reported after G-CSF administration in healthy individuals and cancer patients. Symptoms may include fever, abdominal pain, malaise, and back pain, and may be accompanied by elevated inflammatory markers (e.g., C-reactive protein, white blood cell count). In most cases, aortitis was diagnosed by computed tomography and usually resolved after discontinuation of G-CSF.
Special precautions related to concomitant diseases
Sickle cell trait and sickle cell anemia
Sickle cell crises, sometimes fatal, have been observed in patients with sickle cell trait or sickle cell anemia during filgrastim treatment. Therefore, filgrastim should be administered with caution to patients with sickle cell trait or sickle cell anemia.
Osteoporosis
Patients with concomitant osteoporosis receiving continuous filgrastim therapy for more than 6 months should undergo monitoring of bone mineral density.
Special warnings for oncology patients
Filgrastim should not be used to increase the dosage of cytotoxic chemotherapy beyond established limits.
Risk associated with high-dose chemotherapy
Particular caution should be exercised when treating patients receiving high-dose chemotherapy, as the efficacy of treatment has not been established in these cases, while higher doses of chemotherapeutic agents have shown increased toxicity, leading to cardiac, pulmonary, neurological, and dermatological reactions (see the package leaflet of the respective chemotherapeutic agent).
Effect of chemotherapy on erythrocytes and platelets
Filgrastim monotherapy does not prevent thrombocytopenia and anemia caused by myelosuppressive chemotherapy. Due to the possibility of using higher doses of chemotherapeutic agents (e.g., full doses according to the regimen), patients may be at increased risk of developing thrombocytopenia and anemia; therefore, regular monitoring of platelet count and hematocrit is recommended. Single-agent or combination chemotherapy regimens that may cause severe thrombocytopenia should be used with particular caution.
The use of stem cells mobilized by filgrastim reduces the severity and duration of thrombocytopenia after myelosuppressive or myeloablative chemotherapy.
Myelodysplastic syndrome and acute myeloid leukemia in patients with breast or lung cancer
In an observational post-marketing study, myelodysplastic syndrome (MDS) and acute myeloid leukemia (AML) were associated with the use of pegfilgrastim, an alternative G-CSF, in combination with chemotherapy and/or radiotherapy in patients with breast or lung cancer. A similar association between filgrastim and MDS/AML has not been observed. However, patients with breast cancer and patients with lung cancer should be monitored for signs and symptoms of MDS/AML.
Other special warnings
The effect of filgrastim in patients with markedly reduced numbers of myeloid precursor cells has not been studied. Filgrastim increases neutrophil counts primarily by affecting neutrophil precursor cells. Therefore, in patients with low numbers of precursor cells (e.g., after intensive radiotherapy or chemotherapy, or with tumor infiltration of bone marrow), the degree of neutrophil increase may be reduced.
Vascular complications, including veno-occlusive disease and fluid imbalance, have occasionally been observed in patients receiving high-dose chemotherapy followed by autologous bone marrow transplantation.
Cases of fatal graft-versus-host reaction have been reported in patients who received granulocyte colony-stimulating factor after allogeneic bone marrow transplantation.
Increased bone marrow hematopoietic activity in response to granulocyte colony-stimulating factor treatment, manifesting as transiently positive bone scans, has been reported. This should be considered when interpreting bone scan results.
Special warnings for patients undergoing stem cell mobilization
Mobilization
A prospective randomized comparison of two recommended mobilization methods (filgrastim alone or filgrastim in combination with myelosuppressive chemotherapy) in the same patient population has not been conducted. Individual patient characteristics across different studies and variability in laboratory determination of CD34+ cell counts make direct comparison of study results difficult. Therefore, it is difficult to recommend an optimal method. The mobilization method should be selected based on the patient's treatment goal.
Prior to administration of cytotoxic agents
In patients who have previously received active myelosuppressive therapy, adequate mobilization of stem cells to the recommended minimum level (≥ 2.0×10⁶ CD34+ cells/kg) or accelerated platelet recovery may not occur. Some cytostatic agents have specific toxicity toward hematopoietic precursor cells and may negatively affect their mobilization. Agents such as melphalan, carmustine, and carboplatin, if administered over a prolonged period before stem cell mobilization attempts, may reduce mobilization efficacy. However, the use of melphalan, carboplatin, or carmustine in combination with filgrastim has proven effective in stem cell activation. If stem cell transplantation is planned, mobilization should be scheduled early in the treatment course. Particular attention should be paid to the number of stem cells mobilized in these patients prior to high-dose chemotherapy. If mobilization results are insufficient according to the above criteria, alternative treatments not requiring precursor cells should be considered.
Assessment of mobilized peripheral blood stem cells
When assessing the number of stem cells mobilized by filgrastim in patients, particular attention should be paid to the method of quantitative determination. Results of flow cytometric analysis of CD34+ cell counts vary depending on the specific methodology; therefore, caution should be exercised when applying recommendations based on studies conducted in other laboratories. Statistical analysis of the relationship between the number of infused CD34+ cells and the speed of platelet recovery after high-dose chemotherapy shows a complex but consistent correlation. The recommended minimum number of stem cells is ≥ 2.0×10⁶ CD34+ cells/kg, based on published data on adequate hematological recovery. A higher number of precursor cells appears to lead to faster recovery, while a lower number results in slower blood count normalization.
Special warnings for healthy donors undergoing stem cell mobilization
Stem cell mobilization does not provide direct clinical benefit to healthy donors and should only be considered for the purpose of allogeneic stem cell transplantation.
Stem cell mobilization in donors should only be performed if they meet standard clinical and laboratory criteria for stem cell donation, particularly hematological parameters and absence of infectious diseases.
The safety and efficacy of filgrastim in healthy donors under 16 years of age or over 60 years of age have not been evaluated.
Transient thrombocytopenia (platelet count < 100×10⁹/L) after filgrastim administration and leukapheresis was observed in 35% of subjects studied. Among them, two cases of thrombocytopenia with platelet count < 50×10⁹/L were associated with the leukapheresis procedure.
If more than one leukapheresis is required, particular attention should be paid to donors whose platelet count before leukapheresis is less than 100×10⁹/L; in general, apheresis should not be performed if platelet count is below 75×10⁹/L. Leukapheresis should not be performed in donors receiving anticoagulants or with coagulation disorders.
Donors receiving G-CSF for stem cell mobilization should be monitored until hematological parameters normalize.
Transient cytogenetic abnormalities have been observed in donors receiving G-CSF. The significance of these changes is unknown. However, a risk of stimulation of a malignant myeloid clone cannot be excluded. Apheresis centers are recommended to register and monitor stem cell donors for at least 10 years to ensure long-term safety monitoring.
Special warnings for recipients of allogeneic stem cells mobilized by filgrastim
Available data indicate that allogeneic stem cell transplantation is associated with a higher risk of acute and chronic graft-versus-host disease compared to bone marrow transplantation.
Special warnings for patients with severe chronic neutropenia
Filgrastim should not be administered to patients with severe congenital neutropenia who develop leukemia or show signs of leukemia development.
Blood cell count determination
Other changes in blood count may occur, including anemia and transient increase in myeloid precursor cells, requiring careful monitoring of cell counts.
Transformation to leukemia or myelodysplastic syndrome
Particular caution should be exercised in diagnosing severe chronic neutropenias. These should be differentiated from other hematological disorders such as aplastic anemia, myelodysplasia, and myeloleukemia. Before starting treatment, a complete blood count with leukocyte differential and platelet count, as well as morphological examination of bone marrow and karyotype, should be performed.
In clinical trials, myelodysplastic syndrome (MDS) or leukemia was observed in only a small number (3%) of patients with severe chronic neutropenia receiving filgrastim. This observation was made only in patients with congenital neutropenia. MDS and leukemia are common complications of this disease; their relationship to filgrastim treatment is not established. In approximately 12% of patients with initially normal cytogenetics, abnormalities including monosomy 7 were detected on repeat testing. It is currently unknown whether long-term filgrastim treatment promotes the development of cytogenetic abnormalities, MDS, or leukemia in patients with severe chronic neutropenia. In such patients, regular (approximately every 12 months) morphological and cytogenetic examination of bone marrow is recommended.
Other special warnings
Transient causes of neutropenia, such as viral infections, should be excluded.
Hematuria and/or proteinuria have been observed in a small number of patients; regular urinalysis should be performed for monitoring.
The safety and efficacy of filgrastim in neonates and patients with autoimmune neutropenia have not been established.
Special warnings for patients with HIV infection
Blood cell count determination
Neutrophil counts should be closely monitored, especially during the first few weeks of filgrastim treatment. In some patients, a therapeutic effect is observed very quickly after the first injection, with a significant increase in neutrophil count. Neutrophil counts should be monitored daily during the first 2–3 days of filgrastim treatment, then at least twice weekly during the first two weeks, and at least once weekly or every two weeks during maintenance therapy. If a dose of 30 million IU (300 μg) filgrastim is administered intermittently, significant fluctuations in neutrophil count may occur over time. To determine the nadir of neutrophil count, blood samples should be taken immediately before the next dose of filgrastim.
Risk associated with use of high-dose myelosuppressive agents
Filgrastim monotherapy does not prevent thrombocytopenia and anemia caused by myelosuppressive chemotherapy. Due to the possibility of using higher doses or quantities of chemotherapeutic agents in combination with filgrastim, patients may be at increased risk of developing thrombocytopenia and anemia; therefore, regular blood cell count monitoring is recommended (see above).
Infections and malignancies causing myelosuppression
Neutropenia may be caused by bone marrow infiltration by opportunistic infection agents (such as Mycobacterium avium complex) or tumors (lymphoma). In addition to filgrastim for neutropenia treatment, such patients should receive specific treatment for the underlying disease. The effect of filgrastim on neutropenia caused by infectious agents or malignant bone marrow tumors has not been sufficiently studied.
Other warnings
Traceability. To improve traceability of granulocyte colony-stimulating factors, the brand name of the prescribed medicinal product and the batch number of the administered product should be clearly documented in the patient's medical record.
Important information on excipients
Sorbitol. The medicinal product contains sorbitol. The additive effect of concomitant administration of medicinal products containing sorbitol (or fructose) and dietary intake of sorbitol (or fructose) should be considered.
For intravenous administration. Filgrastim should not be administered to patients with hereditary fructose intolerance (HFI), except in cases of extreme necessity. HFI may not yet be diagnosed in infants and young children (under 2 years of age). Medicinal products for intravenous administration containing sorbitol/fructose may be life-threatening and should not be prescribed to this patient group, except in cases of acute clinical necessity and lack of alternatives. A detailed history of HFI symptoms should be obtained from each patient before prescribing this medicinal product.
Sodium. One pre-filled syringe contains less than 1 mmol (23 mg) of sodium, i.e., the medicinal product is essentially sodium-free.
Use during pregnancy or breastfeeding
Pregnancy. Data on the use of filgrastim in pregnant women are limited or absent. Animal studies have shown reproductive toxicity. Increased embryo loss was observed in animals at multiple high doses and in the presence of maternal toxicity. There are reports of filgrastim crossing the placental barrier. Filgrastim is not recommended during pregnancy.
Breastfeeding. It is unknown whether filgrastim or its metabolites are excreted in human breast milk. Risk to breastfed newborns/infants cannot be excluded. The decision to discontinue breastfeeding or interrupt/stop filgrastim therapy should be made considering the benefit of breastfeeding for the child and the benefit of treatment for the woman.
Fertility. Animal studies have shown that filgrastim does not affect reproductive function or fertility.
Ability to affect reaction speed when driving or operating machinery
Filgrastim may have a minor effect on the ability to drive or operate machinery. Dizziness may occur after filgrastim administration (see section "Adverse reactions").
Method of Administration and Dosage
Treatment should be conducted in collaboration with an oncology center equipped with appropriate diagnostic equipment and staffed by specialists experienced in hematology and in the use of human granulocyte colony-stimulating factor. Procedures for cell mobilization and apheresis should be performed by specialists with sufficient experience in this field and with adequate monitoring of hematopoietic progenitor cells.
Conventional Cytotoxic Chemotherapy
Dosage
The recommended dose of filgrastim is 0.5 million IU (5 mcg) / kg body weight once daily. The first dose should be administered no sooner than 24 hours after completion of cytotoxic chemotherapy. In randomized clinical trials, a subcutaneous dose of 23 million IU (230 mcg)/m²/day (4.0–8.4 mcg/kg body weight per day) was used.
The medicinal product should be administered daily until neutrophil counts recover to normal levels following the expected nadir. In patients receiving cytotoxic chemotherapy for solid tumors, lymphomas, and lympholeukemias, the expected duration of therapy required to meet these criteria is up to 14 days. After induction and consolidation therapy for acute myeloid leukemia, the duration of treatment may be significantly prolonged (up to 38 days), depending on the type, doses, and regimen of cytotoxic chemotherapy used.
A transient increase in neutrophil count is usually observed within 1–2 days after initiating filgrastim therapy in patients receiving cytotoxic chemotherapy. However, to achieve a stable therapeutic effect, filgrastim therapy should not be interrupted until neutrophil counts have recovered to normal levels following the expected nadir. Premature discontinuation of treatment before the expected nadir in neutrophil count is not recommended.
Method of Administration
The medicinal product can be administered via daily subcutaneous injections or daily short (30-minute) intravenous infusions in 5% glucose solution. Subcutaneous administration is preferred, as clinical trial data from single-dose studies suggest that intravenous administration may result in a shorter duration of filgrastim effect. The clinical relevance of these findings for multiple-dose administration has not been established. The route of administration should be selected based on the individual clinical circumstances.
Subcutaneous Administration
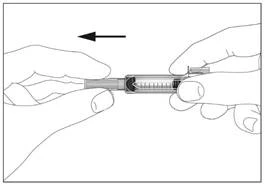
1
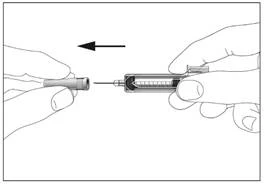
2
Syringe with a safety device preventing needlestick injury and reuse (Fig. 1, 2).
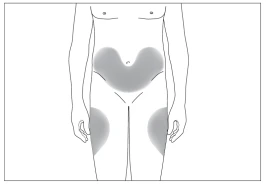
3
4
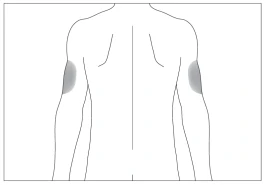
Preferred body sites for subcutaneous injection (Fig. 3, 4).
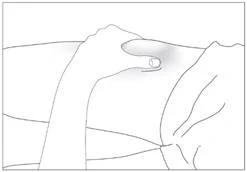
5
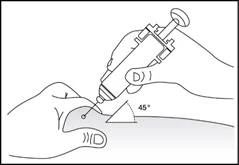
6
Subcutaneous administration of the medicinal product (Fig. 5–6).
- Disinfect the injection site with an alcohol-impregnated wipe. Pinch the skin area between the thumb and index finger without compressing it (Fig. 5).
- Insert the needle fully into the skin (Fig. 6).
- Gently pull back on the syringe plunger to check whether a blood vessel has been punctured. If blood appears in the syringe, withdraw the needle and re-inject at another site.
Filgrastim should be administered daily at the same time.
To avoid pain, it is best to rotate injection sites daily.
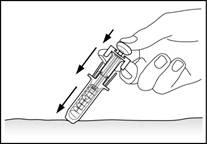
7
Syringes with safety devices
Filgrastim should be injected slowly and evenly, while pinching the skin between fingers, until the full dose is delivered and the syringe plunger stops moving.
Excessive force on the syringe plunger is prohibited.
After administering the medicinal product, withdraw the needle while continuing to press on the plunger, then release the skin (Fig. 7).
After removing the finger from the plunger, the syringe’s safety mechanism will quickly cover the needle to prevent injury.
A separate syringe must be used for each subsequent injection. Re-injection of any remaining filgrastim in the syringe is prohibited.
Myeloablative Therapy Followed by Bone Marrow Transplantation
Dosage
The recommended initial dose is 1.0 million IU (10 mcg) / kg body weight per day. The first dose of filgrastim should be administered no sooner than 24 hours after cytotoxic chemotherapy and no sooner than 24 hours after bone marrow transplantation.
After the nadir in neutrophil count, the daily dose should be adjusted according to changes in neutrophil count (see Table 1).
Table 1
| Neutrophil count |
Filgrastim dose |
| > 1.0×109/L for 3 consecutive days |
Reduce to 0.5 million IU (5 mcg)/kg/day |
| Then, if the absolute neutrophil count remains > 1.0×109/L for the next 3 consecutive days |
Discontinue filgrastim |
| If during treatment the absolute neutrophil count decreases to < 1.0×109/L, increase the filgrastim dose again according to the above scheme. |
|
Method of Administration
The medicinal product can be administered by means of a 30-minute or 24-hour intravenous infusion or by continuous 24-hour subcutaneous infusion. Filgrastim should be diluted in 20 ml of 5 % glucose solution for infusion.
Mobilization of peripheral blood stem cells in patients receiving myelosuppressive or myeloablative therapy followed by autologous transplantation of PBSCs
Dosing
When filgrastim is used as monotherapy for mobilization of PBSCs, the recommended dose is 1.0 million IU (10 mcg)/kg/day for 5–7 consecutive days. Usually, one or two leukapheresis procedures on day 5 or 6 are sufficient. If additional leukapheresis is required, filgrastim administration at the same dose should be continued until the final leukapheresis.
For mobilization of PBSCs after myelosuppressive chemotherapy, the recommended dose is 0.5 million IU (5 mcg)/kg/day, starting on the first day after completion of chemotherapy and continuing until the neutrophil count, following the expected nadir, recovers to normal levels. Leukapheresis should be performed during the period when the absolute neutrophil count rises from less than 0.5×10⁹/L to more than 5.0×10⁹/L. Some patients who have not received intensive chemotherapy may require only one leukapheresis. In other cases, additional leukapheresis procedures are recommended.
Method of Administration
When filgrastim is used as monotherapy for mobilization of PBSCs, it can be administered either as a continuous 24-hour subcutaneous infusion or by subcutaneous injection. For infusion, the medicinal product should be diluted in 20 ml of 5 % glucose solution for infusion. When filgrastim is used for mobilization of PBSCs after myelosuppressive chemotherapy, it should be administered by subcutaneous injection.
Mobilization of peripheral blood stem cells in healthy donors for allogeneic PBSC transplantation
Dosing
For mobilization of PBSCs in healthy donors, filgrastim should be administered at a dose of 1.0 million IU (10 mcg)/kg/day for 4–5 consecutive days. Leukapheresis should begin on day 5 of treatment and, if necessary, continue on day 6 to achieve a yield of 4×10⁶ CD34⁺ cells/kg recipient body weight.
Method of Administration
Filgrastim should be administered by subcutaneous injection.
Severe Chronic Neutropenia
Dosing
Congenital Neutropenia. The recommended initial dose is 1.2 million IU (12 mcg)/kg/day as a single or divided dose.
Idiopathic or Periodic Neutropenia. The recommended initial dose is 0.5 million IU (5 mcg)/kg/day as a single or divided dose.
Dose Adjustment. Filgrastim should be administered daily by subcutaneous injection until the neutrophil count reaches and stably exceeds 1.5×10⁹/L. After achieving the therapeutic effect, the minimal effective dose required to maintain this level should be determined. Long-term daily administration of filgrastim is necessary to maintain the required neutrophil count. After 1 or 2 weeks of treatment, the initial dose may be doubled or halved depending on the therapeutic response. Thereafter, dose adjustments should be made individually every 1–2 weeks to maintain the average neutrophil count within the range of 1.5×10⁹/L to 10×10⁹/L. A more rapid dose escalation regimen may be used in patients with severe infections. In clinical studies, a complete response was observed in 97 % of patients who responded to treatment after administration of doses ≤ 2.4 million IU (24 mcg)/kg/day. The safety of long-term use of filgrastim at doses exceeding 2.4 million IU (24 mcg)/kg/day in patients with severe chronic neutropenia has not been established.
Method of Administration
Congenital, Idiopathic, or Periodic Neutropenia. Filgrastim should be administered by subcutaneous injection.
HIV-Infected Patients
Dosing
For Correction of Neutropenia. The recommended initial dose is 0.1 million IU (1 mcg)/kg/day, with dose escalation up to a maximum of 0.4 million IU (4 mcg)/kg/day until stable normalization of the neutrophil count (absolute neutrophil count > 2.0×10⁹/L). In clinical studies, more than 90 % of patients responded to this dosing regimen, achieving neutrophil recovery within a mean of 2 days.
A small number of patients (less than 10 %) required a dose of up to 1.0 million IU (10 mcg)/kg/day to correct neutropenia.
For Maintenance of Normal Neutrophil Count. After correction of neutropenia, the minimal effective dose of filgrastim required to maintain a normal neutrophil count should be determined. It is recommended to start with 30 million IU (300 mcg) administered every other day. Subsequently, individual dose adjustments may be necessary based on the patient's neutrophil levels to maintain a neutrophil count above 2.0×10⁹/L. In clinical studies, a dose of 30 million IU (300 mcg) administered on days 1 to 7 of the week was sufficient to maintain an absolute neutrophil count above 2.0×10⁹/L with a mean administration frequency of 3 times per week. Long-term use of filgrastim may be required to maintain an absolute neutrophil count above 2.0×10⁹/L.
Method of Administration
Correction of Neutropenia or Maintenance of Normal Neutrophil Count. Filgrastim should be administered by subcutaneous injection.
Special Patient Populations
Elderly Patients
Clinical studies of filgrastim included a limited number of elderly patients, and no specific studies have been conducted in this patient group. Therefore, specific dosing recommendations cannot be made.
Patients with Renal or Hepatic Impairment
Patients with severe renal or hepatic impairment do not require dose adjustment, as studies have demonstrated that their pharmacokinetic and pharmacodynamic parameters are similar to those in healthy volunteers.
Children with Severe Chronic Neutropenia and Oncological Diseases
65 % of patients enrolled in the clinical studies program for the treatment of severe chronic neutropenia were under 18 years of age. Treatment efficacy was evident in this age group, which included the majority of patients with congenital neutropenia. There were no differences in safety profiles between children treated for severe chronic neutropenia. Clinical studies demonstrated comparable efficacy and safety of filgrastim in adults and children receiving cytotoxic chemotherapy.
Dosing recommendations for children are the same as for adults receiving myelosuppressive cytotoxic chemotherapy.
Accidental single freezing of syringes does not affect the stability of filgrastim.
Special Handling Instructions
The medicinal product should be diluted, if necessary, with 5 % (50 mg/ml) glucose solution for infusion. Dilution to a final concentration below 0.2 million IU (2 mcg)/ml is not recommended under any circumstances. The solution should be visually inspected before administration. Only clear solutions without visible particles should be used.
If filgrastim is diluted to a concentration below 1.5 million IU (15 mcg)/ml, human serum albumin should be added to achieve a final concentration of 2 mg/ml. For example, when diluting a total filgrastim dose of less than 30 million IU (300 mcg) to a final volume of 20 ml, 0.2 ml of 20 % (200 mg/ml) human albumin solution should be added.
The medicinal product does not contain preservatives. Therefore, due to the potential risk of microbial contamination, filgrastim syringes are intended for single use only. Filgrastim diluted with 5 % glucose solution is compatible with glass and several plastics, including polyvinyl chloride (PVC), polyolefin (a copolymer of polypropylene and polyethylene), and polypropylene. The chemical and physical stability of the diluted infusion solution is maintained for 24 hours at 2–8 °C.
From a microbiological standpoint, the medicinal product should be used immediately. If filgrastim is not administered immediately after dilution, the time and conditions of storage prior to use are the responsibility of the user and generally should not exceed 24 hours at 2–8 °C, unless the dilution was performed under controlled and validated aseptic conditions.
Children
Clinical trial data indicate that the safety and efficacy of filgrastim treatment do not differ between adults and children receiving cytotoxic chemotherapy. The recommended dose for children and adults receiving myelosuppressive cytotoxic chemotherapy is the same.
Overdose
Symptoms. The effects of filgrastim overdose have not been established.
Treatment. After discontinuation of the medicinal product, the number of circulating neutrophils usually decreases by 50 % within 1–2 days and then returns to normal within 1–7 days.
Adverse Reactions
The most serious adverse reactions that may occur during treatment with filgrastim include: anaphylactic reaction, serious pulmonary reactions (including interstitial pneumonia and acute respiratory distress syndrome), capillary leak syndrome, severe splenomegaly/ruptured spleen, transformation to myelodysplastic syndrome or leukemia in patients with severe chronic neutropenia, graft-versus-host reaction in patients who have undergone allogeneic bone marrow transplantation or peripheral blood stem cell transplantation, and sickle cell crisis in patients with sickle cell anemia.
The most commonly reported adverse reactions were: pyrexia, musculoskeletal pain (including bone pain, back pain, arthralgia, myalgia, limb pain, musculoskeletal pain, chest musculoskeletal pain, neck pain), anemia, vomiting, nausea. In clinical studies involving oncology patients, musculoskeletal pain was mild or moderate in 10% of patients and severe in 3% of patients.
Adverse reactions identified from clinical studies and spontaneous reports are listed in Table 2. Within each frequency category, adverse reactions are listed in descending order of severity.
Table 2
| Body systems |
Adverse reactions |
|||
| Very common (≥1/10) |
Common (≥1/100 and <1/10) |
Uncommon (≥1/1000 and <1/100) |
Rare (≥1/10000 and <1/1000) |
|
| Infections and infestations |
Sepsis Bronchitis Upper respiratory tract infections Urinary tract infections |
|||
| Blood and lymphatic system |
Thrombocytopenia Anemia e |
Splenomegalya Decreased hemoglobine |
Leukocytosisa |
Rupture of spleena Sickle cell anemia with crisis, Extramedullary hematopoiesis |
| Immune system |
Hypersensitivity Drug hypersensitivitya Graft versus host reactionb |
Anaphylactic reaction |
||
| Metabolism and nutrition |
Decreased appetitee Elevated blood lactate dehydrogenase |
Hyperuricemia Elevated blood uric acid |
Decreased blood glucose Pseudogouta (chondrocalcinosis) Fluid imbalance |
|
| Psychiatric |
Insomnia |
|||
| Nervous system |
Headachea |
Dizziness Hypoesthesia Paraesthesia |
||
| Vascular |
Hypertension Hypotension |
Veno-occlusive diseased |
Capillary leak syndromea Aortitis |
|
| Respiratory system |
Haemoptysis Dyspnoea Cougha Oropharyngeal paina, e Nosebleed |
Acute respiratory distress syndromea Respiratory failurea Pulmonary oedemaa Pulmonary haemorrhage Interstitial lung diseasea Lung infiltrationa Hypoxia |
||
| Gastrointestinal tract |
Diarrhoeaa, e Vomitinga, e Nauseaa |
Mouth pain Constipatione |
||
| Hepatobiliary system |
Hepatomegaly Elevated blood alkaline phosphatase |
Elevated blood aspartate aminotransferase Elevated blood gamma-glutamyl transferase |
||
| Skin and subcutaneous tissue |
Alopeciaa |
Rash a Erythema |
Maculopapular rash |
Skin vasculitisa Sweet's syndrome (acute febrile neutrophilic dermatosis) |
| Musculoskeletal and connective tissue |
Musculoskeletal painc |
Muscle spasms |
Osteoporosis |
Decreased bone density Exacerbation of rheumatoid arthritis |
| Renal and urinary system |
Dysuria Hematuria |
Proteinuria |
Glomerulonephritis Urinalysis abnormality |
|
| General and administration site |
Fatiguea Mucosal inflammationa Hyperthermia |
Chest paina Paina Astheniaa Malaisee Peripheral edemae |
Injection site reaction |
|
| Injury, poisoning and procedural complications |
Transfusion reactione |
|||
a See section "Description of selected adverse reactions".
b Cases of graft-versus-host reaction and fatal outcomes have been reported in patients after allogeneic bone marrow transplantation (see section "Description of selected adverse reactions" below).
c Includes bone pain, back pain, arthralgia, myalgia, limb pain, musculoskeletal pain, musculoskeletal chest pain, neck pain.
d Cases occurred during the post-marketing period in patients who underwent bone marrow transplantation or mobilization of HSCs.
e Adverse reactions occurring more frequently in patients receiving filgrastim compared to placebo group, and associated with the consequences of the underlying oncological disease or cytotoxic chemotherapy.
Description of selected adverse reactions
Hypersensitivity
Hypersensitivity reactions, including anaphylaxis, rash, urticaria, angioedema, dyspnea, and hypotension, have been observed during clinical trials and in the post-marketing period, developing at the beginning or during continued administration of filgrastim. Overall, such reactions were more frequent after intravenous administration of filgrastim. Occasionally, re-administration of the drug was associated with recurrence of symptoms, indicating a causal relationship. If serious allergic reactions occur, filgrastim should be discontinued and not restarted.
Respiratory system adverse reactions
During clinical trials and in the post-marketing period, adverse reactions affecting the lungs have been reported, including interstitial lung disease, pulmonary edema, pulmonary infiltrates, sometimes leading to respiratory failure or acute respiratory distress syndrome, which may be fatal (see section "Particular patient populations").
Splenic enlargement and splenic rupture
Cases of splenomegaly and splenic rupture have been reported with filgrastim use. Some cases of splenic rupture were fatal (see section "Particular patient populations").
Capillary leak syndrome
Cases of capillary leak syndrome have been reported with granulocyte colony-stimulating factor administration. These usually occur in patients with progressive malignancies, sepsis, those receiving multiple chemotherapeutic agents, or undergoing apheresis (see section "Particular patient populations").
Cutaneous vasculitis
Cases of cutaneous vasculitis have been reported in patients receiving filgrastim. The mechanism of vasculitis in patients receiving filgrastim is not known. With prolonged use, cutaneous vasculitis occurred in 2% of patients with severe chronic neutropenia.
Leukocytosis
Leukocytosis (white blood cell count > 50×10⁹/L) was observed in 41% of healthy donors; transient thrombocytopenia (platelet count < 100×10⁹/L) after filgrastim administration and leukapheresis was observed in 35% of donors (see section "Particular patient populations").
Sweet’s syndrome
Cases of Sweet’s syndrome (acute febrile neutrophilic dermatosis) have been reported in patients receiving filgrastim.
Pseudogout (chondrocalcinosis)
Pseudogout (chondrocalcinosis) has been reported in cancer patients receiving filgrastim.
Graft-versus-host reaction
Cases of graft-versus-host reaction with fatal outcomes have been reported in patients receiving G-CSF after allogeneic bone marrow transplantation (see sections "Pharmacodynamics", "Particular patient populations").
Pediatric population
Clinical trial data in pediatric patients demonstrate similar efficacy and safety of filgrastim in children and adults receiving cytotoxic chemotherapy, indicating no age-related differences in filgrastim pharmacokinetics. The only consistently reported adverse reaction was musculoskeletal pain, which does not differ from the experience in adults. There is insufficient data to further evaluate the use of filgrastim in children.
Other special patient populations
Elderly patients
No overall differences in safety and efficacy were observed in patients aged over 65 years compared to younger adults (>18 years) receiving cytotoxic chemotherapy; clinical experience did not reveal differences in therapeutic response between elderly and younger adult patients. There is insufficient data to evaluate the use of filgrastim in geriatric patients for other approved indications.
Pediatric patients with severe chronic neutropenia
Cases of decreased bone density and osteoporosis have been reported in pediatric patients with severe chronic neutropenia receiving long-term filgrastim therapy.
Reporting of suspected adverse reactions
Reporting suspected adverse reactions after drug authorization is of great importance. It allows continuous monitoring of the benefit-risk balance of the medicinal product. Healthcare professionals, pharmacists, patients, and their legal representatives should report all cases of suspected adverse reactions and lack of efficacy via the Automated Pharmacovigilance Information System at: https://aisf.dec.gov.ua
Shelf life. 2.5 years.
Storage conditions. Store in the original packaging at a temperature of 2 °C to 8 °C. Keep out of reach of children.
Incompatibilities
The medicinal product must not be diluted with sodium chloride solutions.
The medicinal product must not be mixed with other medicinal products except those specified in the section "Method of administration and dosage".
Filgrastim may adsorb to glass and certain plastics after dilution; this does not occur when dilution is performed as specified in the section "Method of administration and dosage".
Packaging
0.5 mL (30 million IU) in a pre-filled syringe with a safety device for safe administration; packs of 5 pre-filled syringes with a safety device.
0.8 mL (48 million IU) in a pre-filled syringe with a safety device for safe administration; packs of 5 pre-filled syringes with a safety device.
Prescription category. Prescription only.
Manufacturer. Teva Pharmaceutical Industries Ltd.
Manufacturer's address and location of operations. Industrial Zone, 18 Eli Hurvitz Street, P.O. Box 353, Kfar Saba, 4410202, Israel.