Luxturna 5 x 10¹² genomi virali/ml concentrato e solvente per soluzione iniettabile
Spagna
Indice
- Foglio illustrativo: informazioni per l'utente
- Introduzione
- 1. Che cos'è Luxturna e a cosa serve
- 2. Cosa deve sapere prima di iniziare a ricevere Luxturna
- 3. Come viene somministrato Luxturna
- 4. Possibili effetti indesiderati
- 5. Conservazione di Luxturna
- 6. Contenuto della confezione e informazioni aggiuntive
Foglio illustrativo: informazioni per l'utente
Introduzione
Foglio illustrativo: informazioni per il paziente
Luxturna 5 × 1012 genomi vettoriali/ml concentrato e solvente per soluzione iniettabile
voretigene neparvovec
Questo medicinale è sottoposto a monitoraggio addizionale, che permetterà una rapida identificazione di nuove informazioni sulla sua sicurezza. Lei può contribuire segnalando eventuali effetti indesiderati che dovesse manifestare. Nell'ultima parte della sezione 4 è riportata l'informazione su come segnalare tali effetti indesiderati.
Legga attentamente questo foglio illustrativo prima che le venga somministrato questo medicinale, perché contiene informazioni importanti per lei.
- Conservi questo foglio illustrativo: potrebbe essere necessario rileggerlo.
- Se ha domande, si rivolga al suo medico o all'infermiere.
- Se manifesta effetti indesiderati, si rivolga al suo medico o all'infermiere, anche qualora si tratti di effetti indesiderati non elencati nel presente foglio illustrativo. Vedere sezione 4.
Contenuto del foglio illustrativo :
- Che cos’è Luxturna e a che cosa serve
- Cosa deve sapere prima di ricevere Luxturna
- Come viene somministrato Luxturna
- Possibili effetti indesiderati
- Come conservare Luxturna
- Contenuto della confezione e altre informazioni
1. Che cos'è Luxturna e a cosa serve
Luxturna è un medicinale per terapia genica che contiene il principio attivo voretigene neparvovec.
Luxturna è indicato per il trattamento di adulti e bambini con perdita della vista dovuta a una distrofia retinica ereditaria causata da mutazioni nel gene RPE65. Queste mutazioni impediscono all'organismo di produrre una proteina necessaria per la vista, causando un deterioramento visivo e potenzialmente la cecità.
Il principio attivo di Luxturna, voretigene neparvovec, è un virus modificato che contiene una copia del gene RPE65. Dopo l'iniezione, questo gene raggiunge le cellule della retina, lo strato nella parte posteriore dell'occhio che rileva la luce. Ciò consente alla retina di produrre le proteine necessarie per la vista. Il virus utilizzato per somministrare il gene non causa malattie nell'uomo.
Le verrà somministrato Luxturna soltanto se gli esami genetici avranno evidenziato che la sua perdita visiva è causata da mutazioni nel gene RPE65.
2. Cosa deve sapere prima di iniziare a ricevere Luxturna
Non deve ricevere Luxturna
- Se è allergico a voretigene neparvovec o ad uno qualsiasi degli altri componenti di questo medicinale (indicati nella sezione 6)
- Se ha un’infezione all’occhio
- Se ha un’infiammazione all’occhio
Se si trova in una delle situazioni sopra indicate, o se non è sicuro, consulti il medico prima di ricevere Luxturna.
Avvertenze e precauzioni
Prima di ricevere il trattamento con Luxturna:
- Informi il medico se ha segni di infezione oculare o infiammazione oculare, ad esempio se ha arrossamento degli occhi, sensibilità alla luce, gonfiore oculare o dolore agli occhi.
- Informi il medico se ha un’infezione attiva di qualsiasi tipo. Il medico potrebbe decidere di rimandare il trattamento finché l’infezione non sarà scomparsa, poiché questo medicinale potrebbe rendere più difficile combattere l’infezione. Vedere anche la sezione 3.
Dopo aver ricevuto il trattamento con Luxturna:
-
Consulti immediatamente il medico se l’occhio/occhi diventano rossi, se avverte dolore oculare, sensibilità alla luce, vede lampi di luce o corpi mobili (miodesopsie), oppure se nota un peggioramento della vista o una visione offuscata.
-
Deve evitare di viaggiare in aereo o di recarsi ad altezze elevate finché il medico non le indicherà che è possibile farlo. Durante il trattamento con questo medicinale, il medico inserisce una bolla d’aria nell’occhio, che il corpo assorbe lentamente. Fino a quando la bolla non sarà completamente assorbita, viaggiare in aereo o recarsi ad altezze elevate potrebbe causare l’espansione della bolla e provocare danni agli occhi, inclusa la perdita della vista. Consulti il medico prima di viaggiare.
-
Deve evitare di nuotare a causa del rischio aumentato di infezione oculare. Consulti il medico prima di andare a nuotare dopo aver ricevuto il trattamento con Luxturna.
-
Deve evitare attività fisiche intense a causa del rischio aumentato di lesioni all’occhio. Consulti il medico prima di iniziare un’attività fisica intensa dopo aver ricevuto il trattamento con Luxturna.
-
Potrebbe avere disturbi visivi transitori, come sensibilità alla luce e visione offuscata. Informi il medico di qualsiasi disturbo visivo che dovesse manifestarsi. Il medico potrà aiutarla a ridurre il disagio causato da questi effetti transitori.
-
Il principio attivo di Luxturna potrebbe essere temporaneamente escreto attraverso le lacrime. Lei e il suo caregiver dovete mettere le medicazioni e il materiale di scarto che sono entrati in contatto con lacrime e secrezioni nasali in sacchetti sigillati prima di smaltirli. Deve osservare queste precauzioni per 14 giorni.
-
Dopo aver ricevuto il trattamento con Luxturna, non potrà donare sangue, organi, tessuti e cellule per trapianto.
Bambini e adolescenti
Luxturna non è stato studiato nei bambini di età inferiore a quattro anni. I dati disponibili sono limitati.
Altri medicinali e Luxturna
Informi il medico se sta assumendo, ha recentemente assunto o potrebbe dover assumere altri medicinali.
Gravidanza, allattamento e fertilità
Se è in gravidanza o in allattamento, se pensa di essere in gravidanza o se intende rimanere incinta, consulti il medico o l’infermiere prima di ricevere il trattamento con Luxturna.
Gli effetti di questo medicinale sulla gravidanza e sul feto non sono noti. Per precauzione, non deve ricevere Luxturna durante la gravidanza.
Luxturna non è stato studiato nelle donne in allattamento. Non si sa se il medicinale passi nel latte materno. Informi il medico se sta allattando al seno o se prevede di farlo. Il medico la aiuterà a decidere se deve interrompere l’allattamento o non ricevere Luxturna, tenendo conto dei benefici dell’allattamento per il bambino e dei benefici di Luxturna per lei.
Guida di veicoli e uso di macchinari
Potrebbe manifestare disturbi visivi transitori dopo aver ricevuto Luxturna. Non guidi né usi macchinari pesanti finché la sua vista non sarà tornata alla normalità. Consulti il medico prima di riprendere queste attività.
Luxturna contiene sodio
Questo medicinale contiene meno di 1 mmol di sodio (23 mg) per dose, cioè è essenzialmente "privo di sodio".
3. Come viene somministrato Luxturna
Luxturna le verrà somministrato in una sala operatoria da chirurghi esperti nell’esecuzione di interventi chirurgici oculari. Luxturna viene somministrato sotto anestesia. Il suo medico le parlerà dell’anestesia e del modo in cui le verrà somministrato il medicamento.
Il suo medico le eseguirà un intervento chirurgico oculare per rimuovere il gel trasparente presente all’interno dell’occhio, quindi le inietterà Luxturna direttamente nella retina, lo strato sottile sensibile alla luce situato nella parte posteriore dell’occhio. Questa procedura verrà ripetuta nell’altro occhio almeno 6 giorni dopo. Dovrà rimanere in osservazione postoperatoria per alcune ore dopo ciascuna procedura, per monitorare il recupero e osservare eventuali effetti collaterali dell’intervento chirurgico o dell’anestesia.
Prima di iniziare il trattamento con Luxturna, il suo medico potrebbe chiederle di assumere un medicamento che sopprime il sistema immunitario (le difese naturali dell’organismo), in modo che non tenti di combattere Luxturna al momento della somministrazione. È importante che assuma questo medicamento seguendo esattamente le istruzioni fornite dal suo medico. Non interrompa l’assunzione del medicamento senza aver prima consultato il suo medico.
Se riceve una quantità di Luxturna superiore a quella prevista
Poiché questo medicamento viene somministrato da un medico, è poco probabile che le venga somministrata una quantità superiore a quella prevista. Se ciò dovesse accadere, il suo medico tratterà i sintomi in base alle necessità. Informi il suo medico o l’infermiere se dovesse avere problemi alla vista.
Se ha altre domande sull’uso di questo medicamento, chieda al suo medico o all’infermiere.
4. Possibili effetti indesiderati
Come tutti i medicinali, questo medicamento può causare effetti indesiderati, sebbene non tutte le persone li manifestino.
Possono verificarsi i seguenti effetti indesiderati associati a Luxturna:
Frequenti (possono riguardare fino a 1 persona su 10)
- Depositi sotto la retina
Frequenza non nota (non può essere stimata dai dati disponibili)
- Atrofia della (corio)retina
Possono verificarsi i seguenti effetti indesiderati associati alla procedura di iniezione:
Molto frequenti (possono riguardare più di 1 persona su 10)
- Arrossamento degli occhi
- Cataratta (opacità del cristallino)
- Aumento della pressione oculare
Frequenti (possono riguardare fino a 1 persona su 10)
- Lacrimazione della retina
- Dolore agli occhi
- Gonfiore degli occhi
- Distacco della retina
- Emorragia nella parte posteriore dell’occhio
- Dolore o aumento del fastidio all’occhio
- Visione offuscata dovuta a un foro nella retina
- Assottigliamento della superficie dell’occhio (dellen)
- Irritazione oculare
- Infiammazione oculare
- Sensazione di corpo estraneo nell’occhio
- Fastidio oculare
- Anomalie nella parte posteriore dell’occhio
- Nausea (voglia di vomitare), vomito, dolore addominale, dolore alle labbra
- Modifiche dell’attività elettrica del cuore
- Cefalea, capogiri
- Eruzioni cutanee, gonfiore del viso
- Ansia
- Problemi associati al posizionamento di un tubo endotracheale
- Rottura della ferita chirurgica
Non noti (la frequenza non può essere stimata dai dati disponibili)
- Opacizzazione della sostanza gelatinosa presente all’interno dell’occhio (opacità vitree)
- Atrofia della (corio)retina
Il danno ai tessuti oculari può essere accompagnato da emorragia, infiammazione e maggiore rischio di infezione. Nei giorni successivi all’intervento chirurgico si verifica una riduzione della vista che generalmente migliora; informi il medico se la vista non dovesse tornare normale.
Segnalazione degli effetti indesiderati
Se dovesse manifestarsi qualsiasi tipo di effetto indesiderato, consulti il medico o l’infermiere, anche se si tratta di effetti indesiderati che non sono elencati in questo foglio illustrativo. Può inoltre segnalarli direttamente attraverso il sistema nazionale di notifica indicato nell’Appendice V. Segnalando gli effetti indesiderati, lei può contribuire a fornire maggiori informazioni sulla sicurezza di questo medicamento.
5. Conservazione di Luxturna
Luxturna sarà conservato dai professionisti sanitari presso il loro centro sanitario.
Il concentrato e il solvente devono essere trasportati e conservati congelati a temperatura ≤ ‑65 ºC. Una volta scongelato, il medicamento non deve essere ricongelato e deve essere tenuto a temperatura ambiente (al di sotto di 25 ºC).
Non utilizzi questo medicamento dopo la data di scadenza indicata sull'etichetta e sulla confezione dopo SCAD.
6. Contenuto della confezione e informazioni aggiuntive
Composizione di Luxturna
- Il principio attivo è voretigene neparvovec. Ogni ml di concentrato contiene 5 × 1012 genomi virali (vg). Il concentrato (flaconcino monodose da 2 ml con volume estraibile di 0,5 ml) richiede una diluizione 1:10 prima della somministrazione.
- Ogni dose di soluzione diluita contiene 1,5 × 1011 genomi virali di voretigene neparvovec in un volume somministrabile di 0,3 ml.
- Gli altri eccipienti del concentrato sono cloruro di sodio (vedere “Luxturna contiene sodio” alla fine della sezione 2 di questo foglio illustrativo), diidrogenofosfato di sodio monoidrato (per regolare il pH), diidrogenofosfato di sodio diidrato (per regolare il pH), polossamero 188 e acqua per preparazioni iniettabili.
- Il solvente contiene cloruro di sodio (vedere la fine della sezione 2), diidrogenofosfato di sodio monoidrato (per regolare il pH), diidrogenofosfato di sodio diidrato (per regolare il pH), polossamero 188 e acqua per preparazioni iniettabili.
Questo medicinale contiene organismi geneticamente modificati.
Aspetto di Luxturna e contenuto della confezione
Luxturna è un concentrato limpido e incolore per soluzione per iniezione sottoretinica, fornito in un flaconcino di plastica trasparente. Il solvente è un liquido limpido e incolore fornito in un flaconcino di plastica trasparente.
Ogni bustina in alluminio contiene una scatola di cartone che include 1 flaconcino da 0,5 ml di concentrato e 2 flaconcini di solvente (ognuno contiene 1,7 ml).
Titolare dell'autorizzazione all'immissione in commercio
Novartis Europharm Limited
Vista Building
Elm Park, Merrion Road
Dublino 4
Irlanda
Responsabile della produzione
Novartis Pharma GmbH
Sophie-Germain-Strasse 10
90443 Norimberga
Germania
È possibile richiedere ulteriori informazioni su questo medicinale rivolgendosi al rappresentante locale del titolare dell'autorizzazione all'immissione in commercio:
Belgio/Belgio/Belgio Novartis Pharma N.V. Tel/Tel: +32 2 246 16 11 | Lituania SIA Novartis Baltics Lituania filiale Tel: +370 5 269 16 50 |
| Lussemburgo Novartis Pharma N.V. Tel/Tel: +32 2 246 16 11 |
Repubblica Ceca Novartis s.r.o. Tel: +420 225 775 111 | Ungheria Novartis Hungária Kft. Tel.: +36 1 457 65 00 |
Danimarca Novartis Healthcare A/S Tlf: +45 39 16 84 00 | Malta Novartis Pharma Services Inc. Tel: +356 2122 2872 |
Germania Novartis Pharma GmbH Tel: +49 911 273 0 | Paesi Bassi Novartis Pharma B.V. Tel: +31 88 04 52 111 |
Estonia SIA Novartis Baltics Estonia filiale Tel: +372 66 30 810 | Norvegia Novartis Norge AS Tlf: +47 23 05 20 00 |
Grécia Novartis (Hellas) A.E.B.E. Tel: +30 210 281 17 12 | Austria Novartis Pharma GmbH Tel: +43 1 86 6570 |
Spagna Novartis Farmacéutica, S.A. Tel: +34 93 306 42 00 | Polonia Novartis Poland Sp. z o.o. Tel.: +48 22 375 4888 |
Francia Novartis Pharma S.A.S. Tél: +33 1 55 47 66 00 | Portogallo Novartis Farma ‑ Prodotti Farmaceutici, S.A. Tel: +351 21 000 8600 |
Croazia Novartis Hrvatska d.o.o. Tel. +385 1 6274 220 | Romania Novartis Pharma Services Romania SRL Tel: +40 21 31299 01 |
Irlanda Novartis Ireland Limited Tel: +353 1 260 12 55 | Slovenia Novartis Pharma Services Inc. Tel: +386 1 300 75 50 |
Islanda Vistor hf. Sími: +354 535 7000 | Repubblica Slovacca Novartis Slovakia s.r.o. Tel: +421 2 5542 5439 |
Italia Novartis Farma S.p.A. Tel: +39 02 96 54 1 | Finlandia Novartis Finland Oy Puh/Tel: +358 (0)10 6133 200 |
Cipro Novartis Pharma Services Inc. Tel: +357 22 690 690 | Svezia Novartis Sverige AB Tel: +46 8 732 32 00 |
Lettonia SIA Novartis Baltics Tel: +371 67 887 070 |

Data dell'ultima revisione di questo foglio illustrativo:
Altre fonti di informazioni
Questo foglio illustrativo è disponibile in formato audio e in caratteri ingranditi sul sito web: http://www.voretigeneneparvovec.support
Informazioni dettagliate su questo medicinale sono disponibili sul sito web dell’Agenzia Europea per i Medicinali: http://www.ema.europa.eu.
Questa informazione è destinata esclusivamente ai professionisti sanitari:
Precauzioni da adottare prima della manipolazione o della somministrazione del medicinale
Questo medicinale contiene organismi geneticamente modificati. Deve essere utilizzato un equipaggiamento di protezione personale (comprensivo di camice da laboratorio, occhiali di sicurezza e guanti) durante la preparazione o la somministrazione di voretigene neparvovec.
La pressione intraoculare deve essere adeguatamente controllata e monitorata prima e dopo la somministrazione del medicinale.
Dopo la somministrazione, ai pazienti deve essere raccomandato di segnalare immediatamente qualsiasi sintomo che possa suggerire endoftalmite o distacco della retina e devono essere trattati in modo appropriato.
Preparazione prima della somministrazione
Ogni confezione contenente 1 flaconcino di concentrato e 2 flaconcini di solvente è destinata a un solo uso.
Luxturna deve essere ispezionato visivamente prima della somministrazione. Se vengono rilevate particelle, torbidità o decolorazione, il flaconcino monodose non deve essere utilizzato.
La preparazione di Luxturna deve essere effettuata entro le 4 ore precedenti l'inizio della procedura di somministrazione, in condizioni asettiche e secondo la procedura raccomandata riportata di seguito.
Far scongelare a temperatura ambiente un flaconcino monodose di concentrato e due flaconcini di solvente. Una volta scongelati i 3 flaconcini (1 flaconcino di concentrato e 2 flaconcini di solvente), deve essere avviata la diluizione. Mescolare delicatamente i flaconcini capovolgendoli cinque volte.
Ispezionare visivamente la presenza di particelle o anomalie. Qualsiasi anomalia o particella visibile deve essere segnalata al Titolare dell'Autorizzazione all'Immissione in Commercio e il prodotto non deve essere utilizzato.
Trasferire 2,7 ml di solvente proveniente dai due flaconcini scongelati, dispensandoli con una siringa da 3 ml in un flacone sterile di vetro da 10 ml vuoto.
Per la diluizione, prelevare 0,3 ml di concentrato scongelato con una siringa da 1 ml e aggiungerli al flacone sterile da 10 ml contenente il solvente. Capovolgere delicatamente il flacone almeno cinque volte per ottenere un'adeguata miscelazione. Ispezionare visivamente la presenza di particelle. La soluzione diluita deve essere limpida o leggermente opalescente. Etichettare il flacone di vetro da 10 ml contenente il concentrato diluito come segue: "Luxturna diluito".
Non preparare le siringhe se il flacone presenta danni o se sono visibili particelle. Preparare le siringhe per iniezione prelevando 0,8 ml della soluzione diluita in una siringa sterile da 1 ml. Ripetere la stessa procedura per preparare una siringa di riserva. Le siringhe piene di prodotto devono essere trasportate in sala operatoria in un contenitore appositamente destinato a tale scopo.
Misure da adottare in caso di esposizione accidentale
Deve essere evitata l'esposizione accidentale. Devono essere seguite le normative locali sulla biosicurezza per la preparazione, somministrazione e manipolazione di voretigene neparvovec.
- Deve essere utilizzato un equipaggiamento di protezione personale (comprensivo di camice da laboratorio, occhiali di sicurezza e guanti) durante la manipolazione o la somministrazione di voretigene neparvovec.
- Deve essere evitata l'esposizione accidentale a voretigene neparvovec, inclusi il contatto con la pelle, gli occhi e le membrane mucose. Qualsiasi ferita aperta deve essere coperta prima di manipolare questo medicinale.
- Ogni fuoriuscita di voretigene neparvovec deve essere trattata con un agente virucida, come ipoclorito di sodio allo 1%, e asciugata con materiali assorbenti.
- Tutti i materiali che potrebbero essere venuti a contatto con voretigene neparvovec (es. flaconcino, siringa, ago, garze, guanti, maschere o bendaggi) devono essere smaltiti secondo le normative locali sulla biosicurezza.
Esposizione accidentale
- In caso di esposizione occupazionale accidentale (es. schizzi negli occhi o sulle membrane mucose), sciacquare con acqua pulita per almeno 5 minuti.
- In caso di esposizione su cute lesa o lesione da puntura con ago, lavare accuratamente l'area interessata con acqua e sapone e/o con un disinfettante.
Precauzioni da adottare per lo smaltimento del medicinale
Questo medicinale contiene organismi geneticamente modificati. Lo smaltimento del medicinale non utilizzato e di tutti i materiali che sono entrati in contatto con esso deve essere effettuato in conformità con le normative locali sui rifiuti farmaceutici.
Posologia
Il trattamento deve essere iniziato e somministrato da un chirurgo della retina esperto in chirurgia maculare.
I pazienti riceveranno una dose singola di voretigene neparvovec di 1,5 × 1011 genomi vettoriali in ciascun occhio. Ciascuna dose sarà somministrata nello spazio subretinico in un volume totale di 0,3 ml. La somministrazione deve essere effettuata singolarmente per ciascun occhio in giorni separati, con un intervallo minimo di almeno sei giorni tra ciascuna procedura chirurgica.
Regime immunomodulante
Prima di iniziare il regime immunomodulante e prima della somministrazione di voretigene neparvovec, il paziente deve essere esaminato per rilevare sintomi di malattia infettiva attiva di qualsiasi natura; in caso di infezione, l'inizio del trattamento deve essere rinviato fino al recupero del paziente.
Si raccomanda di iniziare il regime immunomodulante 3 giorni prima della somministrazione di voretigene neparvovec nel primo occhio, seguendo il calendario descritto di seguito (Tabella 1). L'inizio del regime immunomodulante per il secondo occhio deve seguire lo stesso schema e deve sostituire il regime immunomodulante del primo occhio.
Tabella 1 Regime immunomodulante pre e postoperatorio per ciascun occhio
Preoperatorio | 3 giorni prima della somministrazione di Luxturna | Prednisone (o equivalente) 1 mg/kg/die (fino a un massimo di 40 mg/die) |
Postoperatorio | 4 giorni (incluso il giorno della somministrazione) | Prednisone (o equivalente) 1 mg/kg/die (fino a un massimo di 40 mg/die) |
Continuare per 5 giorni | Prednisone (o equivalente) 0,5 mg/kg/die (fino a un massimo di 20 mg/die) | |
Continuare per 5 giorni con una dose ogni due giorni | Prednisone (o equivalente) 0,5 mg/kg ogni due giorni (fino a un massimo di 20 mg/die) |
Popolazioni speciali
Pazienti di età avanzata
Non è stata stabilita la sicurezza e l'efficacia di voretigene neparvovec in pazienti ≥ 65 anni. I dati sono limitati. Tuttavia, non è necessario un aggiustamento della dose nei pazienti di età avanzata.
Insufficienza epatica e renale
Non è stata stabilita la sicurezza e l'efficacia di voretigene neparvovec in pazienti con insufficienza epatica o renale. Non è necessario un aggiustamento della dose in questi pazienti (vedere sezione 5.2).
Popolazione pediatrica
Non è stata stabilita la sicurezza e l'efficacia di voretigene neparvovec in bambini di età inferiore a 4 anni. I dati sono limitati. Non è necessario un aggiustamento della dose nei pazienti pediatrici.
Modalità di somministrazione
Uso subretinico.
Luxturna è una soluzione sterile concentrata per iniezione subretinica che richiede scongelamento e diluizione prima della somministrazione.
Questo medicinale non deve essere somministrato mediante iniezione intravitreale.
Luxturna è un flaconcino monodose destinato a un’unica somministrazione in un singolo occhio. Il prodotto viene somministrato mediante iniezione subretinica dopo aver effettuato una vitrectomia in ciascun occhio. Non deve essere somministrato troppo vicino alla fovea per preservare l’integrità foveale.
La somministrazione di voretigene neparvovec deve essere effettuata in ambiente operatorio, in condizioni asettiche controllate. Prima della procedura, al paziente deve essere somministrata un’adeguata anestesia. La pupilla dell’occhio in cui verrà somministrata l’iniezione deve essere dilatata e, prima dell’intervento chirurgico, deve essere somministrato un antibiotico a spettro ampio per via oftalmica, secondo la pratica medica abituale.
Somministrazione
Seguire i passaggi descritti di seguito per somministrare voretigene neparvovec ai pazienti:
- Una volta diluito Luxturna, il prodotto deve essere ispezionato visivamente prima della somministrazione. Se si osservano particelle, torbidità o decolorazione, il medicinale non deve essere utilizzato.
- Collegare la siringa contenente il prodotto diluito al tubo di estensione e alla cannula per iniezione subretinica. Il prodotto deve essere iniettato lentamente attraverso il tubo di estensione e la cannula per iniezione subretinica per eliminare eventuali bolle d’aria dal sistema.
- Il volume di prodotto disponibile per l’iniezione viene confermato nella siringa allineando la punta del pistone con la linea che indica 0,3 ml.
- Al termine della vitrectomia, Luxturna viene somministrato mediante iniezione subretinica utilizzando una cannula per iniezione subretinica inserita per via pars plana.
- Sotto visualizzazione diretta, la punta della cannula per iniezione subretinica viene posta a contatto con la superficie della retina. Il sito di iniezione raccomandato dovrebbe trovarsi lungo l’arcata vascolare superiore, ad almeno 2 mm di distanza dal centro della fovea. Viene iniettata lentamente una piccola quantità di prodotto finché non si osserva una bolla subretinica iniziale, quindi il volume rimanente viene iniettato lentamente fino al raggiungimento dei 0,3 ml totali (Figura 1).
Figura 1: Punta della cannula per iniezione subretinica posizionata nel sito di iniezione raccomandato (vista del chirurgo)
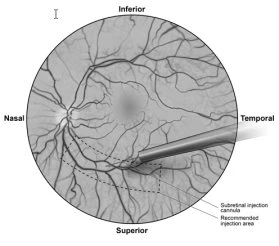
- Al termine dell’iniezione, la cannula per iniezione subretinica viene rimossa dall’occhio.
- Dopo l’iniezione, qualsiasi prodotto non utilizzato deve essere smaltito. Non deve essere conservata alcuna siringa di riserva.
- Deve essere effettuato con attenzione uno scambio fluido-aria, evitando il drenaggio di liquido nelle vicinanze della retinotomia creata per l’iniezione subretinica.
- Nel periodo postoperatorio, il paziente deve essere posizionato in posizione supina immediatamente dopo l’intervento e mantenuto in tale posizione per 24 ore dopo la dimissione.