Luxturna 5 x 10¹² vector genomes/ml concentrate and solvent for solution for injection
Spain
Table of Contents
Patient Information Leaflet
Introduction
Package Leaflet: Information for the Patient
Luxturna 5 × 1012 vector genomes/ml concentrate and solvent for injectable solution
voretigene neparvovec
This medicinal product is subject to additional monitoring, which will allow for rapid identification of new safety information. You can help by reporting any side effects you may experience. Information on how to report side effects is provided at the end of section 4.
Read this entire leaflet carefully before you are administered this medicine, as it contains important information for you.
- Keep this leaflet, as you may need to refer to it again.
- If you have any questions, consult your doctor or nurse.
- If you experience any side effects, consult your doctor or nurse, even if they are not listed in this leaflet. See section 4.
Leaflet Contents:
- What Luxturna is and what it is used for
- What you need to know before receiving Luxturna
- How Luxturna is administered
- Possible side effects
- How to store Luxturna
- Contents of the pack and other information
1. What Luxturna is and what it is used for
Luxturna is a gene therapy product containing the active substance voretigene neparvovec.
Luxturna is used to treat adults and children with vision loss due to inherited retinal dystrophy caused by mutations in the RPE65 gene. These mutations prevent the body from producing a protein essential for vision, leading to vision loss and potential blindness.
The active ingredient in Luxturna, voretigene neparvovec, is a modified virus containing a copy of the RPE65 gene. After injection, this gene reaches the retinal cells, the layer at the back of the eye that detects light. This enables the retina to produce the proteins necessary for vision. The virus used to deliver the gene does not cause disease in humans.
You will only be given Luxturna if genetic testing confirms that your vision loss is caused by mutations in the RPE65 gene.
2. What you need to know before receiving Luxturna
Do not receive Luxturna
- If you are allergic to voretigene neparvovec or to any of the other ingredients of this medicine (listed in section 6)
- If you have an eye infection
- If you have inflammation in the eye
If you are affected by any of the above conditions, or if you are unsure, consult your doctor before receiving Luxturna.
Warnings and precautions
Before receiving treatment with Luxturna:
- Inform your doctor if you have signs of ocular infection or ocular inflammation, for example, if you have red eyes, light sensitivity, eye swelling, or eye pain.
- Inform your doctor if you have an active infection of any kind. Your doctor may delay your treatment until the infection resolves, because this medicine may impair your body's ability to fight infections. See also section 3.
After receiving treatment with Luxturna:
-
Contact your doctor immediately if your eye(s) become red, if you experience eye pain, light sensitivity, see flashes or floaters, or if you notice worsening or blurred vision.
-
You must avoid air travel or other travel to high altitudes until your doctor advises otherwise. During treatment with this medicine, a bubble of air is inserted into your eye, which your body slowly absorbs. Until the bubble is completely absorbed, air travel or travel to high altitudes may cause the bubble to expand and lead to eye damage, including vision loss. Consult your doctor before traveling.
-
You should avoid swimming due to an increased risk of eye infection. Consult your doctor before swimming after receiving treatment with Luxturna.
-
You should avoid strenuous physical activity due to an increased risk of eye injury. Consult your doctor before resuming strenuous physical activity after receiving treatment with Luxturna.
-
You may experience transient visual disturbances, such as light sensitivity and blurred vision. Inform your doctor of any visual disturbances you experience. Your doctor can help reduce any discomfort caused by these temporary effects.
-
The active substance in Luxturna may be temporarily excreted in tears. You and your caregiver must place dressings and waste materials that have been in contact with tears and nasal secretions into sealed bags before disposal. You should follow these precautions for 14 days.
-
You will not be able to donate blood, organs, tissues, or cells for transplantation after treatment with Luxturna.
Children and adolescents
Luxturna has not been studied in children under four years of age. Data are limited.
Other medicines and Luxturna
Inform your doctor if you are taking, have recently taken, or might need to take any other medicines.
Pregnancy, breastfeeding, and fertility
If you are pregnant or breastfeeding, think you may be pregnant, or are planning to become pregnant, consult your doctor or nurse before receiving treatment with Luxturna.
The effects of this medicine on pregnancy and the fetus are unknown. As a precaution, you should not receive Luxturna while pregnant.
Luxturna has not been studied in breastfeeding women. It is not known whether it passes into breast milk. Inform your doctor if you are breastfeeding or plan to breastfeed. Your doctor will help you decide whether to discontinue breastfeeding or not receive Luxturna, taking into account the benefits of breastfeeding for your baby and the benefits of Luxturna for you.
Driving and using machines
You may experience transient visual disturbances after receiving Luxturna. Do not drive or operate heavy machinery until your vision has recovered. Consult your doctor before resuming these activities.
Luxturna contains sodium
This medicine contains less than 1 mmol of sodium (23 mg) per dose, i.e., essentially "sodium-free".
3. How Luxturna is administered
Luxturna will be administered to you in an operating room by surgeons experienced in performing eye surgery. Luxturna is given under anesthesia. Your doctor will discuss anesthesia with you and explain how the medicine will be administered.
Your doctor will perform eye surgery to remove the clear gel that fills the inside of the eye, and then inject Luxturna directly into the retina, the thin light-sensitive layer at the back of the eye. This procedure will be repeated in the other eye at least 6 days later. You will need to remain under postoperative observation for several hours after each procedure to monitor your recovery and watch for any side effects from the surgery or anesthesia.
Before starting treatment with Luxturna, your doctor may ask you to take a medication that suppresses your immune system (the body's natural defenses) so that it does not try to fight against Luxturna when it is administered. It is important that you take this medication exactly as instructed by your doctor. Do not stop taking the medication without first consulting your doctor.
If you receive more Luxturna than you should
Since this medicine is administered by a doctor, it is unlikely that you will receive more than the intended dose. If this does happen, your doctor will treat any symptoms as necessary. Inform your doctor or nurse if you experience any vision problems.
If you have any further questions about the use of this medicine, ask your doctor or nurse.
4. Possible adverse effects
Like all medicines, this medicine can cause adverse effects, although not everyone experiences them.
The following adverse effects may occur with Luxturna:
Frequent (may affect up to 1 in 10 people)
- Deposits under the retina
Frequency not known (cannot be estimated from available data)
- Atrophy of the (chori)retina
The following adverse effects may occur related to the injection procedure:
Very frequent (may affect more than 1 in 10 people)
- Redness of the eyes
- Cataract (clouding of the lens)
- Increased pressure in the eye
Frequent (may affect up to 1 in 10 people)
- Tear in the retina
- Eye pain
- Swelling of the eyes
- Retinal detachment
- Bleeding in the back of the eye
- Pain or increased discomfort in the eye
- Blurred vision due to a hole in the retina
- Thinning of the surface of the eye (dellen)
- Eye irritation
- Eye inflammation
- Sensation of a foreign body in the eye
- Eye discomfort
- Abnormalities in the back of the eye
- Nausea, vomiting, abdominal pain, pain in the lips
- Change in the electrical activity of the heart
- Headache, dizziness
- Skin rash, swelling of the face
- Anxiety
- Problems associated with placement of a breathing tube in the trachea
- Surgical wound rupture
Not known (frequency cannot be estimated from available data)
- Clouding of the gel-like substance inside the eye (vitreous opacities)
- Atrophy of the (chori)retina
Damage to eye tissues may be accompanied by bleeding, inflammation, and an increased risk of infection. Vision loss may occur in the days following surgery, which usually improves; inform your doctor if vision does not return.
Reporting of adverse effects
If you experience any type of adverse effect, consult your doctor or nurse, even if it is a possible adverse effect not listed in this leaflet. You may also report them directly through the national reporting system detailed in Appendix V. By reporting adverse effects, you can help provide more information on the safety of this medicine.
5. Storage of Luxturna
Luxturna will be stored by healthcare professionals at their healthcare facility.
The concentrate and solvent must be transported and stored frozen at a temperature ≤ ‑65 °C. Once thawed, the medicinal product must not be refrozen and should be kept at room temperature (below 25 °C).
Do not use this medicinal product after the expiry date stated on the label and carton after EXP.
6. Contents of the pack and other information
Composition of Luxturna
- The active substance is voretigene neparvovec. Each ml of concentrate contains 5 × 1012 vector genomes (vg). The concentrate (single-dose vial of 2 ml with an extractable volume of 0.5 ml) requires dilution at a ratio of 1:10 before administration.
- Each dose of diluted solution contains 1.5 × 1011 vector genomes of voretigene neparvovec in an administrable volume of 0.3 ml.
- The other components of the concentrate are sodium chloride (see "Luxturna contains sodium" at the end of section 2 of this leaflet), monosodium dihydrogen phosphate monohydrate (to adjust pH), monosodium dihydrogen phosphate dihydrate (to adjust pH), poloxamer 188, and water for injections.
- The diluent contains sodium chloride (see end of section 2), monosodium dihydrogen phosphate monohydrate (to adjust pH), monosodium dihydrogen phosphate dihydrate (to adjust pH), poloxamer 188, and water for injections.
This medicinal product contains genetically modified organisms.
Appearance of Luxturna and contents of the pack
Luxturna is a clear, colourless concentrate for solution for subretinal injection supplied in a transparent plastic vial. The diluent is a clear, colourless liquid supplied in a transparent plastic vial.
Each aluminium pouch contains a cardboard box containing 1 vial of 0.5 ml concentrate and 2 vials of diluent (each containing 1.7 ml).
Marketing Authorisation Holder
Novartis Europharm Limited
Vista Building
Elm Park, Merrion Road
Dublin 4
Ireland
Manufacturer
Novartis Pharma GmbH
Sophie-Germain-Strasse 10
90443 Nuremberg
Germany
For further information about this medicinal product, please contact the local representative of the Marketing Authorisation Holder:
Belgium/Belgium/Belgium Novartis Pharma N.V. Tel/Tel: +32 2 246 16 11 | Lithuania SIA Novartis Baltics Lithuania branch Tel: +370 5 269 16 50 |
| Luxembourg/Luxembourg Novartis Pharma N.V. Tel/Tel: +32 2 246 16 11 |
Czech Republic Novartis s.r.o. Tel: +420 225 775 111 | Hungary Novartis Hungária Kft. Tel.: +36 1 457 65 00 |
Denmark Novartis Healthcare A/S Tlf: +45 39 16 84 00 | Malta Novartis Pharma Services Inc. Tel: +356 2122 2872 |
Germany Novartis Pharma GmbH Tel: +49 911 273 0 | Netherlands Novartis Pharma B.V. Tel: +31 88 04 52 111 |
Estonia SIA Novartis Baltics Estonia branch Tel: +372 66 30 810 | Norway Novartis Norge AS Tlf: +47 23 05 20 00 |
Greece Novartis (Hellas) S.A. Tel: +30 210 281 17 12 | Austria Novartis Pharma GmbH Tel: +43 1 86 6570 |
Spain Novartis Farmacéutica, S.A. Tel: +34 93 306 42 00 | Poland Novartis Poland Sp. z o.o. Tel.: +48 22 375 4888 |
France Novartis Pharma S.A.S. Tél: +33 1 55 47 66 00 | Portugal Novartis Farma ‑ Pharmaceutical Products, S.A. Tel: +351 21 000 8600 |
Croatia Novartis Hrvatska d.o.o. Tel. +385 1 6274 220 | Romania Novartis Pharma Services Romania SRL Tel: +40 21 31299 01 |
Ireland Novartis Ireland Limited Tel: +353 1 260 12 55 | Slovenia Novartis Pharma Services Inc. Tel: +386 1 300 75 50 |
Iceland Vistor hf. Tel: +354 535 7000 | Slovakia Novartis Slovakia s.r.o. Tel: +421 2 5542 5439 |
Italy Novartis Farma S.p.A. Tel: +39 02 96 54 1 | Finland Novartis Finland Oy Puh/Tel: +358 (0)10 6133 200 |
Cyprus Novartis Pharma Services Inc. Tel: +357 22 690 690 | Sweden Novartis Sverige AB Tel: +46 8 732 32 00 |
Latvia SIA Novartis Baltics Tel: +371 67 887 070 |

Date of the most recent review of this leaflet:
Other sources of information
This leaflet is available in audio file format and in large print on the website: http://www.voretigeneneparvovec.support
Detailed information on this medicine is available on the European Medicines Agency website: http://www.ema.europa.eu.
This information is intended for healthcare professionals only:
Precautions to be taken before handling or administering the medicine
This medicine contains genetically modified organisms. Personal protective equipment (including laboratory coat, safety goggles, and gloves) must be used when preparing or administering voretigene neparvovec.
Intraocular pressure should be adequately controlled and monitored before and after administration of the medicine.
After administration, patients should be advised to immediately report any symptoms suggestive of endophthalmitis or retinal detachment and should be appropriately managed.
Preparation prior to administration
Each package containing 1 vial of concentrate and 2 vials of solvent is for single use only.
Luxturna must be visually inspected before administration. If particles, cloudiness, or discoloration are observed, the single-dose vial must not be used.
Preparation of Luxturna must be performed within 4 hours prior to the start of the administration procedure, under aseptic conditions and according to the following recommended procedure.
Allow one single-dose vial of concentrate and two vials of solvent to thaw at room temperature. Once the three vials (1 concentrate vial and 2 solvent vials) are thawed, dilution should be initiated. Gently invert the vials five times to mix the contents.
Visually inspect for any visible particles or abnormalities. Any observed abnormality or visible particles must be reported to the Marketing Authorization Holder, and the product must not be used.
Transfer 2.7 ml of solvent from the two thawed vials and dispense using a 3 ml syringe into a sterile 10 ml glass vial.
For dilution, withdraw 0.3 ml of thawed concentrate using a 1 ml syringe and add to the sterile 10 ml vial containing the solvent. Gently invert the vial at least five times to ensure adequate mixing. Inspect for visible particles. The diluted solution should be clear or slightly opalescent. Label the 10 ml glass vial containing the diluted concentrate as follows: "Diluted Luxturna".
Syringes must not be prepared if the vial shows any damage or if visible particles are observed. Prepare injection syringes by withdrawing 0.8 ml of the diluted solution into a sterile 1 ml syringe. Repeat the same procedure to prepare a spare syringe. The filled syringes must be transported to the operating room in a designated container.
Measures to be taken in case of accidental exposure
Accidental exposure must be avoided. Local biosafety regulations must be followed for the preparation, administration, and handling of voretigene neparvovec.
- Personal protective equipment (including laboratory coat, safety goggles, and gloves) must be worn when handling or administering voretigene neparvovec.
- Accidental exposure to voretigene neparvovec, including contact with skin, eyes, and mucous membranes, must be avoided. Any open wounds should be covered before handling this medicine.
- Any spillage of voretigene neparvovec should be treated with a virucidal agent, such as 1% sodium hypochlorite, and dried with absorbent materials.
- All materials that may have come into contact with voretigene neparvovec (e.g., vial, syringe, needle, cotton swabs, gloves, masks, or dressings) must be disposed of in accordance with local biosafety regulations.
Accidental exposure
- In case of accidental occupational exposure (e.g., splashes into the eyes or onto mucous membranes), rinse thoroughly with clean water for at least 5 minutes.
- In case of exposure to broken skin or needle-stick injury, the affected area should be thoroughly cleaned with water and soap and/or a disinfectant.
Precautions to be taken for disposal of the medicine
This medicine contains genetically modified organisms. Disposal of unused medicine and of all materials that have come into contact with it must be carried out in accordance with local pharmaceutical waste regulations.
Dosage
Treatment must be initiated and administered by a retinal surgeon experienced in macular surgery.
Patients will receive a single dose of voretigene neparvovec of 1.5 × 10¹¹ vector genomes in each eye. Each dose will be administered into the subretinal space in a total volume of 0.3 ml. Administration must be performed individually in each eye on separate days, with a minimum interval of at least six days between each surgical procedure.
Immunomodulatory regimen
Prior to initiating the immunomodulatory regimen and prior to administration of voretigene neparvovec, patients must be examined for signs of active infectious disease of any kind, and if such infection is present, treatment initiation must be postponed until after the patient has recovered.
It is recommended to initiate the immunomodulatory regimen 3 days before administration of voretigene neparvovec in the first eye, following the schedule described below (Table 1). The immunomodulatory regimen for the second eye should follow the same schedule and replace the regimen for the first eye.
Table 1 Pre- and post-operative immunomodulatory regimen for each eye
Preoperative |
3 days before administration of Luxturna |
Prednisone (or equivalent) 1 mg/kg/day (up to a maximum of 40 mg/day) |
Postoperative |
4 days (including day of administration) |
Prednisone (or equivalent) 1 mg/kg/day (up to a maximum of 40 mg/day) |
Continue for 5 days |
Prednisone (or equivalent) 0.5 mg/kg/day (up to a maximum of 20 mg/day) |
|
Continue for 5 days with alternate-day dosing |
Prednisone (or equivalent) 0.5 mg/kg every other day (up to a maximum of 20 mg/day) |
Special populations
Elderly patients
The safety and efficacy of voretigene neparvovec in patients ≥ 65 years of age have not been established. Data are limited. However, no dose adjustment is required in elderly patients.
Hepatic and renal impairment
The safety and efficacy of voretigene neparvovec in patients with hepatic or renal impairment have not been established. No dose adjustment is necessary in these patients (see section 5.2).
Paediatric population
The safety and efficacy of voretigene neparvovec in children under 4 years of age have not been established. Data are limited. No dose adjustment is required in paediatric patients.
Method of administration
Subretinal use.
Luxturna is a sterile, concentrated solution for subretinal injection that requires thawing and dilution prior to administration.
This medicinal product must not be administered by intravitreal injection.
Luxturna is a single-use vial for a single administration in one eye. The product is administered via subretinal injection following a vitrectomy in each eye. It must not be administered close to the fovea in order to preserve foveal integrity.
The administration of voretigene neparvovec must be performed in an operating room under controlled aseptic conditions. Prior to the procedure, appropriate anaesthesia should be administered to the patient. The pupil of the eye to be injected must be dilated, and a broad-spectrum ophthalmic antibiotic should be administered prior to surgery in accordance with standard medical practice.
Administration
Follow the steps described below to administer voretigene neparvovec to patients:
- After dilution, Luxturna must be inspected visually before administration. If particulate matter, cloudiness, or discoloration is observed, the medicinal product must not be used.
- Connect the syringe containing the diluted product to the extension tube and the subretinal injection cannula. The product should be slowly injected through the extension tube and subretinal injection cannula to eliminate any air bubbles in the system.
- The volume of product available for injection should be confirmed in the syringe by aligning the tip of the plunger with the 0.3 ml mark.
- After completion of the vitrectomy, Luxturna is administered via subretinal injection using a subretinal injection cannula inserted through the pars plana.
- Under direct visualization, the tip of the subretinal injection cannula is placed in contact with the retinal surface. The recommended injection site should be located along the superior vascular arcades, at least 2 mm from the center of the fovea. A small amount of product is slowly injected until an initial subretinal bleb is observed, after which the remaining volume is slowly injected until the total volume of 0.3 ml is administered (Figure 1).
Figure 1 Tip of subretinal injection cannula placed at the recommended injection site (surgical view)
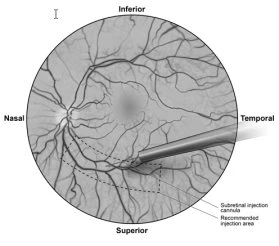
- At the end of the injection, the subretinal injection cannula is withdrawn from the eye.
- After injection, any unused product must be discarded. The spare syringe must not be stored.
- A careful fluid-to-air exchange should be performed, avoiding drainage of fluid near the retinotomy created for the subretinal injection.
- In the postoperative period, the patient’s head should be placed in a supine position immediately and maintained in this position for 24 hours after discharge.