Libertti®
Ucrania
Contenido
INSTRUCCIONES PARA USO MÉDICO DEL MEDICAMENTO LIBERATTI®
Composición:
Principios activos: etinilestradiol, drospirenona;
1 tableta recubierta con película (rosada) contiene: etinilestradiol 0,02 mg, drospirenona 3 mg;
Excipientes: lactosa monohidrato, almidón pregelatinizado (de maíz), povidona K-30, croscarmelosa sódica, polisorbato 80, estearato de magnesio; recubrimiento: Opadry® II rosa (alcohol polivinílico parcialmente hidrogenado, dióxido de titanio (E 171), macrogol 3350, talco (E 553b), óxido de hierro amarillo (E 172), óxido de hierro rojo (E 172), óxido de hierro negro (E 172));
1 tableta recubierta con película, placebo (blanca) contiene: lactosa anhidra, povidona K-30, estearato de magnesio; recubrimiento: Opadry® II blanco (alcohol polivinílico parcialmente hidrogenado, dióxido de titanio (E 171), macrogol 3350, talco (E 553b)).
Forma farmacéutica. Tabletas recubiertas con película.
Propiedades físico-químicas principales: tabletas redondas rosadas, recubiertas con película, sin grabado; placebo: tabletas redondas blancas, recubiertas con película, sin grabado.
Grupo farmacoterapéutico. Anticonceptivos hormonales para uso sistémico. Estrógenos y progestágenos en combinaciones fijas. Código ATC G03A A12.
Propiedades farmacológicas.
Farmacodinamia.
Índice Pearl de fracaso anticonceptivo: 0,41 (intervalo de confianza bilateral del 95 % superior (IC): 0,85). Índice Pearl total (fracasos anticonceptivos + errores del paciente): 0,80 (IC bilateral del 95 % superior: 1,30).
La acción anticonceptiva del medicamento Liberatti® se basa en la interacción de varios factores, siendo los más importantes la supresión de la ovulación y el cambio en la secreción cervical.
En un estudio clínico de 3 ciclos, al comparar la supresión de la ovulación de la combinación de drospirenona 3 mg/etinilestradiol 0,02 mg en régimen de 24 días frente al de 21 días, el régimen de 24 días se asoció con una mayor supresión del desarrollo folicular. Tras errores intencionados en la dosificación durante el tercer ciclo de tratamiento, la mayoría de las mujeres con régimen de 21 días mostraron actividad ovárica, incluyendo ovulación, en comparación con las mujeres con régimen de 24 días. La actividad ovárica regresó al nivel previo al inicio del tratamiento en el ciclo posterior a la terapia en el 91,8 % de las mujeres con régimen de 24 días.
El medicamento Liberatti® es un anticonceptivo oral combinado que contiene etinilestradiol y el progestágeno drospirenona. En dosis terapéuticas, la drospirenona presenta propiedades antiandrógenas y antimineralocorticoides moderadas. No posee actividad estrogénica, glucocorticoide ni antiglucocorticoide. Por lo tanto, la drospirenona tiene un perfil farmacológico similar al de la progesterona natural.
De acuerdo con datos de estudios clínicos, las propiedades antimineralocorticoides moderadas del fármaco producen un efecto antimineralocorticoide moderado.
Se realizaron dos estudios multicéntricos, doble ciego, aleatorizados y controlados con placebo para evaluar la eficacia y seguridad del fármaco en mujeres con acne vulgaris de grado moderado. Tras 6 meses de tratamiento, en comparación con placebo, el fármaco demostró un efecto estadísticamente significativo en la reducción del 15,6 % (49,3 % frente al 33,7 %) del número de lesiones inflamatorias, del 18,5 % (40,6 % frente al 22,1 %) del número de lesiones no inflamatorias y del 16,5 % (44,6 % frente al 28,1 %) del número total de lesiones. Asimismo, un mayor porcentaje de pacientes, del 11,8 % (18,6 % frente al 6,8 %), presentaron piel «clara» o «casi clara», evaluada mediante la escala ISGA (Evaluación Global del Investigador).
Farmacocinética.
Drospirenona
Absorción. La drospirenona administrada por vía oral se absorbe rápidamente y completamente. La concentración máxima (Cmax) en suero sanguíneo, de 38 ng/ml, se alcanza aproximadamente entre 1 y 2 horas tras la administración oral única. La biodisponibilidad es del 76-85 %. La ingestión simultánea de alimentos no afecta la biodisponibilidad de la drospirenona.
Distribución. Tras la administración oral, la concentración de drospirenona en suero sanguíneo disminuye con un periodo de semivida terminal medio de aproximadamente 31 horas. La drospirenona se une a la albúmina sérica, sin unirse a la globulina que une hormonas sexuales (SHBG) ni a la globulina que une corticoides (CBG). Solo el 3-5 % de su concentración total en suero sanguíneo se encuentra en forma libre. El aumento de SHBG inducido por el etinilestradiol no afecta la unión de la drospirenona a las proteínas séricas. El volumen medio de distribución de la drospirenona es de 3,7 ± 1,2 l/kg.
Metabolismo. La drospirenona se metaboliza ampliamente tras la administración oral. Los principales metabolitos en plasma son las formas ácidas de drospirenona, formadas por apertura del anillo lactónico, y el 4,5-dihidro-drospirenona-3-sulfato, formado por hidratación seguida de sulfatación. La drospirenona también es sustrato del metabolismo oxidativo catalizado por CYP3A4. In vitro, la drospirenona puede inhibir débil o moderadamente las enzimas del citocromo P450: CYP1A1, CYP2C9, CYP2C19 y CYP3A4.
Eliminación. La velocidad de aclaramiento metabólico de la drospirenona desde el suero sanguíneo es de aproximadamente 1,5 ± 0,2 ml/min/kg. La drospirenona se excreta en forma inalterada solo en cantidades muy pequeñas. Los metabolitos se excretan por orina y heces en una proporción de 1,2 a 1,4. El periodo de semivida de los metabolitos en orina y heces es de aproximadamente 40 horas.
Estado de equilibrio. Durante el ciclo de tratamiento, la concentración máxima en estado de equilibrio de drospirenona en suero sanguíneo, de aproximadamente 70 ng/ml, se alcanza tras 8 días de tratamiento. Los niveles de drospirenona en sangre se acumulan 3 veces como consecuencia de la relación entre el periodo de semivida terminal y el intervalo de dosificación.
Grupos especiales de pacientes
Mujeres con alteraciones de la función renal. La concentración en estado de equilibrio de drospirenona en suero sanguíneo en mujeres con insuficiencia renal leve (aclaramiento de creatinina de 50-80 ml/min) fue comparable a la observada en mujeres con función renal normal (aclaramiento de creatinina > 80 ml/min). El nivel de drospirenona en suero sanguíneo fue en promedio un 37 % más alto en mujeres con insuficiencia renal moderada (aclaramiento de creatinina de 30-50 ml/min) en comparación con mujeres con función renal normal. El uso de drospirenona demostró una buena tolerabilidad en todos los grupos de pacientes. Se ha demostrado que la administración de drospirenona no tiene un efecto clínicamente significativo sobre la concentración de potasio en suero sanguíneo.
Mujeres con alteraciones de la función hepática.
En un estudio con dosis única, el aclaramiento de drospirenona por vía oral se redujo aproximadamente en un 50 % en pacientes con insuficiencia hepática moderada en comparación con voluntarios adultos sanos con función hepática normal. La alteración observada en el aclaramiento de drospirenona en pacientes con insuficiencia hepática moderada no provocó diferencias evidentes respecto a la concentración de potasio en suero sanguíneo. Incluso en presencia de diabetes mellitus y tratamiento concomitante con espironolactona (dos factores que pueden provocar hiperkalemia), no se observó un aumento de la concentración de potasio en suero sanguíneo por encima del límite superior normal (LSN). Por lo tanto, la drospirenona es bien tolerada por pacientes con insuficiencia hepática leve y moderada (clase B según la clasificación de Child-Pugh).
Grupos étnicos. No se observaron diferencias clínicamente relevantes en la farmacocinética de drospirenona o etinilestradiol entre mujeres de nacionalidad japonesa y europeas.
Etinilestradiol
Absorción. Tras la administración oral, el etinilestradiol se absorbe rápidamente y completamente. La concentración máxima en suero, de 33 pg/ml, se alcanza entre 1 y 2 horas tras la administración oral única. La biodisponibilidad absoluta, debido a la conjugación presistémica y al metabolismo en el primer paso hepático, es de aproximadamente el 60 %. La ingestión simultánea de alimentos reduce la biodisponibilidad del etinilestradiol en aproximadamente el 25 % de los sujetos estudiados, sin cambios en el resto.
Distribución. El nivel de etinilestradiol en suero sanguíneo disminuye de forma bifásica, con una fase terminal que presenta un periodo de semivida de aproximadamente 24 horas. El etinilestradiol se une fuertemente, aunque de forma no específica, a las albúminas séricas (aproximadamente el 98,5 %) e induce un aumento en la concentración de SHBG y CBG en suero sanguíneo. El volumen aparente de distribución es de aproximadamente 5 l/kg.
Metabolismo. El etinilestradiol se metaboliza ampliamente en el tracto gastrointestinal (TGI) y durante el primer paso hepático. El etinilestradiol se metaboliza principalmente mediante hidroxilación del anillo aromático, formando un amplio espectro de metabolitos hidroxilados y metilados, presentes tanto en forma libre como conjugados con glucurónidos y sulfatos. El aclaramiento metabólico del etinilestradiol es de aproximadamente 5 ml/min/kg.
In vitro, el etinilestradiol es un inhibidor reversible de CYP2C19, CYP1A1 y CYP1A2, y también, según su mecanismo de acción, un inhibidor de CYP3A4/5, CYP2C8 y CYP2J2.
Eliminación. El etinilestradiol casi no se excreta en forma inalterada. Los metabolitos del etinilestradiol se excretan por orina y bilis en una proporción de 4:6. El periodo de semivida de los metabolitos es de casi 1 día.
Estado de equilibrio. El estado de equilibrio se alcanza en la segunda mitad del ciclo de tratamiento, momento en el cual la concentración de etinilestradiol en suero sanguíneo aumenta entre 2,0 y 2,3 veces.
Datos preclínicos de seguridad
En animales de laboratorio, los efectos de la drospirenona y el etinilestradiol se limitaron a los asociados con la acción farmacológica conocida. En particular, los estudios de toxicidad reproductiva en animales mostraron efectos embriotóxicos y fetotóxicos específicos de especie. Con exposiciones superiores a las de los usuarios del fármaco, en algunas especies animales se observaron efectos sobre la diferenciación sexual. Los estudios de evaluación del riesgo ecológico mostraron que el etinilestradiol y la drospirenona podrían representar una amenaza potencial para el medio acuático (ver sección «Precauciones especiales de eliminación»).
Características clínicas.
Indicaciones.
Anticoncepción oral.
Contraindicaciones.
No se deben utilizar anticonceptivos hormonales combinados (AHC) si existe al menos una de las condiciones siguientes. Si cualquiera de estas condiciones aparece por primera vez durante el tratamiento con AHC, el medicamento debe suspenderse inmediatamente.
- Presencia o riesgo de tromboembolismo venoso (TEV):
- TEV actual, incluyendo tratamiento con anticoagulantes, o antecedentes (por ejemplo, trombosis venosa profunda [TVP] o embolia pulmonar [EP]);
- predisposición hereditaria o adquirida conocida a TEV, tal como resistencia a la proteína C activada (incluyendo la mutación del factor V de Leiden), deficiencia de antitrombina-III, deficiencia de proteína C, deficiencia de proteína S;
- intervenciones quirúrgicas mayores con inmovilización prolongada (ver sección «Precauciones especiales»);
- alto riesgo de TEV debido a la presencia de múltiples factores de riesgo (ver sección «Precauciones especiales»).
- Presencia o riesgo de tromboembolismo arterial (TEA):
- TEA actual o antecedentes (por ejemplo, infarto de miocardio) o síntomas precursores (por ejemplo, angina de pecho);
- accidente cerebrovascular actual o antecedentes, o síntomas precursores (por ejemplo, ataque isquémico transitorio [AIT]);
- predisposición hereditaria o adquirida conocida a TEA, tal como hiperhomocisteinemia o anticuerpos contra fosfolípidos (anticuerpos contra cardiolipina, anticoagulante lúpico);
- antecedentes de migraña con síntomas neurológicos focales;
- alto riesgo de TEA debido a la presencia de múltiples factores de riesgo (ver sección «Precauciones especiales») o a la presencia de un solo factor de riesgo grave, como:
- diabetes mellitus con complicaciones vasculares;
- hipertensión arterial grave;
- dislipoproteinemia grave.
- Enfermedad hepática grave actual o antecedentes, mientras los índices de función hepática no hayan regresado a valores normales.
- Insuficiencia renal grave o insuficiencia renal aguda.
- Tumores hepáticos actuales o antecedentes (benignos o malignos).
- Tumores malignos conocidos o sospechosos (por ejemplo, de órganos genitales o glándulas mamarias) dependientes de hormonas sexuales.
- Hemorragia vaginal de etiología desconocida.
- Hipersensibilidad conocida a los principios activos o a cualquiera de los excipientes del medicamento.
El medicamento Liberatti® está contraindicado concomitantemente con medicamentos que contengan ombitasvir/paritaprevir/ritonavir, dasabuvir, glecaprevir/pibrentasvir y sofosbuvir/velpatasvir/voxilaprevir (ver secciones «Interacción con otros medicamentos y otras formas de interacción» y «Precauciones especiales»).
Precauciones especiales.
Este medicamento puede representar un peligro para el medio ambiente (ver sección «Propiedades farmacológicas»). Cualquier medicamento no utilizado o residuos deben eliminarse de acuerdo con los requisitos locales.
Interacción con otros medicamentos y otras formas de interacción.
Se debe consultar la información del medicamento que se administra concomitantemente para detectar posibles interacciones.
- Efecto de otros medicamentos sobre Liberatti®
Pueden producirse interacciones con medicamentos que inducen enzimas microsomales. Esto provocará un aumento en el aclaramiento de hormonas sexuales, lo que a su vez puede causar alteraciones en el patrón de sangrado menstrual y/o pérdida de eficacia anticonceptiva.
Tratamiento
La inducción enzimática puede observarse tras varios días de tratamiento. La máxima inducción enzimática generalmente se alcanza tras varias semanas. Tras la interrupción del tratamiento, la inducción enzimática puede persistir aproximadamente 4 semanas.
Tratamiento a corto plazo
Las mujeres que toman medicamentos que inducen enzimas deben utilizar temporalmente un método de barrera u otro método anticonceptivo adicional al anticonceptivo oral combinado (AOC). El método de barrera debe utilizarse durante todo el período de tratamiento con el medicamento inductor y durante 28 días adicionales tras la suspensión del tratamiento. Si el tratamiento comienza durante el período de toma de las últimas tabletas del envase de AOC, se debe iniciar inmediatamente la toma de tabletas del siguiente envase de AOC sin el intervalo habitual sin tabletas.
Tratamiento a largo plazo
A las mujeres sometidas a tratamiento prolongado con sustancias que inducen enzimas hepáticas se recomienda utilizar un método anticonceptivo de barrera u otro método anticonceptivo no hormonal adecuado.
Las siguientes interacciones se han documentado según datos publicados.
Sustancias activas que aumentan el aclaramiento de AOC (disminución de la eficacia de AOC por inducción enzimática), por ejemplo:
barbitúricos, bosentán, carbamazepina, fenitoína, primidona, rifampicina; medicamentos utilizados en infección por VIH: ritonavir, nevirapina y efavirenz; también posiblemente felbamato, griseofulvina, oxcarbazepina, topiramato y medicamentos a base de plantas que contienen extracto de hipérico (Hypericum perforatum).
Sustancias activas con efecto variable sobre el aclaramiento de AOC
La administración concomitante de AOC con numerosas combinaciones de inhibidores de la proteasa del VIH e inhibidores no nucleósidos de la transcriptasa inversa, incluyendo combinaciones con inhibidores del virus de la hepatitis C (VHC), puede aumentar o disminuir la concentración plasmática de estrógenos o progestágenos. El efecto combinado de estos cambios puede ser clínicamente relevante en algunos casos.
Por lo tanto, para detectar posibles interacciones y cualquier otra recomendación, se debe consultar la información sobre el uso médico del medicamento para el tratamiento de VIH/VHC que se administra concomitantemente. En caso de dudas, las mujeres deben utilizar adicionalmente un método de barrera durante el tratamiento con inhibidores de proteasa o inhibidores no nucleósidos de la transcriptasa inversa.
Sustancias activas que disminuyen el aclaramiento de AOC (inhibidores enzimáticos)
La relevancia clínica de la interacción potencial con inhibidores enzimáticos sigue sin aclararse.
La administración concomitante de inhibidores potentes de CYP3A4 puede aumentar las concentraciones plasmáticas de estrógeno, progestágeno o de ambos componentes.
En un estudio con dosis múltiples de la combinación drospirenona (3 mg/día)/etinilestradiol (0,002 mg/día) y el inhibidor potente de CYP3A4 ketoconazol, administrado concomitantemente durante 10 días, el valor de AUC(0-24h) de drospirenona y etinilestradiol aumentó 2,7 y 1,4 veces, respectivamente.
Etoricoxib en dosis de 60 mg/día a 120 mg/día demostró un aumento de las concentraciones plasmáticas de etinilestradiol de 1,4 a 1,6 veces, respectivamente, cuando se administró concomitantemente con un anticonceptivo hormonal combinado (AHC) que contiene 0,035 mg de etinilestradiol.
- Efecto del medicamento Liberatti® sobre otros medicamentos
Los AOC pueden afectar el metabolismo de ciertas sustancias activas. Por consiguiente, la concentración en plasma y tejidos puede aumentar (por ejemplo, ciclosporina) o disminuir (por ejemplo, lamotrigina).
Basándose en estudios de interacción in vivo en mujeres voluntarias que utilizaron omeprazol, simvastatina y midazolam como sustratos marcadores, se ha establecido que es poco probable una interacción clínicamente relevante de la drospirenona a la dosis de 3 mg con otras sustancias activas metabolizadas por el citocromo P450.
Datos clínicos indican que el etinilestradiol inhibe el aclaramiento de sustratos de CYP1A2, lo que a su vez provoca un aumento leve (por ejemplo, teofilina) o moderado (por ejemplo, tizanidina) de sus concentraciones plasmáticas.
Interacciones farmacodinámicas
La administración concomitante con medicamentos que contienen ombitasvir/paritaprevir/ritonavir y dasabuvir con o sin ribavirina, glecaprevir/pibrentasvir y sofosbuvir/velpatasvir/voxilaprevir aumenta el riesgo de elevación de los niveles de alanina aminotransferasa (ALT) (ver secciones «Contraindicaciones» y «Precauciones especiales»).
Por lo tanto, las mujeres que toman Liberatti® deben pasar temporalmente a un método anticonceptivo alternativo (por ejemplo, anticonceptivos que contengan solo progestágeno o métodos no hormonales) antes de iniciar el tratamiento con esta combinación de medicamentos. El uso de Liberatti® puede reanudarse 2 semanas después de finalizar el tratamiento con dicha combinación.
En pacientes con función renal normal, la administración concomitante de drospirenona con inhibidores de la enzima convertidora de angiotensina (ECA) o con antiinflamatorios no esteroideos (AINE) no mostró un efecto significativo sobre los niveles séricos de potasio. Sin embargo, no se ha estudiado la administración concomitante de Liberatti® con antagonistas de la aldosterona o diuréticos ahorradores de potasio. En este caso, se debe evaluar el nivel de potasio sérico durante el primer ciclo de tratamiento (ver también sección «Precauciones especiales»).
Otras formas de interacción
Análisis de laboratorio
La administración de esteroides anticonceptivos puede influir en los resultados de ciertos análisis de laboratorio, tales como parámetros bioquímicos de la función hepática, tiroides, glándulas suprarrenales y riñón, en la concentración en plasma de proteínas transportadoras, como el glucocorticosteroide unidor, en la concentración en plasma de fracciones de lípidos/lipoproteínas, en los parámetros del metabolismo de carbohidratos, coagulación y fibrinólisis. Habitualmente, estos cambios permanecen dentro de los límites normales. La drospirenona aumenta la actividad de renina y aldosterona en plasma, inducida por su actividad anti-mineralocorticoide moderada.
Características de uso.
La decisión de prescribir el medicamento Liberatti® debe tomarse considerando los factores de riesgo individuales actuales de la mujer, incluyendo los factores de riesgo de desarrollar TEV, así como el riesgo de TEV asociado al uso de Liberatti® en comparación con otros ACO (ver secciones «Contraindicaciones» y «Características de uso»).
Advertencia
En caso de presentarse cualquiera de los estados o factores de riesgo indicados a continuación, se debe discutir con la mujer la conveniencia del uso del medicamento Liberatti®.
Si se produce una exacerbación o las primeras manifestaciones de cualquiera de los estados o factores de riesgo mencionados, se recomienda que la mujer consulte a su médico para determinar la necesidad de suspender el uso del medicamento Liberatti®.
En caso de sospecha o confirmación de TEV o TAE, se debe suspender el uso de los ACO. Si se inicia un tratamiento anticoagulante, se debe proporcionar un método anticonceptivo alternativo adecuado debido al efecto teratogénico de los anticoagulantes (cumarinas).
- Trastornos circulatorios
Riesgo de tromboembolismo venoso (TEV)
El uso de cualquier ACO aumenta el riesgo de TEV en las mujeres que los toman, en comparación con las mujeres que no los usan. Los medicamentos que contienen levonorgestrel, norgestimato o noretisterona se asocian con un riesgo menor de TEV. El uso de otros medicamentos, como Liberatti®, puede duplicar el riesgo. La decisión de usar medicamentos distintos de los de menor riesgo de TEV debe tomarse únicamente tras discutirlo con la mujer. Es necesario asegurarse de que la paciente comprenda el riesgo de TEV asociado al uso de Liberatti®, el impacto de sus factores de riesgo individuales y el hecho de que el riesgo de TEV es más alto durante el primer año de uso. Algunos datos indican que el riesgo de TEV puede aumentar al reanudar el uso de un ACO tras una interrupción de 4 semanas o más.
En dos de cada 10.000 mujeres que no toman ACO y que no están embarazadas, se desarrolla un TEV durante un año. Sin embargo, en cada mujer individual el riesgo puede ser considerablemente mayor dependiendo de los factores de riesgo que presente (ver más abajo).
Se ha establecido1 que de cada 10.000 mujeres que usan ACO que contienen drospirenona, entre 9 y 12 desarrollarán TEV durante un año. Esto se compara con una tasa de 6 casos entre mujeres que usan ACO que contienen levonorgestrel.
En ambos casos, el número de casos de TEV por año fue menor que el habitualmente esperado durante el embarazo o en el período posparto.
El TEV puede tener consecuencias fatales en el 1-2 % de los casos.
Número de casos de TEV por cada 10.000 mujeres en un año
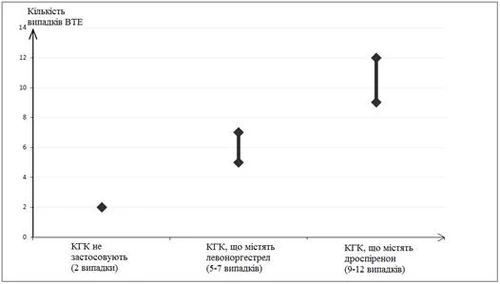
1 Estos valores se obtuvieron a partir de todos los datos de estudios epidemiológicos, considerando los riesgos relativos asociados al uso de diferentes ACO, en comparación con el uso de ACO que contienen levonorgestrel.
2 Promedio de 5-7 casos por 10.000 mujeres-año, basado en el cálculo del riesgo relativo del uso de ACO que contienen levonorgestrel, en comparación con mujeres que no usan ACO (aproximadamente 2,3 - 3,6 casos).
Factores de riesgo de desarrollo de TEV
El riesgo de complicaciones tromboembólicas venosas en mujeres que usan ACO puede ser considerablemente mayor si existen factores de riesgo adicionales, especialmente si son múltiples (ver Tabla 1).
El uso del medicamento Liberatti® está contraindicado en mujeres con múltiples factores de riesgo que puedan aumentar el riesgo de trombosis venosa (ver sección «Contraindicaciones»). Si una mujer tiene más de un factor de riesgo, el aumento del riesgo puede ser mayor que la suma de los riesgos asociados a cada factor individual, por lo que debe considerarse el riesgo global de TEV. Si la relación riesgo-beneficio es desfavorable, no se deben prescribir ACO (ver sección «Contraindicaciones»).
Tabla 1
Factores de riesgo de desarrollo de TEV
| Factor de riesgo |
Nota |
| Obesidad (índice de masa corporal superior a 30 kg/m²) |
El riesgo aumenta considerablemente con el aumento del índice de masa corporal. Requiere especial atención en presencia de otros factores de riesgo. |
| Inmovilización prolongada, intervención quirúrgica mayor, cirugía en extremidades inferiores u órganos pélvicos, intervenciones neuroquirúrgicas o traumatismo grave. Nota: la inmovilización temporal, incluyendo vuelos > 4 horas, también puede ser un factor de riesgo para el desarrollo de TEV, especialmente en mujeres con otros factores de riesgo. |
En tales situaciones se recomienda suspender el uso del medicamento (en caso de cirugía programada, al menos 4 semanas antes) y no reiniciar su uso antes de 2 semanas tras la recuperación completa de la movilidad. Con el fin de evitar un embarazo no deseado, deben utilizarse otros métodos anticonceptivos. Debe considerarse la conveniencia de una terapia antitrombótica si no se ha suspendido previamente el uso del medicamento Libératti®. |
| Antecedentes familiares (TEV en algún familiar cercano o en los padres, especialmente a edad relativamente joven, por ejemplo antes de los 50 años). |
En caso de predisposición hereditaria, se recomienda a las mujeres consultar con un especialista antes de iniciar cualquier ACO. |
| Otras condiciones asociadas con TEV |
Cáncer, lupus eritematoso sistémico, síndrome hemolítico-urémico, enfermedad inflamatoria intestinal crónica (enfermedad de Crohn o colitis ulcerosa) y anemia de células falciformes. |
| Edad |
Particularmente a partir de los 35 años |
No existe un consenso unánime sobre la posible influencia de la varicosis y la tromboflebitis superficial en el desarrollo y progresión del tromboembolismo venoso.
Debe tenerse en cuenta el riesgo aumentado de tromboembolismo durante el embarazo, especialmente durante las 6 semanas posteriores al parto (para obtener información sobre el embarazo o la lactancia, véase la sección «Uso durante el embarazo o la lactancia»).
Síntomas de ETV (trombosis venosa profunda y embolia pulmonar)
Se debe aconsejar a las mujeres que, ante la aparición de los síntomas descritos a continuación, acudan inmediatamente al médico y le informen de que están tomando un ACO.
Los síntomas de la TVP pueden incluir: hinchazón unilateral de la pierna y/o del pie o de una zona a lo largo de la vena en la pierna; dolor o mayor sensibilidad en la pierna, que puede notarse únicamente al estar de pie o al caminar; sensación de calor en la pierna afectada; enrojecimiento o cambio del color de la piel en la pierna.
Los síntomas de la EPA pueden incluir: dificultad repentina para respirar de etiología desconocida o respiración acelerada; tos repentina, posiblemente con sangre; dolor repentino en el pecho; sensación de desmayo o mareo; palpitaciones rápidas o irregulares.
Algunos de estos síntomas (por ejemplo, dificultad para respirar, tos) pueden ser inespecíficos o interpretarse erróneamente como fenómenos más comunes o menos graves (por ejemplo, infecciones respiratorias).
Otras manifestaciones de oclusión vascular pueden incluir dolor repentino, hinchazón, abdomen agudo y ligera cianosis de una extremidad.
En la oclusión de los vasos oculares, los síntomas iniciales pueden ser visión borrosa sin dolor, que puede progresar hasta la pérdida de la visión. A veces, la pérdida de la visión se desarrolla casi instantáneamente.
Riesgo de tromboembolismo arterial (ATE)
Según datos de estudios epidemiológicos, el uso de cualquier ACO se asocia con un riesgo aumentado de ATE (infarto de miocardio) o eventos cerebrovasculares (ACV, accidente isquémico transitorio, ictus). Los eventos tromboembólicos arteriales pueden tener consecuencias fatales.
Factores de riesgo para el desarrollo de ATE
Con el uso de ACO, el riesgo de complicaciones tromboembólicas arteriales o eventos cerebrovasculares aumenta en mujeres con factores de riesgo (véase la tabla 2). El uso del medicamento Libératti® está contraindicado si la mujer presenta un factor de riesgo grave o múltiples factores de riesgo que puedan aumentar el riesgo de trombosis arterial (véase la sección «Contraindicaciones»). Si la mujer tiene más de un factor de riesgo, el aumento del riesgo puede ser mayor que la suma de los riesgos asociados con cada factor individual, por lo que debe considerarse el riesgo global. Si la relación beneficio/riesgo es desfavorable, no se debe prescribir un ACO (véase la sección «Contraindicaciones»).
Tabla 2
Factores de riesgo para el desarrollo de ATE
| Factor de riesgo |
Observación |
| Aumento de la edad |
Particularmente a partir de los 35 años |
| Tabaquismo |
Se recomienda a las mujeres que utilizan ACO que eviten fumar. A las mujeres de 35 años o más que continúen fumando, se les recomienda encarecidamente utilizar otro método anticonceptivo. |
| Hipertensión arterial |
|
| Obesidad (índice de masa corporal superior a 30 kg/m²) |
El riesgo aumenta considerablemente con el incremento del índice de masa corporal. Requiere especial atención en caso de que la mujer presente otros factores de riesgo. |
| Antecedentes familiares (EPV en algún pariente o progenitor, especialmente a edades relativamente jóvenes, por ejemplo antes de los 50 años) |
En caso de predisposición hereditaria, se recomienda a las mujeres consultar con un especialista antes de iniciar cualquier ACO. |
| Migraña |
El aumento de la frecuencia o de la gravedad de la migraña durante el uso de ACO (posibles síntomas prodromales previos a eventos cerebrovasculares) puede ser motivo para la interrupción inmediata del ACO. |
| Otras afecciones relacionadas con reacciones adversas vasculares |
Diabetes mellitus, hiperhomocisteinemia, valvulopatías cardíacas, fibrilación auricular, dislipoproteinemia y lupus eritematoso sistémico. |
Síntomas de ETA
Se debe aconsejar a las mujeres que, si aparecen los síntomas descritos a continuación, consulten inmediatamente a su médico y le informen de que están tomando un ACO.
Los síntomas de un trastorno cerebrovascular pueden incluir: entumecimiento repentino de la cara, debilidad u hormigueo en los brazos o piernas, especialmente en un solo lado del cuerpo; dificultad repentina para caminar, mareo, pérdida del equilibrio o de la coordinación; confusión repentina, alteración del habla o de la comprensión; empeoramiento repentino de la visión en uno o ambos ojos; dolor de cabeza repentino, intenso o prolongado sin causa aparente; pérdida de conciencia o desmayo con o sin convulsiones.
La naturaleza transitoria de los síntomas puede indicar un accidente isquémico transitorio (AIT).
Los síntomas de infarto de miocardio pueden incluir: dolor, molestia, opresión o pesadez en el pecho, brazo o por debajo del esternón; sensación de molestia que se irradia a la espalda, mandíbula, garganta, brazo o estómago; sensación de plenitud, indigestión o acidez gástrica; sudoración excesiva, náuseas, vómitos o mareo; debilidad extrema, ansiedad o dificultad para respirar; latidos cardíacos rápidos o irregulares.
Tumores
Los resultados de algunos estudios epidemiológicos sugieren un aumento adicional del riesgo de cáncer cervical con el uso prolongado de ACO (> 5 años), aunque esta afirmación sigue siendo controvertida, ya que no se ha determinado completamente si los resultados de los estudios tienen en cuenta factores de riesgo concomitantes, como el comportamiento sexual y otros factores, por ejemplo, la infección por el virus del papiloma humano.
Un metaanálisis basado en 54 estudios epidemiológicos indica un ligero aumento del riesgo relativo (RR = 1,24) de cáncer de mama en mujeres que usan ACO. Este riesgo aumentado disminuye gradualmente durante los 10 años posteriores a la interrupción del uso de ACO. Dado que el cáncer de mama es raro en mujeres menores de 40 años, el aumento en el número de casos diagnosticados en mujeres que actualmente usan o recientemente han usado ACO es insignificante en comparación con el riesgo general de cáncer de mama. Los resultados de estos estudios no proporcionan pruebas de una relación causal. El aumento del riesgo puede deberse tanto a una detección más temprana del cáncer de mama en mujeres que usan ACO como a un efecto biológico de los ACO, o a una combinación de ambos factores. Se ha observado una tendencia a que el cáncer de mama detectado en mujeres que alguna vez han tomado ACO sea clínicamente menos agresivo que en aquellas que nunca han usado ACO.
En casos aislados, se han observado en mujeres que usan ACO tumores hepáticos benignos y, más raramente, malignos, que en algunos casos han provocado hemorragia intraabdominal potencialmente mortal. En caso de presentación de quejas de dolor intenso en la región epigástrica, aumento del tamaño del hígado o signos de hemorragia intraabdominal, se debe considerar la posibilidad de un tumor hepático asociado al uso de ACO en el diagnóstico diferencial.
El uso de ACO en dosis altas (50 µg de etinilestradiol) reduce el riesgo de cáncer de endometrio y de ovario. Aún queda por confirmar si estos datos son aplicables también a los ACO de baja dosis.
Otras afecciones
El componente progestágeno del medicamento Libératti® es un antagonista de la aldosterona con propiedades conservadoras de potasio. En la mayoría de los casos, no se espera un aumento significativo del nivel de potasio. Durante los estudios clínicos, en algunos pacientes con insuficiencia renal leve a moderada y que tomaban simultáneamente medicamentos conservadores de potasio, se observó un ligero aumento de los niveles séricos de potasio durante el tratamiento con drospirenona. Por lo tanto, se recomienda el control del nivel de potasio durante el primer ciclo de tratamiento en pacientes con insuficiencia renal. A estos pacientes también se les recomienda mantener el nivel de potasio en suero por debajo del límite superior normal antes de iniciar el tratamiento, especialmente si se usan simultáneamente medicamentos conservadores de potasio (véase la sección «Interacción con otros medicamentos y otras formas de interacción»).
Las mujeres con hipertrigliceridemia o antecedentes familiares de esta alteración constituyen un grupo de riesgo para el desarrollo de pancreatitis durante el uso de ACO.
Aunque se ha informado de un ligero aumento de la presión arterial en muchas mujeres que toman ACO, un aumento clínicamente significativo de la presión arterial se observa solo en casos aislados. La interrupción inmediata del ACO solo es necesaria en estos casos aislados. En caso de hipertensión arterial prolongada o imposibilidad de controlar la presión arterial con medicamentos antihipertensivos, se debe interrumpir el uso de ACO en las mujeres que los toman. Si es apropiado, el uso de ACO puede reanudarse tras alcanzar la normotensión mediante tratamiento antihipertensivo.
Se ha informado de la aparición o empeoramiento de las siguientes afecciones durante el embarazo y con el uso de ACO, aunque su relación con el uso de estrógenos/progestágenos no está completamente aclarada: ictericia y/o prurito asociados con colestasis, formación de cálculos biliares, porfiria, lupus eritematoso sistémico, síndrome hemolítico-urémico, corea de Sydenham, herpes gestacional, pérdida de audición asociada con otosclerosis.
Los estrógenos exógenos pueden provocar la aparición o empeoramiento de síntomas de edema angioneurótico hereditario o adquirido.
Los trastornos agudos o crónicos de la función hepática pueden requerir la interrupción del uso de ACO hasta que los indicadores de función hepática regresen a la normalidad y se excluya una relación causal con el ACO.
Debe interrumpirse el uso de ACO en caso de recurrencia de ictericia colestásica y/o prurito asociado con colestasis que haya ocurrido previamente durante el embarazo o con el uso previo de hormonas sexuales.
Aunque los ACO pueden afectar la resistencia periférica a la insulina y la tolerancia a la glucosa, no hay datos que indiquen la necesidad de modificar el régimen terapéutico en mujeres diabéticas que toman ACO de baja dosis (< 0,05 mg de etinilestradiol). Sin embargo, las mujeres con diabetes deben ser examinadas cuidadosamente durante el uso de ACO, especialmente al inicio del tratamiento.
También se han observado casos de empeoramiento de la depresión endógena, epilepsia, enfermedad de Crohn y colitis ulcerosa durante el uso de ACO.
El estado de ánimo deprimido y la depresión son reacciones adversas bien conocidas que pueden ocurrir durante el uso de anticonceptivos hormonales (véase la sección «Reacciones adversas»). La depresión puede ser un estado grave y es un factor de riesgo bien conocido de conducta suicida y suicidio. Se debe aconsejar a las mujeres que consulten a su médico ante cambios de ánimo y aparición de síntomas de depresión, incluso poco después del inicio del tratamiento.
A veces puede aparecer melasma, especialmente en mujeres con antecedentes de melasma gestacional. Las mujeres predispuestas al melasma deben evitar la exposición directa al sol o a la radiación ultravioleta durante el uso de ACO.
Las tabletas rosadas y blancas contienen monohidrato de lactosa. Esto debe tenerse en cuenta en caso de trastornos hereditarios raros de intolerancia a la galactosa, deficiencia de lactasa de Lapp o malabsorción de glucosa-galactosa, o si se sigue una dieta sin lactosa. Si se tiene intolerancia a ciertos azúcares, se debe consultar al médico antes de tomar este medicamento.
Aumento de los niveles de ALT
Durante los estudios clínicos en pacientes que recibían tratamiento para la hepatitis viral C con medicamentos que contienen ombitasvir/paritaprevir/ritonavir y dasabuvir, con o sin ribavirina, el aumento de los niveles de transaminasas (ALT) por encima de 5 veces el límite superior normal se observó con mayor frecuencia en mujeres que usaban medicamentos que contenían etinilestradiol, como los ACO. También se han observado aumentos de ALT con el uso de medicamentos antivirales contra la hepatitis C que contienen glecaprevir/pibrentasvir y sofosbuvir/velpatasvir/voxilaprevir (véase las secciones «Contraindicaciones» e «Interacción con otros medicamentos y otras formas de interacción»).
Consultas/exámenes médicos
Antes de iniciar o reanudar el uso del medicamento Libératti®, se recomienda obtener un historial médico completo (incluyendo antecedentes familiares), realizar un examen médico completo y descartar el embarazo. Es necesario medir la presión arterial y realizar un examen médico, teniendo en cuenta las contraindicaciones (véase la sección «Contraindicaciones») y las precauciones especiales (véase la sección «Precauciones de uso»). Se debe informar a la mujer sobre la información relativa al tromboembolismo venoso y arterial, incluyendo el riesgo asociado con el uso de Libératti® en comparación con otros ACO, los síntomas de TEV y ETA, los factores de riesgo conocidos y las medidas que deben tomarse ante sospecha de trombosis.
Se recomienda a las pacientes leer cuidadosamente el prospecto del medicamento y seguir las recomendaciones que contiene.
La frecuencia y naturaleza de los exámenes deben basarse en las normas vigentes de la práctica médica, teniendo en cuenta las características individuales de cada mujer.
Se debe advertir a las pacientes que los anticonceptivos hormonales no protegen contra la infección por VIH (SIDA) ni contra ninguna otra enfermedad de transmisión sexual.
Reducción de la eficacia
La eficacia de los ACO puede reducirse si se omite la toma de una tableta (véase la sección «Posología y forma de administración»), en caso de trastornos gastrointestinales (véase la sección «Posología y forma de administración») o con la administración concomitante de otros medicamentos (véase la sección «Interacción con otros medicamentos y otras formas de interacción»).
Alteraciones del ciclo
Durante el uso de ACO pueden presentarse hemorragias irregulares (manchado o sangrado de escape), especialmente durante los primeros meses. Si estas hemorragias continúan después de tres ciclos menstruales, deben considerarse graves.
Si las secreciones sanguinolentas irregulares persisten o aparecen tras un período de sangrados regulares, deben considerarse causas no hormonales de hemorragia y realizarse las medidas diagnósticas adecuadas, incluyendo estudios para descartar tumores y embarazo. Entre las medidas diagnósticas puede incluirse una curetaje.
En algunas mujeres puede no ocurrir la hemorragia de privación durante el período de descanso en la toma del medicamento. Si los ACO se han tomado según las instrucciones de la sección «Posología y forma de administración», el embarazo es improbable. Sin embargo, si el ACO se ha tomado de forma irregular antes de la ausencia del primer sangrado de privación o si no hay sangrado de privación durante dos ciclos, debe descartarse el embarazo antes de continuar con el uso de ACO.
Uso durante el embarazo o la lactancia.
Embarazo. Este medicamento está contraindicado durante el embarazo.
Si se produce un embarazo durante el uso de Libératti®, su administración debe interrumpirse inmediatamente. Sin embargo, los resultados de estudios epidemiológicos no indican un aumento del riesgo de malformaciones congénitas en hijos de madres que tomaron ACO antes del embarazo, ni tampoco una acción teratogénica por la toma accidental de ACO durante el embarazo.
Los estudios en animales mostraron reacciones adversas durante el embarazo y la lactancia (véase la sección «Propiedades farmacológicas»). A partir de estos estudios en animales, no puede excluirse la posibilidad de reacciones adversas debidas al efecto hormonal de los principios activos. Sin embargo, la experiencia general con el uso de ACO durante el embarazo no indica un efecto adverso en humanos.
Los datos disponibles sobre el uso del medicamento durante el embarazo son demasiado limitados para poder concluir sobre un efecto negativo del medicamento Libératti® en el curso del embarazo, la salud del feto y del recién nacido. Hasta la fecha, no existen datos epidemiológicos adecuados.
Al reanudar el uso del medicamento Libératti®, debe tenerse en cuenta el aumento del riesgo de TEV durante el período posparto (véanse las secciones «Precauciones de uso» y «Posología y forma de administración»).
Período de lactancia. Los ACO pueden afectar la lactancia, ya que pueden reducir la cantidad de leche materna y alterar su composición. Por esta razón, no se recomienda tomar ACO durante la lactancia. Pequeñas cantidades de esteroides anticonceptivos y/o sus metabolitos pueden pasar a la leche materna durante el uso de ACO. Estas cantidades pueden afectar al niño.
Fertilidad. El medicamento Libératti® está indicado para la prevención del embarazo. Para obtener información sobre la recuperación de la fertilidad, véase la sección «Propiedades farmacológicas».
Capacidad para afectar la velocidad de reacción al conducir vehículos de motor o utilizar otras máquinas.
No se han realizado estudios sobre el efecto del medicamento Libératti® sobre la velocidad de reacción al conducir vehículos de motor o al trabajar con otras máquinas. No se ha observado ningún efecto sobre la capacidad para conducir automóviles o trabajar con maquinaria en mujeres que usan ACO.
Vía de administración y dosis.
Vía de administración: vía oral.
Las tabletas deben tomarse diariamente según el orden indicado en el blíster, aproximadamente a la misma hora, con una pequeña cantidad de líquido. El medicamento se toma a razón de 1 tableta al día durante 28 días consecutivos. La toma de las tabletas de cada nuevo envase debe comenzar al día siguiente de finalizar el envase anterior.
El envase incluye un soporte para el blíster, en el que se puede colocar el blíster cuando sea necesario llevarlo consigo, así como una escala calendario.
El blíster contiene 24 tabletas con principios activos (tabletas rosadas) y 4 tabletas de placebo (tabletas blancas) que no contienen principios activos.
- Inicio del tratamiento con Liberatti®
Si no se han utilizado anticonceptivos hormonales previamente (mes anterior), se debe comenzar a tomar las tabletas el primer día del ciclo natural (es decir, el primer día del sangrado menstrual). También se puede comenzar entre el día 2 y 5 del ciclo, pero en este caso debe utilizarse un método anticonceptivo adicional (por ejemplo, barrera) durante los primeros 7 días de toma del medicamento.
- Cambio desde otro anticonceptivo oral combinado (AOC), anillo vaginal o parche transdérmico
Se recomienda comenzar a tomar las tabletas de Liberatti® al día siguiente de haber tomado la última tableta hormonal del AOC anterior, pero no más allá del día siguiente al finalizar el periodo de descanso o tras tomar las tabletas de placebo del AOC anterior. En caso de utilizar anillo vaginal anticonceptivo o parche transdérmico, se debe comenzar el tratamiento con Liberatti® el día de la retirada del dispositivo, pero no más allá del día en que debería aplicarse el siguiente uso de estos productos.
- Cambio desde un método basado únicamente en progestágenos («mini-píldora», inyecciones, implantes o sistema intrauterino con progestágeno)
Se puede comenzar el tratamiento con Liberatti® en cualquier momento tras dejar de tomar la «mini-píldora» (en caso de implante o sistema intrauterino, el día de su retirada; en caso de inyección, en lugar de la siguiente inyección). Sin embargo, en todos los casos se recomienda utilizar un método anticonceptivo de barrera adicional durante los primeros 7 días de toma del medicamento.
- Después de un aborto en el primer trimestre de gestación
Se puede comenzar a tomar Liberatti® inmediatamente. En este caso, no es necesario utilizar métodos anticonceptivos adicionales.
- Después del parto o aborto en el segundo trimestre de gestación
En caso de lactancia, véase la sección «Uso durante el embarazo o la lactancia». Se recomienda comenzar a tomar Liberatti® entre los días 21 y 28 tras el parto o aborto en el segundo trimestre de gestación. Si se comienza más tarde, se recomienda utilizar un método anticonceptivo de barrera adicional durante los primeros 7 días de toma de las tabletas. Sin embargo, si ya ha habido relaciones sexuales, antes de iniciar el tratamiento debe descartarse la posibilidad de embarazo o esperar al inicio de la primera menstruación.
Qué hacer en caso de olvidar tomar una tableta
Los olvidos de las tabletas de placebo pueden ignorarse. Sin embargo, deben retirarse del envase para evitar prolongar inadvertidamente el periodo de placebo. Las indicaciones siguientes se refieren únicamente al olvido de tomar tabletas rosadas que contienen principios activos.
Si el retraso en la toma de una tableta rosada no supera las 24 horas, la eficacia anticonceptiva del medicamento no se reduce. La tableta olvidada debe tomarse inmediatamente tan pronto como se recuerde. La siguiente tableta del envase debe tomarse a la hora habitual.
Si el retraso en la toma de una tableta rosada supera las 24 horas, la protección anticonceptiva puede disminuir. En este caso, deben seguirse dos reglas principales:
- La interrupción en la toma de tabletas no debe superar nunca los 4 días.
- La supresión adecuada del eje hipotálamo-hipófisis-ovario se logra con la toma continua de tabletas durante 7 días.
De acuerdo con esto, en la práctica diaria deben seguirse las siguientes recomendaciones:
- Día 1-7
Debe tomarse la última tableta olvidada lo antes posible, incluso si es necesario tomar dos tabletas simultáneamente. Después, se continúa tomando las tabletas a la hora habitual. Además, durante los siguientes 7 días debe utilizarse un método anticonceptivo de barrera, por ejemplo, condón. Si en los últimos 7 días ha habido relaciones sexuales, debe considerarse la posibilidad de embarazo. Cuantas más tabletas se hayan olvidado y más cercano esté el periodo de toma de las tabletas de placebo, mayor será el riesgo de embarazo.
- Día 8-14
Debe tomarse la última tableta olvidada lo antes posible, incluso si es necesario tomar dos tabletas simultáneamente. Después, se continúa tomando las tabletas a la hora habitual. Si la paciente ha tomado correctamente las tabletas durante los 7 días anteriores al olvido, no es necesario utilizar métodos anticonceptivos adicionales. Si no es así, o si se ha olvidado más de una tableta, se recomienda utilizar un método anticonceptivo de barrera adicional durante 7 días.
- Día 15-24
El riesgo de embarazo aumenta al acercarse al periodo de toma de las tabletas de placebo. Sin embargo, si se sigue correctamente el esquema de toma, puede evitarse la disminución de la protección anticonceptiva. Si se sigue una de las recomendaciones siguientes, no será necesario utilizar métodos anticonceptivos adicionales, siempre que se haya tomado correctamente el medicamento durante los 7 días previos al olvido. Si no es así, se recomienda seguir la primera de las recomendaciones siguientes y utilizar métodos anticonceptivos adicionales durante los siguientes 7 días.
- Debe tomarse la última tableta olvidada lo antes posible, incluso si es necesario tomar dos tabletas simultáneamente. Después, se continúa tomando las tabletas a la hora habitual hasta finalizar las tabletas rosadas con principios activos. No es necesario tomar las 4 tabletas de placebo. Debe comenzarse inmediatamente con las tabletas del siguiente envase tras tomar la última tableta rosada. Es poco probable que comience un sangrado similar a la menstruación antes de finalizar todas las tabletas rosadas del segundo envase, aunque durante la toma pueden presentarse sangrados intermenstruales o hemorragias de escape.
- También puede interrumpirse la toma de tabletas del envase actual. En este caso, la interrupción en la toma del medicamento debe ser de 4 días, incluyendo los días en que se olvidaron tabletas; la toma debe reanudarse con el siguiente envase.
Si tras olvidar tomar tabletas no se produce la menstruación esperada durante la primera pausa normal de toma del medicamento, debe considerarse la posibilidad de embarazo. Es necesario consultar al médico antes de comenzar a tomar las tabletas de un nuevo envase.
Recomendaciones en caso de trastornos gastrointestinales
En caso de trastornos gastrointestinales graves, puede producirse una absorción incompleta del medicamento. En este caso, debe utilizarse un método anticonceptivo adicional. Si los vómitos ocurren dentro de las 3-4 horas posteriores a la toma de la tableta de Liberatti®, deben seguirse las mismas recomendaciones que en caso de olvido de una dosis.
Cómo cambiar el momento de la menstruación o retrasarla
Para retrasar la menstruación, debe continuarse la toma de tabletas con principios activos (rosadas) de Liberatti® del nuevo envase sin hacer pausa en la toma de tabletas hormonales. Si se desea, el periodo de toma puede prolongarse hasta finalizar las tabletas rosadas del segundo envase. Durante este tiempo, pueden presentarse hemorragias de escape o sangrados intermenstruales. Normalmente, la toma de Liberatti® se reanuda tras tomar las tabletas de placebo, que no contienen hormonas.
Para cambiar el día de la semana en que comienza la menstruación, se recomienda acortar el periodo de descanso sin tomar tabletas hormonales (tabletas de placebo) tantos días como se desee. Debe tenerse en cuenta que cuanto más corta sea la pausa, más frecuentemente puede ocurrir ausencia de sangrado menstrual, así como hemorragias de escape o sangrados intermenstruales durante la toma de tabletas del segundo envase (como en el caso de retraso de la menstruación).
Información adicional sobre grupos especiales de pacientes
Pacientes de edad avanzada. El medicamento Liberatti® no está indicado para su uso tras la menopausia.
Pacientes con insuficiencia hepática. El medicamento Liberatti® está contraindicado en mujeres con insuficiencia hepática grave (véanse las secciones «Propiedades farmacológicas» y «Contraindicaciones»).
Pacientes con insuficiencia renal. El medicamento Liberatti® está contraindicado en mujeres con insuficiencia renal grave o insuficiencia renal aguda (véanse las secciones «Propiedades farmacológicas» y «Contraindicaciones»).
Niños.
Este medicamento está indicado para su uso bajo prescripción médica únicamente tras la aparición de menstruaciones regulares.
Sobredosis.
Hasta la fecha, no hay datos clínicos sobre sobredosis con las tabletas de Liberatti®. Según la experiencia general con anticonceptivos orales combinados (AOC), en caso de sobredosis pueden presentarse náuseas, vómitos y sangrado de privación. El sangrado de privación puede ocurrir incluso en niñas antes de la menarquía, en caso de uso accidental o no intencional del medicamento. No existe antídoto específico; el tratamiento debe ser sintomático.
Reacciones adversas.
Para reacciones adversas graves en mujeres que utilizan COC, véase también la sección «Precauciones de uso».
En la «Tabla 3» se muestran las reacciones adversas clasificadas según clases de sistemas de órganos del MedDRA. La frecuencia se basa en datos clínicos. Se han utilizado los términos MedDRA más adecuados para describir reacciones específicas, así como sus sinónimos y estados relacionados.
Tabla 3
Frecuencia de reacciones adversas notificadas durante estudios clínicos del medicamento como anticonceptivo oral y para el tratamiento del acné de grado leve a moderado, según terminología y clasificación de sistemas de órganos MedDRA.
| Clases y sistemas orgánicos (MedDRA versión 9.1) |
Frecuentes (≥1/100 a <1/10) |
No frecuentes (≥1/1000 a <1/100) |
Puntuales (≥1/10000 a <1/1000) |
Frecuencia desconocida |
| Infecciones e infestaciones |
candidiasis |
|||
| Trastornos del sistema hematopoyético y del sistema linfático |
anemia, trombocitemia |
|||
| Trastornos del sistema inmunitario |
reacciones alérgicas |
hipersensibilidad |
||
| Trastornos del sistema endocrino |
trastornos endocrinos |
|||
| Trastornos del metabolismo y de la nutrición |
aumento del apetito, anorexia, hiperkalemia, hiponatremia |
|||
| Trastornos psiquiátricos |
labilidad emocional |
depresión, nerviosismo, somnolencia |
anorgasmia, insomnio |
|
| Trastornos del sistema nervioso |
dolor de cabeza |
mareo, parestesia |
vértigo, temblor |
|
| Trastornos oculares |
conjuntivitis, sequedad ocular, alteraciones visuales |
|||
| Trastornos cardíacos |
taquicardia |
|||
| Trastornos vasculares |
migraña, varices, hipertensión arterial |
flebitis, trastornos vasculares, epistaxis, síncope, tromboembolismo venoso (TEV), tromboembolismo arterial (TEA) |
||
| Trastornos gastrointestinales |
náuseas |
dolor abdominal, vómitos, dispepsia, flatulencia, gastritis, diarrea |
aumento del abdomen, trastornos gastrointestinales, sensación de plenitud gastrointestinal, hernia hiatal, candidiasis oral, estreñimiento, sequedad bucal |
|
| Trastornos hepatobiliares |
dolor de vesícula biliar, colecistitis |
|||
| Trastornos de la piel y del tejido subcutáneo |
acné, prurito, erupción cutánea |
melasma, eccema, alopecia, dermatitis acnéiforme, sequedad de la piel, eritema nodoso, hipertricosis, trastornos cutáneos, estrías, dermatitis de contacto, dermatitis fotosensible, piel nodular |
eritema multiforme |
|
| Trastornos del músculo esquelético y del tejido conjuntivo |
dolor de espalda, dolor en extremidades, calambres musculares |
|||
| Trastornos del sistema reproductivo y de las glándulas mamarias |
dolor de las mamas, metrorragia*, amenorrea |
candidiasis vaginal, dolor pélvico, aumento de las mamas, mastopatía fibroquística, hemorragia uterina/vaginal*, secreciones genitales, sofocos, vaginitis, alteraciones del ciclo menstrual, dismenorrea, hipomenorrea, menorragia, sequedad vaginal, prueba de Papanicolaou anómala, disminución del libido |
dispareunia, vulvovaginitis, sangrado poscoital, sangrado de retirada, quiste mamario, hiperplasia mamaria, neoplasia mamaria, pólipo cervical, atrofia endometrial, quiste ovárico, aumento del útero |
|
| Trastornos generales |
astenia, sudoración excesiva, edema (edema generalizado, edema periférico, edema facial) |
malestar general |
||
| Pruebas analíticas |
aumento de peso |
pérdida de peso |
*Los sangrados irregulares suelen desaparecer con la continuación del tratamiento.
Descripción de reacciones adversas individuales
En mujeres que han tomado ACO, se ha observado un riesgo aumentado de desarrollar trastornos trombóticos y tromboembólicos venosos o arteriales, incluyendo infarto de miocardio, accidente cerebrovascular, ataques isquémicos transitorios, trombosis venosa y embolia pulmonar, descritos con mayor detalle en la sección «Precauciones de uso».
Las siguientes reacciones adversas graves se han observado en mujeres que han utilizado ACO, y también se han descrito en la sección «Precauciones de uso»:
- trastornos tromboembólicos venosos;
- trastornos tromboembólicos arteriales;
- hipertensión arterial;
- tumores hepáticos;
- desarrollo o empeoramiento de enfermedades cuya relación con el uso de ACO no está completamente establecida: enfermedad de Crohn, colitis ulcerosa inespecífica, epilepsia, mioma uterino, porfiria, lupus eritematoso sistémico, herpes gestacional, corea de Sydenham, síndrome hemolítico urémico, ictericia colestásica;
- melasma;
- trastornos agudos o crónicos de la función hepática, que pueden requerir la suspensión del ACO hasta que los indicadores de función hepática regresen a la normalidad;
- en mujeres con predisposición hereditaria al angioedema, los estrógenos exógenos pueden provocar o agravar los síntomas de angioedema;
- los estrógenos exógenos pueden causar la aparición o empeoramiento de los síntomas de angioedema hereditario o adquirido.
La frecuencia de diagnóstico de cáncer de mama aumenta ligeramente entre las mujeres que usan ACO. Dado que el cáncer de mama es raro en mujeres menores de 40 años, el aumento en el número de casos diagnosticados de cáncer de mama en mujeres que actualmente usan o recientemente han usado ACO es insignificante en comparación con el riesgo basal de cáncer de mama. La relación con el uso de ACO es desconocida. Véanse también las secciones «Contraindicaciones» y «Precauciones de uso».
Interacciones
Pueden producirse sangrados de escape y/o disminución del efecto anticonceptivo debido a la interacción de otros medicamentos (inductores enzimáticos) con los anticonceptivos orales (véase la sección «Interacción con otros medicamentos y otras formas de interacción»).
Notificación de reacciones adversas sospechadas
La notificación de reacciones adversas tras la autorización del medicamento es de gran importancia. Permite continuar monitorizando la relación beneficio/riesgo del medicamento. Los profesionales médicos y farmacéuticos, así como los pacientes o sus representantes legales, deben informar de cualquier caso sospechoso de reacción adversa y de falta de eficacia del medicamento a través del sistema automatizado de información de farmacovigilancia en el enlace: https://aisf.dec.gov.ua.
Duración del producto. 3 años.
No utilizar después de la fecha de caducidad indicada en el envase.
Condiciones de almacenamiento.
Conservar en el envase original a una temperatura no superior a 30 ºC. Conservar en un lugar fuera del alcance de los niños.
Envase.
Tabletas recubiertas con película, 0,02 mg/3 mg, nº 28 (24 + 4) en blíster; 1 blíster junto con una escala calendario y un soporte para blíster en caja de cartón.
Categoría de dispensación. Bajo receta médica.
Fabricante. Laboratorios Leon Farma, S.A.
Domicilio del fabricante y dirección del lugar de ejercicio de su actividad.
Calle La Vallina S/N, Polígono Industrial Navatejera, Villaquilambre, 24193, España /
Calle La Vallina S/N, Poligono Industrial Navatejera, Villaquilambre, 24193, Spain.
Solicitante.
Sociedad con Responsabilidad Limitada «WORWARDS PHARMA».
Domicilio del solicitante.
Ucrania, 03142, ciudad de Kiev, calle Omeliana Prytsaka, 4.