Liberatti
Ukraine
Table of Contents
INSTRUCTIONS FOR MEDICAL USE OF THE MEDICINAL PRODUCT LIBERATTI®
Composition:
Active substances: ethinylestradiol, drospirenone;
1 film-coated tablet (pink) contains: ethinylestradiol 0.02 mg, drospirenone 3 mg;
Excipients: lactose monohydrate, pregelatinized starch (corn), povidone K-30, sodium croscarmellose, polysorbate 80, magnesium stearate; coating: Opadry® II pink (partially hydrogenated polyvinyl alcohol, titanium dioxide (E 171), macrogol 3350, talc (E 553b), yellow iron oxide (E 172), red iron oxide (E 172), black iron oxide (E 172));
1 film-coated placebo tablet (white) contains: anhydrous lactose, povidone K-30, magnesium stearate; coating: Opadry® II white (partially hydrogenated polyvinyl alcohol, titanium dioxide (E 171), macrogol 3350, talc (E 553b)).
Pharmaceutical form. Film-coated tablets.
Main physicochemical properties: pink, round, film-coated tablets, unmarked; placebo: white, round, film-coated tablets, unmarked.
Pharmacotherapeutic group. Systemic hormonal contraceptives. Fixed combinations of estrogens and progestogens. ATC code G03A A12.
Pharmacological Properties
Pharmacodynamics
Pearl Index of contraceptive failures: 0.41 (upper two-sided 95% confidence interval (CI): 0.85). Overall Pearl Index (contraceptive failures + patient errors): 0.80 (upper two-sided 95% CI: 1.30).
The contraceptive effect of the medicinal product Liberatti® is based on the interaction of several factors, the most important of which are inhibition of ovulation and changes in cervical secretion.
In a 3-cycle clinical study comparing the combination of drospirenone 3 mg/ethinylestradiol 0.02 mg administered in a 24-day regimen versus a 21-day regimen, the 24-day regimen was associated with greater suppression of follicular development. After intentional dosing errors during the third treatment cycle, ovarian activity, including ovulation, was observed in the majority of women in the 21-day regimen group, compared to women in the 24-day regimen group. Ovarian activity returned to pre-treatment levels within the post-treatment cycle in 91.8% of women in the 24-day regimen group.
Liberatti® is a combined oral contraceptive containing ethinylestradiol and the progestogen drospirenone. At therapeutic doses, drospirenone exhibits antiandrogenic and moderate antimineralocorticoid properties. It has no estrogenic, glucocorticoid, or antiglucocorticoid activity. Thus, drospirenone has a pharmacological profile similar to that of natural progesterone.
Clinical study data indicate that the moderate antimineralocorticoid properties of the drug result in a moderate antimineralocorticoid effect.
Two multicenter, double-blind, randomized, placebo-controlled studies were conducted to evaluate the efficacy and safety of the drug in women with moderate acne vulgaris. After 6 months of treatment, compared to placebo, the drug demonstrated a statistically significant reduction of 15.6% (49.3% vs. 33.7%) in the number of inflammatory lesions, 18.5% (40.6% vs. 22.1%) in the number of non-inflammatory lesions, and 16.5% (44.6% vs. 28.1%) in the total number of lesions. Additionally, a higher percentage of patients, 11.8% (18.6% vs. 6.8%), achieved "clear" or "almost clear" skin as assessed by the Investigator’s Static Global Assessment (ISGA) scale.
Pharmacokinetics
Drospirenone
Absorption. Orally administered drospirenone is rapidly and completely absorbed. Maximum serum concentration (Cmax) of 38 ng/mL is reached approximately 1–2 hours after single oral administration. Bioavailability is 76–85%. Concomitant food intake does not affect the bioavailability of drospirenone.
Distribution. After oral administration, drospirenone serum concentration decreases with a mean terminal half-life of approximately 31 hours. Drospirenone binds to serum albumin and does not bind to sex hormone-binding globulin (SHBG) or corticosteroid-binding globulin (CBG). Only 3–5% of its total amount in serum is present in free form. Ethinylestradiol-induced increases in SHBG do not affect the protein binding of drospirenone in serum. The mean volume of distribution of drospirenone is 3.7 ± 1.2 L/kg.
Metabolism. Drospirenone is extensively metabolized after oral administration. The main metabolites in plasma are acid forms of drospirenone formed by opening of the lactone ring, and 4,5-dihydro-drospirenone-3-sulfate formed by hydration followed by sulfation. Drospirenone is also subject to oxidative metabolism catalyzed by CYP3A4. In vitro, drospirenone may weakly or moderately inhibit cytochrome P450 enzymes: CYP1A1, CYP2C9, CYP2C19, and CYP3A4.
Elimination. The metabolic clearance rate of drospirenone from serum is approximately 1.5 ± 0.2 mL/min/kg. Drospirenone is excreted unchanged only in very small amounts. Metabolites are excreted in urine and feces in a ratio of 1.2 to 1.4. The elimination half-life of metabolites in urine and feces is approximately 40 hours.
Steady state. During the treatment cycle, the maximum steady-state serum concentration of drospirenone, approximately 70 ng/mL, is reached after 8 days of administration. Drospirenone serum levels accumulate 3-fold as a result of the relationship between the terminal half-life and the dosing interval.
Special patient groups
Women with renal impairment. Steady-state serum concentration of drospirenone in women with mild renal impairment (creatinine clearance 50–80 mL/min) was comparable to that in women with normal renal function (creatinine clearance >80 mL/min). Serum drospirenone levels were on average 37% higher in women with moderate renal impairment (creatinine clearance 30–50 mL/min) compared to women with normal renal function. Administration of drospirenone demonstrated good tolerability in all patient groups. It has been shown that drospirenone intake has no clinically significant effect on serum potassium concentration.
Women with hepatic impairment.
In a study of single-dose administration, oral clearance of drospirenone was reduced by approximately 50% in patients with moderate hepatic impairment compared to healthy adult volunteers with normal liver function. The observed difference in drospirenone clearance in patients with moderate hepatic impairment did not result in any apparent differences in serum potassium concentration. Even in the presence of diabetes mellitus and concomitant spironolactone therapy (two factors that may provoke hyperkalemia), no increase in serum potassium concentration above the upper limit of normal (ULN) was observed. Therefore, drospirenone is well tolerated in patients with mild to moderate hepatic impairment (Child-Pugh class B).
Ethnic groups. No clinically significant differences in the pharmacokinetics of drospirenone or ethinylestradiol were observed between Japanese women and Europeans.
Ethinylestradiol
Absorption. After oral administration, ethinylestradiol is rapidly and completely absorbed. Maximum serum concentration of 33 pg/mL is reached within 1–2 hours after single oral administration. Absolute bioavailability due to presystemic conjugation and first-pass hepatic metabolism is approximately 60%. Concomitant food intake reduces the bioavailability of ethinylestradiol in approximately 25% of subjects, with unchanged bioavailability in the remainder.
Distribution. Serum ethinylestradiol levels decrease in a biphasic manner, with a terminal phase half-life of approximately 24 hours. Ethinylestradiol binds strongly but non-specifically to serum albumin (approximately 98.5%) and induces an increase in serum concentrations of SHBG and CBG. The apparent volume of distribution is approximately 5 L/kg.
Metabolism. Ethinylestradiol is extensively metabolized in the gastrointestinal tract (GIT) and during first-pass through the liver. Ethinylestradiol is metabolized primarily by hydroxylation of the aromatic ring, forming a wide spectrum of hydroxylated and methylated metabolites present in free form and as conjugates with glucuronides and sulfates. The metabolic clearance of ethinylestradiol is approximately 5 mL/min/kg.
In vitro, ethinylestradiol is a reversible inhibitor of CYP2C19, CYP1A1, and CYP1A2, and, based on its mechanism of action, an inhibitor of CYP3A4/5, CYP2C8, and CYP2J2.
Elimination. Ethinylestradiol is almost not excreted unchanged. Ethinylestradiol metabolites are excreted in urine and bile in a ratio of 4:6. The elimination half-life of metabolites is nearly 1 day.
Steady state. Steady state is reached in the second half of the treatment cycle, when ethinylestradiol serum concentration increases by 2.0–2.3 times.
Preclinical safety data
In laboratory animals, the effects of drospirenone and ethinylestradiol were limited to those associated with known pharmacological actions. In particular, animal studies on reproductive toxicity revealed species-specific embryotoxic and fetotoxic effects. At exposures exceeding those in users of the drug, effects on sexual differentiation were observed in certain animal species. Environmental risk assessment studies indicated that ethinylestradiol and drospirenone may potentially pose a threat to the aquatic environment (see section "Special precautions for safety").
Clinical characteristics.
Indications.
Oral contraception.
Contraindications.
Combined hormonal contraceptives (CHCs) must not be used if any of the conditions listed below is present. If any of these conditions occurs for the first time during CHC use, the medication must be discontinued immediately.
- Presence or risk of venous thromboembolism (VTE):
- Current venous thromboembolism, including anticoagulant therapy, or history of VTE (e.g., deep vein thrombosis (DVT) or pulmonary embolism (PE));
- Known hereditary or acquired predisposition to venous thromboembolism, such as activated protein C resistance (including factor V Leiden mutation), antithrombin-III deficiency, protein C deficiency, protein S deficiency;
- Major surgery with prolonged immobilization (see section "Special precautions");
- High risk of venous thromboembolism due to the presence of multiple risk factors (see section "Special precautions").
- Presence or risk of arterial thromboembolism (ATE):
- Current ATE or history of ATE (e.g., myocardial infarction), or presence of prodromal symptoms (e.g., angina pectoris);
- Current or past cerebrovascular accident, or presence of prodromal symptoms (e.g., transient ischemic attack (TIA));
- Known hereditary or acquired predisposition to ATE, such as hyperhomocysteinemia and antiphospholipid antibodies (anti-cardiolipin antibodies, lupus anticoagulant);
- History of migraine with focal neurological symptoms;
- High risk of ATE due to multiple risk factors (see section "Special precautions") or due to a single serious risk factor such as:
- diabetes mellitus with vascular complications;
- severe arterial hypertension;
- severe dyslipoproteinemia.
- Current or past history of severe liver disease until liver function tests have returned to normal.
- Severe renal insufficiency or acute renal failure.
- Current or past history of liver tumors (benign or malignant).
- Known or suspected hormone-dependent malignancies (e.g., of the genital organs or breast).
- Vaginal bleeding of unknown etiology.
- Hypersensitivity to the active substances or to any of the excipients of the medicinal product.
The medicinal product Libertty® is contraindicated for concomitant use with medicinal products containing ombitasvir/paritaprevir/ritonavir, dasabuvir, glecaprevir/pibrentasvir, and sofosbuvir/velpatasvir/voxilaprevir (see sections "Interaction with other medicinal products and other forms of interaction" and "Special precautions").
Special safety measures.
This medicinal product may pose a risk to the environment (see section "Pharmacological properties"). Any unused medicinal product or waste material should be disposed of in accordance with local requirements.
Interaction with other medicinal products and other forms of interaction.
Information regarding concomitantly administered medicinal products should be reviewed to identify potential interactions.
- Effect of other medicinal products on the medicinal product Libertty®
Interactions are possible with medicinal products that induce microsomal enzymes. This leads to increased clearance of sex hormones, which in turn may result in changes in menstrual bleeding patterns and/or loss of contraceptive efficacy.
Therapy
Enzyme induction may be observed within a few days of treatment initiation. Maximum enzyme induction generally occurs after several weeks. After discontinuation of the inducing agent, enzyme induction may persist for approximately 4 weeks.
Short-term treatment
Women taking enzyme-inducing medicinal products should temporarily use a barrier method or another non-hormonal contraceptive method in addition to the combined oral contraceptive (COC). The barrier method should be used throughout the entire duration of treatment with the enzyme-inducing agent and for an additional 28 days after discontinuation of the agent. If treatment is initiated during the period of taking the last tablets in the COC pack, the next pack of COC tablets should be started immediately after the previous one, without the usual tablet-free interval.
Long-term treatment
Women undergoing long-term therapy with enzyme-inducing substances are advised to use a barrier method or another appropriate non-hormonal contraceptive method.
The following interactions have been reported based on published data.
Substances increasing COC clearance (reduced COC efficacy due to enzyme induction), e.g.:
barbiturates, bosentan, carbamazepine, phenytoin, primidone, rifampicin; HIV medications: ritonavir, nevirapine, and efavirenz; also possibly felbamate, griseofulvin, oxcarbazepine, topiramate, and herbal products containing St. John's wort (Hypericum perforatum).
Substances with variable effects on COC clearance
When used concomitantly with COCs, many combinations of HIV protease inhibitors and non-nucleoside reverse transcriptase inhibitors, including combinations with hepatitis C virus (HCV) inhibitors, may either increase or decrease plasma concentrations of estrogens or progestins. The net effect of these changes may be clinically significant in some cases.
Therefore, information regarding the medical use of the HIV/HCV medicinal product being taken concomitantly should be reviewed to identify potential interactions and any other recommendations. In case of any doubts, women should additionally use a barrier method of contraception during therapy with protease inhibitors or non-nucleoside reverse transcriptase inhibitors.
Substances decreasing COC clearance (enzyme inhibitors)
The clinical significance of potential interactions with enzyme inhibitors remains unclear.
Concomitant use of strong CYP3A4 inhibitors may increase plasma concentrations of estrogen, progestin, or both components.
In a multiple-dose study of drospirenone (3 mg daily)/ethinylestradiol (0.002 mg daily) co-administered with the strong CYP3A4 inhibitor ketoconazole, the AUC(0-24h) of drospirenone and ethinylestradiol increased by 2.7-fold and 1.4-fold, respectively, over 10 days.
Etoricoxib at doses of 60 mg daily to 120 mg daily demonstrated a 1.4- to 1.6-fold increase in ethinylestradiol plasma concentrations, respectively, when co-administered with a combined hormonal contraceptive (CHC) containing 0.035 mg ethinylestradiol.
- Effect of the medicinal product Libertty® on other medicinal products
COCs may affect the metabolism of certain active substances. Consequently, plasma and tissue concentrations may either increase (e.g., cyclosporine) or decrease (e.g., lamotrigine).
Based on in vivo interaction studies in female volunteers using omeprazole, simvastatin, and midazolam as probe substrates, clinically significant interactions between drospirenone 3 mg and other active substances metabolized by cytochrome P450 are unlikely.
Clinical data indicate that ethinylestradiol inhibits the clearance of CYP1A2 substrates, resulting in mild (e.g., theophylline) or moderate (e.g., tizanidine) increases in their plasma concentrations.
Pharmacodynamic interactions
Concomitant use with medicinal products containing ombitasvir/paritaprevir/ritonavir and dasabuvir, with or without ribavirin, glecaprevir/pibrentasvir, and sofosbuvir/velpatasvir/voxilaprevir increases the risk of elevated alanine aminotransferase (ALT) levels (see sections "Contraindications" and "Special precautions").
Therefore, women using the medicinal product Libertty® should temporarily switch to an alternative method of contraception (e.g., progestogen-only contraceptives or non-hormonal methods) before starting therapy with the aforementioned combination of medicinal products. Use of Libertty® may be resumed 2 weeks after completion of therapy with the specified combination.
In patients with normal renal function, concomitant use of drospirenone with angiotensin-converting enzyme (ACE) inhibitors or non-steroidal anti-inflammatory drugs (NSAIDs) did not show a significant effect on serum potassium levels. However, concomitant use of Libertty® with aldosterone antagonists or potassium-sparing diuretics has not been studied. In such cases, serum potassium levels should be monitored during the first treatment cycle (see also section "Special precautions").
Other forms of interaction
Laboratory tests
Use of contraceptive steroids may affect the results of certain laboratory tests, such as biochemical parameters of liver, thyroid, adrenal, and kidney function; plasma concentrations of transport proteins such as corticosteroid-binding globulin; plasma concentrations of lipid/lipoprotein fractions; carbohydrate metabolism parameters; and coagulation and fibrinolysis parameters. These changes are usually within normal limits. Drospirenone increases plasma renin and aldosterone activity due to its moderate anti-mineralocorticoid activity.
Special precautions for use.
The decision to prescribe Liberratí® should be made taking into account the woman's individual current risk factors, including risk factors for venous thromboembolism (VTE), as well as the VTE risk associated with taking Liberratí® compared to other combined oral contraceptives (COCs) (see sections "Contraindications" and "Special precautions for use").
Warning
If any of the conditions or risk factors listed below are present, the appropriateness of using Liberratí® should be discussed with the woman.
In case of exacerbation or at the first signs of any of the listed conditions or risk factors, women are advised to consult a physician to determine whether discontinuation of Liberratí® is necessary.
Combined oral contraceptives should be discontinued in cases of suspected or confirmed venous thromboembolism (VTE) or arterial thromboembolism (ATE). If anticoagulant therapy is initiated, an alternative effective contraceptive method should be provided due to the teratogenic effects of anticoagulants (coumarins).
- Circulatory disorders
Risk of venous thromboembolism (VTE)
The use of any COCs increases the risk of VTE in women who use them compared to women who do not. Medicinal products containing levonorgestrel, norgestimate, or norethisterone are associated with a lower risk of VTE. The use of other medicinal products, such as Liberratí®, may double the risk. The decision to use products other than those with the lowest VTE risk should only be made after discussion with the woman. It is essential to ensure that she understands the VTE risk associated with Liberratí®, the impact of her individual risk factors, and the fact that the VTE risk is highest during the first year of use. According to some data, the risk of VTE may increase when resuming COC use after a break of 4 weeks or longer.
VTE occurs in 2 out of 10,000 women who do not use COCs and are not pregnant over a one-year period. However, for any individual woman, the risk may be significantly higher depending on her individual risk factors (see below).
It has been established\1\ that among 10,000 women using COCs containing drospirenone, 9–12 women will develop VTE within one year. This compares to a rate of 62 among women using COCs containing levonorgestrel.
In both cases, the annual number of VTE cases was lower than typically expected during pregnancy or the postpartum period.
VTE can be fatal in 1–2% of cases.
Number of VTE cases per 10,000 women per year
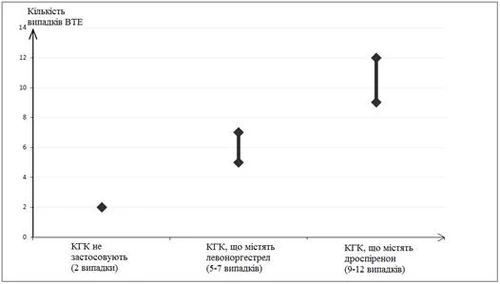
1 These estimates are based on all available epidemiological data, taking into account relative risks associated with different COCs compared to COCs containing levonorgestrel.
2 An average of 5–7 cases per 10,000 woman-years, based on the relative risk of using COCs containing levonorgestrel compared to women not using COCs (approximately 2.3–3.6 cases).
Risk factors for VTE
The risk of venous thromboembolic complications in women using COCs may be substantially increased in the presence of additional risk factors, especially multiple ones (see Table 1).
The use of Liberratí® is contraindicated in women with multiple risk factors that may increase the risk of venous thrombosis (see section "Contraindications"). If a woman has more than one risk factor, the increase in risk may be greater than the sum of risks associated with each individual factor, so the overall VTE risk should be considered. If the benefit-risk balance is unfavorable, COCs should not be prescribed (see section "Contraindications").
Table 1
Risk factors for VTE development
| Risk factor |
Comment |
| Obesity (body mass index over 30 kg/m²) |
Risk increases significantly with higher body mass index. Particular attention is required when other risk factors are present. |
| Long-term immobilization, major surgery, surgery on lower limbs or pelvic organs, neurosurgical procedures, or extensive trauma. Note: temporary immobilization, including flights > 4 hours, may also be a risk factor for VTE, especially in women with other risk factors. |
In such situations, it is recommended to discontinue the use of the medicinal product (at least 4 weeks before planned surgery) and not resume treatment until at least 2 weeks after full recovery of mobility. Alternative contraceptive methods should be used to prevent unintended pregnancy. The need for antithrombotic therapy should be considered if treatment with the medicinal product Liberratì® has not been previously discontinued. |
| Family history (VTE in a close relative or parent, particularly at a relatively young age, e.g., under 50 years). |
In case of hereditary predisposition, women should consult a specialist before using any COCs. |
| Other conditions associated with VTE |
Cancer, systemic lupus erythematosus, hemolytic-uremic syndrome, chronic inflammatory bowel disease (Crohn's disease or ulcerative colitis), and sickle cell anemia. |
| Age |
Especially over 35 years of age |
There is no consensus regarding the possible influence of varicose veins and superficial thrombophlebitis on the development and progression of venous thrombosis.
Particular attention should be paid to the increased risk of thromboembolism during pregnancy, especially within 6 weeks after delivery (information on pregnancy or breastfeeding is provided in the section "Use in pregnancy or breastfeeding").
Symptoms of VTE (venous thromboembolism: deep vein thrombosis and pulmonary embolism)
Women should be advised to seek immediate medical attention and inform their physician that they are taking COCs if any of the symptoms listed below occur.
Symptoms of DVT may include: unilateral swelling of the leg and/or foot or along a vein in the leg; pain or tenderness in the leg, which may occur only when standing or walking; warmth in the affected leg; redness or discoloration of the skin on the leg.
Symptoms of PE may include: sudden unexplained shortness of breath or rapid breathing; sudden cough, possibly with hemoptysis; sudden chest pain; syncope or dizziness; rapid or irregular heartbeat.
Some of these symptoms (e.g., shortness of breath, cough) are nonspecific and may be misinterpreted as more common or less serious conditions (e.g., respiratory tract infections).
Other manifestations of vascular occlusion may include sudden pain, swelling, acute abdomen, and mild cyanosis of a limb.
Ocular vessel occlusion may initially present with painless blurred vision, which may progress to vision loss. In some cases, vision loss develops almost immediately.
Risk of arterial thromboembolism (ATE)
Epidemiological studies have shown that the use of any COCs is associated with an increased risk of ATE (myocardial infarction) and cerebrovascular events (transient ischemic attack [TIA], stroke). Arterial thromboembolic events may be fatal.
Risk factors for ATE
When using COCs, the risk of developing arterial thromboembolic complications or cerebrovascular events increases in women with risk factors (see Table 2). The use of Liberratte® is contraindicated in women who have one serious or multiple risk factors for ATE that may increase the risk of arterial thrombosis (see section "Contraindications"). If a woman has more than one risk factor, the overall risk increase may be greater than the sum of the risks associated with each individual factor, so the total risk should be considered. COCs should not be prescribed if the benefit-risk ratio is unfavorable (see section "Contraindications").
Table 2
Risk factors for ATE
| Increased age |
Especially over the age of 35 years |
| Smoking |
Women using COCs are advised not to smoke. Women aged 35 years and older who continue to smoke are strongly advised to use another method of contraception. |
| Arterial hypertension |
|
| Obesity (body mass index over 30 kg/m²) |
Risk increases significantly with increasing body mass index. Requires particular attention, especially if women have other risk factors. |
| Family history (arterial thromboembolism in a close relative or parent, particularly at a relatively young age, e.g., before 50 years) |
In case of hereditary predisposition, women are advised to consult a specialist before using any COCs. |
| Migraine |
An increase in frequency or severity of migraine during COC use (possible prodromal symptoms preceding cerebrovascular events) may require immediate discontinuation of COC use. |
| Other conditions associated with vascular adverse reactions |
Diabetes mellitus, hyperhomocysteinemia, heart valve disorders, atrial fibrillation, dyslipoproteinemia, and systemic lupus erythematosus. |
Symptoms of ATE
Women should be advised to seek immediate medical attention and inform their physician if they are taking COCs should any of the symptoms listed below occur.
Symptoms of cerebrovascular disorders may include: sudden numbness of the face, weakness or numbness of extremities, especially on one side; sudden difficulty walking, dizziness, loss of balance or coordination; sudden confusion, speech or comprehension disturbances; sudden worsening of vision in one or both eyes; sudden, severe or prolonged headache without apparent cause; loss of consciousness or fainting with or without seizures.
Transient nature of symptoms may indicate transient ischemic attack (TIA).
Symptoms of myocardial infarction may include: pain, discomfort, pressure or heaviness in the chest, arm, or below the sternum; discomfort radiating to the back, jaw, throat, arm, or stomach; sensation of stomach fullness, indigestion, or dyspnea; excessive sweating, nausea, vomiting, or dizziness; extreme weakness, anxiety, or shortness of breath; rapid or irregular heartbeat.
Tumors
Results of some epidemiological studies suggest an additional increased risk of cervical cancer with long-term use of COCs (> 5 years); however, this statement remains controversial, as it has not been definitively established to what extent study results account for confounding risk factors such as sexual behavior and other factors, for example human papillomavirus infection.
A meta-analysis based on 54 epidemiological studies indicates a slight increase in relative risk (RR = 1.24) of breast cancer in women using COCs. This increased risk gradually disappears within 10 years after discontinuation of COC use. Since breast cancer is rare in women under 40 years of age, the increase in the number of diagnosed cases of breast cancer among women who currently or recently used COCs is minimal relative to the overall risk of breast cancer. Results of these studies do not provide evidence of a causal relationship. The increased risk may be due to earlier diagnosis of breast cancer in women using COCs, a biological effect of COCs, or a combination of both factors. There is a tendency for breast cancer diagnosed in women who have ever used COCs to be clinically less advanced than in those who have never used COCs.
In isolated cases, benign and rarely malignant liver tumors have been observed in women using COCs, which in some cases led to life-threatening intra-abdominal bleeding. In case of complaints of severe epigastric pain, hepatomegaly, or signs of intra-abdominal bleeding, the possibility of liver tumor associated with COC use should be considered in differential diagnosis.
Use of COCs at high doses (50 mcg ethinylestradiol) reduces the risk of endometrial and ovarian cancer. It remains to be confirmed whether these data are applicable to low-dose COCs as well.
Other conditions
The progestin component of the medicinal product Liberté® is an aldosterone antagonist with potassium-sparing properties. In most cases, an increase in serum potassium levels is not expected during use. During clinical trials, minor increases in serum potassium levels were observed in some patients with mild to moderate renal insufficiency who were concurrently using potassium-sparing medicinal products during treatment with drospirenone. Therefore, monitoring of serum potassium levels is recommended during the first treatment cycle in patients with renal insufficiency. These patients should also be advised to maintain serum potassium levels not exceeding ULN before initiating treatment, especially when concurrently using potassium-sparing medicinal products (see section "Interaction with other medicinal products and other forms of interaction").
Women with hypertriglyceridemia or a family history of this condition represent a risk group for pancreatitis development during COC use.
Although slight increases in blood pressure have been reported in many women taking COCs, clinically significant hypertension occurs only occasionally. Immediate discontinuation of COCs is required only in these rare cases. In cases of persistent hypertension or inability to control blood pressure with antihypertensive agents, COC use should be discontinued in women taking these agents. If appropriate, COC use may be resumed after normotension is achieved with antihypertensive therapy.
The occurrence or exacerbation of the following conditions has been reported during pregnancy and COC use, but their relationship to estrogen/progestin use has not been definitively established: jaundice and/or pruritus associated with cholestasis, gallstone formation, porphyria, systemic lupus erythematosus, hemolytic-uremic syndrome, Sydenham's chorea, herpes gestationis, hearing loss associated with otosclerosis.
Exogenous estrogens may cause the onset or exacerbation of symptoms of hereditary or acquired angioedema.
Acute or chronic disorders of liver function may require discontinuation of COCs until liver function tests return to normal and a causal relationship with COCs is excluded.
COC use should be discontinued in case of recurrence of cholestatic jaundice and/or cholestatic pruritus previously occurring during pregnancy or prior use of sex hormones.
Although COCs may affect peripheral insulin resistance and glucose tolerance, there are no data indicating the need to modify the therapeutic regimen in diabetic women taking low-dose COCs (< 0.05 mg ethinylestradiol). However, women with diabetes should be carefully monitored during COC use, especially at the beginning of treatment.
Exacerbations of endogenous depression, epilepsy, Crohn's disease, and ulcerative colitis have also been observed during COC use.
Depressed mood and depression are well-known adverse reactions that may occur during use of hormonal contraceptives (see section "Adverse Reactions"). Depression can be a serious condition and is a well-known risk factor for suicidal behavior and suicide. Women should be advised to consult a physician in case of mood changes or onset of depressive symptoms, including shortly after starting treatment.
Chloasma may occasionally occur, particularly in women with a history of chloasma of pregnancy. Women prone to chloasma should avoid direct sunlight or ultraviolet radiation during COC use.
Pink and white tablets contain lactose monohydrate. This should be considered in cases of rare hereditary conditions of galactose intolerance, Lapp lactase deficiency, or glucose-galactose malabsorption, or when on a lactose-free diet. If you have been diagnosed with intolerance to certain sugars, consult your physician before taking this medicinal product.
Elevation of ALT levels
In clinical trials involving patients receiving antiviral therapy for hepatitis C with medicinal products containing ombitasvir/paritaprevir/ritonavir with or without dasabuvir, ALT elevations exceeding 5 times ULN were observed significantly more frequently in women using medicinal products containing ethinylestradiol, such as COCs. ALT elevations were also observed with antiviral medicinal products for HCV containing glecaprevir/pibrentasvir and sofosbuvir/velpatasvir/voxilaprevir (see sections "Contraindications" and "Interaction with other medicinal products and other forms of interaction").
Consultations/Medical examination
Prior to initiation or resumption of the medicinal product Liberté®, a complete medical history (including family history), a full medical examination, and exclusion of pregnancy are recommended. Blood pressure measurement and medical examination should be performed, taking into account contraindications (see section "Contraindications") and special precautions (see section "Special precautions for use"). Women should be informed about venous and arterial thrombosis, including the risk associated with use of Liberté® compared to other COCs, symptoms of VTE and ATE, known risk factors, and actions to take in case of suspected thrombosis.
Patients should be advised to carefully read the package leaflet and follow the recommendations provided therein.
The frequency and nature of examinations should be based on current medical practice guidelines, taking into account individual characteristics of each woman.
Patients should be informed that hormonal contraceptives do not protect against HIV infection (AIDS) or any other sexually transmitted diseases.
Reduced efficacy
The efficacy of COCs may be reduced in case of missed tablet intake (see section "Dosage and administration"), gastrointestinal disorders (see section "Dosage and administration"), or concomitant use of other medicinal products (see section "Interaction with other medicinal products and other forms of interaction").
Cycle disturbances
Irregular bleeding (spotting or breakthrough bleeding) may occur during COC use, particularly during the first few months. If such bleeding persists after three menstrual cycles, it should be considered significant.
If irregular bleeding persists or reappears after a period of regular bleeding, non-hormonal causes of bleeding and appropriate diagnostic measures, including evaluation to exclude tumors and pregnancy, should be considered. Diagnostic procedures may include curettage.
In some women, withdrawal bleeding may not occur during the tablet-free interval. If COCs are taken according to instructions in the section "Dosage and administration," pregnancy is unlikely. However, if COC intake was irregular prior to the absence of the first withdrawal bleed, or if withdrawal bleeding is absent for two consecutive cycles, pregnancy must be excluded before continuing COC use.
Use during pregnancy or breastfeeding.
Pregnancy. This medicinal product is contraindicated during pregnancy.
If pregnancy occurs during use of Liberté®, treatment must be discontinued immediately. However, results of epidemiological studies do not indicate an increased risk of congenital malformations in children whose mothers used COCs prior to pregnancy, nor a teratogenic effect from inadvertent COC use during pregnancy.
Animal studies have shown adverse effects during pregnancy and lactation (see section "Pharmacological properties"). Based on these animal studies, adverse effects due to the hormonal activity of active substances cannot be excluded. However, overall experience with COC use during pregnancy does not indicate an adverse effect in humans.
Available data on use of the drug during pregnancy are too limited to draw conclusions regarding any negative impact of Liberté® on pregnancy outcome or fetal and neonatal health. To date, there are no appropriate epidemiological data.
When resuming use of the medicinal product Liberté®, the increased risk of VTE during the postpartum period should be considered (see sections "Special precautions for use" and "Dosage and administration").
Breastfeeding period. COCs may affect breastfeeding, as they may reduce the quantity and alter the composition of breast milk. Therefore, COCs are not recommended during breastfeeding. Small amounts of contraceptive steroids and/or their metabolites may pass into breast milk during COC use. These amounts may affect the infant.
Fertility. The medicinal product Liberté® is indicated for prevention of pregnancy. Information on fertility recovery can be found in the section "Pharmacological properties."
Ability to influence reaction speed when driving vehicles or operating machinery.
No studies on the effect of the medicinal product Liberté® on reaction speed during driving vehicles or operating machinery have been conducted. No effect on the ability to drive or operate machinery has been observed in women using COCs.
Dosage and Administration
Route of administration: oral.
The tablets should be taken daily at approximately the same time each day, in the order indicated on the blister pack, with a small amount of liquid. The medication is taken as one tablet per day for 28 consecutive days. The next pack should be started the day after the previous pack is finished, without any break.
The packaging includes a blister card holder for carrying the blister when needed, as well as a calendar scale.
The blister pack contains 24 active tablets (pink tablets) and 4 placebo tablets (white tablets) that do not contain active ingredients.
- Initiating treatment with Liberrat®
If hormonal contraceptives have not been used in the previous cycle (last month), the first tablet should be taken on the first day of the natural menstrual cycle (i.e., the first day of menstrual bleeding). It is also possible to start on days 2–5 of the cycle; however, in this case, an additional contraceptive method (e.g., barrier method) must be used for the first 7 days of tablet intake.
- Switching from another combined oral contraceptive (COC), vaginal ring, or transdermal patch
It is recommended to start taking Liberrat® tablets the day after taking the last active tablet of the previous COC, but no later than the day after the tablet-free interval or after the placebo tablets of the previous COC. For users of a contraceptive vaginal ring or transdermal patch, treatment with Liberrat® should begin on the day of device removal, but no later than the day when the next application of the device would have been due.
- Switching from a progestogen-only method (‘mini-pill’, injections, implants, or intrauterine system containing progestogen)
Treatment with Liberrat® can be started on any day after discontinuation of the ‘mini-pill’ (in the case of an implant or intrauterine system, on the day of removal; in the case of injections, instead of the next scheduled injection). However, in all cases, it is recommended to use an additional barrier method of contraception for the first 7 days of tablet intake.
- After first-trimester abortion
Treatment with Liberrat® may be started immediately. In this case, there is no need to use additional contraceptive methods.
- After childbirth or second-trimester abortion
In case of breastfeeding, see section "Use during pregnancy or breastfeeding". It is recommended to start taking Liberrat® between days 21–28 after childbirth or second-trimester abortion. If treatment is started later, an additional barrier method of contraception should be used for the first 7 days of tablet intake. However, if sexual intercourse has already occurred, pregnancy should be ruled out before starting the medication, or the woman should wait until the first menstrual period occurs.
What to do if a tablet is missed
A missed placebo tablet can be disregarded. However, such tablets should be removed from the pack to avoid unintentional prolongation of the placebo phase. The following instructions apply only to missed pink active tablets.
If the delay in taking a pink active tablet does not exceed 24 hours, contraceptive protection is not reduced. The missed tablet should be taken as soon as possible, even if this means taking two tablets at the same time. The next tablet should be taken at the usual time.
If the delay in taking a pink active tablet exceeds 24 hours, contraceptive protection may be reduced. In such cases, the following two main rules should be followed:
- The interval between tablet intakes must never exceed 4 days.
- Adequate suppression of the hypothalamic-pituitary-ovarian system is achieved only with continuous intake of tablets for 7 days.
Accordingly, the following practical recommendations should be followed:
- Days 1–7
Take the last missed tablet as soon as possible, even if this means taking two tablets at the same time. Continue taking tablets at the usual time. Additionally, use a barrier method of contraception (e.g., condom) for the next 7 days. If sexual intercourse occurred within the previous 7 days, consider the possibility of pregnancy. The greater the number of missed tablets and the closer to the placebo phase, the higher the risk of pregnancy.
- Days 8–14
Take the last missed tablet as soon as possible, even if this means taking two tablets simultaneously. Continue taking tablets at the usual time. If the woman has taken tablets correctly for the 7 days prior to the missed dose, no additional contraceptive methods are needed. If this is not the case, or if more than one tablet has been missed, a barrier method of contraception should be used for the next 7 days.
- Days 15–24
The risk of pregnancy increases as the placebo phase approaches. However, if the dosing schedule is followed correctly, contraceptive protection can be maintained. If one of the following recommendations is followed and tablets were taken correctly for the 7 days prior to the missed dose, additional contraceptive methods are not required. If not, the first of the following recommendations should be followed, and a barrier method of contraception should be used for the next 7 days.
- Take the last missed tablet as soon as possible, even if this means taking two tablets at the same time. Continue taking tablets at the usual time until the end of the pink active tablets. The 4 placebo tablets should not be taken. Start the next pack immediately after the last active tablet. Withdrawal bleeding is unlikely to occur before finishing all active tablets from the second pack, although breakthrough bleeding or spotting may occur during tablet intake.
- Alternatively, stop taking tablets from the current pack. In this case, the tablet-free interval should be 4 days, including the days of missed tablets; treatment should resume with the next pack.
If the expected withdrawal bleeding does not occur during the first normal tablet-free interval after a missed tablet, pregnancy is possible. A healthcare provider should be consulted before starting a new pack.
Recommendations in case of gastrointestinal disturbances
In cases of severe gastrointestinal disturbances, incomplete absorption of the medication may occur. In such cases, additional contraceptive methods should be used. If vomiting occurs within 3–4 hours after taking a Liberrat® tablet, follow the same recommendations as for a missed tablet.
How to change or delay menstruation
To delay menstruation, continue taking active (pink) tablets from a new pack without interruption, skipping the placebo tablets. The treatment may be extended up to the end of the active tablets in the second pack. Breakthrough bleeding or spotting may occur during this time. Usually, treatment with Liberrat® resumes after taking the non-hormonal placebo tablets.
To shift the timing of menstruation to another day of the week, shorten the tablet-free interval by the desired number of days. It should be noted that the shorter the interval, the more likely it is that withdrawal bleeding will not occur and that breakthrough bleeding or spotting may occur during intake of tablets from the second pack (as in the case of delayed menstruation).
Additional information for special patient groups
Elderly patients. Liberrat® is not indicated for use after menopause.
Patients with hepatic impairment. Liberrat® is contraindicated in women with severe hepatic impairment (see sections "Pharmacological properties" and "Contraindications").
Patients with renal impairment. Liberrat® is contraindicated in women with severe renal impairment or acute renal failure (see sections "Pharmacological properties" and "Contraindications").
Children.
This medicinal product is indicated for use only after regular menstruation has been established, and only on a physician's prescription.
Overdose.
There are no clinical data on overdose with Liberrat® tablets. Based on general experience with combined oral contraceptives (COCs), overdose may result in nausea, vomiting, and withdrawal bleeding. Withdrawal bleeding may occur even in premenarche girls in cases of accidental or unintentional intake of the medication. There is no specific antidote; treatment should be symptomatic.
Adverse reactions
For serious adverse reactions in women using COCs, see also section "Special precautions".
Table 3 lists adverse reactions by MedDRA organ system classes. Frequencies are based on clinical trial data. The most appropriate MedDRA preferred terms have been used to describe specific reactions and their synonyms and related conditions.
Table 3
Frequency of adverse reactions reported during clinical trials of the drug as an oral contraceptive and for the treatment of mild acne, according to MedDRA system organ classes and preferred terms.
| System organ classes (MedDRA version 9.1) |
Common (≥1/100 to <1/10) |
Uncommon (≥1/1000 to <1/100) |
Rare (≥1/10000 to <1/1000) |
Frequency unknown |
| Infections and infestations |
candidiasis |
|||
| Blood and lymphatic system disorders |
anaemia, thrombocythaemia |
|||
| Immune system disorders |
allergic reactions |
hypersensitivity |
||
| Endocrine disorders |
endocrine disorders |
|||
| Metabolism and nutrition disorders |
increased appetite, anorexia, hyperkalaemia, hyponatraemia |
|||
| Psychiatric disorders |
emotional lability |
depression, nervousness, somnolence |
anorgasmia, insomnia |
|
| Nervous system disorders |
headache |
dizziness, paraesthesia |
vertigo, tremor |
|
| Eye disorders |
conjunctivitis, dry eyes, visual disturbance |
|||
| Cardiac disorders |
tachycardia |
|||
| Vascular disorders |
migraine, varicose veins, hypertension |
phlebitis, vascular disorders, epistaxis, syncope, venous thromboembolism (VTE), arterial thromboembolism (ATE) |
||
| Gastrointestinal disorders |
nausea |
abdominal pain, vomiting, dyspepsia, flatulence, gastritis, diarrhoea |
abdominal distension, gastrointestinal disorders, gastrointestinal fullness, hiatal hernia, oral candidiasis, constipation, dry mouth |
|
| Hepatobiliary disorders |
gallbladder pain, cholecystitis |
|||
| Skin and subcutaneous tissue disorders |
acne, pruritus, rash |
chloasma, eczema, alopecia, acneiform dermatitis, dry skin, nodular erythema, hypertrichosis, skin disorders, striae, contact dermatitis, photo-sensitive dermatitis, nodular skin |
multiform erythema |
|
| Musculoskeletal and connective tissue disorders |
back pain, limb pain, muscle cramps |
|||
| Reproductive system and breast disorders |
breast tenderness, metrorrhagia*, amenorrhoea |
vaginal candidiasis, pelvic pain, breast enlargement, fibrocystic mastopathy, uterine/vaginal bleeding*, genital discharge, hot flushes, vaginitis, menstrual cycle disturbance, dysmenorrhoea, hypomenorrhoea, menorrhagia, vaginal dryness, abnormal Pap smear, decreased libido |
dyspareunia, vulvovaginitis, postcoital bleeding, withdrawal bleeding, breast cyst, breast hyperplasia, breast neoplasm, cervical polyp, endometrial atrophy, ovarian cyst, uterine enlargement |
|
| General disorders |
asthenia, increased sweating, oedema (generalised oedema, peripheral oedema, facial oedema) |
malaise |
||
| Investigations |
weight increased |
weight decreased |
*Irregular bleeding usually diminishes with continued use.
Description of individual adverse reactions
In women taking COCs, an increased risk of venous or arterial thrombotic and thromboembolic events has been observed, including myocardial infarction, stroke, transient ischaemic attacks, venous thrombosis, and pulmonary embolism, which are described in detail in the section "Special precautions for use".
The following serious adverse reactions have been observed in women using COCs and are also described in the section "Special precautions for use":
- venous thromboembolic disorders;
- arterial thromboembolic disorders;
- arterial hypertension;
- liver tumours;
- development or exacerbation of conditions for which a definite link with COC use has not been established: Crohn's disease, ulcerative colitis, epilepsy, uterine fibroids, porphyria, systemic lupus erythematosus, herpes gestationis, Sydenham's chorea, haemolytic-uraemic syndrome, cholestatic jaundice;
- chloasma;
- acute or chronic disorders of liver function, which may require discontinuation of COC use until liver function parameters return to normal;
- in women with hereditary predisposition to angioedema, exogenous estrogens may induce or exacerbate symptoms of angioedema;
- exogenous estrogens may cause the onset or worsening of symptoms of hereditary or acquired angioedema.
The incidence of breast cancer diagnosis is slightly increased among women using COCs. Since breast cancer is rare in women under 40 years of age, the increase in the number of diagnosed cases of breast cancer among women currently or recently using COCs is small relative to the overall risk of breast cancer. The relationship with COC use is unknown. See also sections "Contraindications" and "Special precautions for use".
Interactions
Breakthrough bleeding and/or reduced contraceptive efficacy may occur due to interactions between other medicinal products (enzyme inducers) and oral contraceptives (see section "Interaction with other medicinal products and other forms of interaction").
Reporting of suspected adverse reactions
Reporting suspected adverse reactions after medicine authorization is important. It allows continued monitoring of the benefit-risk balance of the medicine. Healthcare professionals and patients, as well as their legal representatives, should report any suspected adverse reactions and lack of efficacy through the automated pharmacovigilance information system at the following link: https://aisf.dec.gov.ua.
Shelf life. 3 years.
Do not use after the expiry date stated on the packaging.
Storage conditions.
Store in the original packaging at a temperature not exceeding 30 °C. Keep out of reach of children.
Packaging.
Film-coated tablets, 0.02 mg/3 mg, № 28 (24 + 4) in a blister; 1 blister with a calendar strip and blister holder in a cardboard box.
Prescription status. Prescription only.
Manufacturer. Laboratorios Leon Farma, S.A.
Manufacturer's address.
Calle La Vallina S/N, Poligono Industrial Navatejera, Villaquilambre, 24193, Spain.
Marketing Authorization Holder.
LLC "WORWARTS PHARMA".
Address of the Marketing Authorization Holder.
4, Omelyan Prytsaka St., Kyiv, 03142, Ukraine.