Fulvestrant-Vista
Ucrania
Contenido
INSTRUCCIONES PARA USO MÉDICO DEL MEDICAMENTO Fulvestrant-Vista (Fulvestrant-Vista)
Composición:
Principio activo: fulvestrant;
1 jeringa precargada (5 ml) contiene fulvestrant 250 mg;
Excipientes: etanol 96 %, alcohol bencílico (E1519), benzoato de bencilo, aceite de ricino, refinado.
Forma farmacéutica. Solución inyectable.
Propiedades físicas y químicas principales: líquido viscoso transparente, incoloro a amarillo.
Grupo farmacoterapéutico. Antagonistas hormonales y agentes similares. Agentes antiestrógenos. Código ATC L02B A03.
Propiedades farmacodinámicas.
Farmacodinamia.
Mecanismo de acción y efectos farmacodinámicos.
El fulvestranto es un antagonista competitivo de los receptores de estrógenos (RE), con una afinidad comparable a la del estradiol. El fulvestranto bloquea el efecto trófico de los estrógenos sin mostrar actividad agonista parcial (efecto estrogénico). Su mecanismo de acción está relacionado con la regulación negativa de los niveles de proteínas de los receptores de estrógenos. Estudios clínicos en mujeres posmenopáusicas con cáncer de mama primario han demostrado que el fulvestranto reduce significativamente los niveles de proteínas de RE en tumores RE positivos en comparación con placebo. También se observó una reducción significativa en la expresión de los receptores de progesterona, lo que concuerda con la ausencia de efectos agonistas estrogénicos. Asimismo, se ha demostrado que el fulvestranto en una dosis de 500 mg suprime en mayor grado que en dosis de 250 mg los RE y el marcador de proliferación Ki67 en tumores de mama durante el tratamiento neoadyuvante en mujeres posmenopáusicas.
Seguridad clínica y eficacia del medicamento en fases avanzadas del cáncer de mama. Dos estudios clínicos de Fase 3 se realizaron en conjunto con 851 mujeres posmenopáusicas con cáncer de mama en estadios avanzados que habían experimentado recidiva durante o después de terapia hormonal adyuvante, o progresión tras terapia hormonal por enfermedad avanzada. El 77 % de la población estudiada tenía cáncer de mama con receptores de estrógenos positivos. Estos estudios compararon la seguridad y eficacia de la administración mensual de fulvestranto a dosis de 250 mg frente a la administración diaria de 1 mg de anastrozol (inhibidor de la aromatasa). En conjunto, el fulvestranto en dosis mensual de 250 mg fue al menos tan eficaz como el anastrozol respecto a la supervivencia libre de progresión de la enfermedad, la respuesta objetiva y el tiempo hasta el fallecimiento. No hubo diferencias estadísticamente significativas entre los dos grupos de tratamiento en ninguno de estos puntos finales. El punto final principal fue la supervivencia libre de progresión de la enfermedad. Un análisis combinado de ambos estudios mostró que la progresión se observó en el 83 % de las pacientes tratadas con fulvestranto en comparación con el 85 % de las pacientes tratadas con anastrozol. El análisis combinado reveló una razón de riesgos para el fulvestranto 250 mg frente al anastrozol respecto al parámetro de supervivencia libre de progresión de 0,95 (IC del 95 %: 0,82–1,10). La tasa de respuesta objetiva para el fulvestranto 250 mg fue del 19,2 % frente al 16,5 % para el anastrozol. La mediana del tiempo hasta el fallecimiento fue de 27,4 meses en las pacientes tratadas con fulvestranto y de 27,6 meses en las tratadas con anastrozol. La razón de riesgos del fulvestranto 250 mg frente al anastrozol respecto al tiempo hasta el fallecimiento fue de 1,01 (IC del 95 %: 0,86–1,19). Efecto sobre el endometrio en la posmenopausia. Datos preclínicos indican ausencia de efecto estimulante del fulvestranto sobre el endometrio en la posmenopausia. Un estudio de dos semanas con voluntarias sanas posmenopáusicas que recibieron etinilestradiol 20 µg/día mostró que, en comparación con el tratamiento previo con placebo, el tratamiento previo con fulvestranto 250 mg redujo significativamente el efecto estimulante sobre el endometrio, medido por ecografía mediante el espesor endometrial. El tratamiento neoadyuvante durante hasta 16 semanas en pacientes con cáncer de mama que recibieron fulvestranto 500 mg o fulvestranto 250 mg no provocó cambios clínicamente relevantes en el espesor endometrial, lo que indica ausencia de efecto agonista. Hasta la fecha, no hay evidencia de efectos adversos sobre el endometrio en pacientes tratadas por cáncer de mama. No existen datos disponibles sobre la estructura morfológica del endometrio. En dos estudios cortos (1 y 12 semanas) con pacientes premenopáusicas con enfermedades ginecológicas benignas, no se observaron diferencias estadísticamente significativas en el espesor endometrial entre los grupos tratados con fulvestranto y placebo, confirmado mediante ecografía.
Efecto sobre el hueso.
No existen datos a largo plazo sobre el efecto del fulvestranto sobre el hueso. El tratamiento neoadyuvante durante hasta 16 semanas en pacientes con cáncer de mama tratadas con fulvestranto 500 mg o 250 mg no provocó cambios clínicamente relevantes en los niveles séricos de marcadores de remodelación ósea.
Población pediátrica.
El fulvestranto no está indicado para el tratamiento de niños.
En un estudio abierto de Fase 2 se evaluó la seguridad, eficacia y farmacocinética del fulvestranto en 30 niñas de 1 a 8 años con pubertad precoz progresiva asociada al síndrome de McCune-Albright (MAS). Las niñas recibieron mensualmente 4 mg/kg de fulvestranto por vía intramuscular. En este estudio de 12 meses se evaluaron diversos puntos finales relacionados con la eficacia del medicamento en el MAS. Los resultados mostraron una reducción en la frecuencia de hemorragias vaginales y una disminución en la velocidad de maduración del índice óseo. Las concentraciones mínimas en estado de equilibrio del fulvestranto en niños fueron comparables a las observadas en adultos (véase la sección «Farmacocinética»). No surgieron nuevos problemas de seguridad durante este pequeño estudio, aunque aún no están disponibles datos a cinco años.
Farmacocinética.
Absorción.
Tras la administración de fulvestranto mediante inyección intramuscular de liberación prolongada, este se absorbe lentamente, alcanzando la concentración máxima en plasma (Cmax) aproximadamente a los 5 días. Con un régimen de dosis de 500 mg, los niveles de exposición en estado de equilibrio o cercanos a él se alcanzan durante el primer mes de tratamiento (valores medios [coeficiente de variación (CV)]: AUC 475 [33,4 %] ng·día/mL, Cmax 25,1 [35,3 %] ng/mL, Cmin 16,3 [25,9 %] ng/mL, respectivamente). En estado de equilibrio, las concentraciones plasmáticas de fulvestranto permanecen dentro de un rango relativamente estrecho, con una diferencia aproximada de tres veces entre las concentraciones máxima y mínima. Tras la administración intramuscular en un rango de dosis de 50 a 500 mg, la exposición es aproximadamente proporcional a la dosis.
Distribución.
El fulvestranto se distribuye amplia y rápidamente. El elevado volumen de distribución aparente en estado de equilibrio (Vdss), que oscila entre 3 y 5 L/kg, indica una distribución predominantemente extravascular. El fulvestranto se une extensamente (99 %) a las proteínas plasmáticas. Los principales componentes de unión son las fracciones de lipoproteínas de muy baja densidad (VLDL), lipoproteínas de baja densidad (LDL) y lipoproteínas de alta densidad (HDL). No se han realizado estudios sobre interacciones por unión competitiva a proteínas. No se ha establecido el papel de la globulina que une hormonas sexuales (SHBG).
Biotransformación.
El metabolismo del fulvestranto no se ha estudiado completamente, pero incluye una combinación de múltiples vías metabólicas posibles, similares a las de los esteroides endógenos. Los metabolitos identificados (incluyendo 17-cetona, sulfona, 3-sulfato, 3- y 17-glucurónido) son menos activos o muestran una actividad similar al fulvestranto en modelos antiestrógenos. Datos procedentes de estudios con microsomas hepáticos humanos y enzimas recombinantes humanos indican que CYP3A4 es el único isoenzima del sistema P450 implicado en la oxidación del fulvestranto; sin embargo, se considera que in vivo predominan vías independientes de P450. Datos in vitro indican que el fulvestranto no inhibe los isoenzimas CYP450.
Eliminación.
El fulvestranto se elimina principalmente en forma metabolizada. La vía principal de eliminación es fecal, con menos del 1 % excretado en orina. El fulvestranto tiene un aclaramiento elevado, de 11 ± 1,7 mL/min/kg, lo que indica un alto coeficiente de extracción hepática. El periodo de semivida final (t1/2) tras administración intramuscular está determinado por la velocidad de absorción y se ha estimado en 50 días.
Categorías especiales de pacientes.
No se han detectado diferencias en el perfil farmacocinético del fulvestranto según la edad (rango de 33 a 89 años), peso corporal (40 a 127 kg) o ascendencia racial.
Alteración de la función renal.
La alteración leve y moderada de la función renal no tuvo un impacto clínicamente significativo sobre la farmacocinética del fulvestranto.
Alteración de la función hepática.
La farmacocinética del fulvestranto se evaluó en un estudio clínico con dosis única en mujeres con alteración hepática leve a moderada (clase A y B según la clasificación de Child-Pugh). Se administraron dosis altas mediante inyección intramuscular durante un período breve. En comparación con sujetos sanos, en mujeres con alteración hepática se observó un aumento del AUC de casi 2,5 veces. Se espera que este aumento de la exposición sea bien tolerado en pacientes que reciben fulvestranto.
Población pediátrica.
La farmacocinética del fulvestranto se evaluó en 30 niñas con pubertad precoz progresiva asociada al síndrome de McCune-Albright. Las pacientes pediátricas (de 1 a 8 años) recibieron fulvestranto a dosis de 4 mg/kg por vía intramuscular mensualmente. La media geométrica (desviación estándar) de la concentración mínima en estado de equilibrio (Cmin,ss) y del AUCss fue de 4,2 (0,9) ng/mL y 3680 (1020) ng*hora/mL, respectivamente. Aunque los datos son limitados, las concentraciones mínimas en estado de equilibrio del fulvestranto en niños corresponden a las concentraciones observadas en adultos.
Características clínicas.
Indicaciones.
Fulvestrant-Vista está indicado:
- como monoterapia para el tratamiento del cáncer de mama localmente avanzado o metastásico con receptores positivos a estrógenos en mujeres en posmenopausia:
- que previamente no hayan recibido terapia hormonal, o
- en caso de recidiva de la enfermedad durante o después de terapia adyuvante antiestrógena o progresión de la enfermedad durante terapia antiestrógena;
- en combinación con palbociclib para el tratamiento del cáncer de mama positivo para receptores hormonales (HR-positivo), negativo para el receptor del factor de crecimiento epidérmico humano 2 (HER2-negativo), localmente avanzado o metastásico en mujeres que hayan recibido terapia endocrina previa (véase la sección «Farmacodinamia»).
En mujeres en premenopausia o perimenopausia, el tratamiento combinado con palbociclib debe administrarse junto con un análogo del hormona liberadora de gonadotropina (GnRH).
Contraindicaciones.
Hipersensibilidad al principio activo o a cualquiera de los excipientes.
Embarazo o lactancia (véase la sección «Uso durante el embarazo o la lactancia»).
Insuficiencia hepática grave (véanse las secciones «Farmacocinética» y «Precauciones de uso»).
Interacción con otros medicamentos y otras formas de interacción.
Los estudios clínicos de interacción con midazolam (un sustrato del CYP3A4) demostraron que fulvestrant no inhibe el CYP3A4. Los estudios clínicos de interacción con rifampicina (inductor del CYP3A4) y ketoconazol (inhibidor del CYP3A4) no mostraron cambios clínicamente relevantes en el aclaramiento de fulvestrant. Por lo tanto, en pacientes que reciben simultáneamente fulvestrant e inhibidores o inductores del CYP3A4, no se requiere ajuste de la dosis.
Características de aplicación.
Fulvestrant-Vista debe administrarse con precaución a pacientes con alteración hepática de leve a moderada (ver las secciones «Farmacocinética», «Contraindicaciones» y «Vía de administración y dosis»).
Fulvestrant-Vista debe administrarse con precaución a pacientes con alteración renal grave (depuración de creatinina inferior a 30 ml/min).
Debido a la vía de administración intramuscular, Fulvestrant-Vista debe administrarse con precaución en pacientes con diátesis hemorrágica, trombocitopenia o aquellos que estén tomando anticoagulantes.
Se han observado reacciones tromboembólicas en mujeres con cáncer de mama en evolución, registradas durante estudios clínicos con fulvestrant (ver sección «Reacciones adversas»). Esto debe tenerse en cuenta al prescribir el medicamento a pacientes que pertenezcan a grupos de riesgo.
Se han notificado reacciones relacionadas con el sitio de inyección tras la administración de fulvestrant, incluyendo ciática, neuralgia, dolor neuropático y neuropatía periférica. Debido a la proximidad del nervio ciático, se debe tener precaución al administrar Fulvestrant-Vista en el cuadrante superior externo de la nalga (ver secciones «Vía de administración y dosis» y «Reacciones adversas»). No existen datos de estudios a largo plazo sobre el efecto de fulvestrant sobre el hueso. Debido al mecanismo de acción de fulvestrant, existe un riesgo potencial de desarrollo de osteoporosis. No se ha investigado la seguridad ni la eficacia del uso de fulvestrant (como monoterapia o en combinación con palbociclib) en pacientes con enfermedad visceral grave. Al combinar el medicamento Fulvestrant-Vista con palbociclib, véase también la ficha técnica resumida del medicamento palbociclib.
Efecto sobre las pruebas de estradiol mediante anticuerpos.
Debido a la similitud estructural entre fulvestrant y estradiol, fulvestrant puede interferir en los análisis del nivel de estradiol basados en reacciones con anticuerpos, provocando resultados falsamente elevados.
Sustancias auxiliares.
Este medicamento contiene un 10 % v/v de etanol (alcohol), es decir, hasta 1 g por dosis, lo que equivale a 20 ml de cerveza o 8 ml de vino por dosis. Puede ser perjudicial para pacientes con alcoholismo. Debe tenerse precaución en su uso durante el embarazo y la lactancia, en niños, pacientes con enfermedad hepática y en pacientes con epilepsia.
Este medicamento contiene aceite de ricino, que puede provocar reacciones alérgicas graves. Fulvestrant-Vista contiene 1 g de alcohol bencílico por cada dosis, lo que equivale a 100 mg/ml. El alcohol bencílico puede provocar reacciones alérgicas.
Grandes volúmenes deben administrarse con precaución y solo en caso de necesidad extrema, especialmente en mujeres embarazadas o en período de lactancia, así como en pacientes con alteraciones hepáticas o renales, debido al riesgo de acumulación y toxicidad (acidosis metabólica).
Niños.
Fulvestrant-Vista no se recomienda para su uso en niños y adolescentes, ya que su seguridad y eficacia en este grupo de edad no han sido establecidas (ver sección «Farmacodinamia»).
Uso durante el embarazo o la lactancia.
Mujeres en edad fértil.
Las pacientes en edad fértil deben utilizar métodos anticonceptivos eficaces durante el tratamiento con Fulvestrant-Vista y durante 2 años tras la última dosis.
Embarazo.
Fulvestrant-Vista está contraindicado durante el embarazo (ver sección «Contraindicaciones»). Se ha demostrado que fulvestrant atraviesa la barrera placentaria tras una administración intramuscular única en animales. Los estudios en animales han mostrado toxicidad reproductiva, incluyendo un aumento en la frecuencia de anomalías y mortalidad fetal. Si una paciente queda embarazada durante el tratamiento con Fulvestrant-Vista, debe informársele sobre el riesgo potencial para el feto y el riesgo potencial de interrupción del embarazo.
Período de lactancia.
Debe suspenderse la lactancia durante el tratamiento con Fulvestrant-Vista. Fulvestrant se excreta en la leche de animales lactantes. Actualmente no se sabe si fulvestrant pasa a la leche materna humana. Debido al riesgo de reacciones adversas graves provocadas por fulvestrant en lactantes alimentados con leche materna, la lactancia está contraindicada durante el uso de este medicamento (ver sección «Contraindicaciones»).
Fertilidad.
No se ha estudiado el efecto de fulvestrant sobre la fertilidad en humanos.
Capacidad para afectar la velocidad de reacción al conducir vehículos o manejar mecanismos.
Fulvestrant-Vista no afecta o tiene un efecto insignificante sobre la capacidad para conducir vehículos o manejar maquinaria. Sin embargo, dado que durante el tratamiento con fulvestrant se han notificado casos muy frecuentes de astenia, los pacientes que experimenten esta reacción adversa durante la conducción de vehículos o el manejo de maquinaria deben extremar las precauciones.
Vía de administración y dosis.
Dosificación.
Mujeres adultas (incluyendo personas de edad avanzada).
La dosis recomendada es de 500 mg con un intervalo de 1 mes, administrándose una dosis adicional de 500 mg 2 semanas después de la primera inyección.
Cuando se utilice el medicamento Fulvestrant-Vista en combinación con palbociclib, véase también la ficha técnica resumida del medicamento palbociclib.
A las mujeres en período pre/perimenopáusico se les debe administrar agonistas del hormona liberadora de la hormona luteinizante (LHRH) antes y durante la terapia combinada con el medicamento Fulvestrant-Vista y palbociclib, de acuerdo con las normas locales de práctica clínica.
Categorías especiales de pacientes.
Alteración de la función renal.
No es necesario ajustar la dosis en pacientes con alteración renal de leve a moderada (aclaramiento de creatinina ≥30 ml/min). La eficacia y seguridad del medicamento no han sido evaluadas en pacientes con alteración renal grave (aclaramiento de creatinina <30 ml/min); por lo tanto, se debe administrar el medicamento con precaución en estos pacientes (véase la sección «Propiedades farmacodinámicas»).
Alteración de la función hepática.
No es necesario ajustar la dosis en pacientes con alteración hepática de leve a moderada. Sin embargo, se debe administrar el medicamento Fulvestrant-Vista con precaución en estos pacientes debido al posible aumento en la exposición al fulvestrant. No existen datos disponibles en pacientes con alteración hepática grave (véanse las secciones «Farmacocinética» y «Precauciones de uso»).
Vía de administración.
Fulvestrant-Vista debe administrarse mediante dos inyecciones intramusculares consecutivas y lentas (1-2 minutos por inyección), de 5 ml cada una, en cada nalga (región glútea).
Instrucciones para la administración.
El medicamento debe administrarse de acuerdo con las normas locales para la realización de inyecciones intramusculares de gran volumen.
NOTA. Debido a la proximidad del nervio ciático, se debe tener precaución al administrar el medicamento Fulvestrant-Vista en el cuadrante superior externo de la región glútea (véase la sección «Precauciones de uso»).
Advertencia: no esterilizar en autoclave la aguja de seguridad (aguja hipodérmica recubierta con capuchón «BD SafetyGlideTM Safety Hypodermic Needle») antes de su uso.
Durante todo el tiempo de uso y eliminación, las manos deben mantenerse detrás de la aguja.
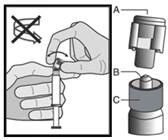
Para cada una de las dos jeringas:
|
Figura 1
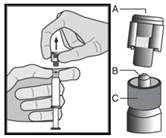
|
|
Figura 2
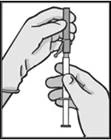
|
|
Figura 3
|
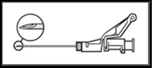
(1–2 minutos/inyección) en el área glútea. Para comodidad del usuario, el bisel de la aguja debe estar orientado hacia el brazo de la palanca (ver fig. 4). |
Figura 4
|
NOTA. Durante la activación, mantener la aguja orientada lejos de uno mismo y de otras personas. Escuchar el clic y verificar visualmente que la punta de la aguja quede completamente cubierta. |
Figura 5
|
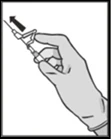
Desecho.
Las jeringas precargadas están destinadas únicamente para uso único.
Este medicamento puede representar un peligro para el medio acuático.
Todos los productos médicos no utilizados o residuos deben desecharse de acuerdo con los requisitos locales.
Niños.
La seguridad y eficacia de fulvestrant en niños desde el nacimiento hasta los 18 años no han sido establecidas. La información disponible actualmente se presenta en la sección «Propiedades farmacológicas», sin embargo, no pueden hacerse recomendaciones sobre la dosificación.
Sobredosis.
Existen informes aislados sobre casos de sobredosis con fulvestrant en humanos. En caso de sobredosis, se recomienda un tratamiento sintomático y de apoyo. En estudios en animales, dosis altas de fulvestrant no provocaron otros efectos distintos de los directa o indirectamente relacionados con la acción antiestrógena.
Reacciones adversas.
Resumen del perfil de seguridad.
Monoterapia.
Esta sección contiene información basada en todas las reacciones adversas notificadas procedentes de estudios clínicos, estudios poscomercialización o notificaciones espontáneas. En la muestra combinada de datos de monoterapia con fulvestranto, las reacciones adversas notificadas con mayor frecuencia fueron reacciones en el sitio de inyección, astenia, náuseas y aumento de los niveles de enzimas hepáticas (ALT [alanina aminotransferasa], AST [aspartato aminotransferasa], FA [fosfatasa alcalina]).
Los datos sobre frecuencia se resumen a partir de todos los eventos notificados, independientemente de la evaluación del investigador sobre la relación causal. La mediana de duración del tratamiento con fulvestranto 500 mg en la muestra combinada de datos fue de 6,5 meses. Lista de reacciones adversas en forma de tabla.
Las reacciones adversas que figuran a continuación se clasifican según la frecuencia y por clasificación de sistemas de órganos (CSO). La agrupación por frecuencia se realizó según las siguientes normas aceptadas: muy frecuentes (>1/10), frecuentes (de >1/100 a <1/10), raras (de >1/1 000 a <1/100). Dentro de cada grupo de frecuencia, las reacciones adversas se enumeran en orden decreciente de gravedad.
Reacciones adversas por clasificación de sistemas de órganos y frecuencia notificadas con el medicamento en pacientes durante el tratamiento con fulvestranto en monoterapia
Tabla 1
| Reacciones adversas clasificadas según la frecuencia y por órganos y sistemas afectados |
||
| Infecciones e infestaciones |
Frecuentes |
Infecciones del tracto urinario |
| Trastornos del sistema inmunitario |
Muy frecuentes |
Reacciones de hipersensibilidad |
| Poco frecuentes |
Reacciones anafilácticas |
|
| Trastornos del metabolismo y nutrición |
Frecuentes |
Anorexia |
| Trastornos del sistema nervioso |
Frecuentes |
Cefalea |
| Trastornos vasculares |
Muy frecuentes |
Bochornos |
| Frecuentes |
Tromboembolismo venoso |
|
| Trastornos gastrointestinales |
Muy frecuentes |
Náuseas |
| Frecuentes |
Vómitos, diarrea |
|
| Trastornos hepáticos y de las vías biliares |
Muy frecuentes |
Aumento de los niveles de enzimas hepáticas (ALT, AST, ALP) |
| Frecuentes |
Aumento del nivel de bilirrubina |
|
| Poco frecuentes |
Insuficiencia hepática, hepatitis, aumento de los niveles de GGT |
|
| Trastornos de la piel y del tejido subcutáneo |
Muy frecuentes |
Erupciones cutáneas |
| Trastornos del sistema musculoesquelético y del tejido conectivo |
Muy frecuentes |
Dolor articular y muscular |
| Frecuentes |
Dolor de espalda |
|
| Trastornos del sistema reproductor y de las mamas |
Frecuentes |
Sangrado vaginal |
| Poco frecuentes |
Candidiasis vaginal, leucorrea |
|
| Trastornos generales y condiciones en el sitio de administración |
Muy frecuentes |
Astenia, reacciones en el sitio de inyección |
| Frecuentes |
Neuropatía periférica, ciática |
|
| Poco frecuentes |
Hemorragias en el sitio de inyección, hematomas en el sitio de inyección, neuralgia |
|
| Trastornos sanguíneos y del sistema linfático |
Frecuentes |
Disminución del número de plaquetas |
a Incluye reacciones adversas al medicamento para las cuales la relación con fulvestranto no puede evaluarse con precisión debido a la enfermedad subyacente.
b El término «reacciones en el lugar de inyección» no incluye los términos «hemorragia en el lugar de inyección» y «hematoma en el lugar de inyección», «ciática», «neuralgia», «neuropatía periférica».
c La reacción no se observó durante los ensayos clínicos amplios.
La frecuencia se calculó utilizando el límite superior del intervalo de confianza del 95 % para la estimación del punto. Se calculó como 3/560 (donde 560 es el número de pacientes en los ensayos clínicos amplios), lo que corresponde a la categoría de frecuencia «poco frecuente».
d Incluye artromialgias y, con menor frecuencia, dolor músculo-esquelético, mialgias y dolor en las extremidades.
e Existen algunas diferencias en la frecuencia de reacciones adversas en las categorías correspondientes por órganos y sistemas entre el estudio de seguridad y el estudio FALCON.
f No se observaron reacciones adversas durante el estudio FALCON.
Descripción de reacciones adversas individuales.
La descripción siguiente se basa en el análisis de seguridad del grupo de pacientes que recibieron al menos una dosis de fulvestranto y del grupo de pacientes que recibieron al menos una dosis de anastrozol en el estudio de Fase 3 FALCON.
Dolor articular y dolor músculo-esquelético.
Según los datos del estudio FALCON, el número de pacientes que informaron dolor articular y dolor músculo-esquelético fue de 65 (31,2 %) y 48 (24,1 %) con fulvestranto y anastrozol, respectivamente. De las 65 pacientes que recibieron fulvestranto, el 40 % (26/65) experimentaron dolor articular y músculo-esquelético durante el primer mes de tratamiento, y el 66 % (43/65) durante los primeros 3 meses de tratamiento. Ninguna de las pacientes informó casos de grado ≥3 según CTCAE ni casos que requirieran reducción de la dosis del medicamento, suspensión temporal o interrupción del tratamiento debido a estas reacciones adversas.
Terapia combinada con palbociclib.
El perfil general de seguridad del fulvestranto cuando se utiliza en combinación con palbociclib se basa en datos de 517 pacientes con cáncer de mama localmente avanzado o metastásico HR-positivo y HER2-negativo procedentes del estudio aleatorizado PALOMA3 (ver sección «Farmacodinamia»). Las reacciones adversas más frecuentes (≥20 %) de cualquier grado notificadas en pacientes que recibieron fulvestranto en combinación con palbociclib fueron neutropenia, leucopenia, infecciones, fatiga, náuseas, anemia, estomatitis, diarrea y trombocitopenia. Las reacciones adversas más frecuentes (≥2 %) de grado ≥III fueron neutropenia, leucopenia, anemia, infecciones, aumento de los niveles de AST, trombocitopenia y fatiga.
En la tabla siguiente se muestran los datos sobre las reacciones adversas observadas en el estudio PALOMA3.
La duración media del tratamiento con fulvestranto fue de 11,2 meses en el grupo de fulvestranto + palbociclib y de 4,9 meses en el grupo de fulvestranto + placebo. La duración media del tratamiento con palbociclib en el grupo de fulvestranto + palbociclib fue de 10,8 meses.
Reacciones adversas según datos del estudio PALOMA3 (N=517)
Tabla 2
| Clase de órganos Frecuencia Término de uso preferente |
Fulvestranto + palbociclib (N=345) |
Fulvestranto + placebo (N=172) |
||
| Todos los grados n (%) |
≥Grado III n (%) |
Todos los grados n (%) |
≥Grado III n (%) |
|
| Enfermedades infecciosas y parasitarias |
||||
| Muy frecuentes |
||||
| Infeccionesb |
163 (47,2) |
11 (3,2) |
54 (31,4) |
5 (2,9) |
| Del sistema sanguíneo y linfático |
||||
| Muy frecuentes |
||||
| Neutropeniac |
287 (83,2) |
228 (66,1) |
7 (4,1) |
1 (0,6) |
| Leucopeniad |
183 (53,0) |
105 (30,4) |
9 (5,2) |
2 (1,2) |
| Anemiae |
102 (29,6) |
12 (3,5) |
22 (12,8) |
3 (1,7) |
| Trombocitopeniaf |
78 (22,6) |
8 (2,3) |
0 (0,0) |
0 |
| No frecuentes |
||||
| Neutropenia febril |
3 (0,9) |
3 (0,9) |
1 (0,6) |
1 (0,6) |
| Alteraciones del metabolismo y nutrición |
||||
| Muy frecuentes |
||||
| Pérdida de apetito |
55 (15,9) |
3 (0,9) |
14 (8,1) |
1 (0,6) |
| Del sistema nervioso |
||||
| Frecuentes |
||||
| Disgeusia |
23 (6,7) |
0 |
5 (2,9) |
0 |
| De los órganos de la visión |
||||
| Frecuentes |
||||
| Aumento de la lagrimeación |
22 (6,4) |
0 |
2 (1,2) |
0 |
| Visión borrosa |
20 (5,8) |
0 |
3 (1,7) |
0 |
| Ojos secos |
13 (3,8) |
0 |
3 (1,7) |
0 |
| Del aparato respiratorio, tórax y mediastino |
||||
| Frecuentes |
||||
| Hemorragia nasal |
23 (6,7) |
0 |
3 (1,7) |
0 |
| Del tubo digestivo |
||||
| Muy frecuentes |
||||
| Náuseas |
117 (33,9) |
0 |
48 (27,9) |
1 (0,6) |
| Estomatitisc |
97 (28,1) |
2 (0,6) |
22 (12,8) |
0 |
| Diárrhea |
81 (23,5) |
0 |
33 (19,2) |
2 (1,2) |
| Vómitos |
65 (18,8) |
2 (0,6) |
26 (15,1) |
1 (0,6) |
| De la piel y tejido subcutáneo |
||||
| Muy frecuentes |
||||
| Alopecia |
62 (18,0) |
0 |
11 (6,4) |
0 |
| Erupción cutáneah |
58 (16,8) |
2 (0,6) |
11 (6,4) |
0 |
| Frecuentes |
||||
| Piel seca |
21 (6,1) |
0 |
2 (1,2) |
0 |
| Alteraciones generales y reacciones en el lugar de administración |
||||
| Muy frecuentes |
||||
| Cansancio |
142 (41,2) |
8 (2,3) |
50 (29,1) |
2 (1,2) |
| Fiebre |
44 (12,8) |
1 (0,3) |
9 (5,2) |
0 |
| Frecuentes |
||||
| Astenia |
26 (7,5) |
0 |
9 (5,2) |
1 (0,6) |
| Resultados de pruebas de laboratorio |
||||
| Frecuentes |
||||
| Aumento de los niveles de AST |
26 (7,5) |
10 (2,9) |
9 (5,2) |
3 (1,7) |
| Aumento de los niveles de ALT |
20 (5,8) |
6 (1,7) |
6 (3,5) |
0 |
ALT = alanina aminotransferasa; AST = aspartato aminotransferasa; N/n = número de pacientes
a Se indican los términos preferentes (TP) para los fenómenos según MedDRA 17.1.
b Todos los TP pertenecientes a la clase de órganos y sistemas: infecciones e infestaciones.
c La neutropenia incluye los siguientes TP: neutropenia, recuento de neutrófilos disminuido.
d La leucopenia incluye los siguientes TP: leucopenia, recuento de leucocitos disminuido.
e La anemia incluye los siguientes TP: anemia, disminución del nivel de hemoglobina, disminución del nivel de hematocrito.
f La trombocitopenia incluye los siguientes TP: trombocitopenia, recuento de plaquetas disminuido.
g La estomatitis incluye los siguientes TP: estomatitis aftosa, queilitis, glositis, glosodinia, úlceras en la cavidad oral, inflamación de las membranas mucosas, dolor en la cavidad oral, molestias orofaríngeas, dolor orofaríngeo, estomatitis.
h Las erupciones incluyen los siguientes TP: erupción, erupción maculopapular, erupción pruriginosa, erupción eritematosa, erupción papular, dermatitis, dermatitis acnéiforme, erupciones cutáneas tóxicas.
Descripción de reacciones adversas individuales.
Neutropenia.
En los pacientes que recibieron fulvestranto en combinación con palbociclib en el estudio PALOMA-3, se notificó neutropenia de cualquier grado en 287 (83,2 %) pacientes, neutropenia de grado III en 191 (55,4 %) pacientes y neutropenia de grado IV en 37 (10,7 %) pacientes. En el grupo que recibió fulvestranto + placebo (n=172), se notificó neutropenia de cualquier grado en 7 (4,1 %) pacientes y neutropenia de grado III en 1 (0,6 %) paciente. No se notificaron casos de neutropenia de grado IV en el grupo que recibió fulvestranto + placebo.
En los pacientes que recibieron fulvestranto en combinación con palbociclib, la mediana del tiempo hasta el primer episodio de neutropenia fue de 15 días (rango: 13-317), y la mediana de duración de la neutropenia de grado ≥III fue de 7 días. En el 0,9 % de los pacientes que recibieron fulvestranto en combinación con palbociclib se notificaron casos de neutropenia febril. Notificación de reacciones adversas sospechosas.
La notificación de reacciones adversas sospechosas tras la autorización del medicamento es importante. Permite continuar con el seguimiento de la relación beneficio/riesgo del medicamento. Los profesionales sanitarios deben informar sobre cualquier reacción adversa sospechosa a través del sistema nacional de notificación.
Periodo de validez.
3 años.
Las fluctuaciones de temperatura fuera del rango de 2 a 8 °C deben estar limitadas. Se debe evitar el almacenamiento a temperaturas superiores a 30 °C y no exceder un período de 28 días en el que la temperatura media de almacenamiento del producto sea inferior a 25 °C (pero superior a 2-8 °C). Tras cualquier fluctuación de temperatura, el medicamento debe devolverse inmediatamente a las condiciones recomendadas de almacenamiento (almacenamiento y transporte en nevera a una temperatura de 2 a 8 °C). Las fluctuaciones de temperatura tienen un efecto acumulativo sobre la calidad del medicamento, y el período de 28 días no debe contarse tras el periodo de validez de dos años del medicamento Fulvestranto-Vista. Las temperaturas inferiores a 2 °C no dañan el medicamento siempre que no se almacene a temperaturas inferiores a -20 °C.
Condiciones de almacenamiento.
Mantener y transportar a una temperatura de 2 a 8 °C.
Mantener fuera del alcance y de la vista de los niños.
Conservar las jeringas precargadas en su envase original para protegerlas de la luz.
Incompatibilidades.
Dado que no existen estudios de compatibilidad, este medicamento no debe mezclarse con otros medicamentos.
Envase.
Una jeringa precargada con control de primera apertura en una caja de cartón con aguja segura (BD SafetyGlide), o dos jeringas precargadas con control de primera apertura en una caja de cartón con dos agujas seguras (BD SafetyGlide).
Categoría de dispensación.
Medicamento sujeto a prescripción médica.
Fabricante.
LABORATORIOS FARMALAN S.A.
Dirección del fabricante y lugar de actividad.
Calle La Valiña s/n – Navatejera, Villacilambre, León, 24193, España.