Ticagrelor Holsten
Polonia
Contenido
- 1. NOMBRE DEL MEDICAMENTO
- 2. COMPOSICIÓN CUALITATIVA Y CUANTITATIVA
- 3. FORMA FARMACÉUTICA
- 4.2 Posología y forma de administración
- 4.3 Contraindicaciones
- 4.4 Advertencias y precauciones especiales de uso
- 4.5 Interacciones con otros medicamentos y otras formas de interacción
- 4.6 Fertilidad, embarazo y lactancia
- 4.7 Efecto sobre la capacidad para conducir y utilizar máquinas
- 4.8 Reacciones adversas
- 4.9 Sobredosificación
- 5.2 Propiedades farmacocinéticas
- 5.3 Datos preclínicos sobre seguridad
- 6.2 Incompatibilidades farmacéuticas
- 6.3 Período de validez
- 6.4 Precauciones especiales de conservación
- 6.5 Tipo y contenido del envase
- 6.6 Medidas especiales de precaución para la eliminación
- 7. TITULAR DEL AUTORIZACIÓN DE COMERCIALIZACIÓN
- 8. NÚMEROS DE AUTORIZACIONES PARA LA COMERCIALIZACIÓN
- 9. FECHA DE PRIMERA CONCESIÓN DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
- 10. FECHA DE APROBACIÓN O DE MODIFICACIÓN PARCIAL DEL TEXTO
CARACTERÍSTICAS DEL PRODUCTO FARMACÉUTICO
1. NOMBRE DEL MEDICAMENTO
Ticagrelor Holsten, 60 mg, comprimidos recubiertos
2. COMPOSICIÓN CUALITATIVA Y CUANTITATIVA
Cada comprimido recubierto contiene 60 mg de ticagrelor.
Para la lista completa de excipientes, ver el punto 6.1.
3. FORMA FARMACÉUTICA
Comprimido recubierto.
Comprimidos redondos (8,1 x 8,1 mm), convexos por ambas caras, de color rosa, marcados con el número «60» en un lado y lisos en el otro lado.
4. DATOS CLÍNICOS DETALLADOS
4.1 Indicaciones
El medicamento Ticagrelor Holsten, en combinación con ácido acetilsalicílico (AAS), está indicado para la prevención de eventos cardiovasculares en adultos:
- con síndrome coronario agudo (SCA), o
- con antecedentes de infarto de miocardio (infarto de miocardio) y alto riesgo de eventos cardiovasculares (ver secciones 4.2 y 5.1).
4.2 Posología y forma de administración
Posología
Los pacientes que tomen el medicamento Ticagrelor Holsten deben tomar diariamente también una dosis de mantenimiento baja de ácido acetilsalicílico (AAS) de 75-150 mg, salvo que esté contraindicado individualmente.
Síndromes coronarios agudos
El tratamiento con Ticagrelor Holsten debe iniciarse con una dosis única de carga de 180 mg (2 comprimidos de 90 mg) y continuar con una dosis de 90 mg dos veces al día.
En pacientes con síndrome coronario agudo (SCA), la duración del tratamiento con Ticagrelor Holsten de 90 mg dos veces al día debe ser de 12 meses, a menos que existan indicaciones clínicas para interrumpir el tratamiento (ver sección 5.1).
La suspensión del AAS puede considerarse tras 3 meses en pacientes con SCA que hayan sido sometidos a una intervención coronaria percutánea (ICP) y que presenten un riesgo elevado de sangrado. En tal caso, debe continuarse la administración de ticagrelor como único tratamiento antiagregante durante 9 meses (ver sección 4.4).
Infarto de miocardio previo
La dosis recomendada de Ticagrelor Holsten es de 60 mg dos veces al día cuando se requiera un tratamiento prolongado en pacientes con infarto de miocardio previo (hace al menos un año) y con alto riesgo de eventos cardiovasculares (ver sección 5.1). El tratamiento puede iniciarse sin interrupción, como continuación del tratamiento inicial de un año con Ticagrelor Holsten 90 mg u otro inhibidor de los receptores de la adenosina difosfato (ADP) en pacientes con SCA y alto riesgo de eventos cardiovasculares. El tratamiento también puede iniciarse hasta 2 años después del infarto de miocardio o dentro del año siguiente a la interrupción del tratamiento previo con un inhibidor del receptor de ADP. Los datos sobre la eficacia y seguridad del uso de ticagrelor más allá de los 3 años de tratamiento prolongado son limitados.
Si se necesita un cambio de medicamento, la primera dosis de Ticagrelor Holsten debe administrarse 24 horas después de la última dosis del otro medicamento antiagregante.
Omisión de dosis
También deben evitarse errores en la posología. Si se omite una dosis de Ticagrelor Holsten, el paciente debe tomar solo un comprimido (la siguiente dosis) según el esquema habitual.
Grupos de pacientes especiales
Pacientes de edad avanzada
No se requiere ajuste de dosis en pacientes de edad avanzada (ver sección 5.2).
Alteraciones de la función renal
No es necesario ajustar la dosis en pacientes con alteraciones de la función renal (ver sección 5.2).
Alteraciones de la función hepática
No se han realizado estudios sobre el uso de ticagrelor en pacientes con alteraciones hepáticas graves y, por tanto, su uso está contraindicado en estos pacientes (ver sección 4.3). Solo existen datos limitados sobre el uso del medicamento en pacientes con alteraciones hepáticas moderadas. No se requiere ajuste de dosis, pero debe administrarse ticagrelor con precaución (ver secciones 4.4 y 5.2). En pacientes con alteraciones hepáticas leves, no es necesario ajustar la dosis (ver sección 5.2).
Niños y adolescentes
No se ha establecido la seguridad ni la eficacia del uso de ticagrelor en niños menores de 18 años.
El uso de ticagrelor no es adecuado en niños para la indicación de anemia falciforme (ver secciones 5.1 y 5.2).
Forma de administración
Vía oral.
Ticagrelor Holsten puede administrarse durante las comidas o independientemente de las mismas.
En caso de pacientes que tengan dificultad para tragar el comprimido/comprimidos enteros, los comprimidos pueden triturarse hasta obtener un polvo fino, mezclarse con media taza de agua y beberse inmediatamente. A continuación, debe enjuagarse el vaso con agua (media taza adicional) y beberse también el contenido.
La mezcla de comprimido(s) triturado(s) con agua también puede administrarse a través de una sonda nasogástrica (CH8 o mayor). Es importante enjuagar la sonda nasogástrica con agua (media taza: mínimo 50 ml, máximo 125 ml) tras la administración de la mezcla.
4.3 Contraindicaciones
- Hipersensibilidad al principio activo o a cualquiera de los excipientes mencionados en el apartado 6.1 (véase el apartado 4.8).
- Hemorragia patológica activa.
- Antecedentes de hemorragia intracraneal (véase el apartado 4.8).
- Alteración hepática grave (véase los apartados 4.2, 4.4 y 5.2).
- Administración concomitante de ticagrelor y potentes inhibidores del enzima CYP3A4 (por ejemplo, ketoconazol, claritromicina, nefazodona, ritonavir y atazanavir), ya que puede provocar un aumento significativo de la exposición al ticagrelor (véase el apartado 4.5).
4.4 Advertencias y precauciones especiales de uso
Riesgo de hemorragia
En pacientes con riesgo aumentado de hemorragia, se debe considerar la relación riesgo-beneficio respecto a la prevención de eventos cardiovasculares (ver secciones 4.8 y 5.1). Cuando existan indicaciones clínicas para el uso de ticagrelor, debe administrarse con precaución en los siguientes grupos de pacientes:
- Pacientes con tendencia a hemorragias (por ejemplo, debido a lesiones recientes, cirugías, trastornos de la coagulación, hemorragia gastrointestinal activa o reciente) o con riesgo aumentado de traumatismos. El uso de ticagrelor está contraindicado en pacientes con hemorragia patológica activa, antecedentes de hemorragia intracraneal y en pacientes con alteraciones graves de la función hepática (ver sección 4.3).
- Pacientes que reciben simultáneamente medicamentos que pueden aumentar el riesgo de hemorragia (por ejemplo, antiinflamatorios no esteroideos (AINE), anticoagulantes orales y/o fármacos fibrinolíticos) administrados en las 24 horas previas a la dosis de ticagrelor.
En dos estudios aleatorizados controlados (TICO y TWILIGHT) con pacientes con síndrome coronario agudo (SCA) sometidos a angioplastia coronaria con stent liberador de fármaco, se demostró que la suspensión de ácido acetilsalicílico (AAS) tras 3 meses de doble terapia antiagregante con ticagrelor y AAS, seguida de continuación con ticagrelor como único antiagregante durante 9 y 12 meses respectivamente, redujo el riesgo de hemorragia sin aumentar el riesgo observado de eventos cardiovasculares adversos graves (MACE, por sus siglas en inglés) en comparación con la continuación de la doble terapia antiagregante. La decisión de suspender el AAS tras 3 meses y continuar con ticagrelor como único antiagregante durante 9 meses en pacientes con riesgo aumentado de hemorragia debe tomarse tras una evaluación clínica individualizada, considerando el riesgo de hemorragia frente al riesgo de eventos trombóticos (ver sección 4.2).
La transfusión de plaquetas no revirtió el efecto antiagregante del ticagrelor en voluntarios sanos y es poco probable que sea clínicamente beneficiosa en pacientes con hemorragia.
Dado que la administración conjunta de desmopresina con ticagrelor no acorta el tiempo de sangrado estándar, es dudoso que la desmopresina sea eficaz en el tratamiento de hemorragias clínicas (ver sección 4.5).
El tratamiento con agentes antifibrinolíticos (ácido aminocaprónico o ácido tranexámico) y/o tratamiento con factor VIIa recombinante puede aumentar la hemostasia. El ticagrelor puede reiniciarse si se ha identificado y controlado la causa de la hemorragia.
Intervenciones quirúrgicas
Se debe informar a los pacientes que deben notificar a sus médicos y dentistas que están tomando ticagrelor antes de cualquier cirugía programada o administración de nuevos medicamentos.
En pacientes del estudio PLATO sometidos a revascularización quirúrgica coronaria (CABG), se observó mayor frecuencia de hemorragias en el grupo tratado con ticagrelor en comparación con clopidogrel cuando el fármaco se interrumpió un día antes de la cirugía; sin embargo, cuando se interrumpió dos o más días antes, la frecuencia de hemorragias graves fue similar en ambos grupos (ver sección 4.8). Si un paciente va a someterse a una cirugía programada y el efecto antiagregante no es deseable, se debe suspender el ticagrelor 5 días antes del procedimiento (ver sección 5.1).
Pacientes con antecedentes de accidente cerebrovascular isquémico
Los pacientes con SCA y antecedentes de accidente cerebrovascular isquémico pueden ser tratados con ticagrelor durante un máximo de 12 meses (estudio PLATO).
Los pacientes con infarto de miocardio previo y antecedentes de accidente cerebrovascular isquémico no fueron incluidos en el estudio PEGASUS. Por tanto, debido a la falta de datos, no se recomienda tratar a estos pacientes por más de un año.
Alteraciones de la función hepática
El uso de ticagrelor está contraindicado en pacientes con alteraciones graves de la función hepática (ver secciones 4.2 y 4.3). La experiencia con ticagrelor en pacientes con insuficiencia hepática moderada es limitada; por tanto, se recomienda precaución en estos pacientes (ver secciones 4.2 y 5.2).
Pacientes con riesgo de eventos de bradicardia
El monitoreo del ECG mediante Holter mostró una mayor frecuencia de pausas ventriculares, en su mayoría asintomáticas, durante el tratamiento con ticagrelor en comparación con clopidogrel. Los pacientes con riesgo aumentado de episodios de bradicardia (por ejemplo, sin marcapasos, con síndrome del seno enfermo, bloqueo auriculoventricular de segundo o tercer grado, o con síncope relacionado con bradicardia) fueron excluidos de los principales estudios de seguridad y eficacia del ticagrelor. Por tanto, debido a la experiencia clínica limitada, el ticagrelor debe usarse con precaución en estos pacientes (ver sección 5.1).
Además, debe tenerse precaución al administrar ticagrelor junto con medicamentos que inducen bradicardia. Sin embargo, no hubo evidencia de efectos adversos clínicamente significativos en el estudio PLATO con la administración concomitante de uno o más medicamentos que inducen bradicardia (es decir, 96% de betabloqueantes, 33% de antagonistas del calcio diltiazem y verapamilo, y 4% de digoxina) (ver sección 4.5).
En el estudio PLATO, en el subgrupo sometido a monitoreo Holter, se observaron más pausas ventriculares >3 segundos en pacientes tratados con ticagrelor que con clopidogrel durante la fase aguda del SCA. El aumento en la detección de pausas ventriculares mediante Holter durante el tratamiento con ticagrelor fue más pronunciado en pacientes con insuficiencia cardíaca crónica que en la población general durante la fase aguda del SCA, pero no en el seguimiento mensual con ticagrelor ni en comparación con clopidogrel. No se observaron consecuencias adversas clínicas asociadas a esta disparidad (incluyendo síncope o implantación de marcapasos) en este grupo de pacientes (ver sección 5.1).
Desde la comercialización del producto, se han notificado casos de bradiarritmias y bloqueos AV en pacientes tratados con ticagrelor, principalmente en pacientes con SCA, donde la isquemia miocárdica y los medicamentos concomitantes que reducen la frecuencia cardíaca o afectan la conducción cardíaca son factores potencialmente contribuyentes. Antes de ajustar el tratamiento, se debe evaluar el estado clínico del paciente y los medicamentos concomitantes como posibles causas.
Disnea
Los pacientes tratados con ticagrelor han notificado disnea. Esta suele ser leve a moderada y frecuentemente remite sin necesidad de suspender el fármaco. En pacientes con asma/enfermedad pulmonar obstructiva crónica (EPOC), puede aumentar el riesgo absoluto de disnea durante el tratamiento con ticagrelor. El ticagrelor debe usarse con precaución en pacientes con antecedentes de asma y/o EPOC. El mecanismo de la disnea no ha sido aclarado. Si un paciente presenta nuevos episodios de disnea, prolongación de los episodios o empeoramiento de los síntomas durante el tratamiento con ticagrelor, se debe realizar una evaluación diagnóstica completa y, si el paciente tolera mal este estado, se debe interrumpir el tratamiento con ticagrelor. Más información detallada en la sección 4.8.
Apnea central del sueño
Desde la comercialización del producto, se han notificado casos de apnea central del sueño, incluyendo respiración de Cheyne-Stokes, en pacientes tratados con ticagrelor. Si se sospecha apnea central del sueño, se debe considerar una evaluación clínica adicional.
Aumento de la creatinina
Durante el tratamiento con ticagrelor puede aumentar la concentración de creatinina. El mecanismo de este fenómeno no ha sido establecido. Se deben realizar pruebas periódicas de función renal según la práctica clínica habitual. En pacientes con SCA se recomienda evaluar la función renal también un mes después del inicio del tratamiento con ticagrelor, prestando especial atención a pacientes con edad ≥75 años, alteraciones de la función renal de moderada a grave y aquellos que toman antagonistas del receptor de angiotensina (ARB).
Aumento del ácido úrico
Durante el tratamiento con ticagrelor puede desarrollarse hiperuricemia (ver sección 4.8). Se debe tener precaución en pacientes con antecedentes de hiperuricemia o gota.
Como medida de precaución, se desaconseja el uso de ticagrelor en pacientes con nefropatía por ácido úrico.
Púrpura trombocitopénica trombótica (TTP)
Durante el tratamiento con ticagrelor se han notificado muy raramente casos de púrpura trombocitopénica trombótica (TTP). Esta se caracteriza por trombocitopenia y anemia hemolítica microangiopática, asociada con síntomas neurológicos, alteraciones renales o fiebre. La TTP es una enfermedad potencialmente mortal que requiere tratamiento rápido, incluyendo plasmaféresis.
Interferencias en pruebas funcionales de plaquetas para el diagnóstico de trombocitopenia inducida por heparina (HIT)
En la prueba funcional de activación plaquetaria inducida por heparina (HIPA), utilizada para diagnosticar la HIT, los anticuerpos contra el complejo factor plaquetario 4/heparina en el suero del paciente activan las plaquetas de donantes sanos en presencia de heparina.
En pacientes que toman ticagrelor se han notificado resultados falsos negativos en pruebas funcionales de plaquetas (incluyendo la prueba HIPA) destinadas a diagnosticar la HIT. Esto se debe a la inhibición del receptor P2Y12 en las plaquetas de donantes sanos por el ticagrelor presente en el suero/plasma del paciente. Es necesaria información sobre el tratamiento concomitante con ticagrelor para interpretar correctamente los resultados de estas pruebas funcionales utilizadas para diagnosticar la HIT.
En pacientes que desarrollen trombocitopenia inducida por heparina, se debe evaluar la relación beneficio-riesgo del tratamiento continuado con ticagrelor, considerando tanto el estado protrombótico de la HIT como el riesgo aumentado de hemorragia durante el tratamiento concomitante con anticoagulantes y ticagrelor.
Otros
Basado en la relación observada en el estudio PLATO entre la dosis de mantenimiento de ácido acetilsalicílico y la eficacia relativa del ticagrelor en comparación con clopidogrel, no se recomienda la administración concomitante de ticagrelor y ácido acetilsalicílico en dosis de mantenimiento altas (>300 mg) (ver sección 5.1).
Interrupción prematura del tratamiento
La interrupción prematura de cualquier tratamiento antiagregante, incluido el medicamento Ticagrelor Holsten, puede aumentar el riesgo de muerte cardiovascular, infarto de miocardio o accidente cerebrovascular debido a la enfermedad subyacente. Por tanto, debe evitarse la interrupción prematura del tratamiento.
Sodio
Ticagrelor Holsten contiene menos de 1 mmol de sodio (23 mg) por dosis, es decir, es esencialmente "exento de sodio".
4.5 Interacciones con otros medicamentos y otras formas de interacción
Ticagrelor es principalmente un sustrato del isoenzima CYP3A4, así como también un inhibidor leve de este.
Ticagrelor es asimismo un sustrato de la glucoproteína P (P-gp) y un inhibidor débil de P-gp, pudiendo aumentar la exposición a sus sustratos.
Influencia de otros medicamentos y productos sobre la acción de ticagrelor
Inhibidores de CYP3A4
- Inhibidores potentes de CYP3A4: la administración concomitante de ketoconazol junto con ticagrelor provocó un aumento de 2,4 veces en la C*max* y de 7,3 veces en el AUC de ticagrelor. La C*max* y el AUC del metabolito activo se redujeron en un 89% y un 56%, respectivamente. Se prevé que otros inhibidores potentes de CYP3A4 (claritromicina, nefazodona, ritonavir, atazanavir) produzcan un efecto similar; por tanto, la administración concomitante de inhibidores potentes de CYP3A4 junto con ticagrelor está contraindicada (ver sección 4.3).
- Inhibidores moderados de CYP3A4: la administración conjunta de diltiazem y ticagrelor provocó un aumento del 69% en la C*max* de ticagrelor y un incremento de 2,7 veces en el AUC, así como una reducción del 38% en la C*max* del metabolito activo, sin afectar su AUC. Ticagrelor no influyó en las concentraciones plasmáticas de diltiazem. Se prevé que otros inhibidores moderados de CYP3A4 (por ejemplo, amprenavir, aprepitant, eritromicina y fluconazol) puedan tener un efecto similar y también pueden administrarse conjuntamente con ticagrelor.
- Se observó un aumento de 2 veces en la exposición a ticagrelor tras el consumo diario de grandes cantidades de zumo de pomelo (3 x 200 ml). No se espera que este incremento en la exposición a ticagrelor sea clínicamente relevante en la mayoría de los pacientes.
Inductores de CYP3A
La administración concomitante de rifampicina y ticagrelor redujo la C*max* y el AUC de ticagrelor en un 73% y un 86%, respectivamente. La C*max* del metabolito activo no se modificó, mientras que su AUC disminuyó en un 46%. Se prevé que otros inductores de CYP3A (por ejemplo, fenitoína, carbamazepina y fenobarbital) también reduzcan la exposición a ticagrelor. La administración concomitante de ticagrelor junto con inductores potentes de CYP3A puede disminuir las concentraciones y la eficacia de ticagrelor; por tanto, no se recomienda su uso combinado.
Ciclosporina (inhibidor de P-gp y CYP3A)
La administración concomitante de ciclosporina (600 mg) y ticagrelor aumentó la C*max* de ticagrelor en un 2,3 veces y el AUC en un 2,8 veces. En presencia de ciclosporina, el AUC del metabolito activo de ticagrelor aumentó en un 32%, mientras que su C*max* disminuyó en un 15%.
No existen datos sobre la administración concomitante de ticagrelor con otras sustancias activas que sean inhibidores potentes de la glucoproteína P (P-gp) e inhibidores moderados de CYP3A4 (por ejemplo, verapamilo, quinidina), que podrían aumentar la exposición a ticagrelor. Si no puede evitarse el tratamiento combinado, su uso conjunto requiere precaución.
Otros
Los estudios clínicos de interacción demostraron que la administración concomitante de ticagrelor con heparina, enoxaparina y AAS o desmopresina no influyó en la farmacocinética de ticagrelor ni de su metabolito activo, ni en la agregación plaquetaria inducida por ADP, en comparación con la administración de ticagrelor solo. Si está clínicamente indicado, los medicamentos que alteran la hemostasia deben administrarse con precaución en combinación con ticagrelor.
En pacientes con síndrome coronario agudo (SCA) tratados con morfina, se observó un retraso y una reducción en la exposición a inhibidores orales de P2Y*, incluyendo ticagrelor y su metabolito activo (reducción del 35% en la exposición a ticagrelor). Esta interacción puede estar relacionada con una menor motilidad gastrointestinal y también afecta a otros opioides. Su relevancia clínica es desconocida, pero los datos sugieren la posibilidad de una reducción de la eficacia de ticagrelor en pacientes que reciben morfina simultáneamente.
En pacientes con SCA en los que no pueda suspenderse la administración de morfina y en los que se considere críticamente importante una rápida inhibición de P2Y*, puede considerarse la administración de un inhibidor intravenoso de P2Y*.
Influencia de ticagrelor sobre la acción de otros medicamentos
Medicamentos metabolizados por CYP3A4
- Simvastatina: la administración concomitante de ticagrelor con simvastatina provocó un aumento del 81% en la C*max* y del 56% en el AUC de simvastatina, así como un incremento del 64% en la C*max* y del 52% en el AUC del ácido simvastatínico, con casos aislados de aumentos de 2 a 3 veces. La administración conjunta de ticagrelor con simvastatina en dosis superiores a 40 mg/día podría provocar efectos adversos de simvastatina, por lo que debe considerarse al evaluar las posibles ventajas de esta combinación. No se observó influencia de simvastatina sobre las concentraciones plasmáticas de ticagrelor. Ticagrelor podría tener un efecto similar sobre la administración de lovastatina. No se recomienda la administración concomitante de ticagrelor con simvastatina o lovastatina en dosis superiores a 40 mg.
- Atorvastatina: la administración conjunta de atorvastatina y ticagrelor provoca un aumento del 23% en la C*max* y del 36% en el AUC del ácido atorvastatínico. Se observó un aumento similar en la C*max* y el AUC de todos los metabolitos del ácido atorvastatínico. Se considera que este efecto no es clínicamente relevante.
- No puede descartarse un efecto similar sobre otras estatinas metabolizadas por CYP3A4. En el estudio PLATO, los pacientes recibieron diferentes estatinas y, en el 93% de todos los participantes, no se observaron problemas de seguridad relacionados con el uso de estatinas.
Ticagrelor es un inhibidor moderado de CYP3A4. No se recomienda la administración concomitante de ticagrelor con sustratos de CYP3A4 que tengan un estrecho índice terapéutico (por ejemplo, cisaprida y alcaloides del cornezuelo del centeno), ya que ticagrelor puede aumentar la exposición a estos medicamentos.
Sustratos de P-gp (incluyendo digoxina, ciclosporina)
La administración concomitante de ticagrelor aumenta la C*max* y el AUC de digoxina en un 75% y un 28%, respectivamente. Las concentraciones plasmáticas mínimas medias de digoxina aumentaron aproximadamente un 30% tras la administración conjunta con ticagrelor, mientras que las concentraciones máximas individuales aumentaron hasta 2 veces. La presencia de digoxina no influye en la C*max* ni en el AUC de ticagrelor ni de su metabolito activo. Por tanto, se recomienda un control clínico adecuado y/o monitorización de parámetros de laboratorio durante la administración concomitante de medicamentos con estrecho índice terapéutico dependientes de P-gp, como digoxina y ticagrelor.
Ticagrelor no influyó en las concentraciones sanguíneas de ciclosporina. No se ha estudiado el efecto de ticagrelor sobre otros sustratos de P-gp.
Medicamentos metabolizados por CYP2C9
La administración concomitante de ticagrelor y tolbutamida no provocó cambios en las concentraciones plasmáticas de ninguno de los dos medicamentos, lo que sugiere que ticagrelor no es un inhibidor de CYP2C9 y es poco probable que modifique el metabolismo de medicamentos como la warfarina o la tolbutamida a través de CYP2C9.
Rosuvastatina
Ticagrelor puede afectar la excreción renal de rosuvastatina, aumentando el riesgo de acumulación de esta. Aunque el mecanismo exacto no se conoce, en algunos casos, la administración concomitante de ticagrelor y rosuvastatina ha provocado deterioro de la función renal, aumento de la actividad de la CPK (creatinina quinasa) y rabdomiólisis.
Medicamentos anticonceptivos orales
La administración concomitante de ticagrelor con levonorgestrel y etinilestradiol provocó un aumento del 20% en la exposición a etinilestradiol, pero no influyó en la farmacocinética de levonorgestrel. No se prevé un efecto clínicamente significativo sobre la eficacia de los anticonceptivos orales en caso de administración conjunta de levonorgestrel y etinilestradiol con ticagrelor.
Medicamentos que provocan bradicardia
Debido a las pausas ventriculares y bradicardia, generalmente asintomáticas, observadas durante el tratamiento con ticagrelor, debe tenerse precaución al administrar conjuntamente ticagrelor con medicamentos que provoquen bradicardia (ver sección 4.4). Sin embargo, en el estudio PLATO no se observaron pruebas de efectos adversos clínicamente significativos tras la administración concomitante con uno o más medicamentos que provocan bradicardia (es decir, 96% betabloqueantes, 33% inhibidores de los canales de calcio: diltiazem y verapamilo, y 4% digoxina).
Administración concomitante con otros medicamentos
En estudios clínicos, ticagrelor se administró simultáneamente con AAS, inhibidores de la bomba de protones, estatinas, betabloqueantes, inhibidores de la enzima convertidora de angiotensina (ECA) y antagonistas del receptor de angiotensina, durante períodos prolongados por necesidad terapéutica de enfermedades concurrentes, así como con heparina, heparina de bajo peso molecular e inhibidores intravenosos de GpIIb/IIIa durante períodos cortos (ver sección 5.1). No se observaron interacciones clínicamente relevantes durante la administración conjunta de estos medicamentos.
La administración concomitante de ticagrelor con heparina, enoxaparina o desmopresina no influyó en el tiempo de tromboplastina parcial activado (aPTT), el tiempo de coagulación activado (ACT) ni en la determinación de la actividad del factor Xa. Sin embargo, debido a posibles interacciones farmacodinámicas, debe tenerse precaución al administrar conjuntamente ticagrelor con medicamentos que alteren la hemostasia.
Debido a las hemorragias cutáneas anormales observadas con la administración de inhibidores selectivos de la recaptación de serotonina (ISRS) (por ejemplo, paroxetina, sertralina y citalopram), debe tenerse precaución al administrar ISRS junto con ticagrelor, ya que podría aumentar el riesgo de hemorragia.
4.6 Fertilidad, embarazo y lactancia
Mujeres en edad fértil
Las mujeres en edad fértil deben utilizar métodos anticonceptivos adecuados para evitar el embarazo durante el tratamiento con ticagrelor.
Embarazo
No existen o existen datos limitados sobre el uso de ticagrelor durante el embarazo. Los estudios en animales mostraron efectos adversos sobre la reproducción (ver punto 5.3). No se recomienda el uso de ticagrelor durante el embarazo.
Lactancia
Los datos farmacodinámicos y toxicológicos disponibles procedentes de estudios en animales han demostrado que el ticagrelor y sus metabolitos activos pasan a la leche materna (ver punto 5.3). No puede descartarse el riesgo para los recién nacidos/lactantes. Se deberá decidir si se interrumpe la lactancia o se interrumpe/termina el tratamiento con ticagrelor, teniendo en cuenta los beneficios de la lactancia para el niño y los beneficios del tratamiento para la mujer.
Fertilidad
En animales, el ticagrelor no afecta la fertilidad de machos ni hembras (ver punto 5.3).
4.7 Efecto sobre la capacidad para conducir y utilizar máquinas
Ticagrelor no tiene o tiene un efecto insignificante sobre la capacidad para conducir y utilizar máquinas. Se han notificado casos de mareo y confusión en pacientes tratados con ticagrelor. Por este motivo, los pacientes que presenten estos síntomas deben tener precaución al conducir vehículos o utilizar máquinas.
4.8 Reacciones adversas
Resumen del perfil de seguridad
El perfil de seguridad del ticagrelor fue evaluado a partir de los resultados de dos grandes estudios de Fase III (PLATO y PEGASUS), que incluyeron a más de 39 000 pacientes (véase el punto 5.1).
En el estudio PLATO, se observó una mayor frecuencia de interrupción del tratamiento debido a eventos adversos en los pacientes que recibieron ticagrelor en comparación con el grupo que recibió clopidogrel (7,4 % frente a 5,4 %).
En el estudio PEGASUS, se observó una mayor frecuencia de interrupción del tratamiento debido a eventos adversos en los pacientes que recibieron ticagrelor en comparación con los pacientes tratados con AAS en monoterapia (16,1 % en el grupo tratado con ticagrelor a la dosis de 60 mg en combinación con AAS frente a 8,5 % en el grupo que recibió AAS en monoterapia). Las reacciones adversas más frecuentemente notificadas en pacientes tratados con ticagrelor fueron hemorragia y disnea (véase el punto 4.4).
Presentación tabulada de las reacciones adversas
Las siguientes reacciones adversas se han identificado como resultado de estudios o notificadas tras la comercialización del ticagrelor (tabla 1).
Las reacciones adversas se enumeran según la clasificación por órganos y sistemas (MedDRA, por sus siglas en inglés). Dentro de cada grupo de clasificación por órganos y sistemas, las reacciones adversas se presentan en orden decreciente de frecuencia. La frecuencia se define como: muy frecuente (≥1/10), frecuente (≥1/100 a <1/10), poco frecuente (≥1/1 000 a <1/100), rara (≥1/10 000 a <1/1 000), muy rara (<1/10 000) y frecuencia no conocida (no puede determinarse a partir de los datos disponibles).
Tabla 1 – Reacciones adversas presentadas según frecuencia y clasificación por órganos y sistemas (SOC)
| Clasificación de sistemas y órganos | Muy frecuente | Frecuente | Infrecuente | Frecuencia desconocida |
| Neoplasias benignas, malignas e indeterminadas (incluyendo quistes y pólipos) | Hemorragia desde un tumora | |||
| Alteraciones de la sangre y del sistema linfático | Alteraciones sanguíneas, hemorragiasb | Trombocitopenia trombótica | ||
| Alteraciones del sistema inmunológico | Hipersensibilidad, incluyendo edema angioneurótico | |||
| Alteraciones del metabolismo y de la nutrición | Hiperuricemiad | Gotas gotaica/Artritis gotaosa | ||
| Alteraciones psiquiátricas | Desorientación | |||
| Alteraciones del sistema nervioso | Vertigo, síncopes, cefalea | Hemorragia intracraneal | ||
| Alteraciones visuales | Hemorragia intraoculare | |||
| Alteraciones del oído y del laberinto | Vertigo de origen laberíntico | Hemorragia del oído | ||
| Alteraciones cardíacas | Bradiarritmia, bloqueo AVc | |||
| Clasificación de sistemas y órganos | Muy frecuente | Frecuente | Infrecuente | Frecuencia desconocida |
| Alteraciones vasculares | Hipotensión | |||
| Alteraciones del sistema respiratorio, torácicas y mediastínicas | Disnea | Hemorragia del sistema respiratoriof | ||
| Alteraciones gastrointestinales | Hemorragia digestivag, diarrea, náuseas, dispepsia, estreñimiento | Hemorragia intraperitoneal | ||
| Alteraciones de la piel y del tejido subcutáneo | Hemorragias subcutáneas o en la pielh, erupción cutánea, prurito | |||
| Alteraciones musculoesqueléticas y del tejido conjuntivo | Hemorragia musculari | |||
| Alteraciones renales y de las vías urinarias | Hemorragia del sistema urinarioj | |||
| Alteraciones del sistema reproductor y mamas | Hemorragia del sistema reproductork | |||
| Pruebas diagnósticas | Aumento de la creatinina sanguínead | |||
| Lesiones, envenenamientos y complicaciones postoperatorias | Hemorragia postoperatoria, hemorragias traumáticasl |
Descripción de determinados efectos adversos
Hemorragias
Resultados del estudio PLATO sobre hemorragias
El resultado general sobre la frecuencia de hemorragias en el estudio PLATO se presenta en la tabla 2.
Tabla 2 – Análisis de todos los eventos hemorrágicos, valores estimados mediante el método de Kaplan-Meier a los 12 meses (PLATO)
| Ticagrelor 90 mg dos veces al día N=9235 | Clopidogrel N=9186 | Valor p* | |
| Hemorragias graves totales, PLATO | 11,6 | 11,2 | 0,4336 |
| Graves que causan muerte/ponen en peligro la vida, PLATO | 5,8 | 5,8 | 0,6988 |
| Graves no relacionadas con CABG, PLATO | 4,5 | 3,8 | 0,0264 |
| Graves no relacionadas con procedimientos, PLATO | 3,1 | 2,3 | 0,0058 |
| Graves + leves totales, PLATO | 16,1 | 14,6 | 0,0084 |
| Graves + leves no relacionadas con procedimientos, PLATO | 5,9 | 4,3 | <0,0001 |
| Graves, definidas según TIMI | 7,9 | 7,7 | 0,5669 |
| Graves + leves, definidas según TIMI | 11,4 | 10,9 | 0,3272 |
Ticagrelor y clopidogrel no diferían en cuanto a la frecuencia de hemorragias graves que conducían a la muerte o que ponían en peligro la vida según los criterios de PLATO, hemorragias graves en general según PLATO, hemorragias graves según TIMI ni hemorragias menores según TIMI (tabla 2). Sin embargo, se observó un mayor número de hemorragias graves y menores en conjunto según los criterios del estudio PLATO en el grupo tratado con ticagrelor en comparación con el grupo tratado con clopidogrel. Únicamente un número muy reducido de pacientes incluidos en el estudio PLATO presentaron hemorragias que condujeron a la muerte: 20 (0,2 %) en el grupo que recibía ticagrelor y 23 (0,3 %) en el grupo que recibía clopidogrel (véase sección 4.4).
La edad, el sexo, el peso corporal, la raza, la región geográfica, las enfermedades concomitantes, el tratamiento simultáneo, y la historia clínica, incluyendo ictus previo o accidente isquémico transitorio, no constituyeron factores predictivos de hemorragias graves en general ni de hemorragias graves no relacionadas con procedimientos, según los criterios del estudio PLATO. Por consiguiente, no se identificó ningún grupo con un riesgo aumentado de algún tipo específico de hemorragia.
Hemorragia relacionada con la CABG:
En el estudio PLATO, el 42 % de los 1584 pacientes (12 % de la cohorte) en los que se realizó una CABG presentaron una hemorragia grave que conducía a la muerte o que ponía en peligro la vida según los criterios del estudio PLATO, sin que se observara diferencia entre los grupos tratados. Las hemorragias fatales posteriores a la CABG ocurrieron en 6 pacientes de cada grupo de tratamiento (véase sección 4.4).
Hemorragias no relacionadas con la CABG y hemorragias no relacionadas con procedimientos:
Ticagrelor y clopidogrel no diferían en cuanto a hemorragias graves no relacionadas con la CABG, que conducían a la muerte o ponían en peligro la vida, según la definición de hemorragias del estudio PLATO. Sin embargo, las hemorragias graves en general según PLATO, las hemorragias graves según TIMI y las hemorragias graves + menores según TIMI fueron más frecuentes en el grupo tratado con ticagrelor. De forma análoga, cuando se eliminaron todas las hemorragias relacionadas con procedimientos, se observó que la frecuencia de hemorragias fue mayor en el grupo tratado con ticagrelor que en el grupo tratado con clopidogrel (tabla 2). La interrupción del tratamiento debido a hemorragias no relacionadas con procedimientos fue más frecuente en el grupo tratado con ticagrelor (2,9 %) que en el grupo tratado con clopidogrel (1,2 %; p<0,001).
Hemorragias intracraneales:
En el estudio PLATO se observó un mayor número de hemorragias intracraneales no relacionadas con procedimientos con ticagrelor (n=27 hemorragias en 26 pacientes, 0,3 %) en comparación con clopidogrel (n=14 hemorragias, 0,2 %), incluyendo 11 hemorragias fatales con ticagrelor y 1 con clopidogrel. No se observó diferencia en cuanto al número total de hemorragias fatales.
Resultados del estudio PEGASUS respecto a hemorragias
Los resultados generales sobre eventos hemorrágicos en el estudio PEGASUS se presentan en la tabla 3.
Tabla 3 – Análisis de todos los eventos hemorrágicos, valores estimados mediante el método de Kaplan-Meier tras 36 meses (PEGASUS)
| Ticagrelor 60 mg dos veces al día + ASA N=6958 | ASA en monoterapia N=6996 | |||
| Puntos finales de evaluación de seguridad | IC% | Coeficiente de riesgo (95% IC) | IC% | Valor p |
| Categorías de hemorragias definidas según TIMI | ||||
| Hemorragia grave según TIMI | 2,3 | 2,32 (1,68; 3,21) | 1,1 | <0,0001 |
| Que conduce a muerte | 0,3 | 1,00 (0,44; 2,27) | 0,3 | 1,0000 |
| Hemorragia intracraneal (ICH) | 0,6 | 1,33 (0,77; 2,31) | 0,5 | 0,3130 |
| Otra hemorragia grave según TIMI | 1,6 | 3,61 (2,31; 5,65) | 0,5 | <0,0001 |
| Hemorragia grave o menor según TIMI | 3,4 | 2,54 (1,93; 3,35) | 1,4 | <0,0001 |
| Hemorragia grave o menor que requiere atención médica según TIMI | 16,6 | 2,64 (2,35; 2,97) | 7,0 | <0,0001 |
| Categorías de hemorragias definidas en el estudio PLATO | ||||
| Hemorragia grave en el estudio PLATO | 3,5 | 2,57 (1,95; 3,37) | 1,4 | <0,0001 |
| Que conduce a muerte/pone en peligro la vida | 2,4 | 2,38 (1,73; 3,26) | 1,1 | <0,0001 |
| Otra hemorragia grave en el estudio PLATO | 1,1 | 3,37 (1,95; 5,83) | 0,3 | <0,0001 |
| Hemorragia grave o menor en el estudio PLATO | 15,2 | 2,71 (2,40; 3,08) | 6,2 | <0,0001 |
En el estudio PEGASUS, la frecuencia de hemorragias graves según TIMI durante el tratamiento con ticagrelor a una dosis de 60 mg dos veces al día fue mayor que durante el tratamiento con AAS en monoterapia. No se observó un aumento del riesgo de hemorragias en los casos de hemorragias que condujeron a la muerte, y únicamente se observó un ligero aumento en el caso de hemorragias intracraneales, en comparación con el tratamiento con AAS en monoterapia. En el estudio se observaron escasos casos de hemorragias que condujeron a la muerte: 11 (0,3 %) durante el tratamiento con ticagrelor a una dosis de 60 mg y 12 (0,3 %) durante el tratamiento con AAS en monoterapia. El aumento observado en el riesgo de hemorragias graves según TIMI durante el tratamiento con ticagrelor a una dosis de 60 mg se debió principalmente a una mayor frecuencia de otras hemorragias graves según TIMI, relacionadas con eventos adversos del tubo digestivo.
Se observó un aumento de la frecuencia de hemorragias, similar al aumento de hemorragias graves según TIMI, en las categorías de hemorragias graves o menores según TIMI y hemorragias graves en el estudio PLATO, así como en hemorragias graves o menores en el estudio PLATO (véase la tabla 5). La interrupción del tratamiento debido a hemorragias fue más frecuente durante el tratamiento con ticagrelor a una dosis de 60 mg que durante el tratamiento con AAS en monoterapia (6,2 % frente a 1,5 %). La mayoría de estas hemorragias fueron de menor gravedad (clasificadas como hemorragias que requirieron atención médica según TIMI), por ejemplo, epistaxis, equimosis y hematomas.
El perfil de hemorragias asociadas al tratamiento con ticagrelor a una dosis de 60 mg fue consistente en diversas subgrupos previamente definidos (por ejemplo, según edad, sexo, peso corporal, raza, región geográfica, enfermedades concomitantes, tratamiento concomitante e historia de enfermedad) en lo referente a hemorragias graves según TIMI, hemorragias graves o menores según TIMI y hemorragias graves según PLATO.
Hemorragias intracraneales:
Las hemorragias intracraneales (ICH) espontáneas se observaron con una frecuencia similar en pacientes que recibieron ticagrelor a una dosis de 60 mg y AAS en monoterapia (n = 13, 0,2 % en ambos grupos de tratamiento). En el caso de ICH traumáticas y relacionadas con procedimientos, se observó un ligero aumento de su frecuencia en pacientes tratados con ticagrelor a una dosis de 60 mg (n = 15, 0,2 %) en comparación con AAS en monoterapia (n = 10, 0,1 %). Se registraron 6 casos de hemorragia intracraneal que condujeron a la muerte durante el tratamiento con ticagrelor a una dosis de 60 mg y 5 casos de hemorragia intracraneal que condujeron a la muerte durante el tratamiento con AAS en monoterapia. La frecuencia de hemorragias intracraneales fue baja en ambos grupos tratados, teniendo en cuenta la alta carga de enfermedades concomitantes y factores de riesgo cardiovascular en la población estudiada.
Disnea
Los pacientes tratados con ticagrelor notifican disnea, sensación de falta de aliento.
En el estudio PLATO, los eventos adversos notificados como disnea (disnea, disnea en reposo, disnea de esfuerzo, disnea paroxística nocturna o disnea nocturna), cuando se agruparon conjuntamente, fueron notificados por el 13,8 % de los pacientes tratados con ticagrelor y por el 7,8 % de los pacientes tratados con clopidogrel. En el 2,2 % de los pacientes tratados con ticagrelor y en el 0,6 % de los tratados con clopidogrel, los investigadores responsables del estudio PLATO consideraron que la disnea estaba causalmente relacionada con el tratamiento, y hubo algunos casos de disnea grave (0,14 % con ticagrelor; 0,02 % con clopidogrel) (véase sección 4.4). La mayoría de los eventos adversos notificados como disnea tuvieron una intensidad leve a moderada y la mayoría se notificaron como un episodio único al comienzo del tratamiento.
En comparación con clopidogrel, los pacientes con asma/Enfermedad Pulmonar Obstructiva Crónica (EPOC) tratados con ticagrelor pueden tener un mayor riesgo de aparición de disnea no grave (3,29 % con ticagrelor frente a 0,53 % con clopidogrel) y disnea grave (0,38 % con ticagrelor frente a 0,00 % con clopidogrel). En valores absolutos, este riesgo es mayor que en toda la población del estudio PLATO. Se debe tener precaución al utilizar ticagrelor en pacientes con antecedentes de asma y/o EPOC (véase sección 4.4).
Aproximadamente el 30 % de los episodios de disnea desaparecieron en un plazo de 7 días. En el estudio PLATO participaron pacientes con insuficiencia cardíaca congestiva, EPOC o asma en su historial; estos pacientes, así como los pacientes de edad avanzada, notificaron con mayor frecuencia disnea. El 0,9 % de los pacientes en el grupo tratado con ticagrelor interrumpió el tratamiento debido a disnea, en comparación con el 0,1 % en el grupo tratado con clopidogrel. El aumento de la frecuencia de episodios de disnea durante el tratamiento con ticagrelor no está relacionado con una enfermedad cardíaca o pulmonar nueva o empeorada (véase sección 4.4). El ticagrelor no afecta a las pruebas de función pulmonar.
En el estudio PEGASUS, la disnea se notificó en el 14,2 % de los pacientes que recibieron ticagrelor a una dosis de 60 mg dos veces al día y en el 5,5 % de los pacientes que recibieron AAS en monoterapia. De forma similar al estudio PLATO, la mayoría de los casos de disnea notificados tuvieron una intensidad leve a moderada (véase sección 4.4). Los pacientes que notificaron disnea solían ser mayores y con mayor frecuencia tenían antecedentes de disnea, EPOC o asma.
Pruebas diagnósticas
Aumento de la concentración de ácido úrico: En el estudio PLATO, el aumento de la concentración sérica de ácido úrico por encima del límite superior del rango normal ocurrió en el 22 % de los pacientes que recibieron ticagrelor, en comparación con el 13 % de los pacientes que recibieron clopidogrel. Los valores correspondientes en el estudio PEGASUS fueron del 9,1 %, 8,8 % y 5,5 % para ticagrelor a una dosis de 90 mg, ticagrelor a una dosis de 60 mg y placebo, respectivamente. La concentración media sérica de ácido úrico aumentó aproximadamente un 15 % en los pacientes que recibieron ticagrelor, en comparación con un aumento de aproximadamente el 7,5 % en los tratados con clopidogrel. Tras la finalización del tratamiento, se observó una disminución de la concentración de ácido úrico hasta aproximadamente el 7 % en los pacientes tratados con ticagrelor, pero no se observó disminución en el caso de clopidogrel. En el estudio PEGASUS se observó un aumento reversible de la concentración media sérica de ácido úrico del 6,3 % y 5,6 % con ticagrelor a dosis de 90 mg y 60 mg, respectivamente, frente a una disminución del 1,5 % en el grupo placebo. En el estudio PLATO, la frecuencia de gota fue del 0,2 % en el grupo ticagrelor frente al 0,1 % en el grupo clopidogrel. Las frecuencias correspondientes de gota/gota en el estudio PEGASUS fueron del 1,6 %, 1,5 % y 1,1 % para ticagrelor a una dosis de 90 mg, ticagrelor a una dosis de 60 mg y placebo, respectivamente.
Notificación de reacciones adversas sospechosas
Una vez autorizado el medicamento, es importante notificar las reacciones adversas sospechosas. Esto permite una vigilancia continua de la relación beneficio/riesgo del medicamento. El personal médico cualificado debe notificar cualquier reacción adversa sospechosa a través del Departamento de Vigilancia de Reacciones Adversas a Medicamentos de la Oficina de Registro de Medicamentos, Productos Médicos y Productos Biocidas
Al. Jerozolimskie 181C
PL-02 222 Varsovia
Tel.: + 48 22 49 21 301
Fax: + 48 22 49 21 309
Página web: https://smz.ezdrowie.gov.pl
Las reacciones adversas también pueden notificarse al titular del permiso de comercialización.
4.9 Sobredosificación
El ticagrelor es bien tolerado tras la administración de una dosis única de hasta 900 mg. En un estudio con dosis única ascendente, la toxicidad gastrointestinal fue dependiente de la dosis. Entre otros efectos adversos clínicamente significativos que podrían presentarse tras una sobredosificación se incluyen disnea y pausas ventriculares (véase punto 4.8).
En caso de sobredosificación, pueden presentarse los efectos adversos potenciales mencionados anteriormente, por lo que debe considerarse el monitoreo del electrocardiograma (ECG).
Actualmente no se conoce un antídoto que invierta los efectos del ticagrelor, y este no se elimina durante la diálisis (véase punto 5.2). El tratamiento de la sobredosificación debe seguir las prácticas médicas estándar locales. El efecto esperado tras una sobredosificación de ticagrelor es el riesgo de prolongación del tiempo de sangrado, relacionado con la inhibición plaquetaria. Es poco probable que la transfusión de plaquetas sea clínicamente beneficiosa en pacientes con hemorragia (véase punto 4.4). Si ocurre hemorragia, deben considerarse otros tratamientos de apoyo adecuados.
5. PROPIEDADES FARMACOLÓGICAS
5.1 Propiedades farmacodinámicas
Grupo farmacoterapéutico: medicamentos que inhiben la agregación plaquetaria, excepto heparina, código ATC: B01AC24
Mecanismo de acción
Ticagrelor Holsten contiene ticagrelor, que pertenece al grupo químico de las ciclopropiltetrazolopirimidinas (CPTP).
El ticagrelor es un antagonista oral, directo, selectivo y reversible del receptor P2Y12, que previene la activación y agregación plaquetaria dependiente de ADP mediada por el receptor P2Y12.
El ticagrelor no impide la unión de ADP, pero al unirse al receptor P2Y12 bloquea la señalización inducida por ADP. Dado que las plaquetas participan en la iniciación y (o) progresión de complicaciones trombóticas en la aterosclerosis, se ha demostrado que la inhibición de la función plaquetaria reduce el riesgo de eventos cardiovasculares, como muerte, infarto de miocardio o accidente cerebrovascular.
El ticagrelor también aumenta las concentraciones locales de adenosina endógena al inhibir el transportador equilibrante de nucleósidos-1 (ENT-1, por sus siglas en inglés).
Se ha demostrado que el ticagrelor potencia los siguientes efectos dependientes de la adenosina en voluntarios sanos y pacientes con síndrome coronario agudo (SCA): vasodilatación (medida como aumento del flujo coronario en voluntarios sanos y pacientes con SCA; cefalea), inhibición de la función plaquetaria (en sangre total humana en condiciones in vitro) y disnea. Sin embargo, la relación entre el aumento observado en los niveles de adenosina y los efectos clínicos (por ejemplo, morbilidad-mortalidad) no ha sido claramente establecida.
Actividad farmacodinámica
Inicio de acción
En pacientes con enfermedad coronaria estable que reciben ácido acetilsalicílico, el ticagrelor presenta un inicio rápido de acción farmacológica, evidenciado por una inhibición promedio de la agregación plaquetaria (IPA) del aproximadamente 41 % a los 30 minutos tras la administración de una dosis de carga de 180 mg, con un efecto máximo de IPA del 89 % entre 2 y 4 horas después de la dosis, que se mantiene entre 2 y 8 horas. En el 90 % de los pacientes, el grado máximo de inhibición plaquetaria superior al 70 % se observa a las 2 horas tras la administración del fármaco.
Fin de la acción
Si se planea una cirugía de derivación aortocoronaria (CABG), el riesgo de sangrado asociado al uso de ticagrelor es mayor en comparación con clopidogrel si se suspende menos de 96 horas antes del procedimiento.
Datos sobre el cambio de terapia
El cambio de tratamiento de clopidogrel 75 mg a ticagrelor 90 mg dos veces al día produce un aumento del IPA del 26,4 % en valores absolutos, mientras que el cambio de ticagrelor a clopidogrel produce una reducción del IPA del 24,5 % en valores absolutos. Los pacientes pueden cambiar de clopidogrel a ticagrelor sin interrupción del efecto antiplaquetario (véase punto 4.2).
Eficacia y seguridad en estudios clínicos
Los datos clínicos que respaldan la eficacia y seguridad del ticagrelor provienen de dos estudios de fase 3:
- El estudio PLATO [ Inhibición plaquetaria y resultados en pacientes ], en el que se comparó ticagrelor con clopidogrel, administrándose ambos fármacos en combinación con AAS (ácido acetilsalicílico) y otras terapias estándar.
- El estudio PEGASUS TIMI-54 [ Prevención con Ticagrelor de Eventos Trombóticos Secundarios en Pacientes con Síndrome Coronario Agudo de Alto Riesgo ], en el que se comparó ticagrelor en combinación con AAS frente a AAS en monoterapia.
Estudio PLATO (síndrome coronario agudo)
El estudio PLATO incluyó a 18 624 pacientes con síndrome coronario agudo que acudieron dentro de las 24 horas desde el inicio de los síntomas de angina inestable (UA), infarto de miocardio sin elevación del segmento ST (NSTEMI) o infarto de miocardio con elevación del segmento ST (STEMI), y que fueron tratados inicialmente con terapia farmacológica, intervención coronaria percutánea (PCI) o derivación aortocoronaria (CABG).
Eficacia clínica
En combinación con la dosis diaria de AAS, el ticagrelor 90 mg dos veces al día mostró superioridad frente al clopidogrel 75 mg una vez al día en la prevención del punto final compuesto (muerte por causas cardiovasculares, infarto de miocardio o accidente cerebrovascular), siendo la diferencia atribuible principalmente a la reducción en muertes cardiovasculares e infartos de miocardio. Los pacientes recibieron una dosis inicial de clopidogrel de 300 mg (en pacientes sometidos a intervención coronaria percutánea, se permitió una dosis de 600 mg) o ticagrelor 180 mg.
Este resultado se observó tempranamente (reducción absoluta del riesgo [ARR] del 0,6 % y reducción relativa del riesgo [RRR] del 12 % a los 30 días), y la eficacia del tratamiento se mantuvo durante todo el período de 12 meses, alcanzando una ARR del 1,9 % en un año y una RRR del 16 %. Estos resultados indican que la duración adecuada del tratamiento con ticagrelor 90 mg dos veces al día es de 12 meses (véase punto 4.2).
El tratamiento con ticagrelor en lugar de clopidogrel en 54 pacientes con SCA previene un evento cardiovascular; el tratamiento de 91 pacientes previene una muerte por causas cardiovasculares (véase gráfico 1 y tabla 4).
Los mejores resultados con ticagrelor en comparación con clopidogrel se observan de forma consistente en múltiples subgrupos de pacientes, incluyendo peso corporal, sexo, antecedentes de diabetes, episodios isquémicos transitorios o accidente cerebrovascular no hemorrágico o revascularización; tratamiento concomitante con heparina, inhibidores del receptor GpIIb/IIIa e inhibidores de la bomba de protones (véase punto 4.5); diagnóstico clínico final (STEMI, NSTEMI o UA) y estrategia terapéutica planeada en el momento de la aleatorización (tratamiento invasivo o conservador).
La eficacia del tratamiento varió ligeramente según la región, de modo que el cociente de riesgos (HR) para el punto final primario indica beneficios del ticagrelor a nivel mundial, excepto en América del Norte, que representa aproximadamente el 10 % de la población total estudiada, donde el resultado HR favorece más al clopidogrel (interacción presente, p=0,045).
Los análisis factoriales sugieren una posible relación con la dosis de AAS, lo que indica que se observó una reducción en la eficacia del ticagrelor al aumentar las dosis de AAS. Las dosis de AAS para uso crónico junto con ticagrelor deben estar entre 75 y 150 mg (véanse puntos 4.2 y 4.4).
El gráfico 1 muestra la estimación del riesgo acumulado del primer evento del punto final compuesto para la evaluación de la eficacia.
Gráfico 1 – Análisis del punto final compuesto primario clínico de muerte por causas cardiovasculares, infarto de miocardio y accidente cerebrovascular (PLATO)
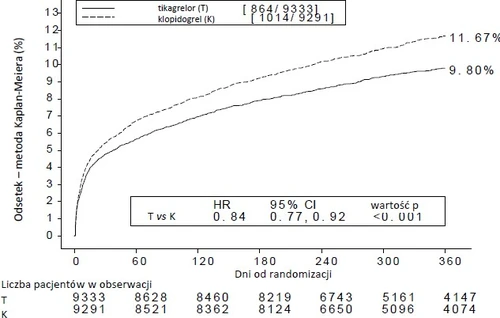
El ticagrelor redujo la frecuencia del punto final compuesto primario en comparación con clopidogrel, tanto en el grupo de pacientes con UA/NSTEMI como en el grupo con STEMI (tabla 4). Por esta razón, el medicamento Ticagrelor Holsten 90 mg dos veces al día, en combinación con AAS en dosis bajas, puede utilizarse en pacientes con SCA (angina inestable, infarto de miocardio sin elevación del segmento ST [NSTEMI] o infarto de miocardio con elevación del segmento ST [STEMI]), incluyendo aquellos tratados farmacológicamente y aquellos sometidos a intervención coronaria percutánea (PCI) o derivación aortocoronaria (CABG).
Tabla 4 – Análisis de los puntos finales primarios y secundarios para la evaluación de la eficacia (PLATO)
| Ticagrelor 90 mg dos veces al día (% pacientes, en los que ocurrió el evento) | Clopidogrel 75 mg una vez al día (% pacientes, en los que ocurrió el evento) | ARRa (%/año) | RRRa (%) (IC del 95 %) | Valor p | |
| N=9333 | N=9291 | ||||
| Fallecimiento por causas CV (cardiovasculares), IAM (infarto de miocardio, excepto IAM silencioso) o ictus | 8,5 | 10,0 | 1,7 | 16 (6, 25) | 0,0025 |
| Plan de tratamiento invasivo | 8,5 | 10,0 | 1,7 | 16 (6, 25) | 0,0025 |
| Plan de tratamiento conservador | 11,3 | 13,2 | 2,3 | 15 (0,3, 27) | 0,0444d |
| Fallecimiento por causas CV | 3,8 | 4,8 | 1,1 | 21 (9, 31) | 0,0013 |
| IAM (excepto IAM silencioso)b | 5,4 | 6,4 | 1,1 | 16 (5, 25) | 0,0045 |
| Ictus | 1,3 | 1,1 |
|
| 0,2249 |
| Fallecimiento por cualquier causa, IAM (excepto IAM silencioso) o ictus | 9,7 | 11,5 | 2,1 | 16 (8, 23) | 0,0001 |
| Fallecimiento por causas CV, IAM total, ictus, SRI, IAM recurrente, AIT u otro EATE | 13,8 | 15,7 | 2,1 | 12 (5, 19) | 0,0006 |
| Fallecimiento por cualquier causa | 4,3 | 5,4 | 1,4 | 22 (11, 31) | 0,0003d |
| Trombosis del stent | 1,2 | 1,7 | 0,6 | 32 (8, 49) | 0,0123d |
Subanálisis genético en el estudio PLATO
La genotipificación respecto a CYP2C19 y ABCB1, realizada en el estudio PLATO en 10 285 pacientes, permitió
determinar la relación entre los grupos genotípicos y los resultados del estudio PLATO.
La superioridad de ticagrelor frente a clopidogrel en la reducción del número de eventos cardiovasculares graves no
fue significativamente dependiente del genotipo CYP2C19 o ABCB1. Al igual que en todo el estudio PLATO, la
frecuencia total de hemorragias graves según la definición de PLATO no difirió entre el grupo de ticagrelor y el de
clopidogrel, independientemente del genotipo CYP2C19 o ABCB1. Las hemorragias graves según la definición
PLATO, no relacionadas con CABG, ocurrieron con mayor frecuencia en el grupo de ticagrelor en comparación con
clopidogrel en pacientes con pérdida de uno o más alelos funcionales de CYP2C19, pero de forma similar al grupo
de clopidogrel en pacientes sin pérdida de alelos funcionales.
Evaluación global de eficacia y seguridad del tratamiento
La evaluación global de la eficacia y seguridad del tratamiento (muerte por causas cardiovasculares, infarto de
miocardio, accidente cerebrovascular o hemorragia grave según la definición PLATO) indica que los beneficios
derivados de la eficacia de ticagrelor, en comparación con clopidogrel, no se pierden debido al número de
hemorragias graves (ARR 1,4 %, RRR 8 %, HR 0,92; p = 0,0257) durante el período de 12 meses tras el episodio
de SCA.
Seguridad clínica
Subgrupo con estudio Holter
Con el fin de evaluar la aparición de pausas ventriculares y otras arritmias durante el estudio PLATO, los
investigadores monitorizaron mediante Holter un subgrupo de cerca de 3000 pacientes, de los cuales aproximadamente
2000 tuvieron registros realizados en la fase aguda del SCA y tras un mes transcurrido. La variable principal observada
fue la aparición de pausas ventriculares ≥ 3 segundos. Se observó un mayor número de pausas ventriculares en el
grupo de ticagrelor (6,0 %) que en el grupo de clopidogrel (3,5 %) en la fase aguda del SCA; y tras un mes,
respectivamente, 2,2 % y 1,6 % (ver sección 4.4). La mayor frecuencia de pausas ventriculares en la fase aguda del
SCA fue más evidente en pacientes tratados con ticagrelor que tenían antecedentes de insuficiencia cardíaca
congestiva (9,2 % frente a 5,4 % en pacientes sin antecedentes de insuficiencia cardíaca congestiva; en el caso de
clopidogrel, 4,0 % en pacientes con insuficiencia cardíaca congestiva y 3,6 % sin ella). Esta disparidad no se observó
tras 1 mes: 2 % frente a 2,1 % en pacientes que recibían ticagrelor, con o sin antecedentes de insuficiencia cardíaca
congestiva, respectivamente; y 3,8 % frente a 1,4 % en el caso de clopidogrel. No se observaron consecuencias
clínicas adversas asociadas a estas alteraciones (incluyendo la necesidad de marcapasos) en este grupo de pacientes.
Estudio PEGASUS (antecedentes de infarto de miocardio)
El estudio PEGASUS TIMI-54 fue un ensayo internacional, multicéntrico, aleatorizado, controlado con placebo, de
grupos paralelos y doble ciego, evaluado según eventos clínicos, que incluyó a 21 162 pacientes y evaluó la prevención
de eventos cardiovasculares mediante el uso de ticagrelor en dos dosis (90 mg dos veces al día o 60 mg dos veces al
día) en combinación con AAS en dosis bajas (75-150 mg) en comparación con AAS en monoterapia en pacientes con
antecedentes de infarto de miocardio y factores de riesgo adicionales para la aparición de tales eventos.
Los pacientes fueron incluidos en el estudio si tenían al menos 50 años, habían sufrido un infarto de miocardio en el
pasado (entre 1 y 3 años antes de la aleatorización) y presentaban al menos uno de los siguientes factores de riesgo
de eventos trombóticos de base aterosclerótica: edad ≥65 años, diabetes que requiriera tratamiento farmacológico,
segundo infarto de miocardio previo, enfermedad coronaria multivaso o insuficiencia renal crónica no terminal.
Los pacientes no fueron incluidos en el estudio si se preveía el uso de un antagonista del receptor P2Y12,
dipiridamol, cilostazol o tratamiento anticoagulante durante el período del estudio; si presentaban trastornos
hemorrágicos o antecedentes de accidente cerebrovascular isquémico, hemorragia intracraneal, tumor del sistema
nervioso central o malformación vascular intracraneal; si habían presentado hemorragia gastrointestinal en los últimos
6 meses o habían sido sometidos a una cirugía mayor en los últimos 30 días.
Eficacia clínica
Gráfico 2 – Análisis del punto final compuesto clínico primario de muerte por causas cardiovasculares,
infarto de miocardio y accidente cerebrovascular (PEGASUS)
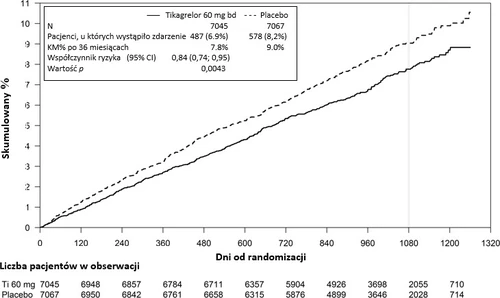
Tabla 5 - Análisis de los puntos finales primarios y secundarios de evaluación de la eficacia (PEGASUS)
| Tikagrelor 60 mg dos veces al día + ASA N = 7045 | ASA en monoterapia N = 7067 | Valor p | ||||
| Característica | Pacientes en los que se produjo el acontecimiento | KM % | HR (95% IC) | Pacientes en los que se produjo el acontecimiento | KM % | |
| Punto final primario | ||||||
| Punto final compuesto de muerte por causas cardiovasculares/IM/ACV | 487 (6,9%) | 7,8% | 0,84 (0,74; 0,95) | 578 (8,2%) | 9,0% | 0,0043 (s) |
| Muerte por causas cardiovasculares | 174 (2,5%) | 2,9% | 0,83 (0,68; 1,01) | 210 (3,0%) | 3,4% | 0,0676 |
| IM | 285 (4,0%) | 4,5% | 0,84 (0,72; 0,98) | 338 (4,8%) | 5,2% | 0,0314 |
| ACV | 91 (1,3%) | 1,5% | 0,75 (0,57; 0,98) | 122 (1,7%) | 1,9% | 0,0337 |
| Tikagrelor 60 mg dos veces al día + ASA N = 7045 | ASA en monoterapia N = 7067 | Valor p | ||||
| Característica | Pacientes en los que se produjo el acontecimiento | KM % | HR (95% IC) | Pacientes en los que se produjo el acontecimiento | KM % | |
| Punto final secundario | ||||||
| Muerte por causas cardiovasculares | 174 (2,5%) | 2,9% | 0,83 (0,68; 1,01) | 210 (3,0%) | 3,4% | |
| Muerte por cualquier causa | 289 (4,1%) | 4,7% | 0,89 (0,76; 1,04) | 326 (4,6%) | 5,2% | |
.
Ambos regímenes de tratamiento con ticagrelor en combinación con AAS (utilizando dosis de ticagrelor de 60 mg dos veces al día y 90 mg dos veces al día) demostraron superioridad frente al AAS en monoterapia en la prevención de eventos cardiovasculares (punto final compuesto: muerte por causas cardiovasculares, infarto de miocardio y accidente cerebrovascular), con un efecto terapéutico constante durante todo el período del estudio, lo que arrojó índices de reducción relativa del riesgo (RRR) del 16% y reducción absoluta del riesgo (ARR) del 1,27% en el grupo de ticagrelor a la dosis de 60 mg, y un RRR del 15% y un ARR del 1,19% en el grupo de ticagrelor a la dosis de 90 mg.
Aunque los perfiles de eficacia de las dosis de 90 mg y 60 mg fueron similares, existen datos que indican que la dosis más baja presenta un perfil de tolerancia y seguridad más favorable respecto al riesgo de hemorragias y disnea. Por ello, solo el medicamento Ticagrelor Holsten a la dosis de 60 mg dos veces al día en combinación con AAS está indicado en la prevención de eventos cardiovasculares (muerte por causas cardiovasculares, infarto de miocardio y accidente cerebrovascular) en pacientes con antecedentes de infarto de miocardio y alto riesgo de eventos trombóticos de base aterosclerótica.
En comparación con el AAS en monoterapia, el uso de ticagrelor a la dosis de 60 mg dos veces al día produjo una reducción estadísticamente significativa en la frecuencia del punto final compuesto primario, compuesto por muerte por causas cardiovasculares, infarto de miocardio y accidente cerebrovascular. Cada uno de los componentes contribuyó a la reducción de la frecuencia del punto final compuesto (muerte por causas cardiovasculares: RRR del 17%, infarto de miocardio: RRR del 16% y accidente cerebrovascular: RRR del 25%).
El coeficiente de RRR del punto final compuesto fue similar desde el día 1 hasta el día 360 (RRR del 17%) y a partir del día 361 (RRR del 16%). Los datos sobre la eficacia y seguridad del uso de ticagrelor más allá de los 3 años de tratamiento prolongado son limitados. No existen datos que indiquen beneficios del tratamiento (sin reducción del punto final compuesto primario referente a muerte cardiovascular, infarto de miocardio y accidente cerebrovascular, con aumento de hemorragias graves) con ticagrelor 60 mg dos veces al día en pacientes clínicamente estables más de 2 años después del infarto de miocardio o cuando ha transcurrido más de un año desde la interrupción del tratamiento previo con un inhibidor del ADP (ver sección 4.2).
Seguridad clínica
La frecuencia de interrupción del tratamiento con ticagrelor 60 mg debido a hemorragias y disnea fue mayor en pacientes mayores de 75 años (42%) que en pacientes más jóvenes (entre 23 y 31%), con una diferencia respecto al placebo superior al 10% (42% frente a 29%) en pacientes mayores de 75 años.
Niños y adolescentes
En un estudio aleatorizado de Fase III, controlado con placebo, en grupos paralelos y doble ciego (HESTIA 3), 193 niños y adolescentes (entre 2 y menos de 18 años) con anemia de células falciformes fueron asignados aleatoriamente a recibir placebo o ticagrelor en dosis de entre 15 mg y 45 mg dos veces al día, según la masa corporal.
El ticagrelor produjo una inhibición de las plaquetas con una mediana del 35% antes de la administración de la dosis y del 56% a las 2 horas tras la dosis en estado de equilibrio.
En comparación con el placebo, no se observó beneficio del tratamiento con ticagrelor respecto a la frecuencia de episodios vasoclusivos.
La Agencia Europea de Medicamentos ha eximido del requisito de presentar resultados de estudios del medicamento Ticagrelor Holsten en todas las subpoblaciones pediátricas en el síndrome coronario agudo (SCA) y en pacientes con antecedentes de infarto de miocardio (la información sobre el uso del producto en niños y adolescentes se proporciona en la sección 4.2).
5.2 Propiedades farmacocinéticas
El ticagrelor presenta farmacocinética lineal, y la exposición al ticagrelor y a su metabolito activo
(AR-C124910XX) es aproximadamente dependiente de la dosis, en el rango de hasta 1260 mg.
Absorción
La absorción del ticagrelor es rápida, con una mediana del tiempo tmax de aproximadamente 1,5 horas. La formación del principal metabolito circulante AR-C124910XX (también activo) a partir del ticagrelor es rápida, con una mediana del tmax de aproximadamente 2,5 horas. Tras la administración de una dosis única oral de 90 mg de ticagrelor en ayunas a voluntarios sanos, la Cmáx es de 529 ng/ml y el AUC es de 3451 ng*h/ml. Para el metabolito, los coeficientes relativos a la sustancia madre son de 0,28 para la Cmáx y de 0,42 para el AUC.
La farmacocinética del ticagrelor y del AR-C124910XX en pacientes con infarto de miocardio previo fue esencialmente similar a la observada en la población de pacientes con SCA. Según el análisis de farmacocinética poblacional en el estudio PEGASUS, la mediana de la Cmáx del ticagrelor fue de 391 ng/ml y el AUC fue de 3801 ng*h/ml en estado de equilibrio tras la administración de ticagrelor a una dosis de 60 mg. En el caso del ticagrelor a una dosis de 90 mg, la Cmáx fue de 627 ng/ml y el AUC fue de 6255 ng*h/ml en estado de equilibrio.
La biodisponibilidad absoluta media del ticagrelor se estimó en un 36%. La ingesta de una comida rica en grasas provoca un aumento del AUC del ticagrelor del 21% y una reducción de la Cmáx del metabolito activo del 22%, pero no provocó cambios en la Cmáx del ticagrelor ni en el AUC del metabolito activo. Se considera que estos pequeños cambios tienen una importancia clínica mínima, por lo que el ticagrelor puede administrarse durante las comidas o independientemente de ellas. Tanto el ticagrelor como el metabolito activo son sustratos de la glucoproteína P (P-gp).
El ticagrelor en forma de comprimidos triturados mezclados con agua, administrados por vía oral o mediante sonda nasogástrica, tiene una biodisponibilidad comparable a la del comprimido administrado entero en cuanto al AUC y la Cmáx del ticagrelor y del metabolito activo. La exposición inicial (0,5 y 1 hora tras la administración) del ticagrelor administrado en forma de comprimido triturado mezclado con agua fue mayor que la del administrado en forma de comprimido entero (no triturado), con un perfil de concentraciones esencialmente idéntico en tiempos posteriores (de 2 a 48 horas).
Distribución
El volumen de distribución en estado de equilibrio es de 87,5 l. El ticagrelor y el metabolito activo se unen en gran medida a las proteínas del plasma humano (>99,0%).
Biotransformación
La CYP3A4 es el principal enzima responsable del metabolismo del ticagrelor y de la formación del metabolito activo, y sus interacciones con otros sustratos de la isoenzima CYP3A incluyen tanto la activación como la inhibición.
El principal metabolito del ticagrelor, AR-C124910XX, también es activo, como se ha determinado en estudios in vitro, en los que se une al receptor plaquetario de ADP P2Y12.
La exposición sistémica total al metabolito activo representa aproximadamente entre el 30% y el 40% de la exposición al ticagrelor.
Eliminación
La principal vía de eliminación del ticagrelor es el metabolismo hepático. Tras la administración de ticagrelor marcado radiactivamente, la recuperación media de la radioactividad fue de aproximadamente el 84% (57,8% en heces y 26,5% en orina). El ticagrelor y el metabolito activo recuperados en orina representaron cada uno menos del 1% de la dosis administrada. La principal vía de eliminación del metabolito activo es probablemente la excreción biliar. El periodo medio de eliminación fue de aproximadamente 7 horas para el ticagrelor y de 8,5 horas para el metabolito activo.
Grupos de pacientes especiales
Personas de edad avanzada
En los análisis farmacocinéticos realizados en poblaciones de pacientes de edad avanzada (≥75 años) con SCA, se observó una mayor exposición al ticagrelor (aproximadamente un 25% más para la Cmáx y el AUC) y al metabolito activo en comparación con pacientes más jóvenes. Se considera que estas diferencias no son clínicamente relevantes (ver sección 4.2).
Niños y adolescentes
Existen datos limitados disponibles en niños y adolescentes con anemia de células falciformes (ver secciones 4.2 y 5.1).
En el estudio HESTIA, se administró ticagrelor en forma de comprimidos orodispersables de 15 mg destinados a niños, en dosis de 15, 30 y 45 mg dos veces al día, a pacientes de entre 2 y menos de 18 años, con pesos de ≥12 a ≤24 kg, >24 a ≤48 kg y >48 kg, respectivamente. El análisis de farmacocinética poblacional mostró que el AUC medio osciló entre 1095 ng*h/ml y 1458 ng*h/ml, y la Cmáx media entre 143 ng/ml y 206 ng/ml en estado de equilibrio.
Sexo
En mujeres se observó una mayor exposición al ticagrelor y al metabolito activo que en hombres. Se considera que estas diferencias no son clínicamente relevantes.
Alteraciones de la función renal
En pacientes con alteraciones graves de la función renal (clearance de creatinina <30 ml/min), la exposición al ticagrelor fue aproximadamente un 20% menor y la exposición al metabolito activo fue aproximadamente un 17% mayor que en pacientes con función renal normal.
En pacientes con enfermedad renal terminal sometidos a hemodiálisis, los valores de AUC y Cmáx del ticagrelor a una dosis de 90 mg administrado en un día sin diálisis fueron un 38% y un 51% mayores, respectivamente, que en pacientes con función renal normal. Un aumento similar de la exposición se observó cuando el ticagrelor se administró inmediatamente antes de la diálisis (49% y 61%, respectivamente), lo que indica que el ticagrelor no se elimina mediante diálisis. La exposición al metabolito activo aumentó en menor medida (AUC en un 13-14% y Cmáx en un 17-36%). El efecto del ticagrelor en la inhibición de la agregación plaquetaria fue independiente de la diálisis en pacientes con enfermedad renal terminal y similar al de pacientes con función renal normal (ver sección 4.2).
Alteraciones de la función hepática
La Cmáx y el AUC del ticagrelor fueron un 12% y un 23% mayores, respectivamente, en pacientes con alteraciones leves de la función hepática en comparación con sujetos sanos emparejados, aunque el efecto del ticagrelor en la inhibición de la agregación plaquetaria fue similar en ambos grupos. No es necesario ajustar la dosis en pacientes con alteraciones moderadas de la función hepática. No se han realizado estudios sobre el uso de ticagrelor en pacientes con insuficiencia hepática grave, ni existen datos disponibles sobre su farmacocinética en pacientes con alteraciones moderadas de la función hepática. En pacientes con aumentos basales moderados o graves en uno o dos tests de función hepática, la concentración plasmática de ticagrelor fue media similar o ligeramente mayor que en pacientes sin aumento basal de estos parámetros. No es necesario ajustar la dosificación en pacientes con alteraciones moderadas de la función hepática (ver secciones 4.2 y 4.4).
Diferencias raciales
En pacientes de origen asiático se observa una biodisponibilidad media un 39% mayor en comparación con pacientes de raza caucásica. En pacientes que se autoidentifican como negros, la biodisponibilidad del ticagrelor es un 18% menor que en pacientes de raza caucásica. En estudios de farmacología clínica en japoneses se observó una exposición al ticagrelor (Cmáx y AUC) aproximadamente un 40% mayor (y un 20% mayor tras ajuste por peso corporal) en comparación con personas de raza caucásica. La exposición en pacientes que se autoidentifican como latinos fue similar a la de pacientes de raza caucásica.
5.3 Datos preclínicos sobre seguridad
Los datos preclínicos procedentes de estudios farmacológicos convencionales del ticagrelor y su metabolito principal, relativos a la seguridad farmacológica, estudios de toxicidad tras dosis única y múltiple, así como de potencial genotoxicidad, no han mostrado riesgos inaceptables de efectos adversos en humanos.
Tras una exposición comparable a las condiciones clínicas en varias especies animales, se observó irritación del tracto gastrointestinal (ver punto 4.8).
En ratas hembra tratadas con altas dosis de ticagrelor se observó un aumento en el número de casos de tumores uterinos (adenocarcinomas) y un incremento en la incidencia de adenomas hepáticos. El mecanismo de formación de tumores uterinos en ratas probablemente se deba a una alteración del equilibrio hormonal, que puede provocar el desarrollo de tumores en esta especie. El mecanismo de formación de adenomas hepáticos probablemente sea un aumento específico en roedores de la actividad enzimática hepática. Por tanto, se considera poco probable que estos casos de carcinogenicidad tengan relevancia para el ser humano.
En ratas se observaron pequeñas anomalías en el desarrollo tras la administración a hembras preñadas de dosis tóxicas (margen de seguridad 5,1). En fetos de conejo se observó un ligero retraso en la maduración hepática y en el desarrollo del sistema esquelético cuando a las hembras preñadas se les administraron altas dosis sin signos de toxicidad en las madres (margen de seguridad 4,5).
Estudios en ratas y conejos mostraron efectos tóxicos sobre la reproducción, con una ligera reducción del aumento de peso corporal en hembras preñadas, menor supervivencia de recién nacidos, menor peso al nacer y retraso en el crecimiento.
El ticagrelor provocó ciclos irregulares (en su mayoría alargados) en ratas hembra, aunque no afectó a la fertilidad total de ratas macho ni hembra. Estudios farmacocinéticos realizados con ticagrelor marcado radiactivamente demostraron que tanto el principio activo como sus metabolitos atraviesan la leche de rata (ver punto 4.6).
6. DATOS FARMACÉUTICOS
6.1 Lista de excipientes
Núcleo de la tableta
Hipromelosa (E464)
Mannitol (E421)
Celulosa microcristalina (E460)
Carboximetilalmidón sódico
Estearato de magnesio (E470b)
Recubrimiento de la tableta
Hipromelosa (E464)
Dióxido de titanio (E171)
Macrogol 400 (E1521)
Talc (E553b)
Óxido de hierro rojo (E172)
6.2 Incompatibilidades farmacéuticas
No aplicable.
6.3 Período de validez
3 años
6.4 Precauciones especiales de conservación
Este medicamento no requiere condiciones especiales de conservación en cuanto a temperatura; conservar en el envase original para protegerlo de la luz.
6.5 Tipo y contenido del envase
Blísters transparentes de lámina de PVC/PVDC/Aluminio y/o blísters transparentes de lámina
PVC/PE/PVDC/Aluminio en cajas de cartón.
Blísters de lámina (con símbolos de sol/luna o sin ellos) en cajas de cartón con 14, 15, 20, 28, 30,
56, 60, 90, 98, 100, 168, 195, 196 y 200 comprimidos recubiertos.
No todos los tamaños de envase tienen por qué comercializarse.
6.6 Medidas especiales de precaución para la eliminación
Cualquier residuo no utilizado del medicamento o sus desechos debe eliminarse de acuerdo con
la normativa local.
7. TITULAR DEL AUTORIZACIÓN DE COMERCIALIZACIÓN
Holsten Pharma GmbH
Hahnstraße 31-35
60528 Frankfurt am Main
Alemania
8. NÚMEROS DE AUTORIZACIONES PARA LA COMERCIALIZACIÓN
Autorización nº:
9. FECHA DE PRIMERA CONCESIÓN DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Y FECHA DE RENOVACIÓN DE LA AUTORIZACIÓN
Fecha de primera concesión de la autorización de comercialización:
10. FECHA DE APROBACIÓN O DE MODIFICACIÓN PARCIAL DEL TEXTO
FICHA TÉCNICA